

Abstract
Clostridium difficile is an anaerobic, Gram-positive, and spore-forming bacterium that is the leading worldwide infective cause of hospital-acquired and antibiotic-associated diarrhea. Several studies have reported associations between humoral immunity and the clinical course of C. difficile infection (CDI). Host humoral immune responses are determined using conventional enzyme-linked immunosorbent assay (ELISA) techniques. Herein, we report the first use of a novel protein microarray assay to determine systemic IgG antibody responses against a panel of highly purified C. difficile-specific antigens, including native toxins A and B (TcdA and TcdB, respectively), recombinant fragments of toxins A and B (TxA4 and TxB4, respectively), ribotype-specific surface layer proteins (SLPs; 001, 002, 027), and control proteins (tetanus toxoid and Candida albicans). Microarrays were probed with sera from a total of 327 individuals with CDI, cystic fibrosis without diarrhea, and healthy controls. For all antigens, precision profiles demonstrated <10% coefficient of variation (CV). Significant correlation was observed between microarray and ELISA in the quantification of antitoxin A and antitoxin B IgG. These results indicate that microarray is a suitable assay for defining humoral immune responses to C. difficile protein antigens and may have potential advantages in throughput, convenience, and cost.
INTRODUCTION
Clostridium difficile is the leading worldwide infective cause of hospital-acquired and antibiotic-associated diarrhea, imposing a considerable financial burden on health service providers in both Europe and the United States (1–3). Infection causes a spectrum of clinical presentations ranging from an asymptomatic carrier state to severe fulminant colitis and death (4). Following successful treatment, an estimated 20% to 30% of patients with primary C. difficile infection (CDI) develop recurrence of symptoms, caused by either relapse of the original infection or reinfection with a new strain (5).
This anaerobic and spore-forming bacterium exerts its major pathological effects through two proinflammatory and cytotoxic protein exotoxins, TcdA (toxin A) and TcdB (toxin B) (6). Nontoxin virulence factors, such as surface layer proteins (SLPs) and cell wall proteins, have also been described and may play a role in disease expression (7–9).
The majority of healthy adults have detectable antibodies to C. difficile TcdA and TcdB in their sera that are thought to arise from colonization in infancy or from repeated exposure to C. difficile in adulthood from the environment (10, 11). Several clinical studies suggest that adaptive humoral immune responses, in particular to TcdA and TcdB, may influence clinical outcomes of CDI (12). Most notably, a landmark study in 2000 reported that a low IgG titer to TcdA, but not TcdB, at the time of infection is associated with development of symptomatic disease (13). More recently, the same group demonstrated an association between median IgG titers to TcdA and 30-day all-cause mortality (14). Several reports have also assessed antibody responses following infection and have shown protection against recurrence associated with antibody responses to TcdA, TcdB, and several nontoxin antigens (Cwp66, Cwp84, FliC, FliD, and the surface layer proteins) (15–18). In contrast, other studies have reported that humoral immune responses did not influence the clinical course of CDI (18–21). These conflicting reports may be attributed to heterogeneity in study design and subject populations. Although the role of humoral immunity remains incompletely understood, vaccination strategies using inactivated toxins or recombinant toxin fragments are currently the subject of intense investigation (22, 23). More recently, the possibility of adding other vaccine targets, such as surface-associated proteins and polysaccharides, to toxin combinations is gaining traction and could be of added value in the prevention of C. difficile colonization and disease transmission (22, 23). It is likely that the design of these next-generation multicomponent vaccines targeting colonization, persistence, and toxin production will stimulate the requirement for evaluating humoral immune responses to multiple antigens.
The enzyme-linked immunosorbent assay (ELISA) is the traditional method of accurately quantifying antibodies with different specificities in epidemiologic research and vaccine development as well as in the diagnosis of allergies and autoimmune and infectious diseases. We and other groups (13, 15, 18–20) have independently developed and used a traditional standardized ELISA format for the purposes of determining human specific IgG responses against C. difficile antigens (toxins). However, ELISA-based tests can be time consuming and require large quantities of both sample and reagents, thus limiting their potential for high-throughput use (23, 24). ELISA offers only monoplex data, or results of a single protein per assay (typically TcdA, TcdB, or SLPs) and from a single C. difficile strain. Additional concerns include the lack of a uniform standard for calibration purposes and, thus, no generally accepted way of expressing ELISA units as well as poor consistency between protocols and reagents, including, notably, the quality and source of antigens. Moreover, the linear region of the dynamic range is highly platform dependent.
