

Abstract
Diabetes leads to complications in select organ systems primarily by disrupting the vasculature of the target organs. These complications include both micro- (cardiomyopathy, retinopathy, nephropathy, and neuropathy) and macro-(atherosclerosis) angiopathies. Bone marrow angiopathy is also evident in both experimental models of the disease as well as in human diabetes. In addition to vascular disruption, bone loss and increased marrow adiposity have become hallmarks of the diabetic bone phenotype. Emerging evidence now implicates enhanced marrow adipogenesis and changes to cellular makeup of the marrow in a novel mechanistic link between various secondary complications of diabetes. In this review, we explore the mechanisms of enhanced marrow adipogenesis in diabetes and the link between changes to marrow cellular composition, and disruption and depletion of reparative stem cells.
Keywords: diabetes, stem cells, adipogenesis, osteoblasts, angiogenesis, repair
Abbreviations
- AGE
advanced glycation end product
- BADGE
bisphenol-A-diglycidyl ether
- C/EBP
CCAAT/enhancer-binding protein
- DAG
diacylglycerol
- ERK1/2
extracellular signal response kinase
- FABP4
fatty acid binding protein-4
- FADH2
flavin adenine dinucleotide
- FoxO1
forkhead box protein O1
- GAPDH
glyceraldehyde-3 phosphate
- GSK3β
glycogen synthase kinase 3β
- IGF-1
insulin-like growth factor-1
- IP3
inositol triphosphate
- LEF/TCF
lymphoid enhancer factor/T cell factor
- MPC
mesenchymal progenitor cell
- mTOR
mammalian target of rapamycin
- NADH
nicotinamide adenine dinucleotide
- PI3K
phosphatidylinositol-4; 5-biphosphate 3-kinase
- PKA
protein kinase A
- PKB
protein kinase B
- PKC
protein kinase C
- PLC
phospholipase C
- PPARγ
peroxisome proliferator-activated receptor γ
- RAGE
receptor for advanced glycation end product
- ROS
reactive oxygen species
- Runx2
runt-related transcription factor 2
- SETB1
SET domain bifurcated-1
- TCA
tricarboxylic acid
- TNF-α
tumor necrosis factor-α
- TZD
thiazolidinedione derivatives
- Wnt
wingless-type MMTV integration site family
Diabetes and Its Complications
Diabetes is an incredibly prevalent disease, afflicting an estimated 220 million people in North America and 347 million people worldwide.1,2 Prevalence estimates have increased sharply since 1980 and are predicted to continue rising.3 As diabetes is a significant cause of morbidity and mortality, the economic burden is truly staggering and estimated to reach $17 billion a year by 2020 in Canada, and $116 billion in the United States.4,5 The main concern here is that nearly three-fourths of all diabetic patients suffer from at least one secondary complication of the disease.6 These secondary complications stem from the effects of sustained levels of hyperglycemia on the vascular system of select organs.7,8 Vascular endothelial cells lining blood vessel walls are the first to encounter high levels of circulating glucose.7-9 Sustained uptake of glucose by vessel endothelial cells results in impaired cellular function, resulting in microvascular and macrovascular changes.8,9 One of the earliest defects apparent in target organs of diabetic complications is a diminished capacity for vasodilation due to the unbalanced production of vasodilators and vasoconstrictors.7-9 Decreased levels of the vasodilator nitric oxide, coupled increased production of the powerful vasoconstrictor endothelin-1, results in impaired vasoregulation. This functional alteration is accompanied by sustained structural remodelling of the vessels in target organs manifesting as retinopathy, nephropathy, neuropathy, cardiomyopathy, and accelerated atherosclerosis.7-9 Initiation as well as the progression of these complications also entails an impaired repair/regenerative mechanism.10,11 Vascular repair is largely dependent on the proliferation, mobilization and differentiation of bone marrow-derived progenitor cells.12 The angiogenic potential (reparative function) of these precursor cells is diminished in vasculopathies and may be resultant from diabetes-induced changes to the cellular composition of the marrow where these stem/progenitor cells reside.13-15
Diabetic Marrow Dysfunction: Consequences of Enhanced Adipogenesis and Impaired Osteoblast-Genesis
Bone marrow is a rich source of stem cells. At least two different stem cell populations reside in the marrow: hematopoietic stem cells and multipotential stem cells (also known as mesenchymal/mesodermal stem cells, mesenchymal/marrow stromal cells; MSCs). Both of these stem cell types consist of a hierarchy of cells. MSCs are believed to give rise to endothelial cells, mesenchymal progenitor cells (MPCs; cells restricted to the mesenchymal lineage), adipocytes and osteoblasts. MSC progeny also create a cellular environment to maintain stem cell self-renewal in the marrow (Fig. 1).
