

Abstract
Although most sequenced members of the industrially important ketol-acid reductoisomerase (KARI) family are Class I enzymes, structural studies to date have focused primarily on the Class II KARIs, which arose through domain duplication. Here, we present five new crystal structures of Class I KARIs. These include the first structure of a KARI with a 6-residue β2αB (cofactor specificity determining) loop and an NADPH phosphate binding geometry distinct from that of the 7- and 12-residue loops. We also present the first structures of naturally occurring KARIs that utilize NADH as cofactor. These results show insertions in the specificity loops that confounded previous attempts to classify them according to loop length. Lastly, we explore the conformational changes that occur in Class I KARIs upon binding of cofactor and metal ions. The Class I KARI structures indicate that the active sites close upon binding NAD(P)H, similar to what is observed in the Class II KARIs of rice and spinach and different from the opening of the active site observed in the Class II KARI of E. coli. This conformational change involves a decrease in the bending of the helix that runs between the domains and a rearrangement of the nicotinamide binding site.
Keywords: Crystal structure, cofactor binding, conformational change, KARI, AHAIR
Introduction
The bifunctional enzyme ketol-acid reductoisomerase (KARI, EC 1.1.1.86, also known as acetohydroxyacid isomeroreductase (AHIR or AHAIR)), catalyzes the second step in the biosynthesis of the branched-chain amino acids (BCAAs) valine, leucine, and isoleucine [1]. The enzyme converts 2-(S)-acetolactate (2SAL) into (R)-dihydroxyisovalerate (RDHIV) in an ordered two-step reaction, where a Mg2+-dependent alkyl migration is followed by reduction using a hydride from NAD(P)H (Figure 1a) [1]. This unusual reaction has inspired much work to characterize the order of binding, metal ion requirements, and NAD(P)H specificity [1-7].
Figure 1.

a) The two-step reaction catalyzed by KARI. b) The four substrate analogues which have been crystallized in the active site of KARIs (including in this study). PDB ligand IDs are shown in parentheses.
Because animals lack the BCAA pathway, there is a sizable market for these amino acids produced in microorganisms for human and animal dietary supplements [8-11], and inhibitors of KARIs such as N-hydroxy-N-isopropyloxamate (IpOHA) are an active area of research as potential herbicides and antibiotics [12-16]. Beyond this, the BCAA pathway has been re-engineered in microbes for production of isobutanol, a promising second-generation biofuel and chemical feedstock [17].
KARIs are divided into two classes based on their length and oligomerization state. Class I KARIs include all fungal KARIs and are ∼340 amino acid residues in length. Class II KARIs include all plant KARIs and are ∼490 residues long. Bacterial KARIs can be either Class I or Class II [18]. KARIs are composed of two types of domains, an N-terminal Rossmann domain and one or two C-terminal knotted domains. Two intertwined knotted domains are required for function, and in the short-chain or Class I KARIs, each polypeptide chain has one knotted domain. As a result, dimerization of two monomers forms two complete KARI active sites (Figure 2a). In the long-chain or Class II KARIs, a duplication of the knotted domain has occurred, and, as a result, the protein does not require dimerization to complete its active site. As first proposed by Ahn and co-workers [18], this domain duplication suggests that Class II KARIs evolved from a primordial Class I KARI. This pair of knotted domains is extensively intertwined to form a figure-of-eight knot. Dimerization of the Class I KARIs also forms this knot, albeit with an additional disconnection due to being comprised of two chains. The Class II KARI from spinach was the first deeply knotted protein identified [19] and remains the most deeply embedded knot observed in a protein crystal structure [20].
Figure 2.
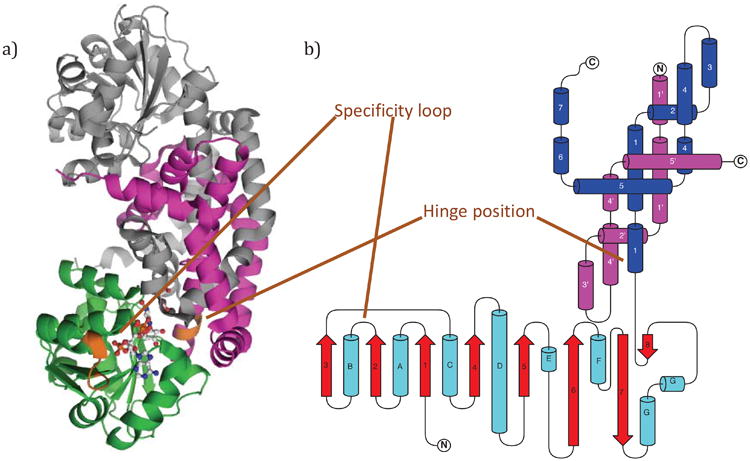
a) The crystal structure of the Ia_KARI_holo dimer showing bound NADPH and inhibitor (white) and Mg2+ ions (cyan). One monomer is shown in grey; the other is shown in green (Rossmann domain) and magenta (knotted domain). b) The topology of Class I KARIs is illustrated schematically, with cyan helices and red β-strands; the knotted domain has blue helices. Also shown is the knotted domain of the dimeric partner (magenta), which has been truncated for simplicity. The figure was constructed using TopDraw [50].
Class I and Class II KARIs are also distinguished by the length of the Rossmann fold's β2αB-loop, which binds the adenine ribose of NAD(P)(H) and has been shown to be the primary determinant of whether an enzyme prefers NADH or NADPH [2, 21]. From a set of 558 Class II KARI sequences downloaded from Pfam [22], all but 20 have a 12-residue β2αB-loop. In contrast, only five of 2,860 Class I KARIs do. Class I KARIs instead favor 6- and 7-residue loops (Supplemental Information 1). Prior to this study, no KARI from the significant proportion of Class I homologs having a 6-residue β2αB-loop had been crystallized, nor had any of the Class I KARIs with 12-residue β2αB-loops. This loop, which we will hereafter term the “specificity loop,” has been the target of our efforts to engineer the cofactor specificity of KARIs [21, 23]. The lack of crystal structures for KARIs having 6-residue loops, however, prevented a thorough understanding of the loop structure-function relationship.