Recently, protein microarrays, a miniaturized version of a sandwich ELISA, have evolved as a promising tool for quantifying specific antibodies directed against various microbial antigens in human sera, and they may be an attractive alternative to conventional ELISA in determining antigen-specific antibody responses (25–31). Microarray assays have potentially important advantages compared with standard ELISA formats. These include a much increased capacity for multiplexing detection of a range of specific antibodies due to the flexibility of array printing of multiple antigens per array over a single protein; vastly reduced requirements for antigens, serum, and reagents; increased assay robustness due to increased technical replication within each assay; multiple internal quality control measures; and improved quality control capabilities. The unique capabilities of microarray, including parallelism, high-throughput, and miniaturization, are ideally suited to comprehensive investigation of the humoral immune response to the entire proteome of an infectious agent consisting of thousands of potential antigens in a patient-specific manner (29). Microarray technology can also be applied to the development of improved serodiagnostic tests, discovery of subunit vaccine antigen candidates, epidemiological research, and vaccine development, in addition to providing novel insights into infectious disease and the immune system (29).
We have developed and validated a novel customized microarray platform that enables the simultaneous quantification of systemic IgG immune responses to a 7-plex panel of highly purified C. difficile-specific virulence factors, including whole toxins A and B, recombinant fragments of toxin A (TxA4) and toxin B (TxB4), ribotype-specific surface layer proteins, and suitable control proteins. We compared the performance of the microarray technique with a conventional ELISA using an established panel of sera.
MATERIALS AND METHODS
Microbial proteins and serum samples.
Highly purified whole toxins A and B (toxinotype 0, strain VPI 10463, ribotype 087) in addition to recombinant toxin fragments TxA4 and TxB4 (comprising central and receptor binding domains and both based on toxinotype 0 sequences) were obtained from Public Health England, United Kingdom (provided by Clifford Shone). Purified PCR ribotype-specific native whole SLPs (001, 002, 027) were provided by Dublin City University, Eire (provided by Christine Loscher). Positive controls incorporated on each plate included tetanus toxoid and lysates from Candida albicans containing the cytoplasm and cell wall. Negative controls included spotted printing buffer (phosphate-buffered saline [PBS] Trehalose Tween) and no serum (blank) on each array.
Banked sera from adult patients with CDI (n = 150; median age, 67 years [range, 19 to 98 years]), a group of patients with cystic fibrosis (CF) without diarrhea (n = 17, 2 of which patients were found to be asymptomatic carriers; median age, 28 years [range,19 to 49 years]), and healthy controls (n = 67; median age, 36 years [range, 22 to 65 years]) were used to investigate the ability of the microarray assay to detect the presence or absence of IgG directed against C. difficile microbial and control antigens. Adult healthy donors were recruited from within the hospital and university workforce setting. All the patients in the CDI group had diarrhea (defined as a change in bowel habit with 3 or more unformed stools per day for at least 48 h) and positive stool C. difficile toxin test. Asymptomatic carriers were defined as those who did not have diarrhea but had a positive stool culture for C. difficile. The diagnosis of CF had previously been made on the basis of a positive sweat test and/or demonstration of 2 known CF mutations and typical clinical features of disease (without a history of CDI). All subjects provided written informed consent under approvals granted by the Nottingham Research Ethics Committee.
Preparation and processing of arrayed antigens.
Microbial antigens were diluted to 200 μg/ml in printing buffer (PBS Trehalose Tween) in a 384-well plate (Genetix) and spotted in quadruplicate in a 16 × 16 array format onto poly-l-lysine-coated glass slides (Electron Microscopy Sciences) using a BioRobotics MicroGrid II arrayer (MicroGrid 610, Digilab, Malborough, MA, USA) in addition to 15 human serial IgG dilutions (range, 50 μg/ml to 3.05 ng/ml) to create a calibration curve. The slides were blocked with 5% bovine serum albumin (BSA) diluted in PBS-Tween (PBST; PBS containing 0.05% Tween 20) wash buffer for 1 h at room temperature with shaking. After washing 5 times for 3 min each with PBST, all slides were incubated with sera diluted 1:500 in antibody diluent (Dako) for 1 h. Following washing, the slides were incubated with biotinylated antihuman IgG (Vector Laboratories) diluted 1:20,000 in antibody diluent for 1 h. After further washing, slides were incubated with Streptavidin Cy5 (eBioscience) diluted 1:2,000 in 5% BSA for 15 min. After a final wash with PBST followed by distilled water, slides were dried by centrifugation at 500 × g for 4 min. Unless stated otherwise, all wash steps were carried out at room temperature with shaking. Slides were scanned using a GenePix 4200AL scanner, a photomultiplier tube with 450 nm, and 100% power. The resultant tagged image file format (TIFF) images were processed with Axon Genepix Pro-6 microarray image analysis software (Molecular Services, Inc.) to obtain fluorescence data for each feature and generate GenePix results format (gpr) files. Protein signals were finally determined with background subtraction using the RPPanalyzer, a module within the R statistical language on CRAN (http://cran.r-project.org/) (32).