Figure 1.
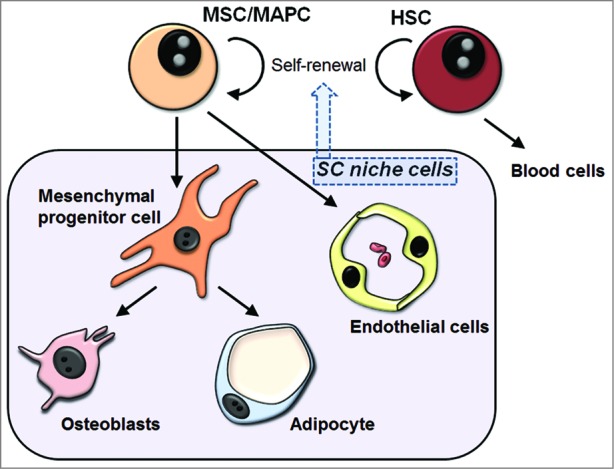
Schematic illustrating bone marrow niche components. Bone marrow contains at least two different stem cell types: hematopoietic stem cells and mesenchymal stem cells. Self-renewal and differentiation activity of these stem cells is regulated by the surrounding microenvironment including cell types at various differentiation states. These niche cells include endothelial cells, osteoblasts, adipocytes and mesenchymal progenitor cells (cells restricted to the mesenchymal lineage). HSC, hematopoietic stem cells; MAPC, multipotential adult progenitor cell; MSC, mesenchymal/multipotential stem cell; SC, stem cell.
Long-standing diabetes leads to cellular changes in the bone marrow, the functional significance of which is just being realized. These cellular changes include enhanced adipogenesis of MPCs as observed in both type 1 and 2 models of diabetes.16,17 In the insulin-deficient form of the disease, this leads to diminished bone density, with human studies and streptozotocin-induced diabetic animal models noting a decrease in trabecular bone mass and a reciprocal increase in the adiposity of the marrow.16-20 Alternatively, analyses of bones of type 2 diabetics have generally observed unchanged or increased bone mineral density, though clinically, both diabetic populations have a substantially increased risk of fractures in comparison to non-diabetics.18,21-24 Additionally, there is increasing evidence that some diabetic medications may negatively impact bone density and marrow adiposity.1,25-29 Diabetes also induces microvascular remodeling in the bone marrow manifesting as impaired angiogenic ability, vascular endothelial cell dysfunction, and a reduction in stem cell number.30,31 These findings suggest that disruption of the bone marrow microenvironment, enhanced adipogenesis/suppressed osteoblastogenesis, may be responsible for detrimental effects on stem cell function and differentiation. If true, this provides a novel mechanistic link to impairment of endogenous repair in diabetes (Fig. 2). Indeed, both type 1 and type 2 diabetes is associated with lower circulating number of endothelial progenitor cells (cells that play a critical role in vascular regeneration) when compared with healthy subjects.15,32-34 Furthermore, the number of endothelial progenitor cells correlates with glycemic control.35 There are a number of possible mechanisms at play here: (1) diabetes may cause depletion of resident stem/progenitor cells in the marrow through alteration of the marrow stem cell environment, (2) diabetes may alter the mobilization of stem/progenitor cells, and (3) high levels of glucose in the circulation may reduce the number of cells that have mobilized. In fact, there is experimental evidence for all three possibilities. We and others have recently shown that diabetes leads to reduced number of stem cells in the bone marrow.31,36 These stem cells can be distinguished from hematopoietic stem cells by their ability to differentiate into endothelial and mural lineages and to regenerate functional vessels.7,36,37 Studies have also shown that diabetes leads to reduced mobilization of stem cells from the marrow.38,39 A number of signaling mechanisms have been identified underlying this abnormality. And finally, we have shown that high levels of glucose decrease endothelial and mesenchymal progenitor cell numbers acutely (within 24 h of culture).37 However, cells