

Abstract
Background & Aims
Multigene panels are commercially available tools for hereditary cancer risk assessment that allow for next-generation sequencing of numerous genes in parallel. However, it is not clear if these panels offer advantages over traditional genetic testing. We investigated the number of cancer predisposition gene mutations identified by parallel sequencing in individuals with suspected Lynch syndrome.
Methods
We performed germline analysis with a 25-gene next-generation sequencing panel using DNA from 1260 individuals who underwent clinical genetic testing for Lynch syndrome from 2012 through 2013. All subjects had a history of Lynch syndrome-associated cancer and/or polyps. We classified all identified germline alterations for pathogenicity and calculated the frequencies of pathogenic mutations and variants of uncertain significance (VUS). We also analyzed data on patients’ personal and family history of cancer, including fulfillment of clinical guidelines for genetic testing.
Results
Of the 1260 subjects, 1112 met National Comprehensive Cancer Network (NCCN) criteria for Lynch syndrome testing (88%; 95% confidence interval [CI], 86%–90%). Multigene panel testing identified 114 probands with Lynch syndrome mutations (9.0%; 95% CI, 7.6%−10.8%) and 71 with mutations in other cancer predisposition genes (5.6%; 95% CI, 4.4%−7.1%). Fifteen individuals had mutations in BRCA1 or BRCA2; 93% of these met the NCCN criteria for Lynch syndrome testing and 33% met NCCN criteria for BRCA1 and BRCA2 analysis (P=.0017). An additional 9 individuals carried mutations in other genes linked to high lifetime risks of cancer (5 had mutations in APC, 3 had bi-allelic mutations in MUTYH, and 1 had a mutation in STK11); all of these patients met NCCN criteria for Lynch syndrome testing. Four hundred seventy-nine individuals had ≥1 VUS (38%; 95% CI, 35%–41%).
Conclusions
In individuals with suspected Lynch syndrome, multigene panel testing identified high-penetrance mutations in cancer predisposition genes, many of which were unexpected based on patients’ histories. Parallel sequencing also detected a high number of potentially uninformative germline findings, including VUS.
Keywords: hereditary nonpolyposis colorectal cancer, HNPCC, colon cancer genetics, inherited cancer
INTRODUCTION
Hereditary cancer syndromes are classically characterized by markedly increased lifetime risks of multiple cancers, typically at young ages. Identifying individuals with specific inherited predispositions to cancer thus greatly impacts risk counseling for affected patients and their families, including the type and timing of cancer surveillance and potential recommendations for prophylactic surgery. Timely implementation of appropriate, enhanced cancer prevention strategies can have a profound impact on decreasing cancer incidence and mortality in such patients.1–4 Two of the most common inherited cancer syndromes are Lynch syndrome (LS), caused by mutations in one of the DNA mismatch repair (MMR) genes, and hereditary breast/ovarian cancer (HBOC), caused by germline mutations in BRCA1 or BRCA2.1–3 LS is the most common inherited cause of colorectal cancer (CRC) and is also associated with markedly increased risks of endometrial, ovarian, gastric, pancreatic, small bowel, urinary tract, and other cancers.1, 2, 5, 6
The traditional model of hereditary cancer risk assessment involves identifying individuals whose histories fulfill clinical criteria for a specific syndrome, followed by targeted germline testing only on the gene(s) associated with that syndrome.7 Although clinical guidelines and prediction models can help direct the use of genetic testing for LS, 30–50% of families fulfilling stringent clinical criteria for LS will ultimately have normal germline testing for MMR gene mutations.1, 8–12 Furthermore, there is increasing recognition that the wide phenotypic spectrum of LS cancers can overlap with other hereditary cancer syndromes.13–16 Thus, traditional, criteria-based genetic testing may not be the ideal hereditary cancer risk assessment strategy in individuals with suspected LS.
With recent advances in next-generation sequencing (NGS) technologies, multigene panel testing has emerged as an alternative strategy for hereditary cancer risk assessment, in which numerous cancer susceptibility genes are analyzed in parallel.7, 17 Whether panel testing offers meaningful advantages over targeted criteria-based genetic testing practices, however, is unknown. This study’s aim was to determine the frequency of non-LS gene mutations detected by a multigene hereditary cancer panel among individuals undergoing clinical genetic testing for LS.
METHODS
Study Population
3057 individuals with a history of LS-associated cancer and/or colorectal polyps whose clinicians submitted germline DNA to a CLIA-approved commercial laboratory (Myriad Genetic Laboratories, Inc., Salt Lake City, Utah, USA) for clinical genetic testing for all 5 genes underlying LS (MLH1, MSH2, MSH6, PMS2, and EPCAM) between 2012–2013 were consecutively ascertained. Upon completion of clinical LS testing, samples were anonymized for research-based multigene panel testing. 1615 subjects were excluded since their testing originated from states with legislation mandating destruction of biospecimens after completion of clinical genetic testing. Another 182 subjects were excluded due to technical factors (insufficient remaining DNA after clinical testing; DNA extracted from a non-blood sample) to give an overall cohort of 1260 individuals for this cross-sectional analysis. The study was approved by the Dana-Farber Cancer Institute’s institutional review board.
Clinical Data
As part of routine clinical LS genetic testing, subjects’ clinicians completed a test request form for each individual describing basic demographics (gender, ancestry), cancer/polyp history, ages at diagnosis, and family history of cancer.