Statistical analysis.
Antibody levels were calculated using GraphPad Prism 6.0 software. As data collected for antibody measurements were not normally distributed, nonparametric tests were employed, with medians and ranges calculated. For the comparison of multiple groups, the Kruskal-Wallis one-way analysis of variance was used with Dunn's posttest. Correlation was evaluated using the Spearman rank correlation coefficient test. P values of <0.05 were considered to represent statistically significant differences.
RESULTS
Quality control measures.
Internal quality control (QC) measures on each array were devised to support interassay normalization, assay performance, and data acquisition machine performance monitoring. These measures included the addition of a replicated serial dilution of human IgG to verify the function of the detection system and provide a standard curve of human IgG against which antibody responses could be calibrated. Antigens from 2 known human pathogens (tetanus toxoid and Candida albicans), of which the majority of normal individuals would be expected to have some existing protective antibody response, were incorporated onto each array. These positive-control antigens were examined for each array as an indicator of sample integrity. Figure 1 shows a plot of the responses seen in 327 serum samples for each of the 2 control antigens. Strong responses are seen to tetanus toxoid and Candida albicans. Negative controls were also incorporated onto each array as a further internal QC measure.
FIG 1.
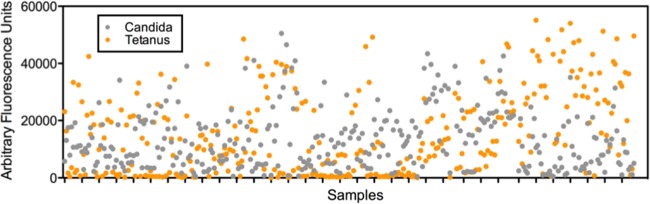
Testing sample integrity of all serum samples using two positive-control antigens: tetanus toxoid and Candida albicans antigen. Strong responses are seen to both antigens from all the samples.
Microarray intraassay and interassay precision.
Microarray intraassay and interassay variabilities were calculated using the sera of 7 patients. Identical samples were assayed on each of 2 slides at 2 independent time points. All antigens were spotted in replicates of 5 on each array. In the case of intraassay variation, all 7 test and 2 control antigens fell within acceptable limits of precision (coefficient of variation [CV] <10%) (toxin A, 7.76%; toxin B, 6.39%; SLP001, 7.44%; SLP002, 5.19%; SLP027, 7.64%; TxA4, 7.03%; TxB4, 3.71%; tetanus, 4.21%; Candida albicans, 8.28%). The interassay coefficient of variation for each antigen was calculated as 7.76%, 6.39%, 7.44%, 5.19%, 7.64%, 7.03%, 3.71%, 4.21%, and 8.26%, respectively.
Correlation between microarray and ELISA results.
Due to the lack of validated and commercially available quantitative and standardized IgG ELISAs targeting toxins A and B or other C. difficile-associated antigens, we compared specific IgG antitoxin A and antitoxin B antibody measurements generated by microarray versus previously obtained in-house indirect ELISA readings using the same patient test sera (21). The Spearman correlation coefficient was used to assess the level of agreement between the two platforms, and results are visually represented in Fig. 2. When comparing the microarray performance with the in-house ELISAs, a good correlation coefficient was observed for toxin A (r = 0.7051; P < 0.0001), with a moderately good correlation for toxin B (r = 0.5809; P < 0.0001).
FIG 2.
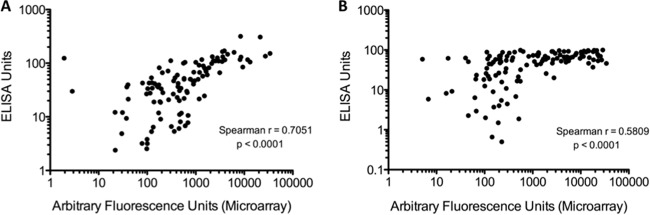
Correlation between microarray and enzyme-linked immunosorbent assay (ELISA) IgG anti-toxin A (A) and IgG anti-toxin B (B) antibody levels in patients with C. difficile infection and in patients with cystic fibrosis without a history of diarrhea. Each dot represents a serum sample from an individual patient. Spearman correlation coefficient tests revealed significant agreement between both assay results.