The other major gap in our knowledge of KARIs is how the Class I enzymes change conformation upon binding the Mg2+ ions, substrate, and cofactor. Two crystallographic studies by Guddat and co-workers on substrate-induced rearrangements of Class II KARIs reported different mechanisms of induced fit in plant KARIs [24] and E. coli KARI [25]. Though both enzymes show an ordered binding in which Mg2+ and NADPH bind before substrate [1, 26], the structural effects of binding are dramatically different. In the plant enzymes, the hinge between the Rossmann domain and the knotted domain moves freely in the apo state, an observation supported by H-D exchange mass spectroscopy [5]. Binding of magnesium and NADPH draws the domains together, closing the active site, and rearranges the α3+1 helix to preorganize the enzyme for substrate binding. In contrast, in the bacterial enzyme the active site is closed in the apo crystal structure, but binding of magnesium and NADPH opens the interface between the domains and allows the substrate to bind. Nothing, however, is known about the conformational changes in Class I KARIs, which may be complicated by the dimeric nature of the protein.
In this study, we present five new Class I KARI structures from four previously un-crystallized enzymes. Three of these structures give insight into the unexpected diversity of the specificity loop. With the other two structures, we demonstrate that the mechanism of induced fit in the bacterial Class I KARIs we studied involves closure of the interdomain hinge, coupled with a rearrangement in the nicotinamide amide-binding portion of the active site. This behavior is opposite that observed in the bacterial Class II KARI previously described by Wong et al. [25] and more closely resembles the behavior of the plant KARIs [24]. Taken together, these structures enhance our understanding of the structural and functional diversity of this enzyme class.
Experimental
Cloning, Expression, and Purification of KARIs
Cloning, expression and purification of Aa_KARI (Alicyclobacillus acidocaldarius), Ua_KARI (Uncultured archaeon), and Ia_KARI (Ignisphaera aggregans) were described previously [2, 21]. Av_KARI was isolated from Azotobacter vinelandii strain AvOP (ATCC BAA-1303). Cells growth and Av_KARI purification by anion exchange and gel filtration chromatography was performed as published earlier [27].
Crystallization and Data Collection of Aa_KARI, Ua_KARI, and Ia_KARI
N-hydroxy-N-isopropyloxamate (IpOHA) was prepared as described [21]. High-throughput screening of crystallization conditions was conducted at the Beckman Molecular Observatory at the California Institute of Technology. For Aa_KARI in the presence of NADPH, the best condition was 1M sodium potassium tartrate, 200 mM sodium chloride, 100 mM imidazole pH 8. For Ia_KARI in the presence of NADPH and with IpOHA as inhibitor, the best condition was a 0.1 M bis-tris, pH 6, solution with 22% polyethylene glycol monomethylether 5000. The apo crystal of Ia_KARI was obtained from the same conditions, but with 200 μM NADH instead of NADPH. For Ua_KARI in the presence of NADH and with IpOHA as inhibitor, the best condition was a 0.1 M bis-tris, pH 5, solution with 20% w/v polyethylene glycol 1500 as precipitant. For cryo-protection, the crystals were soaked in a mother liquor containing 25% glycerol prior to flash-freezing in liquid nitrogen. Diffraction data were collected using a Dectris Pilatus 6M detector on beamline 12-2 at the Stanford Synchrotron Radiation Laboratory SSRL at 100 K. Diffraction datasets were integrated with XDS [28] and scaled using SCALA [29].
Crystallization and Data Collection of Av_KARI
Av_KARI was crystallized under strictly anaerobic conditions using 0.25 M NaCl, 28 % PEG3350 and 0.1 M Bis-Tris, pH 5.5. Partial dehydration of the observed crystals was achieved by equilibrating the crystals against 40 % PEG 3350 for 12 h, followed by subsequent flash-freezing in liquid nitrogen. Diffraction experiments were carried out at 100K at the Swiss-Light-Source PXI beamline X06SA (Paul-Scherrer-Institute, Villigen/Switzerland). Data were integrated using iMOSFLM [30] and scaled in SCALA [29].
Structure solution
For Aa_KARI_holo the structure of S. exigua KARI (Se_KARI_holo, PDB code 4KQW) was used for molecular replacement with MOLREP [31]; Ia_KARI_holo and Ua_KARI_holo used the structure of the DDV mutant of that enzyme (Se_KARI_DDV, PDB code 4KQX) and molecular replacement was performed using Phaser [32]. For Ia_KARI_apo, the structure of Ia_KARI_holo was used for molecular replacement with Phaser; the structure was split into two PDB files containing the Rossmann and knotted domains, which were fit as separate ensembles. Molecular replacement for Av_KARI_met used Pa_KARI_apo (PDB code 1NP3) [18] for molecular replacement with MOLREP, with determination and refinement of iron-sites performed using SHELX [33]. After molecular replacement, several iterations of automated refinement with Refmac5 [34] (CCP4 suite [35]) and manual refinement in Coot were performed [36].
Structure analysis
For the whole-enzyme alignments (i.e. Figure 3a), the alignment was performed using PyMOL [37]; for the domain-specific alignments, LSQ superposition was performed using Coot [36]. For analysis of helix bending, the HELANAL-Plus web server [38] was used, with the helices of each structure assigned by STRIDE. From the first half of the α1-helix (that is, before the break in helicity) the l, m, n direction cosines of the first and last i/i-3 pair were extracted, and the angle between them was computed.
Figure 3.

A structure-guided alignment of the specificity loops (including four residues on each side) of some of the KARIs discussed in this paper, annotated with the length, the class of the KARI, the kingdom of origin, and the cofactor preference. Residues contacting the phosphate are highlighted in orange.
Ia_KARI thermostability determination
T50 is defined as the temperature at which 50% of the initial activity is retained after 1 h incubation. Thirty-μL aliquots of purified enzyme were transferred to PCR tubes. Each tube was assigned a specific incubation temperature on the block of an Eppendorf master cycler PCR machine. The measurements were conducted in duplicates. The tubes were incubated in their slots for 1 h, and then quenched on ice. Residual activity was determined with the activity assay described previously [2].
Results and Discussion
We present five crystal structures representing four Class I KARIs, with resolutions of 1.15–2.50 Å (Table 1). We previously described three of these KARIs in our work on cofactor specificity engineering. The cofactor specificity of Aa_KARI from Alicyclobacillus acidocaldarius was inverted by the introduction of three mutations in its specificity loop [21]. Ua_KARI from an uncultured organism and Ia_KARI from Ignisphaera aggregans were correctly predicted to utilize NADH, based on their loop sequences [2]. The fourth KARI, from Azotobacter vinelandii, crystallized serendipitously as an impurity of another protein purified from A. vinelandii lysate. Table 1 places these enzymes and the structures reported here in the context of the previously available KARI structural data.
Table 1.