recover from glucose toxicity with sustained exposure. In addition, the differentiation capacity of blood and bone marrow-derived stem cells to produce endothelial and mesenchymal progenitor cells is not altered by the presence of high levels of glucose. Experimental evidence also shows that stem/progenitor cells isolated from diabetic mice are able to restore vascular homeostasis.28,29 Taken together, these studies suggest that changing the cellular microenvironment in the marrow directly leads to dysfunction and reduction of stem cells in diabetes. The aim of this review is to elucidate the mechanisms underlying the increase in bone marrow adipogenesis observed in diabetes and examine the bidirectional relationship between bone adiposity and disease progression.
Figure 2.

Effect of diabetes in target organs systems. Diabetes leads to structural and functional changes in target organs resulting in loss of blood vessel integrity and vasoregulation. Continued damage to blood vessels leads to a reduction in blood flow to target organs and loss of vascular cells. Vessel degeneration and ischemia play critical roles in the development of secondary complications of diabetes including retinopathy, nephropathy, and cardiomyopathy. In the bone marrow, diabetes changes the cellular composition by increasing adipogenesis and reducing osteoblastogenesis. These are believed to alter the stem cell niche resulting in stem cell dysfunction and depletion. The end result would be impaired repair and regeneration of vasculature in target organs of secondary complications.
Mechanisms of Enhanced Marrow Adipogenesis in Diabetes
There is a wealth of knowledge on the process and factors involved in adipogenesis. The current understanding of adipogenesis has largely emerged from in vitro studies using cell lines such as the preadipocyte 3T3-L1 and 3T3-F442A cells.40,41 Although recently, studies conducted in human cells have also emerged. The process of differentiation in murine cell lines appears to be similar to the signaling cascade that drives adipogenesis in human bone marrow cells, with the principle actors being peroxisome proliferator-activated receptor γ (PPARγ) and the CCAAT/enhancer-binding protein (C/EBPα, -β, and -δ) transcription factors.42,43 It should be noted that there are reports of differences between murine cell lines and human MSC/MPCs. For example, Yu and colleagues suggested that human marrow cells primarily express PPARγ1 isoform upon differentiation with PPARγ2 increases being noted at later time point, which is believed to be in contrast to murine cells.44 PPARγ2 isoform does appear to be the minor species, comprising only 15% of all PPARγ expression within adipose tissue, although it has been shown to be the predominant isoform in regulating adipogenesis.45,46 Furthermore, knocking down the expression of C/EBPα prevents PPARγ2 induction and adipogenesis in human marrow cells.44 The expression profile during terminal adipogenic differentiation in human MSCs is also similar to that of 3T3-L1 preadipocytes, characterized by early expression of C/EBPβ and -δ and followed by C/EBPα and PPARγ expression.42 In addition, we have shown specific induction of PPARγ2 in human marrow MPCs following addition of adipogenic differentiation media and the levels parallel C/EBPα.36,37 Therefore, examining the expression of these transcription factors offer insight into paracrine factors regulating adipogenesis in diabetes.
