

Abstract
Traditional chelator-based radio-labeled nanoparticles and positron emission tomography (PET) imaging are playing vital roles in the field of nano-oncology. However, their long-term in vivo integrity and potential mismatch of the biodistribution patterns between nanoparticles and radio-isotopes are two major concerns for this approach. Here, we present a chelator-free zirconium-89 (89Zr, t1/2 = 78.4 h) labeling of mesoporous silica nanoparticle (MSN) with significantly enhanced in vivo long-term (>20 days) stability. Successful radio-labeling and in vivo stability are demonstrated to be highly dependent on both the concentration and location of deprotonated silanol groups (−Si–O–) from two types of silica nanoparticles investigated. This work reports 89Zr-labeled MSN with a detailed labeling mechanism investigation and long-term stability study. With its attractive radio-stability and the simplicity of chelator-free radio-labeling, 89Zr-MSN offers a novel, simple, and accurate way for studying the in vivo long-term fate and PET image-guided drug delivery of MSN in the near future.
Keywords: zirconium-89, mesoporous silica nanoparticle, chelator-free radio-labeling, positron emission tomography
Positron emission tomography (PET) is a highly sensitive and noninvasive imaging modality, which is playing a vital role in nano-oncology for assessing the quantitative tumor uptake and studying the pharmacokinetic (PK) profiles of radio-labeled nanoparticles in live animals.1,2 To date, chelator-based radio-labeling is still the most widely used technique for the synthesis of various kinds of radio-labeled nanoparticles. For example, copper-free click chemistry has been developed to attach fluorine-18 (18F, t1/2 = 109 min) to porous silica nanoparticles.3,4 By using either NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) or DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraaceticacid) as the chelator, copper-64 (64Cu, t1/2 = 12.7 h)-labeled carbon nanotubes,5 quantum dots,6 superparamagnetic iron oxide nanoparticles (SPIONs),7 porous silica nanoparticles,8,9 to name a few, have also been reported and used for tumor-targeted cancer imaging and biodistribution studies.
Despite the high sensitivity and no limitation in tissue penetration of signal, radio-labeled nanoparticles still provoke concerns of their in vivo (long-term) integrity (or stability).10 For example, dissociation of free 64Cu (or 64Cu-DOTA) and significant bladder uptake have been reported during the in vivo PET imaging of 64Cu-DOTA-PEG-Au.11 It is worthy to mention that the nanoparticle itself is not a positron emitter, and PET imaging only detects signals from the radio-isotopes (which usually are linked to the nanoparticle with a selected chelator as the bridge). Most free radio-isotopes (or isotopes stabilized by chelators) do not share the same PK profile with the nanoparticle of interest. Potential detachment of these radio-isotopes from nanoparticles in vivo could easily lead to erroneous interpretation of PET imaging results.
To address these concerns, there is an emerging concept of synthesizing intrinsically radio-labeled nanoparticles for more accurate investigation of the nanoparticle’s PK profile in vivo.12 Successful synthesis of intrinsically radio-labeled nanoparticle lies in the strong interaction between rationally selected nanoparticles and radio-isotopes.12 Interesting combinations, such as 64Cu-labeled porphysome,13 arsenic-72 (72As, t1/2 = 26 h), and germanium-69 (69Ge, t1/2 = 39.1 h)-labeled SPION,14,15 [64Cu]CuS nanoparticles,1618F-labeled upconversion nanoparticles and hydroxyapatite,17,18etc., have been reported over the last several years. Although still in the early stages, design and synthesis of intrinsically radio-labeled nanoparticles have shown an attractive potential in offering an easier, faster, more stable, and more specific radio-labeling technique for the next generation of nano-oncology.
Zirconium-89 (89Zr, t1/2 = 78.4 h) is a radio-isotope with a relatively low positron energy (β+avg = 395.5 keV), which is highly suitable for long blood circulating monoclonal antibody (mAb)-based PET imaging.19 Importantly, translational research of 89Zr-based agents is now under active development with at least five clinical trials ongoing in the USA alone (many more in Europe).20 Desferrioxamine B (DFO), a hexadentate ligand with three hydroxamate groups that provide six oxygen donors for metal binding, is currently the preferred chelator for labeling of 89Zr4+.21−25 However, significant bone uptake of 89Zr (3–15 %ID/g) was still observed for various 89Zr-DFO-mAb studies within 7 days postinjection (p.i.),22−25 indicating the release of free 89Zr and limited stability of DFO-based 89Zr labeling.
Mesoporous silica nanoparticles (MSNs) with controllable mesoporous structure, high specific surface area, and large pore volume have attracted increasing interest as drug delivery nanosystems.26 The presence of abundant silanol groups (−Si–OH) on the silica surface has been well-known for many decades.27 However, this has not been considered for potential radio-labeling until now.
In this work, we hypothesize that numerous deprotonated silanol groups (−Si–O–) inside the meso-channels or on the surface of MSN could function as inherent hard oxygen donors for stable radio-labeling of 89Zr. By using an amorphous dense silica nanoparticle as a control, our systematic research demonstrated a significantly enhanced stability of 89Zr-MSN in mice over 3 weeks (∼7 t1/2 of 89Zr). This study provides a chelator-free labeling mechanism for mesoporous silica nanoparticles to study their long-term in vivo stability and fate.
Results and Discussion
MSN Synthesis and Characterization
Zr4+ is a hard Lewis acid and thus prefers hard Lewis bases as donor groups. It has been known for many decades that, during the hydrolysis and condensation of tetraethyl orthosilicate (TEOS), abundant silanol groups (−Si–OH) form on the surface of amorphous silica particles.27 Here, we demonstrate that it is the deprotonated silanol groups (−Si–O–) that function as the hard Lewis bases for the successful chelator-free 89Zr labeling of amorphous silica nanoparticles.