Published KARI structures (above the line) and the new ones presented here (below). Each is assigned a name for discussion in this paper. For each, the metals and bound ligands are given. Cofactor IDs are as follows: NDP = NADPH, NAP = NADP+, NAD = NAD+, APX = 2′-monophosphate adenosine diphosphate ribose; see Figure 1b for structures of substrate analogues. KARI class and β2αB-loop (specificity loop) length are also given for each enzyme.
| PDB ID | Name | Host | Resn. (Å) | Metal | Co-factor | Analogue | Class | β2αB -loop |
|---|---|---|---|---|---|---|---|---|
| 1YVE [39] | So_KARI_holo | S. oleracea | 1.65 | 2×Mg2+ | NDP | HIO | II | 7 |
| 1QMG [40] | So_KARI_prod | S. oleracea | 1.60 | 2×Mn2+ | APX | DMV | II | 7 |
| 1NP3 [18] | Pa_KARI_apo | P. aeruginosa | 2.00 | I | 7 | |||
| 1YRL [49] | Ec_KARI_apo | E. coli | 2.60 | II | 12 | |||
| 3FR7 [24] | Os_KARI_met | O. sativa | 1.55 | 2×Mg2+ | II | 7 | ||
| 3FR8 [24] | Os_KARI_holo | O. sativa | 2.80 | 1×Mg2+ | NDP | II | 7 | |
| 3ULK[25] | Ec_KARI_holo | E. coli | 2.30 | 2×Mg2+ | NDP | II | 12 | |
| 4KQW [21] | Se_KARI_holo | S. exigua | 1.38 | 2×Mg2+ | NAP | TLA | I | 7 |
| 4KQX [21] | Se_KARI_DDV | S. exigua | 1.80 | 2×Mg2+ | NAD | HIO | I | 7 |
|
| ||||||||
| 4TSK | Aa_ KARI_ holo | A. acidocaldarius | 2.50 | 2×Mg2+ | NAP | TLA | I | 6 |
| 4XDY | Ua_KARI_holo | Uncultured archaeon | 1.54 | 2×Mg2+ | NAD | HIO | I | 12 |
| 4XDZ | Ia_KARI_holo | I. aggregans | 1.15 | 2×Mg2+ | NDP | 40E | I | 7 |
| 4XEH | Ia_KAR_apo | I. aggregans | 1.39 | I | 7 | |||
| 4XIY | Av_KARI_met | A. vinelandii | 2.50 | 1×Mg2+, 1×Fe2+ | I | 7 | ||
For some proteins, including Ia_KARI in this study, multiple structures are available with different bound cofactors, metals, and substrate analogues. Refinement statistics of the structures reported here are provided in Supplemental Information 2. All of these KARIs have the same fold as previously solved Class I KARIs (Figure 2). Their active sites, particularly the metal-binding sites, are also in agreement with those of previous structures (see, e.g. Biou et al. [39]).
Various nomenclatures have been used to discuss the structures of KARIs. In this paper, we will refer to the N-terminal domain as the Rossmann domain and the C-terminal domain as the knotted domain. The topology of the Class I enzyme is shown and labeled in Figure 2b. Secondary structural elements are numbered starting from the first β-strand that makes up the canonical Rossmann fold; secondary structure prior to this strand is not conserved. In the Rossmann domain, β-strands are numbered 1 to 8, and α-helices are assigned the letters from A to G. In the knotted domain, which is entirely α-helical, helices are assigned numbers from 1 to 8. Loops between secondary structural elements are named based on the flanking secondary structural elements, as in β2αB-loop or α3α4-loop. We refer to the β2αB-loop of the Rossmann domain as the specificity loop. Supplemental Information 3, which has an alignment of the KARI sequences discussed herein, is also annotated with the secondary structure labels used. In Class I KARIs, the secondary structural elements of the dimeric partner are denoted with an apostrophe, as in α3′-helix. In Class II KARIs, the duplicated elements are denoted with ‘+1’, as in α3+1-helix. Lastly, we refer to the proteins themselves with two-part names (i.e. Aa_KARI) and the structures with three-part names (Aa_KARI_holo) indicating their complexation state.
Structural diversity of the specificity loop: structure of Aa_KARI's 6-residue specificity loop
Until recently, KARI family enzymes were believed to be exclusively NADPH-dependent [2]. Because NADH utilization is advantageous in the industrial production of amino acids and biofuels [21, 23], engineering KARI cofactor specificity has been of considerable interest. Previous studies by our group aimed at reversing KARI cofactor specificity focused extensively on the specificity loop of the Rossmann domain due to its key role in binding the adenine ribose of the cofactor. Structures of enzymes with a 12-residue specificity loop [25] and multiple structures with 7-residue specificity loops [21, 39, 40] with bound NADPH were available. However, no structure of a KARI having a 6-residue specificity loop was available, with or without NADPH. In a previous study, we found that whereas similar mutations reversed the specificity of KARIs with 7- and 12-residue specificity loops, a modified approach was required for the 6-residue specificity loop enzymes [21]. Ambiguities in the alignments of the different length of the loop sequences limited our ability to propose a structural model to explain the different sets of mutations (see the alignment in Figure 3). For that reason, we selected for structural analysis the KARI from A. acidocaldarius (Aa_KARI), a mildly thermophilic enzyme that had been engineered for NADH preference in our previous study [21]. Aa_KARI crystallized readily, and we obtained a crystal that diffracted to 2.5 Å.
The overall structure of Aa_KARI_holo is very similar to that of the other Class I KARIs that have been published, with RMSDs of 0.48 Å and 1.56 Å against Se_KARI_holo [21] and Pa_KARI_apo [18], respectively. Figure 4 shows the specificity loop of Aa_KARI_holo (part c) as well as other KARIs, including ones that will be discussed later in this paper. While the side-chains of three residues of the 7- and 12-member specificity loops form hydrogen bonds to the NADP(H) phosphate (e.g. Arg58, Ser61, and Ser63 in Se_KARI_holo), only two residues do so for the 6-member specificity loop of Aa_KARI_holo, Arg48 and Ser52. However, the rearrangement of the loop brings the αB-helix closer to the adenine ring, allowing the terminal serine residue's side chain to hydrogen bond not only with the phosphate but also with O2′ and O3′ of the ribose, creating a hydrogen bond between the backbone N-H group of Ser52 and the phosphate oxygen. This interaction between the phosphate and the dipole of the αB-helix has not been observed previously in a KARI.
Figure 4.