Sensitivity and specificity.
Sensitivity and specificity of individual and panels of antibody response to C. difficile-specific antigens were calculated for both the CDI and CF groups using selected age-matched samples and the same panel of 7 antigens (toxin A, toxin B, SLP001, SLP002, SLP027, TxA4, and TxB4) based upon the same cutoff 95th percentile of the control samples. CF samples had a sensitivity and specificity of 75% and 100%, respectively, while the sensitivity and specificity for CDI were 25% and 100%, respectively.
Serum antibody reactivity profiles using microarray.
A total of 327 serum samples were tested by microarray for the presence of specific IgG antibody. The microarray assay was able to detect specific antibody responses to all C. difficile antigens, including recombinant toxin fragments tested (example response to native antigens demonstrated in Fig. 3). The signals from positive-control proteins (tetanus toxoid and Candida albicans) were similar in healthy control individuals, patients with CF, and patients with CDI. Each array also included negative controls (buffer only and no serum or blank) which gave no signal. The reactivity of these spots was routinely subtracted from all signals obtained from specific antigens. The microarrays detected significantly higher levels of specific antibodies in the CF group across all C. difficile antigens tested compared with healthy controls and with CDI sera (Fig. 4). In the CDI group, antibody responses to whole (Fig. 4A and B) and recombinant toxins A and B (Fig. 5) did not differ compared with the healthy control group, but infected patients did exhibit significantly lower anti-SLP IgG levels (all ribotypes) compared with controls and with patients with CF (Fig. 4C). No statistically significant differences were observed in specific antibody levels to any of the antigens comparing single and relapsing CDI sera (data not presented).
FIG 3.
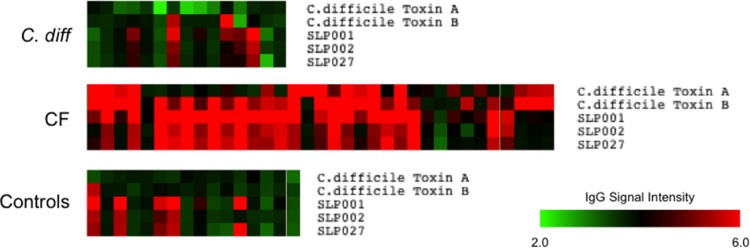
Selected serum IgG responses to C. difficile proteins on microarray. Green (low) to red (high) signal intensity heat map representing the relative IgG response to C. difficile immunoreactive antigens (native toxins A and B and ribotype-specific surface layer proteins 001, 002, 027) in different serum samples from patients infected with C. difficile, CF patients, and healthy controls.
FIG 4.

Anti-toxin A (A) and anti-toxin B (B) IgG responses in healthy controls, in patients with C. difficile infection, and in a group of patients with cystic fibrosis and no history of diarrhea. Differences between groups were calculated using the Kruskall-Wallis test followed by Dunn's posttest for multiple comparisons. Horizontal lines in each graph represent the median. (C) Patients with cystic fibrosis had significantly higher levels of specific IgG antibody levels to toxins A and B compared to healthy controls and patients with C. difficile infection. In the microarray assays, there were significantly lower anti-surface layer protein (SLP) IgG levels across all ribotypes tested (001, 002, 027) in patients with C. difficile infection compared to patients with cystic fibrosis and healthy controls (P values of ***, ≤0.001; **, ≤0.01; *, ≤0.05).
FIG 5.
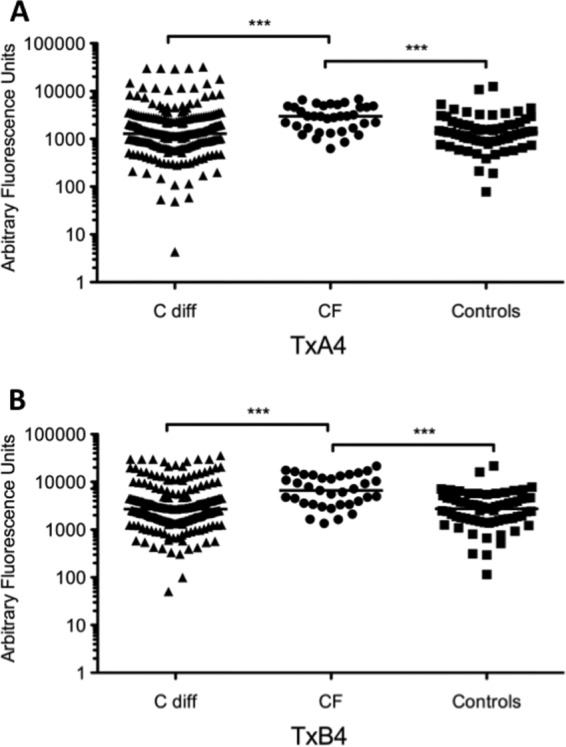
Anti-TxA4 (A) and anti-TxB4 (B) IgG responses in healthy controls, in patients with C. difficile infection, and in a CF group of patients with no history of diarrhea. Differences between groups were calculated using the Kruskall-Wallis test followed by Dunn's posttest for multiple comparisons. Horizontal lines in each graph represent the median. In the microarray assays, CF patients displayed significantly higher IgG levels against both recombinant toxin fragments compared to healthy controls and patients with C. difficile infection (***, P ≤ 0.001).