Consistent with prior studies, the following were considered LS-associated cancers: CRC, endometrial cancer (EC), ovarian cancer, gastric cancer, pancreatic cancer, small intestine cancer, urinary tract cancer, hepatobiliary cancer, sebaceous adenomas/carcinomas, and brain tumors.12 Based on their reported personal/family histories, subjects were assessed as to whether they fulfilled National Comprehensive Cancer Network (NCCN) guidelines for LS testing (Supplementary Methods).9 A numeric estimate of the likelihood of identifying a germline mutation in MLH1, MSH2, or MSH6 was calculated for each subject using the PREMM1,2,6 prediction model (http://premm.dfci.harvard.edu/).12 Each subject was assessed for whether their personal/family histories fulfilled NCCN criteria for HBOC testing for germline BRCA1/2 mutations (Supplementary Methods).18
Germline Sequencing/Interpretation
After completion of clinical LS testing, anonymized genomic DNA samples were PCR-amplified with a custom amplicon library on a Raindance ThunderStorm instrument (RainDance Technologies, Inc., Lexington, MA) for NGS (Supplemental Methods). DNA products were sequenced on an Illumina HiSeq 2500 (Illumina, Inc., San Diego, CA) to detect sequence variations and large rearrangements among twenty-five cancer susceptibility genes with at least 1000x average coverage.
All sequence variations and large rearrangements detected were classified for pathogenicity into the following categories, as previously described: deleterious mutation, suspected deleterious mutation, variant of uncertain clinical significance (VUS), favor polymorphism, and polymorphism (Supplemental Methods).19, 20 Individuals with deleterious or suspected deleterious genomic alterations were collectively defined as having “pathogenic” mutations. Alterations were classified as VUS if data were insufficient to support either a deleterious or benign interpretation.
Genes analyzed with the multigene panel were categorized as high- or moderate-penetrance based on expected lifetime risks of cancer (≥40% versus <40% or unknown) associated with the respective cancer predisposition syndrome (Table 1).21–26 The genes underlying LS, adenomatous polyposis (APC and MUTYH) and hamartomatous polyposis (BMPR1A, PTEN, SMAD4, and STK11) syndromes, BRCA1/2, familial atypical multiple mole melanoma syndrome (CDKN2A and CDK4), hereditary diffuse gastric cancer (CDH1), and Li-Fraumeni syndrome (TP53) were categorized as high-penetrance, whereas the remaining eight genes (ATM, BARD1, BRIP1, CHEK2, NBN, PALB2, RAD51C, and RAD51D) were considered moderate-penetrance. Biallelic MUTYH mutations were considered high-penetrance whereas monoallelic MUTYH mutations were not.26–29
Table 1.
Genes analyzed by a multigene hereditary cancer panel
| High-penetrance genes | Moderate-penetrance genes |
|---|---|
| Lynch syndrome | ATM |
| MLH1 | BARD1 |
| MSH2 | BRIP1 |
| MSH6 | CHEK2 |
| PMS2 | NBN |
| EPCAM | PALB2 |
| Adenomatous polyposis syndromes | RAD51C |
| APC | RAD51D |
| MUTYH (biallelic) | |
| Hamartomatous polyposis syndromes | |
| BMPR1A | |
| PTEN | |
| SMAD4 | |
| STK11 | |
| Hereditary breast/ovarian cancer (HBOC) | |
| BRCA1 | |
| BRCA2 | |
| Familial atypical multiple mole melanoma syndrome | |
| CDKN2A | |
| CDK4 | |
| Hereditary diffuse gastric cancer syndrome | |
| CDH1 | |
| Li-Fraumeni syndrome | |
| TP53 |
Statistical Methods
The primary outcome was detection of pathogenic mutations in ≥1 cancer susceptibility genes on the multigene panel. Subjects’ ages and PREMM1,2,6 scores were described as continuous variables, and mean PREMM1,2,6 scores were compared using the Student’s t-test. All other clinical characteristics were described as categorical variables, and proportions were compared with Fisher’s exact test. All P-values were two-tailed, and P-values <0.05 were considered statistically significant.
RESULTS
Clinical Characteristics
915/1260 (73%) participants were female (Table 2). All subjects had a personal history of ≥1 LS-associated cancer and/or colorectal polyps with a median age at first cancer/polyp diagnosis of 47 years. 790 subjects (63%) had a history of CRC and 172 (14%) had a history of ≥2 primary cancers. 930 subjects (74%) had a family history of any LS-associated cancer, including 726 (58%) with a family history of CRC and 191 (15%) with a family history of EC. Based on reported personal/family histories, the cohort’s mean PREMM1,2,6 score was 11.2% (95% CI: 10.4%–12.0%), and 1112/1260 (88%; 95% CI: 86%–90%) fulfilled NCCN guidelines for LS testing.
Table 2.
Characteristics of 1260 individuals undergoing clinical testing for Lynch syndrome
| Total cohort (N=1260) N (%) |
|
|---|---|
| Female | 915 (73) |
| Median age (years) at first cancer diagnosis [IQR] | 47 [39 – 55.5]† |
| Personal history* | |
| Colorectal cancer, any age | 790 (63) |
| Colorectal cancer, age <50 | 434 (34) |
| Endometrial cancer | 292 (23) |
| Ovarian cancer | 84 (7) |
| Multiple primary cancers | 172 (14) |
| Colorectal polyps | 280 (22) |
| Family history* | |
| Any Lynch cancer | 930 (74) |
| Colorectal cancer | 726 (58) |
| Endometrial cancer | 191 (15) |
| Ovarian cancer | 142 (11) |
| Breast cancer | 294 (23) |
| No/unknown family history | 161 (13) |
| Met NCCN Lynch criteria | 1112 (88) |
Age data missing for 56 subjects
Personal and family history classifications are not mutually exclusive
Germline Findings
182/1260 (14.4%; 95% CI: 12.6–16.5%) subjects were found to carry ≥1 pathogenic mutation with the multigene panel (Supplemental Table 1), including 114 (9.0%; 95% CI: 7.6–10.8%) with a LS mutation and 71 (5.6%; 95% CI: 4.4–7.1%) with a non-LS mutation (3 subjects carried both a LS and non-LS mutation; Figure 1A, Table 3). Of the 182 mutation carriers identified, 137 (75%; 95% CI: 68–81%) had ≥1 high-penetrance gene mutations.