A vast number of factors have been shown to modulate adipogenesis. Some of these modulating factors that are pertinent to the diabetic context include insulin,47,48 insulin-like growth factor-1 (IGF-1),49 extracellular proteins including collagen and fibronectin,50,51 and tumor necrosis factor-α (TNF-α).52,53 Shifts in the expression or function of these and other effectors disrupt the homeostatic balance between adipogenic and osteogenic differentiation of MPCs.54-59 While the general consensus is that diabetic hyperglycemia is associated with increased adipogenesis in the marrow, inhibition of fat cell formation and promotion of osteoblastic differentiation following the administration of exogenous glucose has also been reported.60,61 Shilpa and colleagues found that culturing 3T3-L1 preadipocytes in extremely high glucose levels of 105 mM resulted in diminished adipogenesis, with downregulation of PPARγ and C/EBPα relative to cells cultured in 25 mM glucose concentration.61 The extreme hyperglycemic conditions emulated by the 105 mM glucose condition was found to increase cellular stress, leading to the induction of inflammatory cytokines, such as TNF-α, known to inhibit adipocyte differentiation and potentially induce dedifferentiation.61,62 This glucose level was considerably greater than the 25 mM concentration used to mimic hyperglycemia in most other studies, which may account for contrasting results.36,63
The enhanced adiposity of the bone marrow observed in diabetes models and human diabetes appears to be a multifactorial consequence of augmented insulin signaling, hyperlipidemia, elevated blood glucose levels, and heightened oxidative stress. Recently however, novel signaling mechanisms have been highlighted that enhance adipogenic differentiation.
PI3K-PKB pathway
High levels of blood glucose have been demonstrated to increase adipocyte formation, lipid accumulation, and the expression of PPARγ in MPCs.64 It has been suggested that hyperglycemia mediates its effects through changes in post-receptor insulin signaling, which may be implicated in the development of insulin resistance.64 High levels of glucose increases the activity of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and the subsequent phosphorylation of protein kinase B (PKB), both of which are involved in the insulin signaling cascade. PKB-facilitated de-repression of the pparγ gene though forkhead box protein O1 (FoxO1) nuclear export leads to the induction of PPARγ and C/EBPα expression, resulting in increased adiposity of the bone marrow.64-66 PKB induced by hyperglycemia is also able to activate mammalian target of rapamycin (mTOR), which leads to increased expression of C/EBPα, and other adipocyte-specific factors in pre-adipocytes, as well as muscle satellite cells, leading to the formation of intramuscular adipose depots.67-69
Reactive oxygen species
The cellular production of reactive oxygen species (ROS) has been shown to be elevated in diabetic patients, largely due to increased glucose levels and metabolism.70-73 The predominant mechanism underlying the heightened oxidative stress in diabetes involves dysfunction of the mitochondrial electron transport system.74 In hyperglycemic cells, more glucose becomes oxidized through the tricarboxylic acid (TCA) cycle, which results in an increased number of electron donors, NADH and FADH2, being fed into the electron transport chain. This leads to an increase in the mitochondrial membrane voltage gradient until a specific threshold limit is reached, at which point further electron transfer inside complex III is halted, causing a backlog of electrons in coenzyme Q. Coenzyme Q dissipates this excess negative charge through the donation of single electrons to molecular oxygen, leading to the formation of superoxide.