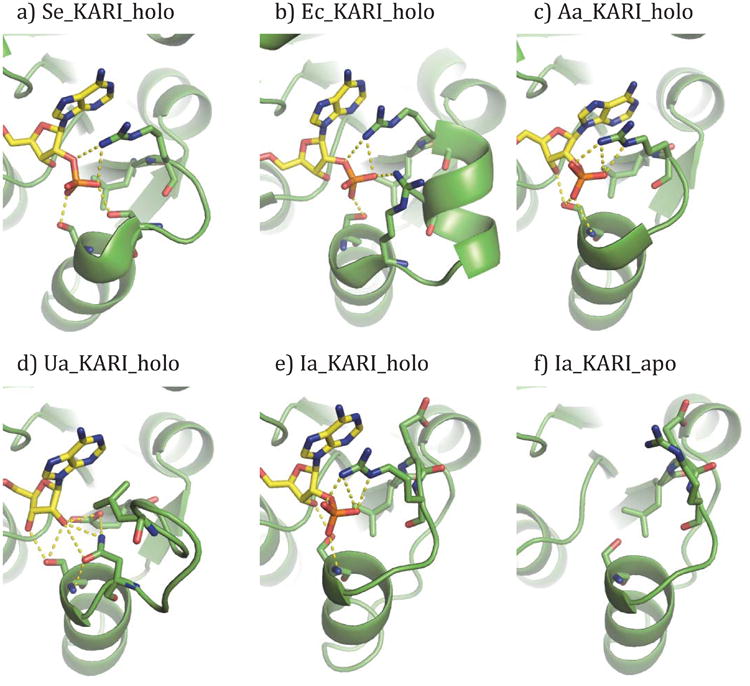
The β2αB-loops of a) Se_KARI_holo (PDB 4KQW) [21], b) Ec_KARI_holo (PDB 3ULK) [25], c) Aa_KARI_holo, d) Ua_KARI_holo, e-f) Ia_KARI_holo and _apo. Polar interactions (i.e. hydrogen bonds) of 3.5 Å or less to the 2′ and 3′ moieties of the ribose are shown with dashed lines, as are selected interactions between amino acid residues.
In our previous study we found that reversing the cofactor specificity of KARIs having 7- and 12-residue specificity loops from NADPH to NADH required installation of aspartate residues at the ultimate and antepenultimate loop positions [21]. Existing structures showed that these two residues provide the primary hydrogen bonding interactions with the NADPH phosphate [21, 25]. The 6-residue specificity loops, in contrast, only required a single aspartate along with mutation of the arginine to proline at the second position of the loop to switch cofactor preference to NADH. Having obtained the structure of the first KARI with a 6-residue specificity loop, we can now explain why. Only the ultimate residue of Aa_KARI_holo's specificity loop forms a hydrogen bond with the phosphate. There is no residue corresponding to the interaction provided by the antepenultimate position. Presumably to compensate for this missing interaction, the contact between the arginine at the second position of the loop and the phosphate is more extensive, such that mutation of this residue is also critical in order to abolish phosphate binding and switch cofactor specificity.
Structural diversity of the specificity loop: structure of the naturally NADH-preferring Ua_KARI
In the past, it had been assumed that all KARIs were specific for NADPH [23]. However, we recently showed that some native KARIs display strong preference for NADH or at least exhibit equal activity on NADPH and NADH [2]. The KARI with the greatest preference for NADH (in terms of catalytic efficiency, kcat/KM) [2] came from an uncultured archaeon (Ua_KARI) and was also one of the only five known Class I KARIs to possess a 12-residue specificity loop (Supplemental Information 1). However, we could not predict the arrangement of the residues around the NADH based on sequence alone, because the loop sequence was so different from any previously crystallized KARI (Figure 3). It also did not allow us to effectively compare it to the recipe we proposed previously for engineering naturally NADPH-preferring KARIs to use NADH. For this study, we were able to obtain a crystal of Ua_KARI bound to NADH, which diffracted to 1.54 Å.
Ua_KARI_holo possesses the same overall KARI fold as other Class I KARIs, with a 0.62 Å RMSD to Se_KARI_holo. However, the specificity loop of Ua_KARI_holo differs significantly from any other crystallized KARI (Figure 4d). Glu46, the first residue of the β2αB-loop, lies along the N3 edge of the adenine moiety and forms a bi-dentate interaction with the O2′ hydroxyl of the ribose. This residue is structurally homologous to the conserved leucine that makes a similar interaction with the adenine in other KARIs. The position of this glutamate is stabilized by a hydrogen bond interaction with Asn55.
In most KARIs, an arginine in the position immediately following the conserved leucine forms a cation-pi interaction with the adenine and a salt bridge with the phosphate of the cofactor [18]. In Ua_KARI_holo, the arginine is replaced by Leu49, which removes the positive charge. Additionally, the specificity loop is extended by five residues that accommodate the structural differences between arginine and leucine. In E. coli KARI, which also has a 12-residue loop, the five additional residues form a short helix inserted after the arginine. In Ua_KARI_holo, by contrast, there are three residues before Leu49 and two after. This leads to a reorientation of the specificity loop (Figure 4d). As with the canonical NADPH-binding 7- and 12-residue specificity loops, Ua_KARI_holo's specificity loop has two additional residues that provide contacts to the (phospho-) ribose. Ser57 at the end of the specificity loop is highly conserved and, as in Aa_KARI_holo, forms hydrogen bonds with both O2′ and O3′ of the ribose. Though there is no phosphate to hydrogen bond to its backbone amine, this backbone nitrogen hydrogen bonds to the side-chain of another residue, Asn55. The increased size of Asn55 over the consensus serine at this position allows it to make contact with O2′ rather than the absent phosphate, and the nitrogen forms a hydrogen bond with Glu46, as previously noted.
The lack of a short helix within the loop suggests that the ancestral Ua_KARI did not have a canonical 12-residue specificity loop. Ua_KARI may have evolved from a KARI possessing a shorter loop. This is supported by the fact that the KARIs with the sequences most similar to Ua_KARI (other than a 98% identical one from another uncultured archaeon) both had 6-residue specificity loops, as discussed previously [2].
Structure of Ia_KARI: an unusual β2αB-loop in a bi-specific KARI
The I. aggregans KARI from our previous study (Ia_KARI) displayed sub-micromolar KM values for both NADH and NADPH [2], and measurements carried out for this study confirmed that it is highly thermostable, with half-maximal residual activity (T50) at 95 °C. We were unable to obtain a structure of Ia_KARI bound to NADH, but did succeed in co-crystallizing it with NADPH and the transition state analogues N-isopropyloxamate (Figure 1). The ligand's electron density, however, indicated that it had been reduced at the N position (cause unknown) and a water molecule had taken the place of the missing -OH group (Supplemental Information 4).