DISCUSSION
Current knowledge of the complete antigen repertoire recognized by patients during CDI is sparse, limiting a detailed interrogation of immunity and exposure and hindering preclinical vaccine development. The goal of this study was to develop, validate, and implement a novel protein microarray readout assay that allows the accurate, precise, and reproducible quantification of specific antibody responses to a selected panel of C. difficile-specific microbial antigens using a preexisting bank of test sera.
This study represents, to our knowledge, the first report of highly purified anti-SLP 001, 002, and 027 IgG responses in a large cohort of patient sera and extends the usefulness of immunoassay techniques through simultaneous examination of multiple C. difficile-specific antigens, including toxins in one immunoassay layout. We demonstrate that serum C. difficile antigen-specific IgG antibody responses can be detected using this technique and that the magnitude and breadth of response to individual specific microbial antigens differ greatly between individuals and patient groups.
While our assay achieved excellent specificity for the target panel of antigens analyzed, lower detection sensitivity was observed, particularly for the CDI group. Importantly, antibody-based serological assays are hampered by the high likelihood or prior exposure to microorganisms encountered in the environment. In this regard, all populations previously exposed to C. difficile bacterial antigens will produce seropositive responses. Nevertheless, it should be possible to enhance sensitivity for specific antibody detection through probing the microarrays with a larger bank of longitudinal (acute and convalescent) test sera and/or activated B lymphocyte supernatant samples, which secrete antigen-specific antibodies from patients with symptomatic CDI (21) and healthy controls who do not carry C. difficile in their stool. In addition, detection sensitivity may be improved by pooling several antigenic targets specific for C. difficile.
In contrast to previous studies asserting that development of symptomatic CDI may be correlated with low IgG titers to toxin A but not to toxin B, our data do not demonstrate any significant differences in IgG antitoxin A or IgG antitoxin B levels in CDI patients compared to healthy controls. Similarly, other investigators have either reported no disparities or higher serum antitoxin A IgG levels in CDI patients compared to controls (11, 19, 33). Although the carriage rate of C. difficile in the control subjects was not known, the lack of difference may have arisen due to the fact that most of the healthy control subjects in this study were recruited from a pool of hospital and university coworkers. As such, it is likely that natural asymptomatic exposure to toxigenic C. difficile was a more common occurrence and, thus, may be due to transient colonization. Furthermore, we acknowledge that patients in the CDI group were also older compared to individuals in the CF and control groups. However, other investigators have shown that serum antibody levels were not affected by age (13, 15). It will be of interest to determine whether specific qualitative and quantitative differences in T and B cell responses to C. difficile and its antigens account for a higher prevalence of CDI in older populations.
Notably, significantly higher antitoxin and anti-SLP IgG antibody concentrations in patients with CF (with no history of CDI) are likely to be due to more frequent contact with the toxins and SLPs of C. difficile which occur with colonization (especially after admission to hospital) and following antibiotic-mediated disruption of the protective resident microflora. Indeed, 2 of the patients with CF were asymptomatic carriers of C. difficile. Furthermore, this particular small cohort of CF patients also had additional risk factors for C. difficile colonization/infection, including tube feeding (n = 5) and the use of proton pump inhibitors (n = 14).