As seen in Figure 1a, a uniform MSN with an average particle size of ∼150 nm in diameter was synthesized using literature procedures.28 The presence of radial meso-channels throughout each MSN could be easily seen from the transmission electron microscopy (TEM) image. Nitrogen adsorption–desorption isotherms further confirmed an obvious capillary condensation step at P/P0 around 0.4 (Figure 1b), which is clear evidence for the presence of mesopores in MSN. The second adsorption of the isotherms at high relative pressure (P/P0 > 0.8) represents the formation of interstitial pores among the dried MSN agglomerates (also known as textural porosity). The Brunauer–Emmett–Teller (BET) surface area was measured to be 581.5 m2/g, with a high pore volume of 1.3 cm3/g. Pore size distribution showed an average pore size of 4–5 nm (Figure 1b, inset), consistent with the previously reported values.28
Figure 1.

Chelator-free 89Zr labeling of MSN. (a) TEM image of MSN with an average particle size of ∼150 nm. (b) Nitrogen adsorption–desorption isotherms and the corresponding pore size distributions of MSN. (c) Schematic illustration showing the labeling of 89Zr4+ to the deprotonated silanol groups (−Si–O–) from the outer surface and inner meso-channels of MSN. (d) Time-dependent 89Zr labeling yield in HEPES buffer solution (pH 7–8) with varied MSN concentrations (from 2 mg/mL to 2 × 10–4 mg/mL).
According to the Zhuravlev model, for both porous and nonporous silica particles, the concentrations of silanol groups are directly proportional to the specific surface area, with the density (−Si–OH/nm2) found to be in the range of 4–5 (also known as the Kiselev–Zhuravlev constant).27 About 2–3 million −Si–OH groups in each MSN particle were estimated based on our calculation (detailed in Supporting Information, Table S1). Considering that over 95% of the surface in MSN is internal mesopore surface based on a recent hemolysis study of MSN,29 nearly all of the −Si–OH groups are expected to be inside the meso-channels but not on MSN’s outer surface, as schematically shown in Figure 1c.
These silanol groups (−Si–OH) can be fully deprotonated to become −Si–O– when the pH value of aqueous solution is higher than the isoelectric point of silica, which has been known to be in the range of pH 2–3. Our ζ-potential analysis confirmed the negatively charged surface of as-synthesized MSN (−48.4 ± 0.3 mV) in water (pH 7–8), suggesting the presence of −Si–O– groups on MSN surface.
Chelator-Free 89Zr Labeling
Our results showed that about 70% 89Zr labeling yield was achieved for MSN within the first 15 min of incubation (Table S2). The yield continued to increase over time and reached 82.5% at 2 h post-incubation (Figure 1d and Table S2). As expected, 89Zr-labeling was MSN concentration and temperature dependent, where higher concentration and high incubation temperature gave higher labeling yield (Figure 1d, Figure S1, Table S3). We further demonstrated that there was no significant reduction (less than 5%) of drug loading capacity after labeling MSN with 89Zr (Table S4).
To further demonstrate the role of deprotonated silanol groups in 89Zr-labeling, the pH value of MSN in HEPES (0.1 M) was adjusted to 2–3 to ensure protonation of silanol groups (as −Si–OH). Such protonation was confirmed by a positively charged surface (3.6 ± 0.3 mV) of MSN at pH 2–3. As expected, 89Zr labeling yield was significantly reduced even at a MSN concentration of 2 mg/mL (Figure S1 and Table S3). With these findings, we demonstrated that deprotonated silanol groups play a vital role in chelator-free labeling of 89Zr to MSN.
In Vitro Stability Study of 89Zr-MSN
To further investigate how the location and concentration of deprotonated silanol groups could affect the 89Zr labeling yield and stability, dSiO2 was used as a control. Amorphous dSiO2 synthesized by a classic Stöber method is known to have no mesopores inside and a significantly lower surface area (∼40 m2/g for dSiO2 with a size of ∼90 nm in diameter, Figure S2) when compared with that of MSN (∼600 m2/g in this work).30 In stark contrast to MSN, very little mesopore surface but a dominant external surface is expected in these dSiO2 nanoparticles, as schematically shown in Figure 2a.
Figure 2.

Chelator-free 89Zr labeling of dSiO2. (a) Schematic illustration showing the labeling of 89Zr4+ to the deprotonated silanol groups (−Si–O–) from the outer surface of dSiO2. (b) TEM image of dSiO2 with an average particle size of ∼90 nm. (c) Time-dependent 89Zr labeling yield in HEPES buffer solution (pH 7–8) with varied dSiO2 concentrations (from 2 mg/mL to 2 × 10–4 mg/mL).
The TEM image in Figure 2b shows successful synthesis of spherically shaped dSiO2 with an average size of ∼90 nm in diameter. The surface charge of dSiO2 in pH 7–8 water was also found to be highly negative (−38.9 ± 0.3 mV), confirming the presence of abundant deprotonated silanol groups on the outer surface of dSiO2. In comparison with MSN, about 17-fold less −Si–O– groups in each dSiO2 was estimated based on their marked difference in specific surface area (Table S1). By using the same labeling protocol, a similar concentration-dependent 89Zr labeling yield was observed (Figure 2c). As expected, we found significantly lower labeling yield at each time point examined when compared with MSN with the same particle concentration (Tables S2 and S5), again demonstrating the vital role of −Si–O– group concentration during 89Zr labeling.