Because Ia_KARI_holo has a 7-residue specificity loop, we had expected the second loop residue, a glutamate, to make contact with the face of the adenine ring and the antepenultimate and ultimate residues to bind the phosphate. Surprisingly, the binding mode for NADP closely resembles that of the 6-residue loop from Aa_KARI_holo, with a single serine residue at the ultimate position of the loop hydrogen bonding with both the O3′ hydroxyl and the phosphate (Figure 4e). Furthermore, the glutamate at the second position of the loop faces away from the cofactor entirely, while the arginine at the third position forms the typical packing interaction against the adenine ring. Without an NADH co-crystal structure, it is difficult to speculate what functional role this glutamate serves or how it contributes to the enzyme's cofactor bi-specificity. However, it is possible that a rearrangement of the loop might allow it to occupy the position of either of the conserved residues flanking it. Such rearrangements of adenosine ribose-binding loops have been observed in bi-specific glucose-6-phosphate dehydrogenase [41], xylose reductase [42], glyceraldehyde-3-phosphate dehydrogenase [43], and coenzyme A-disulfide reductase [44].
It is interesting that both of the naturally NADH-utilizing KARIs, Ua_KARI and Ia_KARI, had amino acid insertions relative to other structurally similar enzymes. Insertions and deletions in genes represent a major source of sequence and function variation in natural evolution [45, 46], but are rarely used in protein engineering and directed evolution. In our previous work, classifying the binding mode based on the length of the specificity loop was the first step to identifying a protein engineering strategy for engineering KARI cofactor specificity [21]. These results, however, demonstrate that loop length may be an insufficient indicator of binding geometry.
Conformational changes in Class I KARIs: induced fit in Ia_KARI
The Ia_KARI structures with and without bound metals or ligands allowed us to observe the changes in the protein structure upon the binding of NADPH, metals, and N-isopropyloxamate. As in the plant and E. coli Class II KARIs, the two domains of Ia_KARI undergo a rigid body movement, where only the angle between them changes significantly. With metals, cofactor, and inhibitor bound, the Rossmann domains have moved closer together, sealing off the active sites and generating a “closed” state. Either domain of the ligand-bound protein can be superimposed on its respective apo-protein domain, but the full structures do not superimpose. Figure 5 shows a plot of the Cα-Cα distances along the length of the chain for Ia_KARI_holo and Ia_KARI_apo, aligned by Rossmann domain (blue) or knotted domain (red). With either alignment a distinct jump in the Cα-Cα distances indicates a “hinge point” at Glu189. Glu189 is on the α1-helix of the knotted domain rather than the β8α1-loop between the domains, which stays rigid with respect to the N-terminal domain. In the ligand-bound structure, the α1-helix is less bent, with an angle of 4.7° versus 8.7° in the apo structure, as measured using HELANAL-Plus [38]. Figure 6 shows this movement structurally (a and b) and quantitatively (c) for Ia_KARI as well as other structure pairs to be discussed later. However, there is no significant rearrangement in the helix to explain this change in curvature. The changes are subtle and cumulative at the various backbone positions. Only a handful of residues in the protein change rotamer state between the two crystals, most notably His108, Lys131, Asp191, and Glu195, all of which are involved in the binding of the two Mg2+ ions. Similarly, Arg49 and Arg56, which are involved in binding the NADP(H) cofactor, reorient slightly.
Figure 5.

Cα-Cα distances for residues of Ia_KARI_holo aligned against Ia_KARI_apo. Because of the rigid body movement of the interdomain hinge, the Rossmann (blue) and knotted (red) domains have been aligned separately. The nonaligned domains are shown in a lighter color. Points of interest are numbered as follows: 1) αA-loop, 2) Specificity loop, 3) β6αF-loop, 4) Hinge, 5) α3-helix.
Figure 6.
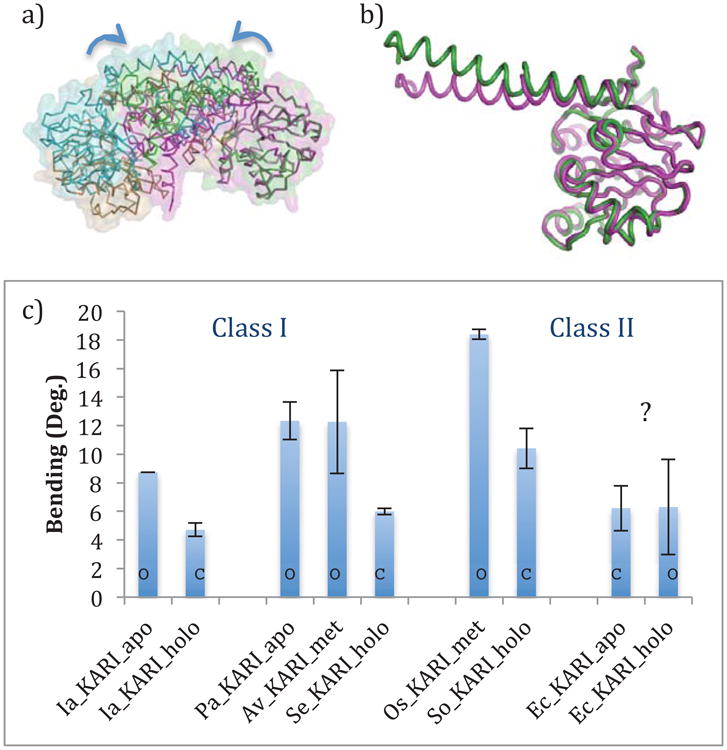
a) An alignment between the apo and holo dimers of Ia_KARI. The structures are aligned based on their rightmost Rossmann domains. The apo structure is shown in green and cyan, and the holo structure is shown in magenta and tan. b) The Rossmann domains and αl-helices of the apo (green) and holo (magenta) crystals of Ia_KARI, showing the change in the bend of the first half of the α1-helix, c) Quantitative analysis of the bending of the first half of the α1-helix. Error bars show standard deviations of the values for multiple chains in the asymmetric unit of the crystal. Bars are denoted with an ‘O’ or a ‘C’ to designate open and closed states of the active site, as assigned visually. Ec_KARI is a clear outlier, both because it goes from a closed to open state upon binding cofactor, and because there is no corresponding change in the α1-helix bending.