While firm conclusions cannot be drawn because of the small number of subjects studied, our present data suggest that the host's ability to mount a robust antibody response to multiple C. difficile-specific protein antigens, as seen in the CF group, may help confer protection from developing symptomatic CDI. Protection from symptomatic CDI may be a higher-order phenomenon related to patterns of antibody response as opposed to being attributable to any single antigenic target. Notably, CDI is rarely seen in CF patients despite the presence of multiple risk factors for infection, including frequent exposures to antibiotics and hospitals. Several studies have also shown that patients with CF are often asymptomatic carriers of C. difficile (34–37), with one recent report indicating that most strains carried by CF patients were nontoxigenic (77% versus 17%) (37). It is also possible that colonization with nontoxigenic C. difficile may protect against colonization with toxin-producing strains and/or that the differences in colonic mucus or the microbiome may also contribute to protection in the CF population. Understanding the role of the gut microbiota in programming the immune phenotype in the context of CF may offer a series of interactive windows that could be aligned to prevent CDI. Further detailed studies that aim to dissect the complex dialogue among the host, immune system, and intestinal microbiota are under way in a larger cohort of CF patients.
Limitations of the present study are the small sample of strain-specific bacterial proteins employed, unequal sample sizes in the different groups studies, lack of age matching, the absence of colonizing/immunizing strain information, the study of only one isotype, the lack of antibody neutralization data, and the absence of ELISA and microarray correlation data for the recombinant toxin fragments and nontoxin antigens examined. Whether or not the immunogenicity of the recombinant toxin fragments and nontoxin antigens contributes to CDI protection remains to be fully determined. We observed a lower correlation between microarray and ELISA in the toxin B assays (r = 0.58; P < 0.0001). This finding was particularly apparent at increasing IgG antitoxin B concentrations and may mean that ELISA lacks accuracy at higher specific antitoxin B IgG concentrations. Discordance between both methods could also be an effect of the low throughput of ELISA that requires the samples to be analyzed in small batches over a longer period of time; by contrast, the high-throughput array platforms permit analysis of large sample cohorts under similar experimental conditions in a much more rapid time frame, likely enhancing result reproducibility. Discrepancies between both technologies may also have arisen due to variations in the quality of sera and toxins over time, especially when new batches/different sources of toxin were tested. These results also suggest that mapping temporal changes in serological responses to C. difficile may be best undertaken using high-throughput methods, such as protein microarray.
In summary, we verify that this initial design and implementation of a protein microarray platform is well suited to identify, quantify, and compare multiple specific antigenic responses following challenge by C. difficile. Given that antigenic variation occurs between different strains, host responses may well vary according to which are the prevalent strains. High-throughput assays will be important in measuring the heterogeneity of host immune responses. Modifications of this microarray approach could be employed to expand the antigen targets to include proteins derived from multiple strains of C. difficile in addition to investigating multiple isotype specificities. The microarray platform could also be adapted to study cytokine/chemokine repertoires in response to infection or vaccination for large collections of individual patient sera. Optimized immunological marker panels are yet to be developed for predicting host responses to C. difficile. Before advancements can be made, more detailed careful studies in larger, well-defined prospective cohorts will be required before this C. difficile antigen-specific microarray assay can be used as a prognostic tool as well as for tailoring interventional strategies. Nevertheless, protein microarrays have the potential to provide a more comprehensive, antigen-specific, humoral immune response profile in vaccinated or infected humans that could find beneficial applications in large-scale seroepidemiological, longitudinal, and serosurveillance analyses.
ACKNOWLEDGMENTS
We thank Melanie Lingaya and Yirga Falcone from the NIHR Nottingham Digestive Diseases Biomedical Research Unit for help with sample storage and preparation.
M.H.W. received grant and research support and acted as a consultant for multiple diagnostic and therapeutic companies. The other authors report no potential conflicts of interest. Conflicts that the authors consider relevant to the content of the manuscript have been disclosed.
This work was supported by the NIHR Nottingham Digestive Diseases Biomedical Research Unit, University of Nottingham, Nottingham Hospitals Charity, and Nottingham University Hospitals NHS Trust Department of Research and Development.
The views expressed are those of the authors and not necessarily those of the National Health Service (NHS), the National Institute for Health Research, or the Department of Health.