Figure 1.

Pathogenic mutations identified with a multigene panel among 1260 individuals with suspected Lynch syndrome. (A) Proportion of mutation carriers with Lynch syndrome mutations (purple), non-Lynch syndrome mutations (blue), or both Lynch and non-Lynch syndrome mutations (dark purple). (B) Distribution of Lynch syndrome mutation carriers by specific gene. (C) Distribution of non-Lynch syndrome mutation carriers by gene type (BRCA1/2, monoallelic MUTYH, other high-penetrance genes, or moderate-penetrance genes).
Table 3.
Clinical characteristics of mutation carriers identified by multigene panel testing
| Personal history* | Family history* | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Female N (%) |
Median age† (yrs) at first cancer/ polyp |
Met NCCN Lynch criteria N (%) |
CRC, any age N (%) |
CRC, age <50 N (%) |
EC N (%) |
Ovarian cancer N (%) |
Breast cancer N (%) |
Multiple primary cancers N (%) |
Colorectal polyps N (%) |
Any Lynch cancer N (%) |
CRC N (%) |
EC N (%) |
Ovarian cancer N (%) |
Breast cancer N (%) |
None/ unknown N (%) |
|
| High-penetrance genes | ||||||||||||||||
| Lynch (N=111) | 75 (68) | 45 | 105 (95) | 81 (73) | 58 (52) | 33 (30) | 3 (3) | 4 (4) | 26 (23) | 12 (11) | 95 (86) | 82 (74) | 29 (26) | 9 (8) | 22 (20) | 4 (4) |
| BRCA1 (N=6) | 4 (67) | 55 | 6 (100) | 3 (50) | 0 | 2 (33) | 1 (17) | 0 | 1 (17) | 3 (50) | 5 (83) | 4 (67) | 2 (33) | 1 (17) | 2 (33) | 0 |
| BRCA2 (N=9) | 4 (44) | 42 | 8 (89) | 6 (67) | 5 (56) | 2 (22) | 0 | 0 | 0 | 2 (22) | 5 (56) | 3 (33) | 0 | 1 (11) | 5 (56) | 2 (22) |
| APC (N=5) | 3 (60) | 44 | 5 (100) | 3 (60) | 2 (40) | 0 | 0 | 0 | 0 | 2 (40) | 5 (100) | 5 (100) | 0 | 1 (20) | 0 | 0 |
| Biallelic MUTYH (N=3) | 2 (67) | 58 | 3 (100) | 3 (100) | 1 (33) | 0 | 0 | 1 (33) | 1 (33) | 1 (33) | 3 (100) | 3 (100) | 0 | 0 | 1 (33) | 0 |
| Moderate-penetrance genes | ||||||||||||||||
| ATM (N=8) | 6 (75) | 47.5 | 7 (88) | 6 (75) | 3 (38) | 3 (38) | 1 (13) | 0 | 2 (25) | 1 (13) | 5 (63) | 4 (50) | 1 (13) | 0 | 0 | 2 (25) |
| CHEK2 (N=5) | 4 (80) | 52 | 4 (80) | 4 (80) | 1 (20) | 1 (20) | 0 | 0 | 0 | 0 | 3 (60) | 3 (60) | 0 | 0 | 1 (20) | 1 (20) |
| BRIP1 (N=2) | 0 | 42 | 2 (100) | 2 (100) | 2 (100) | 0 | 0 | 0 | 0 | 0 | 1 (50) | 1 (50) | 0 | 0 | 0 | 0 |
| BARD1 (N=1) | 0 | 66 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 |
| NBN (N=1) | 1 (100) | 35 | 1 (100) | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PALB2 (N=1) | 1 (100) | 41 | 1 (100) | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 |
| RAD51C (N=1) | 1 (100) | 48 | 1 (100) | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) |
| Dual mutation carriers | ||||||||||||||||
| MSH6 and STK11 (N=1) | 1 (100) | 50 | 1 (100) | 1 (100) | 0 | 1 (100) | 0 | 1 (100) | 1 (100) | 1 (100) | 1 (100) | 1 (100) | 1 (100) | 0 | 0 | 0 |
| MSH2 and ATM (N=1) | 0 | 43 | 1 (100) | 1 (100) | 1 (100) | 0 | 0 | 0 | 1 (100) | 0 | 1 (100) | 1 (100) | 0 | 0 | 1 (100) | 0 |
| MSH2 and monoallelic MUTYH (N=1) | 1 (100) | 47 | 1 (100) | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 |
| Monoallelic MUTYH only (N=26) | 17 (65) | 48.5 | 19 (73) | 12 (46) | 5 (19) | 9 (35) | 2 (8) | 2 (8) | 3 (12) | 5 (19) | 19 (73) | 11 (42) | 4 (15) | 2 (8) | 9 (35) | 2 (8) |
Personal and family history classifications are not mutually exclusive
Age data missing for 4 mutation carriers
Abbrevations: CRC = colorectal cancer; EC = endometrial cancer
Of the 114 LS mutations identified, there were 31 (27%) MLH1 mutations, 40 (35%) MSH2 mutations, 26 (23%) MSH6 mutations, 14 (12%) PMS2 mutations, and 3 (3%) EPCAM mutations (Figure 1B).