Three regions have notably non-rigid-body behavior: the N-terminus of the αA-helix (including the latter half of the Rossmann-identifying GxGxxG motif [47]), the β6αF-loop and the N-terminus of the αF-helix, and the α3α4-loop. Indeed, in the ligand-bound structure, the αA-helix and β6αF-loop interact with the α3-helix not of their own chain but of their dimeric partner. This interaction makes up the opposite side of the active site from the Asp191/Glu195/α1 face closer to the hinge. A rearrangement of the α3+1-helix was previously identified in the plant KARIs and was thought to be involved in binding IpOHA and “capping” the active site [24]. In the Ia_KARI_apo structure, a combination of this rearrangement and an increase in the α1-helix curvature means that the interaction between these regions is absent. The distance between the alpha carbons of Gly134 (on the αF-helix) and Ala250′ (on the dimeric partner's α3-helix) goes from 11.8 Å in the apo structure to 6.6 Å in the ligand-bound structure.
Critically, this closure is centered around the nicotinamide moiety of NADPH, in particular the C7N amide. As shown schematically in Figure 7a, the side-chain of Gln28 hydrogen bonds to the backbone amine of Gly134 in the absence of the cofactor. With cofactor bound, the terminal amide is inserted into this gap, with O7N hydrogen bonding to the backbone amine and Gln28 flipped out of the way. The N7N nitrogen of the amide recruits the α3′-helix, specifically the backbone carbonyl of Ala250′, as well as the side-chain of Ser27. Ser27 hydrogen-bonds to the cofactor, Gly134, and Ala250′, holding the complex together and “capping” the active site. Supplemental Information 5 shows this motion in the structure.
Figure 7.

Hydrogen bonds involved in the recruitment of the α3′α4′-loop upon cofactor binding in (a) Ia_KARI (b) and and Av_KARI_met and Se_KARI_holo. R = adenosine diphosphate ribose phosphate.
Conformational changes in Class I KARIs: induced fit in other class I KARIs
From this pair of structures, it was unclear to what extent the gross conformational change was driven by the smaller-scale rearrangements related to metal binding, or cofactor binding, or some combination thereof. However, we were also able to obtain a structure of another KARI, from A. vinelandii, with metal ions (Mg2+ and Fe2+) bound in the active site, but without a cofactor or a substrate analogue. Because Av_KARI_met is from a mesophile and possesses a 7-residue NADPH-dependent β2αB-loop, we have compared it not to Ia_KARI, but to the structures of Pseudomonas aeruginosa KARI (Pa_KARI_apo), which was crystallized in the apo form [18], and Slackia exigua (Se_KARI_holo), which was crystallized with cofactor, substrate analogue, and metals [21]. Additionally, Av_KARI has higher sequence identity (Supplemental Information 6) to these than to Ia_KARI. Below, residues will be discussed using the numberings in Av_KARI; the corresponding numberings in Pa_KARI and Se_KARI are given in Supplemental Information 7.
The structure of Av_KARI_met aligns very well with that of Pa_KARI_apo, deviating only in the β2αB-loop and at the C-terminus. The former deviation is likely a result of increased flexibility in this loop due to the lack of a cofactor. Both structures are in an “open” state, with their active sites exposed to solvent. Se_KARI_holo is in the “closed” state, and to align Av_KARI_met to Se_KARI_holo, we were forced to align the N-termini and C-termini separately, as in Figure 5. As with Ia_KARI, it is clear that each domain is separate and rigid, with a hinge between them, and again this hinge is the α1-helix. This provides an unambiguous answer to the question above: the bending of the α1-helix is driven not by the binding of metal ions to the acidic residues on this helix, but by the subsequent binding of the cofactor.
It is worth noting that of the six metal-binding residues, only Lys130, Asp190, and Glu226′ undergo rotamer changes upon metal binding that are much more subtle than the changes observed with Ia_KARI. The residues of Pa_KARI_apo are therefore more preorganized for metal binding, although it is not clear whether this is an artifact of crystallization or a function of the protein structure. Nevertheless, this is further evidence that metal ion binding is not a driver of a larger rearrangement.
The gross conformational change between Av_KARI_met and Se_KARI_holo closely resembles that observed in Ia_KARI, as does the correlated reorganization of the α3′α4′-loop and the αF-helix around the nicotinamide amide group. The reorganization in Av_KARI_met/Se_KARI_holo, however, involves a more intricate hydrogen bond network (Figure 7b, Supplemental Information 8). In place of Ala250′, Av_KARI has Ser249′, the side-chain of which is capable of hydrogen bonding. Likewise, Pro135 of the αF-helix, which did not participate in the rearrangement is replaced with His134, is reoriented upon cofactor binding and hydrogen bonds to the side-chains of both Ser249′ and Ser26. Furthermore, Gln27, instead of hydrogen-bonding to the backbone amine of Gly133 in the cofactor-free structure, hydrogen-bonds to the carbonyl of Ala131. This hydrogen bond is not interrupted by the binding of the cofactor. A different network of interactions is present in Pa_KARI_apo, but inspection of the electron density unambiguously shows this to be a modeling error (Supplemental Information 9).
It is interesting that this mechanism (open apo state and closed cofactor-bound state) resembles that of the plant Class II KARIs rather than the one from the bacterial Class II KARIs. The plant KARIs also undergo rearrangement of the α3+1-helix upon binding of NADPH, though the plant KARI conformational change is more complex, involving a reorganization of a long C-terminal tail – absent in Class I KARIs – upon cofactor binding [24]. In contrast, E. coli KARI adopts an open conformation after binding of cofactor and substrate, the reverse of what is observed in the plant KARIs and the bacterial Class I KARIs discussed in this paper. Nonetheless, the conformational change in E. coli KARI also involves rearrangements centered around the nicotinamide amide. In E. coli KARI, the α2+1- and α3+1-helices interact with the β6αF-loop in the absence of cofactor; the binding of nicotinamide into the active site collapses the β6αF-loop (as briefly discussed by Wong et al. [25]) and breaks this interaction (Supplemental Information 10). Until more structural information is available, it is impossible to say whether E. coli KARI is unique in this regard or what evolutionary pathway led it to have this induced fit mechanism opposite that of the other KARIs.
Conclusions
Understanding and controlling the cofactor specificity of KARI enzymes has been a goal for protein engineers for nearly two decades [48]. These efforts have, by necessity, relied heavily on incomplete structural data to guide mutagenesis strategies. With the addition of the structures described in this paper to those previously available, we now have representatives of all known canonical KARI cofactor specificity loop lengths. These data show that the 6-residue specificity loops use a distinct binding arrangement for the phosphate of NADPH, providing a clear structural explanation for results from previous cofactor engineering efforts [21]. Furthermore, structures of two of the naturally NADH-utilizing KARIs show that insertions near the N-terminus of the specificity loop likely determine cofactor specificity, whereas the laboratory engineering of NADH preference involved specific loop substitutions.