REFERENCES
- 1.Davies KA, Longshaw CM, Davis GL, Bouza E, Barbur F, Barna Z, Delmée M, Fitzpatrick F, Ivanova K, Kuijper E, Macovei IS, Mentula S, Mastrantonio P, von Müller L, Oleastro M, Petinaki E, Pituch H, Norén T, Novákova E, Nyč O, Rupnik M, Schmid D, Wilcox MH. 2014. Underdiagnosis of Clostridium difficile across Europe: the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalized patients with diarrhea (EUCLID). Lancet Infect Dis 14:1208–1219. doi: 10.1016/S1473-3099(14)70991-0. [DOI] [PubMed] [Google Scholar]
- 2.Kwon JH, Olsen MA, Dubberke ER. 2015. The morbidity, mortality, and costs associated with Clostridium difficile infection. Infect Dis Clin North Am 29:123–134. doi: 10.1016/j.idc.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Burke KE, Lamont JT. 2014. Clostridium difficile infection: a worldwide disease. Gut Liver 8:1–6. doi: 10.5009/gnl.2014.8.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monaghan T, Boswell T, Mahida YR. 2008. Recent advances in Clostridium difficile-associated disease. Gut 57:850–860. doi: 10.1136/gut.2007.128157. [DOI] [PubMed] [Google Scholar]
- 5.Deshpande A, Pasupuleti V, Thota P, Pant C, Rolston DD, Hernandez AV, Donskey CJ, Fraser TG. 2015. Risk factors for recurrent Clostridium difficile infection: a systematic review and meta-analysis. Infect Control Hosp Epidemiol 36:452–460. doi: 10.1017/ice.2014.88. [DOI] [PubMed] [Google Scholar]
- 6.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 7.Fagan RP, Fairweather NF. 2014. Biogenesis and functions of bacterial S-layers. Nature Rev Microbiol 12:211–222. doi: 10.1038/nrmicro3213. [DOI] [PubMed] [Google Scholar]
- 8.Ryan A, Lynch M, Smith SM, Amu S, Nel HJ, McCoy CE, Dowling JK, Drpaer E, O'Reilly V, McCarthy C, O'Brien J, Ni Eidhin D, O'Connnell MJ, Keogh B, Morton CO, Rogers TR, Fellon PG, O'Neill LA, Kelleher D, Loscher CE. 2011. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog 7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins LE, Lynch M, Marszalowska I, Kristek M, Rochfort K, O'Connell M, Windle H, Kelleher D, Loscher CE. 2014. Surface layer proteins isolated from Clostridium difficile induce clearance responses in macrophages. Microbes Infect 16:391–400. doi: 10.1016/j.micinf.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Bacon AE III, Fekety R. 1994. Immunoglobulin G directed against toxins A and B of Clostridium difficile in the general population and patients with antibiotic-associated diarrhea. Diagn Microbiol Infect Dis 18:205–209. doi: 10.1016/0732-8893(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 11.Viscidi R, Laughon BE, Yolken R, Bo-Linn P, Moench T, Ryder RW, Bartlett JG. 1983. Serum antibody response to toxins A and B of Clostridium difficile. J Infect Dis 148:93–100. doi: 10.1093/infdis/148.1.93. [DOI] [PubMed] [Google Scholar]
- 12.Kelly CP, Kyne L. 2011. The host immune response to Clostridium difficile. J Med Microbiol 60:1070–1079. doi: 10.1099/jmm.0.030015-0. [DOI] [PubMed] [Google Scholar]
- 13.Kyne L, Warny M, Qamar A, Kelly CP. 2000. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med 342:390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 14.Solomon K, Martin AJ, O'Donoghue C, Chen X, Fenelon L, Fanning S, Kelly CP, Kyne L. 2013. Mortality in patients with Clostridium difficile infection correlates with host proinflammatory and humoral immune responses. J Med Microbiol 62:1453–1460. doi: 10.1099/jmm.0.058479-0. [DOI] [PubMed] [Google Scholar]
- 15.Kyne L, Warny M, Qamar A, Kelly CP. 2001. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhea. Lancet 357:189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 16.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, Gerding DN, Kelly CP, Katchar K, Baxter R, Ambrosino D, Molrine D. 2010. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28:965–969. doi: 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]
- 17.Drudy D, Calabi E, Kyne L, Sougioultzis S, Kelly E, Fairweather N, Kelly CP. 2004. Human antibody response to surface layer proteins in Clostridium difficile infection. FEMS Immunol Med Microbiol 41:237–242. doi: 10.1016/j.femsim.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Bauer MP, Nibbering PH, Poxton IR, Kuijper EJ, van Dissel JT. 2014. Humoral immune response as a predictor of recurrence in Clostridium difficile infection. Clin Microbiol Infect 20:1323–1328. doi: 10.1111/1469-0691.12769. [DOI] [PubMed] [Google Scholar]