Superoxide inhibits the action of glycolytic enzyme glyceraldehyde-3 phosphate (GAPDH), which leads to the activation of the advanced glycation end product (AGE) pathway that has been shown to be increasingly stimulated in diabetes.74-76 AGEs are proteins or lipids that become glycosylated following exposure to sugars and accelerate cellular oxidative damage, and have been implicated in both micro- and macro-vascular diabetic complications.72,77,78 Binding of AGE and their receptors, known as RAGE, have been associated with reduced bone formation by osteoblasts and diminished matrix mineralization, in addition to impaired osteoblastogenesis.79-81 AGE-RAGE interactions have also been identified as promoting the apoptosis of osteoblasts and MPCs, contributing to the depletion of the stem cell niche.82,83
Oxidative stress induced by hyperglycemia has also been found to stimulate the PI3K/PKB pathway, which acts to inhibit osteoblastic maturation and stimulate adipogenesis.84 Osteoblasts exposed to ROS resulting from a high glucose environment demonstrate decreased expression of runt-related transcription factor 2 (Runx2) and osteocalcin, with a concomitant increase in the abundance of the adipogenesis-related factors PPARγ, adipsin, and fatty acid binding protein-4 (FABP4).84 ROS is also able to prevent the mineralization of osteoblasts and enhance their accumulation of lipid droplets.
Non-canonical Wnt-PKC pathway
Perhaps the best-studied system in adipogenesis is the wingless-type MMTV integration site family (Wnt)-mediated signaling pathway.85 In humans, the Wnt family is comprised of 19 secreted glycoproteins that affect the differentiation and development of many cell types through autocrine and paracrine processes.86,87 It has been well accepted that activation of the Wnt pathway (β-catenin signaling) constrains progenitor cells to differentiate into osteoblast or myoblasts and prevents development along the adipocytic lineage.85 It is believed that endogenous production of Wnt ligands act to curb the terminal differentiation of preadipocytes and attempt to maintain a stem cell-like phenotype. Crosstalk also appears to exist between the canonical Wnt system and PPARγ. When induced, PPARγ binds the Lymphoid enhancer factor/T cell factor (LEF/TCF)-binding domain of β-catenin and facilitates its phosphorylation by glycogen synthase kinase 3β (GSK3β), directing the factor to the proteasome for degradation.88-91 Following the induction of differentiation within preadipocytes, levels of β-catenin remain elevated, until the expression of PPARγ is heightened, resulting in the post-transcriptional downregulation of β-catenin and terminal differentiation.92
Of the several non-canonical Wnt signaling cascades, the Wnt/Ca2+ pathway is presumed to be the most relevant in the regulation of adipogenesis. Interactions between specific members of the Wnt and Wnt receptor subtypes result in the activation of phospholipase C (PLC).87,93 PLC then leads to the generation of diacylglycerol (DAG) and inositol triphosphate (Ins[1,4,5]P3). Release of intracellular calcium activates protein kinase C (PKC), ultimately leading to phosphorylation of SETB1 (SET domain bifurcated-1) histone methyltransferase. This leads to the creation of a co-repressor complex that inhibits PPARγ through H3-K9 histone methylation and directs the progenitor cell toward osteoblastogenesis through upregulated expression of Runx2, which is requisite for bone cell maturation.85,94 Interestingly, depending on the distinct isoform activated, PKC may have either a positive or negative influence on adipogenesis. PKC isoforms -α, -δ, and -μ are suspected to inhibit maturation.95 The initiation of adipogenesis appears to be reliant on PKC-βI and PKC-γ is believed to be necessary for clonal expansion.95,96 PKC-ε is presumed to be critical for pre-adipocyte commitment and the final acquisition of the adipocytic phenotype, though the mechanisms leading to the effects of these three positive modulators are not yet understood.97,98
We have recently shown that non-canonical Wnt11 is induced by hyperglycemia in MPCs and enhances the adipocytic differentiation.36 While the mechanism remains to be fully elucidated, a current hypothesis is that, through a non-canonical pathway, hyperglycemia induces a switch in Wnt11 signaling that differentially activates the various isoforms of PKC, specifically inducing the phosphorylation and consequent activation of PKC. PKC-ε is translocated from the cytoplasm to the nucleus where it is expressed in spatiotemporal symmetry with C/EBPβ, indicative of a potential interaction.36,95 Through a currently unknown process likely involving the phosphorylation and regulation of key nuclear adipogenic factors, PKC-ε activation results in the acceleration of adipogenic differentiation.