Of the 71 non-LS mutations, 24 (34%; 95% CI: 23–46%) were in high-penetrance genes (Figure 1C), including BRCA1/2 (N=15), APC (N=5), biallelic MUTYH mutations (N=3), and STK11 (N=1). There were 20/71 (28%; 95% CI: 18–40%) non-LS mutations in moderate-penetrance cancer susceptibility genes and another 27 (38%; 95% CI: 27–50%) individuals were monoallelic MUTYH mutation carriers. The three individuals with two germline mutations included one subject with pathogenic MSH2 and ATM mutations, one with MSH6 and STK11 mutations, and one with MSH2 and a monoallelic MUTYH mutation.
The clinical significance of monoallelic MUTYH mutation carriage is a matter of debate.27–34 If monoallelic MUTYH mutation carriers are excluded from the tally of pathogenic mutations in this study, then a total of 156 (12.4% of the overall 1260 patient cohort; 95% CI: 10.6–14.4%) mutation carriers were identified, including 44 (3.5% of the cohort; 95% CI: 2.6–4.7%) with a non-LS mutation, two of whom had both a LS and non-LS mutation.
The 15 BRCA1/2 probands represented 8% of all mutation carriers identified with the multigene panel, and BRCA1/2 mutations were found in 1.2% (15/1260; 95% CI: 0.7–2.0%) of the entire cohort. Eight (53%) BRCA1/2 mutation carriers were female and 7 (47%) were male. Five (33%) of the BRCA1/2 mutations were Ashkenazi founder mutations (three BRCA1 5382insC and two BRCA2 6174delT), though only 1 of the 15 BRCA1/2 probands was identified on the test request form as being of Ashkenazi descent. 9/15 (60%) BRCA1/2 probands had a history of CRC, including 6/7 (86%) male BRCA1/2 carriers. 4/15 (27%) BRCA1/2 probands had a history of EC, 1 (7%) had a history of ovarian cancer, and none had a history of breast or pancreatic cancer. Ten (67%) BRCA1/2 carriers had a family history of any LS cancer, including 7 (47%) with a family history of CRC. Seven (47%) BRCA1/2 carriers had a family history of breast cancer. BRCA1/2 carriers were significantly more likely to fulfill NCCN criteria for LS testing than for HBOC testing (93% vs 33%; P=0.0017).
Nine individuals were found to carry mutations in high-penetrance non-LS cancer susceptibility genes other than BRCA1/2. One had both pathogenic STK11 and MSH6 mutations, with a personal history of CRC, EC, and breast cancer. Of the remaining 8 who carried either germline APC mutations (N=5) or biallelic MUTYH mutations (N=3), all had a family history of CRC and fulfilled NCCN criteria for LS testing, and 6 (75%) had a personal history of CRC. Three (38%) reported prior colorectal polyps, although details on polyp number and histology were not available.
Of the 26 individuals found to carry a monoallelic MUTYH mutation (excluding the proband with both a MSH2 and monoallelic MUTYH mutation), 12 (46%; 95% CI: 27–66%) had a personal history of CRC and 11 (42%; 95% CI: 24–63%) had a family history of CRC.
682 VUS were detected in 479 individuals (38% of the cohort; 95% CI: 35–41%) (Supplemental Table 2). The most common genes in which VUS were discovered were ATM (N=128), APC (N=51), NBN (N=51), and BRIP1 (N=50) (Figure 2).
Figure 2.
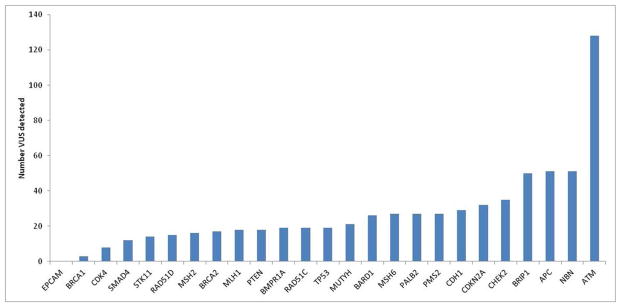
Number of variants of uncertain significance (VUS), per gene, detected with a multigene panel in 1260 individuals undergoing Lynch syndrome testing.
PREMM1,2,6 scores and NCCN criteria
The majority of mutation carriers had a PREMM1,2,6 score ≥5% (the cutoff recommended by NCCN guidelines for consideration of LS evaluation), regardless of whether they carried a LS or a non-LS mutation (Table 4).9 52% of LS carriers had a PREMM1,2,6 score ≥15%, versus 26% of non-LS probands (P=0.001). There was no significant difference between the proportion of LS carriers who fulfilled NCCN criteria for LS testing compared to BRCA1/2 carriers (P=1.00) or other high-penetrance mutation carriers (P=1.00).
Table 4.
PREMM1,2,6 scores and fulfillment of NCCN criteria for Lynch syndrome testing among individuals† with pathogenic mutations identified by a multigene panel
| Mean PREMM1,2,6 score, % (95% CI) | P-value¥ | PREMM1,2,6 score ≥5%, N (%) | P-value¥ | PREMM1,2,6 score ≥15%, N (%) | P-value¥ | Met NCCN Lynch criteria, N (%) | P-value¥ | |
|---|---|---|---|---|---|---|---|---|
| Lynch mutation carriers (N=111)† | 28.6 (23.7–33.5) | - | 96 (86) | - | 58 (52) | - | 105 (95) | - |
| All non-Lynch mutation carriers (N=68)† | 13.4 (9.9–16.8) | 0.00002 | 51 (75) | 0.07 | 18 (26) | 0.001 | 58 (85) | 0.056 |
| BRCA1/2 mutation carriers (N=15) | 12.4 (6.9–17.8) | 0.02 | 12 (80) | 0.45 | 3 (20) | 0.03 | 14 (93) | 1.00 |
| Other high-penetrance gene mutation carriers (N=8)† | 27.8 (10.1–45.5) | 0.93 | 6 (75) | 0.32 | 5 (63) | 0.72 | 8 (100) | 1.00 |
| Moderate-penetrance gene mutation carriers (N=19)† | 11.7 (5.4–18.0) | 0.007 | 14 (74) | 0.17 | 4 (21) | 0.01 | 17 (89) | 0.33 |
| Monoallelic MUTYH mutation carriers (N=26)† | 10.8 (7.0–14.6) | 0.001 | 19 (73) | 0.13 | 6 (23) | 0.009 | 19 (73) | 0.0032 |
P-values for comparison with Lynch carriers
Excluding 3 subjects with both a Lynch mutation and a non-Lynch mutation: one subject with both MSH2 and ATM mutations, one with both MSH 6 and STK11 mutations, and one with both MSH2 and a monoallelic MUTYH mutation
DISCUSSION
Multigene panel testing identified clinically unsuspected mutations in non-LS cancer susceptibility genes in 71/1260 (5.6%) individuals undergoing LS genetic testing, including 3 with both LS and non-LS mutations. In total, 75% of pathogenic mutations identified by the multigene panel were in high-penetrance genes.25 The most common unexpected findings in our cohort were BRCA1/2, APC, and biallelic MUTYH mutations in individuals with clinical features of LS.