- 19.Johnson S, Gerding DN, Janoff EN. 1992. Systemic and mucosal antibody responses to toxin A in patients infected with Clostridium difficile. J Infect Dis 166:1287–1294. doi: 10.1093/infdis/166.6.1287. [DOI] [PubMed] [Google Scholar]
- 20.Sánchez-Hurtado K, Crrretge M, Mutlu E, McIIhagger R, Starr JM, Poxton IR. 2008. Systemic antibody response to Clostridium difficile in colonized patients with and without symptoms and matched controls. J Med Microbiol 57:717–724. doi: 10.1099/jmm.0.47713-0. [DOI] [PubMed] [Google Scholar]
- 21.Monaghan TM, Robins A, Knox A, Sewell HF, Mahida YR. 2013. Circulating antibody and memory B-cell responses to C. difficile toxins A and B in patients with C. difficile-associated diarrhea, inflammatory bowel disease and cystic fibrosis. PLoS One 8:e74452. doi: 10.1371/journal.pone.0074452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leuzzi R, Adamo R, Scarselli M. 2014. Vaccines against Clostridium difficile. Hum Vaccin Immunother 10:1466–1477. doi: 10.4161/hv.28428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghose C, Kelly CP. 2015. The prospect for vaccines to prevent Clostridium difficile infection. Infect Dis Clin North Am 29:145–162. doi: 10.1016/j.idc.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Kricka L. 1993. Trends in immunoassay technologies. J Clin Immunoassay 16:267–271. [Google Scholar]
- 25.Silzel JW, Cercek B, Dodson C, Tsay T, Obremski RJ. 1998. Mass-sensing, multianalyte microarray immunoassay with imaging detection. Clin Chem 44:2036–2043. [PubMed] [Google Scholar]
- 26.Bacarese-Hamilton T, Mezzasoma L, Ardizzoni A, Bistoni F, Crisanti A. 2004. Serodiagnosis of infectious diseases with antigen microarrays. J Appl Microbiol 96:10–17. doi: 10.1046/j.1365-2672.2003.02111.x. [DOI] [PubMed] [Google Scholar]
- 27.Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y, Romero KM, Nguyen TT, Kanantari-Dehaghi M, Crotty S, Baldi P, Villarreal LP, Felgner PL. 2005. Profiling the humoral immune response to infection by using proteome microarrays: highthroughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci U S A 102:547–552. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y, Bruno JF, Luft BJ. 2008. Profiling the humoral immune response to Borrelia burgdorferi infection with protein microarrays. Microb Pathog 45:403–407. doi: 10.1016/j.micpath.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Doolan DL, Mu Y, Unal B, Sundaresh S, Hirst S, Valdez C, Randall A, Molina D, Liang X, Freilich DA, Oloo JA, Blair PL, Aguiar JC, Baldia P, Davies DH, Felgner PL. 2008. Profiling humoral immune responses to P. falciparum infection with protein microarrays. Proteomics 8:4680–4694. doi: 10.1002/pmic.200800194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vigil A, Davies DH, Felgner PL. 2010. Defining the humoral immune response to infectious agents using high-density protein microarrays. Future Microbiol 5:241–251. doi: 10.2217/fmb.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wadia PP, Sahaf B, Miklos DB. 2011. Recombinant antigen microarrays for serum/plasma antibody detection, p 81–104. In Wu CJ. (ed), Protein microarray for disease analysis. Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 32.Mannsperger HA, Gade S, Henjes F, Beissbarth T, Korf U. 2010. RPPanalyzer: analysis of reverse-phase protein array data. Bioinformatics 26:2202–2203. doi: 10.1093/bioinformatics/btq347. [DOI] [PubMed] [Google Scholar]
- 33.Warny M, Vaerman JP, Avesani V, Delmée M. 1994. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun 62:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu TC, McCarthy VP, Gill VJ. 1983. Isolation rate and toxigenic potential of Clostridium difficile isolates from patients with cystic fibrosis. J Infect Dis 148:176. doi: 10.1093/infdis/148.1.176. [DOI] [PubMed] [Google Scholar]
- 35.Peach SL, Borriello SP, Gaya H, Barclay FE, Welch AR. 1986. Asymptomatic carriage of Clostridium difficile in patients with cystic fibrosis. J Clin Path 39:1013–1018. doi: 10.1136/jcp.39.9.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welkon CJ, Long SS, Thompson CM Jr, Gilligan PH. 1985. Clostridium difficile in patients with cystic fibrosis. Am J Dis Child 139:805–808. [DOI] [PubMed] [Google Scholar]
- 37.Bauer MP, Farid A, Bakker M, Hoek RA, Kuijper EJ, van Dissel JT. 2014. Patients with cystic fibrosis have a high carriage of nontoxigenic Clostridium difficile. Clin Microbiol Infect 20:O446–O449. doi: 10.1111/1469-0691.12439. [DOI] [PubMed] [Google Scholar]