These structures also provide insight into the large conformational change associated with KARI cofactor binding and catalysis. The rearrangement observed in these Class I KARIs facilitates hydrogen bond interactions with the nicotinamide group and effectively closes the enzyme active site through motion of the α1-helix. This geometry resembles that observed in plant KARIs [24], but is distinct from that of E. coli KARI, which goes from a closed conformation to an open one upon cofactor binding [25]. The principal study on order of ligand binding in KARIs was conducted using E. coli KARI [1], raising the question of whether the kinetic mechanism for ordered binding has been maintained throughout divergent evolution. The structures provided in the current study answer several outstanding questions about the structural and functional diversity of KARIs and bring us closer to a comprehensive understanding of this enzyme class.
Supplementary Material
Summary.
The Class I KARI subfamily has received less attention than the smaller Class II subfamily, leaving several gaps in our structural knowledge. We present five new structures that elucidate basis of cofactor specificity and ligand-induced conformational changes.
Acknowledgments
We thank Dr. Jens Kaiser and Dr. Pavle Nikolovski for their continued support.
Funding: This publication was supported by the Gordon and Betty Moore Foundation through GBMF2809 to the Caltech Programmable Molecular Technology Initiative, by the Resnick Sustainability Institute at Caltech (J.K.B.C.), and by NIH Grant GM 67626 (M.W.R.). The Molecular Observatory is supported by the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program at Caltech.
Footnotes
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 4TSK, 4XDY, 4XDZ, 4XEH, and 4XIY, as shown in Table 1.
Author Contributions: Jackson Cahn and Sabine Brinkmann-Chen conceived the project; Sabine Brinkmann-Chen prepared and crystallized three proteins, Jackson Cahn one, and Jared Wiig and Thomas Spatzal one; Jackson Cahn solved the protein crystal structures, one in collaboration with Thomas Spatzal and one with Andrew Buller; Yilin Hu and Markus Ribbe provided guidance to Jared Wiig, Oliver Einsle to Thomas Spatzal, and Frances Arnold to Jackson Cahn, Sabine Brinkmann-Chen, and Andrew Buller; Sabine Brinkmann-Chen measured the thermostability of one of the KARIs; Jackson Cahn analyzed the crystal structures and prepared the figures; Jackson Cahn, Sabine Brinkmann-Chen, Andrew Buller, and Frances Arnold wrote the manuscript.
References
- 1.Chunduru SK, Mrachko GT, Calvo KC. Mechanism of ketol acid reductoisomerase - steady-state analysis and metal-ion requirement. Biochemistry. 1989;28(2):486–493. doi: 10.1021/bi00428a012. [DOI] [PubMed] [Google Scholar]
- 2.Brinkmann-Chen S, Cahn JKB, Arnold FH. Uncovering rare NADH-preferring ketol-acid reductoisomerases. Metabolic Engineering. 2014;26:17–22. doi: 10.1016/j.ymben.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Dumas R, et al. Enzymology, structure, and dynamics of acetohydroxy acid isomeroreductase. Accounts of Chemical Research. 2001;34(5):399–408. doi: 10.1021/ar000082w. [DOI] [PubMed] [Google Scholar]
- 4.Dumas R, et al. Evidence for 2 catalytically different magnesium-binding sites in acetohydroxy acid isomeroreductase by site-directed mutagenesis. Biochemistry. 1995;34(18):6026–6036. doi: 10.1021/bi00018a004. [DOI] [PubMed] [Google Scholar]
- 5.Halgand F, et al. Characterization of the conformational changes of acetohydroxy acid isomeroreductase induced by the binding of Mg2+ ions, NADPH, and a competitive inhibitor. Biochemistry. 1999;38(19):6025–6034. doi: 10.1021/bi982412e. [DOI] [PubMed] [Google Scholar]
- 6.Mrachko GT, Chunduru SK, Calvo KC. The pH-dependence of the kinetic parameters of ketol acid reductoisomerase indicates a proton shuttle mechanism for alkyl migration. Archives of Biochemistry and Biophysics. 1992;294(2):446–453. doi: 10.1016/0003-9861(92)90710-e. [DOI] [PubMed] [Google Scholar]
- 7.Tyagi R, et al. Probing the mechanism of the bifunctional enzyme ketol-acid reductoisomerase by site-directed mutagenesis of the active site. FEBS Journal. 2005;272(2):593–602. doi: 10.1111/j.1742-4658.2004.04506.x. [DOI] [PubMed] [Google Scholar]
- 8.Hasegawa S, et al. Improvement of the redox balance increases L-valine production by Corynebacterium glutamicum under oxygen deprivation Conditions. Applied and Environmental Microbiology. 2012;78(3):865–875. doi: 10.1128/AEM.07056-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Lee SY. Fermentative production of branched chain amino acids: a focus on metabolic engineering. Applied Microbiology and Biotechnology. 2010;85(3):491–506. doi: 10.1007/s00253-009-2307-y. [DOI] [PubMed] [Google Scholar]
- 10.Park JH, et al. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proceedings of the National Academy of Sciences. 2007;104(19):7797–7802. doi: 10.1073/pnas.0702609104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogt M, et al. Pushing product formation to its limit: Metabolic engineering of Corynebacterium glutamicum for L-leucine overproduction. Metabolic Engineering. 2014;22(0):40–52. doi: 10.1016/j.ymben.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Dumas R, et al. Interactions of plant acetohydroxy acid isomeroreductase with reaction intermediate analogs - Correlation of the slow, competitive, inhibition-kinetics of enzyme-activity and herbicidal effects. Biochemical Journal. 1994;301:813–820. doi: 10.1042/bj3010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Epelbaum S, Chipman DM, Barak Z. Metabolic effects of inhibitors of two enzymes of the branched-chain amino acid pathway in Salmonella typhimurium. Journal of Bacteriology. 1996;178(4):1187–1196. doi: 10.1128/jb.178.4.1187-1196.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YT, Ta HT, Duggleby RG. Cyclopropane-1,1-dicarboxylate is a slow-, tight-binding inhibitor of rice ketol-acid reductoisomerase. Plant Science. 2005;168(4):1035–1040. [Google Scholar]
- 15.Liu XH, et al. Synthesis, bioactivity, theoretical and molecular docking study of 1-cyano-N-substituted-cyclopropanecarboxamide as ketol-acid reductoisomerase inhibitor. Bioorganic & Medicinal Chemistry Letters. 2007;17(13):3784–3788. doi: 10.1016/j.bmcl.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Liu XH, et al. KARI and its inhibitors. Progress in Chemistry. 2008;20(11):1788–1797. [Google Scholar]