The growing availability of multigene panels provides clinicians with the option of broad-based genetic analysis for hereditary cancer risk assessment, rather than traditional, phenotype-driven genetic testing. The benefits of such comprehensive testing strategies have been debated and are only beginning to be scientifically evaluated.7, 35 Clinical guidelines, such as NCCN criteria, and prediction models, such as PREMM1,2,6, have been developed to select individuals for LS evaluation, based on their personal/family histories.9, 12 Our study, where the vast majority of both LS and non-LS mutation carriers fulfilled NCCN criteria for LS and had a PREMM1,2,6 score ≥5%, demonstrates that such criteria, although very useful for identifying which individuals should be referred for genetic evaluation, ultimately may not be specific for underlying LS.
In the only prior study to specifically examine panel testing in patients with suspected hereditary gastrointestinal cancer, actionable mutations were detected in 42/586 (7.2%) patients, 23 of which were LS mutations.36 All patients in this study, however, were specifically selected by their clinicians to undergo testing with a panel of 13 CRC susceptibility genes, rather than targeted, phenotype-directed testing, suggesting that this was a particularly high-risk cohort. Furthermore, the panel used did not include BRCA1/2 testing, thereby precluding analysis regarding phenotypic overlap between LS and HBOC.
In other recent analyses studying panel testing in women with suspected HBOC, the identification of mutations in high-penetrance genes other than BRCA1/2 was uncommon.37–39 As such, a recent editorial cautioned that identifying unexpected, clinically useful, high-penetrance mutations with multigene panel testing is likely to be rare.35 Panel testing in our cohort, however, found >1 high-penetrance non-LS gene mutation for every 5 LS mutations identified, demonstrating that unexpected actionable findings are not uncommon in patients with LS-like phenotypes.
The identification of pathogenic BRCA1/2 mutations in 8% of mutation carriers and 1.2% of our overall cohort is unexpected, and raises important clinical questions. The carrier rate of BRCA1/2 mutations is known to be particularly high (1.1–2.5%) in Ashkenazi Jewish individuals, but is considerably lower (0.22–0.33%) in the general population.40–43 Since only 2% of our cohort was identified as being of Ashkenazi descent and only 5 BRCA1/2 mutations identified were Ashkenazi founder mutations, it seems unlikely that the unexpected identification of BRCA1/2 mutations in our study can be attributed to simply detecting their background population prevalence. Even if the 5 Ashkenazi founder mutations are excluded from the analysis, the 10 non-founder BRCA1/2 mutations identified in this study is substantially higher (0.8%; 10/1260) than the expected prevalence in the general population.
Prior studies have shown no increased CRC risk in BRCA1/2 probands, and the traditional thinking has thus been that LS and HBOC are phenotypically distinct syndromes, aside from both conferring increased risks of ovarian cancer.44 In this study, however, BRCA1/2 probands had phenotypes that were markedly more “Lynch-like” than “HBOC-like,” suggesting that standard clinical evaluation would not have identified most of these individuals as needing BRCA1/2 testing. Such atypical phenotypes may be more common in men, since 86% of the male BRCA1/2 probands in our study had a history of CRC. Our findings thus raise the hypothesis that a subset of BRCA1/2 probands may have particularly atypical phenotypes that can mimic LS.
The identification of such patients with “unexpected” high-penetrance germline mutations that do not seem concordant with their clinical histories raises the question as to whether hereditary cancer syndromes should be defined based on genotypic data, phenotypic data, or both. Prior to the identification of specific genes linked to familial cancer risks, assessment of an individual’s clinical phenotype was the primary means of diagnosing a particular hereditary cancer syndrome (e.g. fulfillment of Amsterdam criteria for Lynch syndrome).45 With the discovery of specific cancer susceptibility genes linked to particular syndromes and the availability of clinical genetic testing, it has become clear that such criteria are often too stringent and insensitive.1 As such, the current gold standard for diagnosing a hereditary cancer syndrome is now the identification of a germline mutation in the associated gene (e.g. Lynch syndrome is defined by the presence of a germline MMR mutation), and cancer surveillance recommendations are usually made based on genotype more so than family history.9 If multigene panel testing routinely identifies a subset of patients with pathogenic mutations in the setting of highly atypical clinical histories, however, such patients’ management recommendations may need to take into account phenotype as well as genotype. For example, prophylactic total gastrectomy is the current recommendation for CDH1 mutation carriers from hereditary diffuse gastric cancer families, though this recommendation may be overly aggressive in the context of an “incidental” CDH1 mutation in an individual with no personal or family history of diffuse gastric cancer.46 Larger studies with more detailed clinical histories will be needed to address this more definitively.