A DFO challenge study has been reported to demonstrate the stability of 89Zr-labeled nanoparticles.31 As-synthesized 89Zr-MSN and 89Zr-dSiO2 (suspended in HEPES, 0.1 M, pH 7–8) were mixed with increasing concentrations of DFO, ranging from 0.05 to 5 mM, and incubated at 37 °C under constant shaking. At each time point, a 100k filter was used to separate the formation of possible 89Zr-DFO. As shown in Figure 3a,b, nearly negligible 89Zr detachment was detected for both 89Zr-MSN and 89Zr-dSiO2 within 48 h, demonstrating a strong binding affinity of 89Zr to both silica nanoparticles. The stability of 89Zr-MSN and 89Zr-dSiO2 in complete mouse serum was further tested. 89Zr-labeled nanoparticles were mixed with mouse serum and shaken at 37 °C. Again, no obvious detachment of free 89Zr was observed, with 89Zr-MSN showing slightly better serum stability (Figure 3c). Similar high radio-stability (with over 96% 89Zr retained in MSN) has been observed after challenging with EDTA (1 mM) at 37 °C for 7 days (Figure 3d). Taken together, we concluded that both 89Zr-MSN and 89Zr-dSiO2 showed high stability in vitro, which encouraged us to test their long-term stabilities in vivo.
Figure 3.

In vitro stability of 89Zr-labeled silica nanoparticles. Stability of (a) 89Zr-MSN and (b) 89Zr-dSiO2 when challenged with DFO of varied concentrations from 0.05 to 5 mM at 37 °C for 48 h. (c) Stability of 89Zr-MSN (red line) and 89Zr-dSiO2 (blue line) in whole mouse serum at 37 °C for 48 h. (d) Stability study of 89Zr-MSN when challenged with EDTA (1 mM) at 37 °C for 1 week.
In Vivo Long-Term Stability and Biodistribution Studies
With a ∼3 day half-life, 89Zr-based PET imaging holds great potential as a useful tool for long-term monitoring of the dynamic biodistribution, biodegradation, clearance pathway, and rate of nanoparticles (or mAbs) in vivo. However, a major challenge still exists in developing an optimal chelator for 89Zr labeling. Although our in vitro DFO challenge and serum stability studies suggested high stability of 89Zr in both MSN and dSiO2, their ultimate stability needs to be tested in vivo for a long period of time.
To do that, healthy BALB/c mice (n = 3) were intravenously (i.v.) injected with 89Zr-MSN or 89Zr-dSiO2 and monitored for 3 weeks (∼7 t1/2 of 89Zr). Since detached free 89Zr is a well-known osteophilic cation,32 with a fast and retained uptake in bones,33 monitoring the change of bone uptake could be one of the best ways to study the in vivo stability of 89Zr-labeled nanoparticles. Our in vivo PET and maximum intensity projection images of mice injected with free 89Zr-oxalate (in phosphate-buffered saline, PBS) also confirmed high uptake of 89Zr in the bones and joints, as shown in Figure S3.
Considering that MSN with a larger (e.g., >5 nm) pore size has a faster biodegradation rate in simulated body fluid,28 in order to potentially visualize the degradation process of 89Zr-MSN using PET imaging, we used ∼90 nm sized MSN with a high surface area (710.7 m2/g) and 9–10 nm pore size (Figure 4) in the in vivo stability study (Figure 5). Normal dSiO2 with a similar particle size served as the control group.
Figure 4.

(a) TEM image of MSN with an average particle size of ∼90 nm. (b) Nitrogen adsorption–desorption isotherms and the corresponding pore size distributions of MSN. BET surface area was estimated to be 710.7 m2/g, and the average pore size was about 9–10 nm.
Figure 5.

In vivo radiostability and ex vivo biodistribution studies. (a) Schematic illustration of 89Zr-dSiO2. (b) Biodistribution study of 89Zr-dSiO2 on day 21 p.i. (c) TEM image of 89Zr-dSiO2. (d) In vivo serial coronal maximum intensity projection PET images of mice at different time points after i.v. injection of 89Zr-dSiO2. (e) TEM image of 89Zr-MSN. (f) In vivo serial coronal maximum intensity projection PET images of mice at different time points after i.v. injection of 89Zr-MSN. (g) Schematic illustration of 89Zr-MSN. (h) Biodistribution study of 89Zr-MSN on day 21 p.i.
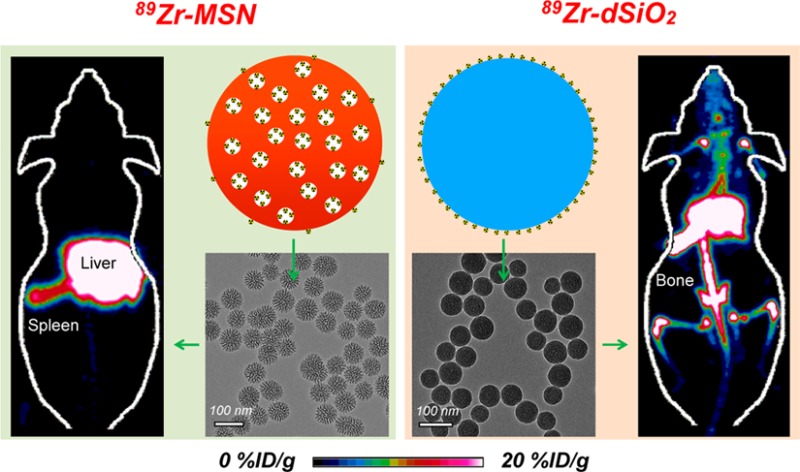
TEM images of 89Zr-dSiO2 and 89Zr-MSN (after decay of radio-activity) showed no obvious morphology change after 89Zr labeling (Figure 5c,e). The maximum intensity projection images showed dominant liver and spleen uptake with no obvious bone uptake within 3 h p.i. in both cases (Figure 5d,f). However, mice injected with 89Zr-dSiO2 started to show significant bone uptake (6.5 ± 5.9 %ID/g; n = 3) from day 1 p.i., which increased to 11.1 ± 7.5 %ID/g on day 3 p.i. (video S1), indicating that the exposed 89Zr from the dSiO2 surface can be vulnerable to attack by the endogenous (protein) ligands. Burst detachment (denoted as stage 1) of free 89Zr from dSiO2 can also be seen in Figure 6a. No further increase in bone uptake and a slow clearance of 89Zr from bone were observed over the next 18 days (denoted as stage 2, Figure 5d, Figure 6a, and Table S6), suggesting a strong binding of the remaining 89Zr on the surface of dSiO2.