Hyper- and hypo-insulinemia
Insulin is one of the factors commonly used to stimulate adipogenic differentiation in cell culture systems, and in vivo models of insulin receptor knockout display impaired adipogenic differentiation and lipid storage capacity.99-101 A hyperinsulinemic state is frequently observed in the development of type 2 diabetes as pancreatic production of insulin surges in an attempt to counteract the ever-increasing resistance of peripheral tissues.102,103 Hyperinsulinemia may be capable of inducing the adipogenesis of cells within the marrow stem cell niche through a signaling cascade involving PKB and mTOR, culminating with activation of C/EBPα and PPARγ.104 Conversely, hypoinsulinemia, a hallmark of type 1 diabetes and an eventual occurrence following β-cell failure in type 2 diabetics, may also indirectly lead to enhanced adipogenesis. Insulin receptor knockout mice display a 2-fold upregulation of the IGF-1 receptor through a yet unknown mechanism.101 Both the IGF and insulin signaling systems converge on a common pathway involving PKB, which may grant IGF partial control of adipogenic differentiation under hypoinsulinemic conditions.105 When combined with the administration of exogenous insulin therapies, the overexpression of IGF-1 receptor may lead to disproportionate fat cell development.
Hyperlipidemia
A large proportion of diabetics are subject to hyperlipidemia, particularly if their condition is poorly controlled.106,107 A study of diabetic mice has observed elevations in the relative quantities of plasma di- and tri-unsaturated fatty acids compared with saturated fats.108 Fatty acids, particularly polyunsaturated fatty acids, have been identified as agonists for PPARγ, and although they possess a relatively low affinity, the substantial elevation of serum lipids in diabetes may be sufficient for activation.47,109,110 Dyslipidemia may prohibit the efficient maturation of osteoblast-like cells and is capable of inducing the trans-differentiation of osteoblast-like cells into adipocytes, further attenuating the density of the bone marrow.111
Diabetic Medications
Another potential contributor to the diminished bone integrity seen in diabetes may be the effects of anti-diabetic medications. While insulin-sensitizing agents are crucial to the maintenance of normoglycemia and avoidance of life-threatening complications, they also have a chronic effect on the bone marrow. The current therapy in the treatment of type 2 diabetes involves metformin, which suppresses hepatic gluconeogenesis to moderate blood glucose levels and increase insulin sensitivity.112 Both in vitro and in vivo, metformin has been found to increase markers of osteogenic differentiation, as well as function.113 The developmental shift in the lineage potential of MPCs induced by this commonly-used drug may account for a portion of the enhanced bone mineral density that has controversially been observed in noninsulin-dependent diabetics.