In order to fully assess the potential benefits and downsides of multigene panel testing compared to traditional hereditary cancer risk assessment strategies, the cost of testing must be taken into consideration. Although rigorous cost-effectiveness analyses were beyond the scope of this study, multigene panel testing offers a lower cost of testing per gene and may also decrease some of the ancillary costs of genetic testing, such as additional physician and counselor visits, by analyzing genes in parallel, rather than sequentially.47 One recent analysis concluded that multigene panel testing was cost-effective as an initial diagnostic test for patients with suspected hereditary CRC syndromes, particularly for panels that include genes associated with high-penetrance CRC syndromes.48 Such potential cost savings, however, must be weighed carefully against the costs (both financial and non-financial) that are likely to arise from the increased identification of VUSs and mutations in moderate-penetrance genes.
The discovery of uninformative and potentially anxiety-provoking results remains a primary limitation of multigene panel testing, and the identification of ≥1 VUS in 38% of our cohort validates such concerns.7, 35 Other results of debatable clinical utility include the detection of mutations in moderate-penetrance cancer susceptibility genes, which may not account for subjects’ clinical phenotypes, and the identification of monoallelic MUTYH mutations in 2.1% of participants.35 The population prevalence of monoallelic MUTYH mutation carriage is estimated to be 1%,28 and prior studies have shown a roughly 2-fold increase in CRC risk among monoallelic carriers with an estimated 7.2% and 5.6% risk of CRC by age 70 for male and female carriers, respectively.27, 31, 32 Recent data have also suggested that monoallelic MUTYH mutation carriers with a first-degree relative with early-onset CRC are at particularly increased CRC risk (12.4% and 9.9% risk of CRC by age 70 for male and female carriers, respectively).27, 30 Other studies, however, have found no significant increase in the risk of CRC or other cancers among monoallelic MUTYH mutation carriers, thus leaving the clinical significance of such findings up for debate.29, 33, 34 While the clinical utility of detecting monoallelic MUTYH carrier status for the proband themselves is thus uncertain, such results at the very least may prompt family members with a history of CRC to be evaluated for biallelic carriage.
Our study’s main strength is its use of a large, consecutive cohort of individuals with clinical histories suggestive of LS, which makes its findings generalizable to other populations of patients with suspected LS. The use of a CLIA-certified laboratory with extensive experience in clinical genetic testing and interpretation of germline cancer susceptibility gene alterations allowed for rapid and comprehensive genetic analysis of a large panel of cancer susceptibility genes. The availability of linked personal/family cancer history data allowed for determination of whether mutation carriers fulfilled various clinical guidelines for hereditary cancer risk assessment.
We recognize that our study has limitations. Data regarding subjects’ personal/family histories of cancer were obtained via clinician report on a test request form, and we were thus unable to confirm its accuracy or completeness. Although this is a potential limitation, the same approach was used to develop the PREMM1,2,6 prediction model for LS risk assessment, and PREMM1,2,6 has been subsequently validated in clinic- and population-based cohorts where clinical data were extensively verified.12 Furthermore, all subjects in this cohort were ascertained from a large commercial laboratory which receives genetic testing referrals from academic medical centers as well as community practices. Given that patients from academic cancer centers may have higher-risk clinical histories than those from smaller practices, we are unable to account for the possibility that the performance of multigene panel testing may vary across different healthcare settings.
The specific frequencies of mutation carriers detected by panel testing are also likely to vary depending on the genes included in a given multigene panel. Although there is a growing array of commercially available multigene panels for hereditary cancer risk assessment, almost all such panels include the same high-penetrance cancer susceptibility genes (i.e. MMR genes, BRCA1/2, APC, MUTYH, STK11, PTEN, CDH1, and TP53), and thus the key findings of our study are likely generalizable to testing performed with other multigene panels.47
Another limitation of our study is that we did not have data on tumor testing results that may have prompted referral for germline testing. NCCN guidelines9 recommend that all CRC specimens undergo MMR IHC or MSI testing as an initial screen for LS. Roughly 20% of the MSI-H/MMR-deficient CRCs identified with such testing will be due to LS, and additional tumor testing for BRAF V600E mutations or MLH1 promoter hypermethylation can help identify the 80% of MSI-H/MMR-deficient cases that are likely sporadic and thus do not need LS germline testing. Without such data, we are unable to extrapolate our study’s findings on multigene panel testing into contemporary LS diagnostic algorithms which rely heavily on MMR IHC and MSI screening of tumor specimens. Multiple studies, however, have found that the uptake and efficacy of universal tumor testing strategies are highly variable, even within large academic medical centers.49–51 Furthermore, most studies examining universal tumor testing have only performed germline LS testing on individuals with MSI-H/MMR-deficient CRC, and thus the mutation rate amongst patients with normal or absent tumor testing results is not well-studied.52–54
Within our cohort of patients with a history of LS-associated cancer/polyps, MSI and MMR IHC tumor testing likely would have identified individuals where targeted germline LS testing would have been indicated, rather than panel testing, although this would still miss the rare individual with both a LS and non-LS mutation. Future research is needed to determine the yield of multigene panel testing in patients for whom MSI, MMR IHC, and other tumor testing results are available. Universal tumor testing algorithms only screen for LS, however, and our results demonstrate that a substantial fraction of patients with Lynch-like clinical histories will actually have other inherited cancer syndromes. Thus, the practice of using tumor testing to distinguish between patients with “familial” and “sporadic” cancers will ultimately miss some individuals with actionable mutations in non-Lynch cancer susceptibility genes.