Figure 6.

Quantitative region of interest analysis of the dynamic uptake change of 89Zr in bone and liver. Time–activity curves of bone (a) and liver (c) upon i.v. injection of 89Zr-dSiO2 or 89Zr-MSN into BALB/c mice over 21 days. Linear fitting of 89Zr in (b) bone during stage 1 and (d) liver during stage 2.
Surprisingly, mice injected with 89Zr-MSN showed an extremely high in vivo stability throughout 3 weeks, with less than 1 %ID/g uptake in bone on day 7 p.i. (video S2), which remains very low at 1.5 ± 0.2 %ID/g at 2 more weeks later (Figure 5h, Figure 6a, and Table S6). The much smaller (but not zero) uptake in bones for 89Zr-MSN may be attributed to the detachment of 89Zr4+ ions, which are on the outer surface and not necessarily inside the pores of the MSNs. Such ions form a small fraction of the total labeled activity (due to much higher internal surface area in each MSN29). Since the coordination between silanol groups and 89Zr4+ ions is expected to be weaker on the outer surface (due to fewer surrounding silanol groups) than inside the meso-channels, the surface-bound 89Zr4+ ions may be rendered more vulnerable to endogenous chelators. Remarkably, the detachment rate of 89Zr from MSN was found to be >20-fold slower than that of 89Zr from dSiO2 (Figure 6b), clearly indicating the vital role of meso-channels in protecting 89Zr from attack by the endogenous chelators. Although 89Zr-labeled MSN using DFO as the chelator has been reported,21 this is the longest in vivo stability study of 89Zr-labeled MSN with such high stability, which could open up a new possibility for studying the long-term fate of MSN-based nanoparticles in live animals.
In addition to the dynamic change of 89Zr uptake in bone, our region of interest (ROI) quantification also provided highly accurate information regarding the accumulation and clearance rate of 89Zr-MSN in mouse liver. Figure 6c shows an increased liver uptake of 89Zr-MSN from 29.7 ± 4.8 %ID/g (n = 3) at 0.5 h p.i. to 39.5 ± 4.5 %ID/g on day 7 p.i. For 89Zr-dSiO2, a significant reduction of 89Zr in liver, from 38.1 ± 14.5 to 27.0 ± 0.3 %ID/g due to the lower stability of 89Zr on the surface of dSiO2, was observed over the first 7 days (Table S7). Of note, such dramatic 89Zr uptake change did not correctly reflect the real distribution change of dSiO2 in the liver (i.e., dSiO2 was not cleared from the mouse liver over this time period, but the radio-isotope 89Zr was) and was the main reason for increased 89Zr bone uptake in mice injected with 89Zr-dSiO2, as shown in Figure 5d and 6a (stage 1).
The hepatic clearance of 89Zr-MSN was also observed with a gradual decrease in liver uptake to 34.2 ± 3.3 %ID/g on day 21 p.i. (Figure 6c and Table S7). Interestingly, we found the same hepatic clearance rate for both 89Zr-dSiO2 and 89Zr-MSN from day 3 to day 21, with their linear fitting slopes both estimated to be −0.01 (Figure 6d). Dominant liver/spleen uptake of 89Zr-MSN and 89Zr-dSiO2 (as expected for i.v. injected nanoparticles) and significantly higher bone uptake of free 89Zr detached from the dSiO2 surface (but not from MSN) were further confirmed in the ex vivo biodistribution studies on day 21 p.i. (Figure 5b,h and Table S8).
Renal clearance of porous silicon (130–180 nm in diameter) after degradation in vivo has been previously reported.34 MSN with an average pore size larger than 5 nm has been reported to have a decent (within 3 days) degradation rate in vitro.28 Neither bladder nor kidney uptake of 89Zr-MSN (average pore size: 9–10 nm, Figure 4 and Figure 5f) fragments was observed in this study. Without PEGylation, 89Zr-MSN with a highly negatively charged surface was expected to be accumulated in liver and spleen shortly.
Although bare (unmodified/non-PEGylated) silica nanoparticles were used here in order to better study the chelator-free radio-labeling mechanism and in vivo long-term stability, suitable surface functionalization is needed before they can be considered as drug delivery systems. Our pilot studies have successfully demonstrated the in vivo tumor active targeting of porous-silica-based nanoparticles (e.g., hollow mesoporous silica nanoparticle or yolk/shell structured nanoparticle), by modifying nanoparticles with polyethylene glycol (PEG) (molecular weight: 5 kDa) and targeting ligands (e.g., TRC105, an anti-CD105 antibody; or YY146, an anti-CD146 antibody) (unpublished data).
We also believe the use of intrinsic deprotonated silanol groups from MSN (or other mesoporous silica-coated nanoparticles) for chelator-free radio-labeling may be applicable to other oxophilic radio-metals, such as titanium-45 (45Ti, t1/2 = 184.8 m), scandium-44 (44Sc, t1/2 = 3.97 h), indium-111 (111In, t1/2 = 2.8 d), yttrium-90 (90Y, t1/2 = 64.0 h), to name a few. Considering that PEGylated MSN can only circulate in the blood for a couple of hours (depending on the particle size, surface charge, and PEG density, etc.), 89Zr with a half-life greater than 3 days might not be the best isotope for short-term PET imaging studies. However, thanks to the general applicability of the as-proposed labeling mechanism, other short-lived oxophilic radio-metals (such as 45Ti and 44Sc) could be used for better matching the biological half-life of PEGylated MSN. Silica-based chelator-free 89Zr labeling can also benefit other applications, such as systematic and long-term in vivo biodegradation and biodistribution tracking of MSN (with varied particle and pore sizes), which could provide highly valuable information regarding the in vivo fate, potential toxicity, drug delivery, and chemotherapeutic efficacy of MSN in the near future.