- 17.Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451(7174):86–89. doi: 10.1038/nature06450. [DOI] [PubMed] [Google Scholar]
- 18.Ahn HJ, et al. Crystal structure of class I acetohydroxy acid isomeroreductase from Pseudomonas aeruginosa. Journal of Molecular Biology. 2003;328(2):505–515. doi: 10.1016/s0022-2836(03)00264-x. [DOI] [PubMed] [Google Scholar]
- 19.Taylor WR. A deeply knotted protein structure and how it might fold. Nature. 2000;406(6798):916–919. doi: 10.1038/35022623. [DOI] [PubMed] [Google Scholar]
- 20.Virnau P, Mallam A, Jackson S. Structures and folding pathways of topologically knotted proteins. Journal of Physics-Condensed Matter. 2011;23 doi: 10.1088/0953-8984/23/3/033101. [DOI] [PubMed] [Google Scholar]
- 21.Brinkmann-Chen S, et al. General approach to reversing ketol-acid reductoisomerase cofactor dependence from NADPH to NADH. Proceedings of the National Academy of Sciences. 2013;110(27):10946–10951. doi: 10.1073/pnas.1306073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Research. 2014;42(D1):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bastian S, et al. Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metabolic Engineering. 2011;13(3):345–352. doi: 10.1016/j.ymben.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Leung EWW, Guddat LW. Conformational changes in a plant ketol-acid reductoisomerase upon Mg2+ and NADPH binding as revealed by two crystal structures. Journal of Molecular Biology. 2009;389(1):167–182. doi: 10.1016/j.jmb.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Wong SH, et al. Bacterial and plant ketol-acid reductoisomerases have different mechanisms of induced fit during the catalytic cycle. Journal of Molecular Biology. 2012;424(3-4):168–179. doi: 10.1016/j.jmb.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 26.Dumas R, et al. Isolation and kinetic properties of acetohydroxy acid isomeroreductase from spinach (Spinacia oleracea) chloroplasts overexpressed in Escherichia coli. Biochemical Journal. 1992;288:865–874. doi: 10.1042/bj2880865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bursey EH, Burgess BK. Characterization of a variant iron protein of nitrogenase that is impaired in its ability to adopt the MgATP-induced conformational change. Journal of Biological Chemistry. 1998;273(27):16927–16934. doi: 10.1074/jbc.273.27.16927. [DOI] [PubMed] [Google Scholar]
- 28.Kabsch W. XDS. Acta Crystallographica Section D-Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans P. Scaling and assessment of data quality. Acta Crystallographica Section D-Biological Crystallography. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 30.Battye TGG, et al. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallographica Section D. 2011;67(4):271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallographica Section D-Biological Crystallography. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ, et al. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40(4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallographica Section D. 2002;58(10 Part 2):1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica Section D. 2011;67(4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D. 2011;67(4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1 2010 [Google Scholar]
- 38.Kumar P, Bansal M. HELANAL-Plus: a web server for analysis of helix geometry in protein structures. Journal of Biomolecular Structure and Dynamics. 2012;30(6):773–783. doi: 10.1080/07391102.2012.689705. [DOI] [PubMed] [Google Scholar]
- 39.Biou V, et al. The crystal structure of plant acetohydroxy acid isomeroreductase complexed with NADPH, two magnesium ions and a herbicidal transition state analog determined at 1.65 angstrom resolution. EMBO Journal. 1997;16(12):3405–3415. doi: 10.1093/emboj/16.12.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomazeau K, et al. Structure of spinach acetohydroxyacid isomeroreductase complexed with its reaction product dihydroxymethylvalerate, manganese and (phospho)-ADP-ribose. Acta Crystallographica Section D-Biological Crystallography. 2000;56:389–397. doi: 10.1107/s0907444900001694. [DOI] [PubMed] [Google Scholar]
- 41.Naylor CE, et al. NADP(+) and NAD(+) binding to the dual coenzyme specific enzyme Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase: different interdomain hinge angles are seen in different binary and ternary complexes. Acta Crystallographica Section D-Biological Crystallography. 2001;57:635–648. doi: 10.1107/s0907444901003420. [DOI] [PubMed] [Google Scholar]
- 42.Kavanagh KL, et al. Structure of xylose reductase bound to NAD(+) and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochemical Journal. 2003;373:319–326. doi: 10.1042/BJ20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Falini G, et al. Dual coenzyme specificity of photosynthetic glyceraldehyde-3-phosphate dehydrogenase interpreted by the crystal structure of A(4) isoform complexed with NAD. Biochemistry. 2003;42(16):4631–4639. doi: 10.1021/bi0272149. [DOI] [PubMed] [Google Scholar]
- 44.Wallen JR, et al. Pyridine nucleotide complexes with Bacillus anthracis coenzyme A-disulfide reductase: A structural analysis of dual NAD(P)H specificity. Biochemistry. 2008;47(18):5182–5193. doi: 10.1021/bi8002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Britten RJ, et al. Majority of divergence between closely related DNA samples is due to indels. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4661–4665. doi: 10.1073/pnas.0330964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tóth-Petróczy Á, Tawfik DS. Protein insertions and deletions enabled by neutral roaming in sequence space. Molecular Biology and Evolution. 2013;30(4):761–771. doi: 10.1093/molbev/mst003. [DOI] [PubMed] [Google Scholar]
- 47.Wierenga RK, De Maeyer MCH, Hol WGJ. Interaction of pyrophosphate moieties with α-helixes in dinucleotide-binding proteins. Biochemistry. 1985;24(6):1346–1357. [Google Scholar]
- 48.Rane MJ, Calvo KC. Reversal of the nucleotide specificity of ketol acid reductoisomerase by site-directed mutagenesis identifies the NADPH binding site. Archives of Biochemistry and Biophysics. 1997;338(1):83–89. doi: 10.1006/abbi.1996.9802. [DOI] [PubMed] [Google Scholar]
- 49.Tyagi R, et al. The crystal structure of a bacterial Class II ketol-acid reductoisomerase: Domain conservation and evolution. Protein Sceince. 2005;14:3089–3100. doi: 10.1110/ps.051791305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bond CS. TopDraw: A sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.