In spite of these limitations, our findings provide novel insight about the evaluation of patients with suspected LS in the era of multigene panel testing. Since clinical criteria for LS analysis appear to identify a substantial number of probands with unexpected actionable mutations in high-penetrance non-LS cancer susceptibility genes, panel testing may ultimately replace targeted genetic testing in patients with suspected LS, except when tumor testing suggests a specific underlying MMR mutation. Increased use of panel testing, however, will undoubtedly lead to more patients being diagnosed with VUS and other germline findings of uncertain clinical utility. Furthermore, with expanded use of panel testing, the question as to how patients with “unexpected,” high-penetrance germline mutations identified by panel testing (e.g. BRCA1/2 mutations in individuals with a clinical history suggestive of hereditary colorectal cancer) should be managed is likely to become an increasingly common dilemma for practicing clinicians.
Supplementary Material
Acknowledgments
Author MBY reports receiving research funding from Myriad Genetic Laboratories, Inc. for other ongoing projects. Author SS does not receive research funding from Myriad, however, she is MBY’s mentor and collaborator on the projects that will be supported by the research grant. Authors BA, RRK, KRB, TJ, PK, BBR, RJW, and ARH are full-time employees of and receive compensation from Myriad Genetic Laboratories, Inc.
Funding support: NIH (National Cancer Institute) K24CA113433 and R01CA132829 (Dr. Syngal).
Employees of Myriad Genetic Laboratories, Inc., served as co-investigators in this study and provided material support, including germline testing and interpretation, as described in the manuscript. The specific co-investigators listed as authors participated in the review and final approval of the submitted manuscript. No grant support was obtained from Myriad Genetic Laboratories, Inc., for the study described in this manuscript.
Footnotes
Dr. Yurgelun and Dr. Syngal had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis
Previous publication:
Preliminary data from this manuscript were presented as an oral abstract at the 50th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 30 – June 3, 2014, and as an oral abstract at the 18th Annual Meeting of the Collaborative Group of the Americas on Inherited Colorectal Cancer, New Orleans, LA, September 15–16, 2014.
Role of funding organizations:
The funding organizations had no role in the design and conduct of the study; the collection, analysis, and interpretation of the data; or the preparation, review, or approval of the manuscript.
Author contributions:
Study concept and design: MBY, BA, RRK, ARH, RJW, SS.
Acquisition of data: RRK, KRB, PK, BBR, RJW.
Analysis and interpretation of data: All authors.
Drafting of the manuscript: MBY, BA, TJ, ARH, SS.
Critical revision of the manuscript for important intellectual content: All authors.
Approval of the manuscript: All authors
Obtained funding: SS.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palomaki GE, McClain MR, Melillo S, et al. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11:42–65. doi: 10.1097/GIM.0b013e31818fa2db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62:812–23. doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson HD, Pappas M, Zakher B, et al. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: a systematic review to update the U.S. Preventive Services Task Force recommendation. Ann Intern Med. 2014;160:255–66. doi: 10.7326/M13-1684. [DOI] [PubMed] [Google Scholar]
- 4.Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–34. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 5.Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958–64. doi: 10.1200/JCO.2011.39.5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–5. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domchek SM, Bradbury A, Garber JE, et al. Multiplex genetic testing for cancer susceptibility: out on the high wire without a net? J Clin Oncol. 2013;31:1267–70. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- 8.Kastrinos F, Steyerberg EW, Balmana J, et al. Comparison of the clinical prediction model PREMM(1,2,6) and molecular testing for the systematic identification of Lynch syndrome in colorectal cancer. Gut. 2013;62:272–9. doi: 10.1136/gutjnl-2011-301265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy DE, Byfield SD, Comstock CB, et al. Underutilization of BRCA1/2 testing to guide breast cancer treatment: black and Hispanic women particularly at risk. Genet Med. 2011;13:349–55. doi: 10.1097/GIM.0b013e3182091ba4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293:1979–85. doi: 10.1001/jama.293.16.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balmana J, Balaguer F, Castellvi-Bel S, et al. Comparison of predictive models, clinical criteria and molecular tumour screening for the identification of patients with Lynch syndrome in a population-based cohort of colorectal cancer patients. J Med Genet. 2008;45:557–63. doi: 10.1136/jmg.2008.059311. [DOI] [PubMed] [Google Scholar]
- 12.Kastrinos F, Steyerberg EW, Mercado R, et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology. 2011;140:73–81. doi: 10.1053/j.gastro.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morak M, Heidenreich B, Keller G, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet. 2014;22:1334–7. doi: 10.1038/ejhg.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mester JL, Moore RA, Eng C. PTEN germline mutations in patients initially tested for other hereditary cancer syndromes: would use of risk assessment tools reduce genetic testing? Oncologist. 2013;18:1083–90. doi: 10.1634/theoncologist.2013-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weissman SM, Bellcross C, Bittner CC, et al. Genetic counseling considerations in the evaluation of families for Lynch syndrome--a review. J Genet Couns. 2011;20:5–19. doi: 10.1007/s10897-010-9325-x. [DOI] [PubMed] [Google Scholar]
- 16.Castillejo A, Vargas G, Castillejo MI, et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer. 2014;50:2241–50. doi: 10.1016/j.ejca.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 17.Stadler ZK, Schrader KA, Vijai J, et al. Cancer genomics and inherited risk. J Clin Oncol. 2014;32:687–98. doi: 10.1200/JCO.2013.49.7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall MJ, Reid JE, Burbidge LA, et al. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer. 2009;115:2222–33. doi: 10.1002/cncr.24200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eggington JM, Bowles KR, Moyes K, et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2013 doi: 10.1111/cge.12315. [DOI] [PubMed] [Google Scholar]