Conclusions
In conclusion, herein we report a chelator-free technique to stably label 89Zr to MSN. Systematic studies demonstrated the vital role of deprotonated silanol groups during the labeling of 89Zr to amorphous porous and nonporous silica nanoparticles. 89Zr labeling yield was concentration- and temperature-dependent, where higher silica concentration and higher incubation temperature gave higher 89Zr labeling yield. In vitro DFO challenge and serum stability studies demonstrated the strong binding affinity of 89Zr to both MSN and dSiO2 with nearly negligible 89Zr detachment within 48 h. Remarkably, 89Zr-MSN exhibited high in vivo stability with very little bone uptake over 3 weeks. The detachment rate of 89Zr from MSN’s meso-channels was further found to be >20-fold slower than that of 89Zr from the dSiO2 surface in vivo, highlighting the vital role of meso-channels in stabilizing 89Zr inside MSN.
Considering that silica (or silicon dioxide) is “generally recognized as safe” by the Food and Drug Administration (ID Code: 14808-60-7),35 which is highly desirable for future clinical translation, and silica coating has been widely used in biomedical applications,36,37 the as-developed labeling technique might provide a simpler yet more reliable method for theranostic application of future silica-based radio-labeled nanoparticles.
Materials and Methods
89Zr Production
89Zr-oxalate was produced according to our previous procedures by the University of Wisconsin—Madison cyclotron group.38 Briefly, natural yttrium-89 (89Y) foil (250 μm, 99.9%) was irradiated with a proton beam to create 89Zr via the 89Y(p,n)89Zr reaction by using a 16 MeV GE PETtrace cyclotron. After isotope separation and purification, 89Zr-oxalate was obtained, which has a specific activity of >20 GBq/μmol of Zr.
Synthesis of Dense Silica Nanoparticles (dSiO2)
Uniform ∼90 nm sized dSiO2 was prepared using a modified Stöber method.39 In a typical synthesis, 35.7 mL of absolute ethanol was mixed with 5 mL of water and 0.8 mL of ammonia and stirred for 5–10 min at room temperature. One milliliter of TEOS was then added, and the mixture was allowed to react at room temperature for 1 h. Afterward, dSiO2 nanoparticles were collected by centrifugation (at 12 500g for 10 min), washed with water/ethanol three times, and resuspended in 20 mL of water before use.
Synthesis of Mesoporous Silica Nanoparticles
A previously reported biphase stratification approach with slight modifications was used for the synthesis of MSN with varied pore sizes.28 In a typical synthesis of ∼150 nm sized MSN with 4–5 nm pore size, 24 mL of hexadecyltrimethylammonium chloride (CTAC, 25 wt %) solution and 0.18 g of triethylamine (TEA) were added to 36 mL of water and stirred gently at 60 °C for 3 h in a 100 mL round-bottom flask. Twenty milliliters of (20% v/v) TEOS in cyclohexane was carefully added to the water–CTAC–TEA solution and kept at 60 °C in a water bath for 12 h (stirring rate was set to 125 rpm). Afterward, milky white samples were collected by centrifugation (at 12 500g for 10 min), which were subjected to the CTAC removal process by stirring in 1 wt % NaCl/methanol solution three times (24 h/time). Complete removal of CTAC was demonstrated by using Fourier transform infrared spectroscopy.
For the synthesis of MSN with 9–10 nm pore size, the upper organic layer was replaced with 20 mL of (5% v/v) TEOS in cyclohexane. The mixture was reacted at 60 °C in a water bath for 60 h before the CTAC removal process.
Chelator-Free 89Zr Labeling Using dSiO2
For 89Zr labeling, 250 μL of dSiO2 (concentration range: 2 mg/mL to 2 × 10–4 mg/mL) in HEPES buffer (pH 7.5, 0.1 M) was directly mixed with 3 mCi (or 111 MBq) of 89Zr-oxalate at 37 °C (or 75 °C). The final pH value of the mixture was adjusted to 7–8 by using 2 M Na2CO3. 89Zr labeling yield was monitored and quantified at different time points (from 30 min to 2 h) by using thin layer chromatography (TLC). 89Zr-dSiO2 could be easily collected by centrifugation (at 21 000g for 10 min).
Chelator-Free 89Zr Labeling Using MSN
For 89Zr labeling, 250 μL of MSN (concentration range: 2 mg/mL to 2 × 10–4 mg/mL) in HEPES buffer (pH 7.5, 0.1 M) was simply mixed with 3 mCi (or 111 MBq) of 89Zr-oxalate at 37 °C (or 75 °C). The final pH value of the mixture was adjusted to 7–8 by using 2 M Na2CO3. 89Zr labeling yield was monitored and quantified at different time points (from 30 min to 2 h) by using radio-TLC. 89Zr-MSN could be easily collected by centrifugation (at 21 000g for 10 min).
DFO Challenge Study
To demonstrate the stability of 89Zr in MSN and dSiO2, DFO-Bz-NCS (p-isothiocyanatobenzyl desferrioxamine B) with varied concentrations was added into 250 μL of 89Zr-MSN (∼200 μCi) or 89Zr-dSiO2 (∼200 μCi) HEPES solution (pH 7–8) at 37 °C under constant shaking (550 rpm) for 48 h. The final DFO concentrations were fixed to be 5, 0.5, and 0.05 mM. At each time point, 10 μL of mixture was taken out and resuspended in 200 μL of HEPES. A 100 kDa filter was used to separate potential 89Zr-DFO from 89Zr-labeled nanoparticles. The 89Zr-DFO radio-activity was measured by using a gamma counter (PerkinElmer).