Thiazolidinedione derivatives (TZDs), also known as glitazones, are a group of medications that act to improve insulin responsiveness within target tissues, concomitantly augmenting hyperglycemia and hyperlipidemia.114-116 TZDs have also been implicated in diabetic bone loss, with a significantly increased risk of fractures and osteoporosis while on these medications being well-documented.25,26,28,29,117-120 The primary mechanism of action of TZDs is through the direct induction and activation of PPARγ, leading to improved insulin sensitivity throughout the body via an unknown mechanism.121,122 The efficacy of TZDs in rectifying systemic insulin resistance through a factor found predominantly in adipocytes accentuates the intimate and complex relationship between fat tissue and diabetes. Through the promotion of PPARγ, a common side effect of this class of drugs is weight gain, with increased adipogenesis leading to increased fat depots primarily within the subcutaneous site, along with the bone marrow.123-127
The stimulation of differentiation has been widely associated with the increased adiposity in bone marrow seen during TZD treatment, though the reported effects of TZDs on osteoblasts and osteoclasts have been contradictory. Some reports have shown that exposure of multipotent cells to TZDs in vitro results in potent activation of adipogenesis, with no negative effects on osteoblast development or function, suggesting that the two cell types do not compete for the same population of precursor cells.128,129 It has also been proposed that rosiglitazone is able to accelerate both fat and bone cell maturation through the PPARγ2 isoform in adipocytes and PPARγ1 in osteoblasts, with a substantial build-up of ROS.130 Yet, others have reported that induction of adipogenesis through PPARγ stimulation does, in fact, necessarily reduce osteoblast differentiation and function, supporting the notion of a shared pool of progenitor cells.123,131-133
A number of prospective and retrospective observational investigations have aimed to determine whether TZD use is associated with a negative effect on bone density in humans. While two small studies reported a minor protective effect of troglitazone in reducing bone turnover, larger-scale surveys tend to purport a significant decrease in bone mass with TZD treatment.28,29,134,135 Several randomized controlled trials have also examined the involvement of TZDs in skeletal quality.128,136-138 Most of these studies found essentially no changes in indicators of bone resorption, with a 10–20% decrease in markers associated with osteoblast function and a reduced quantity of osteoblastic precursors. This translated into a significant reduction in bone density in the experimental groups administered TZDs.137,138
Are Bone Loss and Marrow Adiposity Two Sides of the Same Coin?
It has long been accepted that a reciprocal relationship exists between marrow adiposity and bone mineral density, with bone loss and increased adiposity often coinciding.55,105 As both osteoblasts and adipocytes are generated from the same population of precursors, it would appear that the predominance of one lineage would occur at the expense of the development of the other cell type. Several studies have been undertaken investigating whether the induction of adipogenesis forces the repression of osteoblast generation and activity. In hetero- and homozygous PPARγ knockout systems, embryonic stem cells spontaneously differentiated into osteoblasts, while adipogenesis was inhibited.139,140 In vivo, PPARγ haploinsufficient mice also display heightened levels of osteoblastogenesis, leading to increased bone mass.139 Conversely, deletion of β-catenin in osterix-expressing cells (early osteoblast lineage) leads to a striking reduction in bone mass and an increase in bone marrow adiposity.141 These studies suggest that one mechanism of diabetic bone phenotype may be depletion of available progenitor cells through commitment to one lineage. However, treatment of diabetic mice with the PPARγ antagonist bisphenol-A-diglycidyl ether (BADGE) inhibits adipogenesis without suppression of osteoblast markers and consequent bone loss.142 The mechanism of this disconnect is not fully clear but it may be related to duration of BADGE treatment. In fact, acute exposure of BADGE has been shown to be ineffective in reducing osteocalcin levels in cultured osteoblasts, whereas chronic treatment significantly suppresses the levels.142 Osteocalcin has also been observed to decrease in diabetes, with changes in factors involved in earlier differentiation, such as Runx2, occasionally being observed.16,17,143,144 The changes to osteoblastic gene expression are mediated through activation of protein kinase A (PKA) and extracellular signal response kinase (ERK1/2) signaling mechanisms, which attenuate osteoblast differentiation.145
In diabetes, both acute and chronic hyperglycemia induces osmotic changes in cells as they adapt to the heightened colloid pressure by reducing their volume and augmenting gene expression.143,146 Acute in vitro exposure of osteoblast precursor cells to high glucose levels and hyperosmolarity resulted in increased expression of collagen I, along with downregulation of osteocalcin mRNA.146 With sustained exposure, high levels of glucose are able to induce the upregulation of alkaline phosphatase, along with diminishing production of osteocalcin. The mRNA levels of PPARγ were also found to be increased nearly 2-fold in pre-osteoblasts challenged with high levels of glucose.143,145 In vivo studies have shown similar findings in the bones of diabetic mice, reporting a 40% reduction in osteocalcin expression, though markers of early osteoblast development, such as Runx2, remain unchanged.16,17 Adipogenic genes PPARγ, resistin, and FABP4 were all found to be significantly upregulated, with a 3-fold increase in the quantity of marrow adipocytes being observed. This suggests that an elevated serum glucose level impairs the later stages of osteblastogenesis, while promoting the expression of markers of the adipocytic phenotype.