- 20.Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 21.Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 22.Stoffel EM, Kastrinos F. Familial colorectal cancer, beyond Lynch syndrome. Clin Gastroenterol Hepatol. 2014;12:1059–68. doi: 10.1016/j.cgh.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun N, Ford JM. Genetic testing by cancer site: stomach. Cancer J. 2012;18:355–63. doi: 10.1097/PPO.0b013e31826246dc. [DOI] [PubMed] [Google Scholar]
- 24.Mukherjee B, Delancey JO, Raskin L, et al. Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J Natl Cancer Inst. 2012;104:953–6. doi: 10.1093/jnci/djs221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stadler ZK, Thom P, Robson ME, et al. Genome-wide association studies of cancer. J Clin Oncol. 2010;28:4255–67. doi: 10.1200/JCO.2009.25.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nieuwenhuis MH, Vogt S, Jones N, et al. Evidence for accelerated colorectal adenoma--carcinoma progression in MUTYH-associated polyposis? Gut. 2012;61:734–8. doi: 10.1136/gut.2010.229104. [DOI] [PubMed] [Google Scholar]
- 27.Win AK, Dowty JG, Cleary SP, et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology. 2014;146:1208–11. e1–5. doi: 10.1053/j.gastro.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casper M, Plotz G, Juengling B, et al. MUTYH hotspot mutations in unselected colonoscopy patients. Colorectal Dis. 2012;14:e238–44. doi: 10.1111/j.1463-1318.2012.02920.x. [DOI] [PubMed] [Google Scholar]
- 29.Lubbe SJ, Di Bernardo MC, Chandler IP, et al. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27:3975–80. doi: 10.1200/JCO.2008.21.6853. [DOI] [PubMed] [Google Scholar]
- 30.Win AK, Cleary SP, Dowty JG, et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer. 2011;129:2256–62. doi: 10.1002/ijc.25870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst. 2004;96:1631–4. doi: 10.1093/jnci/djh288. [DOI] [PubMed] [Google Scholar]
- 32.Jones N, Vogt S, Nielsen M, et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology. 2009;137:489–94. 494 e1. doi: 10.1053/j.gastro.2009.04.047. quiz 725–6. [DOI] [PubMed] [Google Scholar]
- 33.Balaguer F, Castellvi-Bel S, Castells A, et al. Identification of MYH mutation carriers in colorectal cancer: a multicenter, case-control, population-based study. Clin Gastroenterol Hepatol. 2007;5:379–87. doi: 10.1016/j.cgh.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 34.Peterlongo P, Mitra N, Chuai S, et al. Colorectal cancer risk in individuals with biallelic or monoallelic mutations of MYH. Int J Cancer. 2005;114:505–7. doi: 10.1002/ijc.20767. [DOI] [PubMed] [Google Scholar]
- 35.Robson M. Multigene panel testing: planning the next generation of research studies in clinical cancer genetics. J Clin Oncol. 2014;32:1987–9. doi: 10.1200/JCO.2014.56.0474. [DOI] [PubMed] [Google Scholar]
- 36.Cragun D, Radford C, Dolinsky JS, et al. Panel-based testing for inherited colorectal cancer: a descriptive study of clinical testing performed by a US laboratory. Clin Genet. 2014 doi: 10.1111/cge.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032–7. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurian AW, Hare EE, Mills MA, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32:2001–9. doi: 10.1200/JCO.2013.53.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tung N, Battelli C, Allen B, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25–33. doi: 10.1002/cncr.29010. [DOI] [PubMed] [Google Scholar]
- 40.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–8. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 41.Metcalfe KA, Poll A, Royer R, et al. Screening for founder mutations in BRCA1 and BRCA2 in unselected Jewish women. J Clin Oncol. 2010;28:387–91. doi: 10.1200/JCO.2009.25.0712. [DOI] [PubMed] [Google Scholar]
- 42.Roa BB, Boyd AA, Volcik K, et al. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996;14:185–7. doi: 10.1038/ng1096-185. [DOI] [PubMed] [Google Scholar]
- 43.McClain MR, Palomaki GE, Nathanson KL, et al. Adjusting the estimated proportion of breast cancer cases associated with BRCA1 and BRCA2 mutations: public health implications. Genet Med. 2005;7:28–33. doi: 10.1097/01.gim.0000151155.36470.ff. [DOI] [PubMed] [Google Scholar]
- 44.Garber JE, Syngal S. One less thing to worry about: the shrinking spectrum of tumors in BRCA founder mutation carriers. J Natl Cancer Inst. 2004;96:2–3. doi: 10.1093/jnci/djh021. [DOI] [PubMed] [Google Scholar]
- 45.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 46.Fitzgerald RC, Hardwick R, Huntsman D, et al. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet. 2010;47:436–44. doi: 10.1136/jmg.2009.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall MJ, Forman AD, Pilarski R, et al. Gene panel testing for inherited cancer risk. J Natl Compr Canc Netw. 2014;12:1339–46. doi: 10.6004/jnccn.2014.0128. [DOI] [PubMed] [Google Scholar]
- 48.Gallego CJ, Shirts BH, Bennette CS, et al. Next-Generation Sequencing Panels for the Diagnosis of Colorectal Cancer and Polyposis Syndromes: A Cost-Effectiveness Analysis. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.59.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol. 2013;31:2554–62. doi: 10.1200/JCO.2012.46.8454. [DOI] [PubMed] [Google Scholar]
- 50.Cragun D, DeBate RD, Vadaparampil ST, et al. Comparing universal Lynch syndrome tumor-screening programs to evaluate associations between implementation strategies and patient follow-through. Genet Med. 2014;16:773–82. doi: 10.1038/gim.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beamer LC, Grant ML, Espenschied CR, et al. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol. 2012;30:1058–63. doi: 10.1200/JCO.2011.38.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308:1555–65. doi: 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez-Carbonell L, Ruiz-Ponte C, Guarinos C, et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut. 2012;61:865–72. doi: 10.1136/gutjnl-2011-300041. [DOI] [PubMed] [Google Scholar]
- 54.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–8. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.