EDTA Challenge Study
To further demonstrate the radio-stability of 89Zr in MSN, as-synthesized 89Zr-MSN was soaked in 1 mM EDTA and kept at 37 °C under constant shaking (550 rpm) for 1 week. Both free 89Zr and 89Zr-MSN were monitored and quantified at different time points (from day 1 to day 7) by using radio-TLC. 89Zr-EDTA migrates along the iTLC strip, while 89Zr-MSN remains at the origin.
Loading MSN and 89Zr-MSN with Hydrophilic Anticancer Drugs
Doxorubicin hydrochloride (DOX) was selected as the model drug to test the drug loading capacity of MSN before and after 89Zr labeling. MSN (size: 150 nm; surface area: 581.5 m2/g; pore volume: 1.31 cm3/g) with a known mass (0.5 mg) was resuspended in 1 mL (0.5 mg/mL) of DOX-PBS solution (total amount of DOX was 0.5 mg). The mixture was kept under constant shaking for 48 h at room temperature. Afterward, MSN(DOX) was collected by centrifugation (at 21 000g for 10 min) and washed with PBS three times. All DOX in the supernatant was carefully collected and quantified based on the DOX (in PBS solution) standard curve. The loading capacity was calculated by the following equation:
To load DOX into 89Zr-MSN, the same amount of MSN (0.5 mg) was first labeled with 89Zr as described previously and then went through the same drug loading process as MSN(DOX). More details about the drug loading can be found in Table S4.
In Vitro Serum Stability Study
To study the in vitro serum stability of both 89Zr-dSiO2 and 89Zr-MSN, 50 μL of 89Zr-MSN (or 89Zr-dSiO2) was mixed with 50 μL of 2x whole mouse serum at 37 °C under constant shaking (550 rpm) for 48 h. At each time point, a small fraction of the mixture (15 μL) was collected, resuspended in 100 μL of PBS, and purified by using a 100 kDa filter. The 89Zr activity of the filtrate and that retained in the filter was measured by using a gamma counter.
In Vivo PET Imaging of 89Zr-dSiO2 and 89Zr-MSN
For in vivo PET imaging, 150 μL (∼400 μCi or 14.8 MBq) of 89Zr-MSN (or 89Zr-dSiO2) in water was i.v. injected into healthy BALB/c mice (n = 3). PET scans at various time points p.i., from 0.5 h to 21 days, were performed by using a microPET/microCT Inveon rodent model scanner (Siemens Medical Solutions USA, Inc.). The images were reconstructed using a maximum a posteriori algorithm, with no scatter correction. ROI analysis of each PET scan was performed using vendor software (Inveon Research Workplace [IRW]) on decay-corrected whole-body images, as we described previously,40 to calculate the percentage injected dose per gram of tissue (%ID/g) values in mouse liver and bone.
Ex Vivo Biodistribution of 89Zr-dSiO2 and 89Zr-MSN
Biodistribution studies were also carried out to confirm that the quantitative uptake values based on PET imaging truly represented the radio-activity distribution in mice. After the last in vivo PET imaging on day 21 p.i., all major organs/tissues were collected and wet-weighed. The radio-activity in the tissue was measured using a gamma counter and presented as %ID/g.
Characterizations
Transmission electron microscopy images were obtained on a FEI T12 microscope operated at an accelerating voltage of 120 kV. Standard TEM samples were prepared by dropping dilute products onto carbon-coated copper grids. Nitrogen adsorption–desorption isotherms were measured at 77 K using a Quantachrome Autosorb-1 system. The samples were pretreated under a vacuum at 393 K for 24 h. Surface areas were determined using the BET method. Pore size distribution data were collected by the Barrett–Joyner–Halenda method of the desorption branch of the isotherm. ζ-Potential analysis was performed on Nano-Zetasizer (Malvern Instruments Ltd.).
Acknowledgments
This work is supported, in part, by the University of Wisconsin—Madison, the National Institutes of Health (NIBIB/NCI 1R01CA169365, P30CA014520, and 5T32GM08349), the National Science Foundation (DGE-1256259), the Department of Defense (W81XWH-11-1-0644), and the American Cancer Society (125246-RSG-13-099-01-CCE).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b00526.
Video S1 (AVI)
Video S2 (AVI)
Temperature- and pH-dependent 89Zr labeling to MSN, maximum intensity projection images of mice injected with free 89Zr-oxalate, labeling yields of 89Zr-MSN and 89Zr-dSiO2, ROI quantification data of 89Zr-dSiO2 and 89Zr-MSN uptake in bone and liver, ex vivo biodistribution data of 89Zr-dSiO2 and 89Zr-MSN on Day 21 postinjection, calculation of number of silanol groups per MSN particle (PDF)
Author Contributions
⊥ F.C. and S.G. contributed equally to this work
The authors declare no competing financial interest.
Supplementary Material
References
- Hong H.; Chen F.; Cai W. Pharmacokinetic Issues of Imaging with Nanoparticles: Focusing on Carbon Nanotubes and Quantum Dots. Mol. Imaging Biol. 2013, 15, 507–520 10.1007/s11307-013-0648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakor A. S.; Gambhir S. S. Nanooncology: The Future of Cancer Diagnosis and Therapy. Ca-Cancer J. Clin. 2013, 63, 395–418 10.3322/caac.21199. [DOI] [PubMed] [Google Scholar]
- Lee S. B.; Kim H. L.; Jeong H. J.; Lim S. T.; Sohn M. H.; Kim D. W. Mesoporous Silica Nanoparticle Pretargeting for PET Imaging Based on a Rapid Bioorthogonal Reaction in a Living Body. Angew. Chem., Int. Ed. 2013, 52, 10549–10552 10.1002/anie.201304026. [DOI] [PubMed] [Google Scholar]
- Kim D. W. Bioorthogonal Click Chemistry for Fluorine-18 Labeling Protocols under Physiologically Friendly Reaction Condition. J. Fluorine Chem. 2015, 174, 142–147 10.1016/j.jfluchem.2014.11.009. [DOI] [Google Scholar]
- Liu Z.; Cai W.; He L.; Nakayama N.; Chen K.; Sun X.; Chen X.; Dai H. In Vivo Biodistribution and Highly Efficient Tumour Targeting of Carbon Nanotubes in Mice. Nat. Nanotechnol. 2007, 2, 47–52 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- Cai W.; Chen K.; Li Z. B.; Gambhir S. S.; Chen X. Dual-Function Probe for PET and Near-Infrared Fluorescence Imaging of Tumor Vasculature. J. Nucl. Med. 2007, 48, 1862–1870 10.2967/jnumed.107.043216. [DOI] [PubMed] [Google Scholar]
- Yang X.; Hong H.; Grailer J. J.; Rowland I. J.; Javadi A.; Hurley S. A.; Xiao Y.; Yang Y.; Zhang Y.; Nickles R. J.; et al. cRGD-Functionalized, DOX-Conjugated, and 64Cu-Labeled Superparamagnetic Iron Oxide Nanoparticles for Targeted Anticancer Drug Delivery and PET/MR Imaging. Biomaterials 2011, 32, 4151–4160 10.1016/j.biomaterials.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Hong H.; Zhang Y.; Valdovinos H. F.; Shi S.; Kwon G. S.; Theuer C. P.; Barnhart T. E.; Cai W. In Vivo Tumor Targeting and Image-Guided Drug Delivery with Antibody-Conjugated, Radiolabeled Mesoporous Silica Nanoparticles. ACS Nano 2013, 7, 9027–9039 10.1021/nn403617j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Ehlerding E. B.; Cai W. Theranostic Nanoparticles. J. Nucl. Med. 2014, 55, 1919–1922 10.2967/jnumed.114.146019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Zhang Y.; Sun J.; Cai W. Molecular Imaging and Therapy of Cancer with Radiolabeled Nanoparticles. Nano Today 2009, 4, 399–413 10.1016/j.nantod.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu Y.; Luehmann H.; Xia X.; Brown P.; Jarreau C.; Welch M.; Xia Y. Evaluating the Pharmacokinetics and in Vivo Cancer Targeting Capability of Au Nanocages by Positron Emission Tomography Imaging. ACS Nano 2012, 6, 5880–5888 10.1021/nn300464r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel S.; Chen F.; Ehlerding E. B.; Cai W. Intrinsically Radiolabeled Nanoparticles: An Emerging Paradigm. Small 2014, 10, 3825–3830 10.1002/smll.201401048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L.; Yang K.; Chen Q.; Liu Z. Organic Stealth Nanoparticles for Highly Effective in Vivo Near-Infrared Photothermal Therapy of Cancer. ACS Nano 2012, 6, 5605–5613 10.1021/nn301539m. [DOI] [PubMed] [Google Scholar]
- Chen F.; Ellison P. A.; Lewis C. M.; Hong H.; Zhang Y.; Shi S.; Hernandez R.; Meyerand M. E.; Barnhart T. E.; Cai W. Chelator-Free Synthesis of a Dual-Modality PET/MRI Agent. Angew. Chem., Int. Ed. 2013, 52, 13319–13323 10.1002/anie.201306306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty R.; Valdovinos H. F.; Chen F.; Lewis C. M.; Ellison P. A.; Luo H.; Meyerand M. E.; Nickles R. J.; Cai W. Intrinsically Germanium-69-Labeled Iron Oxide Nanoparticles: Synthesis and in-Vivo Dual-Modality PET/MR Imaging. Adv. Mater. 2014, 26, 5119–5123 10.1002/adma.201401372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Zhang R.; Huang M.; Lu W.; Song S.; Melancon M. P.; Tian M.; Liang D.; Li C. A Chelator-Free Multifunctional [64Cu]CuS Nanoparticle Platform for Simultaneous Micro-PET/CT Imaging and Photothermal Ablation Therapy. J. Am. Chem. Soc. 2010, 132, 15351–15358 10.1021/ja106855m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Yu M.; Sun Y.; Zhang X.; Zhu X.; Wu Z.; Wu D.; Li F. Fluorine-18-Labeled Gd3+/Yb3+/Er3+ Co-Doped NaYF4 Nanophosphors for Multimodality PET/MR/UCL Imaging. Biomaterials 2011, 32, 1148–1156 10.1016/j.biomaterials.2010.09.071. [DOI] [PubMed] [Google Scholar]
- Jauregui-Osoro M.; Williamson P. A.; Glaria A.; Sunassee K.; Charoenphun P.; Green M. A.; Mullen G. E.; Blower P. J. Biocompatible Inorganic Nanoparticles for [18F]-Fluoride Binding with Applications in PET Imaging. Dalton Trans. 2011, 40, 6226–6237 10.1039/c0dt01618g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Hong H.; Cai W. PET Tracers Based on Zirconium-89. Curr. Radiopharm. 2011, 4, 131–139 10.2174/1874471011104020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchun S.; Dass C. R.; Sriamornsak P. Targeted Therapy for Cancer Using pH-Responsive Nanocarrier Systems. Life Sci. 2012, 90, 381–387 10.1016/j.lfs.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Miller L.; Winter G.; Baur B.; Witulla B.; Solbach C.; Reske S.; Linden M. Synthesis, Characterization, and Biodistribution of Multiple 89Zr-Labeled Pore-Expanded Mesoporous Silica Nanoparticles for Pet. Nanoscale 2014, 6, 4928–4935 10.1039/c3nr06800e. [DOI] [PubMed] [Google Scholar]
- Chang A. J.; Desilva R.; Jain S.; Lears K.; Rogers B.; Lapi S. 89Zr-Radiolabeled Trastuzumab Imaging in Orthotopic and Metastatic Breast Tumors. Pharmaceuticals 2012, 5, 79–93 10.3390/ph5010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts H. J.; Dubois L.; Perk L.; Vermaelen P.; van Dongen G. A.; Wouters B. G.; Lambin P. Disparity between in Vivo EGFR Expression and 89Zr-Labeled Cetuximab Uptake Assessed with PET. J. Nucl. Med. 2009, 50, 123–131 10.2967/jnumed.108.054312. [DOI] [PubMed] [Google Scholar]
- Tinianow J. N.; Gill H. S.; Ogasawara A.; Flores J. E.; Vanderbilt A. N.; Luis E.; Vandlen R.; Darwish M.; Junutula J. R.; Williams S. P.; et al. Site-Specifically 89Zr-Labeled Monoclonal Antibodies for Immunopet. Nucl. Med. Biol. 2010, 37, 289–297 10.1016/j.nucmedbio.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Holland J. P.; Divilov V.; Bander N. H.; Smith-Jones P. M.; Larson S. M.; Lewis J. S. 89Zr-DFO-J591 for Immunopet of Prostate-Specific Membrane Antigen Expression in Vivo. J. Nucl. Med. 2010, 51, 1293–1300 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Chen H.; Shi J. In Vivo Bio-Safety Evaluations and Diagnostic/Therapeutic Applications of Chemically Designed Mesoporous Silica Nanoparticles. Adv. Mater. 2013, 25, 3144–3176 10.1002/adma.201205292. [DOI] [PubMed] [Google Scholar]
- Zhuravlev L. T. The Surface Chemistry of Amorphous Silica. Zhuravlev Model. Colloids Surf., A 2000, 173, 1–38 10.1016/S0927-7757(00)00556-2. [DOI] [Google Scholar]
- Shen D.; Yang J.; Li X.; Zhou L.; Zhang R.; Li W.; Chen L.; Wang R.; Zhang F.; Zhao D. Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres. Nano Lett. 2014, 14, 923–932 10.1021/nl404316v. [DOI] [PubMed] [Google Scholar]
- Slowing I. I.; Wu C. W.; Vivero-Escoto J. L.; Lin V. S. Mesoporous Silica Nanoparticles for Reducing Hemolytic Activity Towards Mammalian Red Blood Cells. Small 2009, 5, 57–62 10.1002/smll.200800926. [DOI] [PubMed] [Google Scholar]
- Lin Y. S.; Haynes C. L. Impacts of Mesoporous Silica Nanoparticle Size, Pore Ordering, and Pore Integrity on Hemolytic Activity. J. Am. Chem. Soc. 2010, 132, 4834–4842 10.1021/ja910846q. [DOI] [PubMed] [Google Scholar]
- Boros E.; Bowen A. M.; Josephson L.; Vasdev N.; Holland J. P. Chelate-Free Metal Ion Binding and Heat-Induced Radiolabeling of Iron Oxide Nanoparticles. Chem. Sci. 2015, 6, 225–236 10.1039/C4SC02778G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijs W. E.; Haisma H. J.; Klok R. P.; van Gog F. B.; Kievit E.; Pinedo H. M.; Herscheid J. D. Zirconium-Labeled Monoclonal Antibodies and Their Distribution in Tumor-Bearing Nude Mice. J. Nucl. Med. 1997, 38, 112–118. [PubMed] [Google Scholar]
- Abou D. S.; Ku T.; Smith-Jones P. M. In Vivo Biodistribution and Accumulation of 89Zr in Mice. Nucl. Med. Biol. 2011, 38, 675–681 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H.; Gu L.; von Maltzahn G.; Ruoslahti E.; Bhatia S. N.; Sailor M. J. Biodegradable Luminescent Porous Silicon Nanoparticles for in Vivo Applications. Nat. Mater. 2009, 8, 331–336 10.1038/nmat2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/ (accessed July 22, 2015).
- Wang K.; He X.; Yang X.; Shi H. Functionalized Silica Nanoparticles: A Platform for Fluorescence Imaging at the Cell and Small Animal Levels. Acc. Chem. Res. 2013, 46, 1367–1376 10.1021/ar3001525. [DOI] [PubMed] [Google Scholar]
- Tallury P.; Payton K.; Santra S. Silica-Based Multimodal/Multifunctional Nanoparticles for Bioimaging and Biosensing Applications. Nanomedicine (London, U. K.) 2008, 3, 579–592 10.2217/17435889.3.4.579. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Hong H.; Severin G. W.; Engle J. W.; Yang Y.; Goel S.; Nathanson A. J.; Liu G.; Nickles R. J.; Leigh B. R.; et al. Immunopet and Near-Infrared Fluorescence Imaging of CD105 Expression Using a Monoclonal Antibody Dual-Labeled with 89Zr and IRdye 800CW. Am. J. Transl. Res. 2012, 4, 333–346. [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Chen H.; Guo L.; He Q.; Chen F.; Zhou J.; Feng J.; Shi J. Hollow/Rattle-Type Mesoporous Nanostructures by a Structural Difference-Based Selective Etching Strategy. ACS Nano 2010, 4, 529–539 10.1021/nn901398j. [DOI] [PubMed] [Google Scholar]
- Hong H.; Yang Y.; Zhang Y.; Engle J. W.; Barnhart T. E.; Nickles R. J.; Leigh B. R.; Cai W. Positron Emission Tomography Imaging of CD105 Expression During Tumor Angiogenesis. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1335–1343 10.1007/s00259-011-1765-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.