Upon enhanced adipogenesis, the interaction between adipocytes and osteoblasts takes on another layer of complexity. Adipocytes secrete numerous proteins collectively referred to as adipokines, which include adiponectin, leptin, resistin, and tumor necrosis factor-α (TNF-α), among others.147 Paradoxically, adiponectin which is secreted nearly exclusively by adipocytes has been found to prevent adipogenic differentiation in bone marrow cultures and increase trabecular bone mass by promoting osteoblastogenesis and repressing osteoclast formation.148-151 Unlike adiponectin, the expression of leptin increases concurrently with adiposity and appears to be unaffected by diabetes.152-154 Leptin appears to have contradictory effects on bone, activating the sympathetic nervous system to accelerate bone loss, as well as stimulating the osteogenic differentiation of marrow MPCs, with the net outcome dependent on its concentration.155-160 Lastly, TNF-α, has also been identified as having detrimental effects on the skeleton. A positive correlation has been found between the expression of TNF-α by adipocytes and both obesity and insulin resistance.161 Local TNF-α signaling leads to enhanced differentiation of osteoclast precursors and increased bone resorption.162,163 We are just starting to understand how these adipokines are altered in diabetes and the subsequent effect of this alteration both systemically and in the marrow. This field of research will bring upon a new era in our understanding of chronic diabetic complications.
Concluding Remarks
With the incidence of diabetes on a rise, it is imperative that we understand the mechanisms of secondary diabetic complications. Sustained hyperglycemia has been demonstrated to lead to increased adiposity of the bone marrow, with a concomitant escalation in the risk of fractures and may potentially be the cause of reduced stem cells for endogenous vascular repair. While the basic process underlying adipogenesis is well-elucidated, the specific factors promoting and inhibiting the C/EBP-PPAR signaling pathway are numerous and complex, with their potential roles in preventing diabetes-induced bone marrow adipogenesis relatively unexplored. The exploitation of marrow adipose biology may soon become a central treatment strategy in diabetes, precluding complications or preventing the disease itself.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The authors would like to acknowledge support from the Canadian Diabetes Association (OG-3-13-4034-ZK to ZAK) and the Lawson Health Research Institute (ZAK). ZAK is a recipient of a New Investigator Award from the Heart and Stroke Foundation of Canada (Great-West Life and London Life New Investigator Award).
References
- 1. Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, et al.; Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Blood Glucose). National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet 2011; 378:31-40; PMID:21705069; http://dx.doi.org/ 10.1016/S0140-6736(11)60679-X [DOI] [PubMed] [Google Scholar]
- 2. Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med 1997; 14(Suppl 5):S1-85; PMID:9450510; http://dx.doi.org/ 10.1002/(SICI)1096-9136(199712)14:5±≤S7::AID-DIA522≥3.3.CO;2-I [DOI] [PubMed] [Google Scholar]
- 3. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004; 27:1047-53; PMID:15111519; http://dx.doi.org/ 10.2337/diacare.27.5.1047 [DOI] [PubMed] [Google Scholar]
- 4. An economic tsunami, the cost of diabetes in Canada. Toronto, Ontario: Canadian Diabetes Association, 2009. [Google Scholar]
- 5. 2011 National Diabetes Fact Sheet, National Center for Chronic Disease Prevention and Health Promotion. American Diabetes Association; 2011. [Google Scholar]
- 6. Williams R, Van Gaal L, Lucioni C; CODE-2 Advisory Board . Assessing the impact of complications on the costs of Type II diabetes. Diabetologia 2002; 45:S13-7; PMID:12136406; http://dx.doi.org/ 10.1007/s00125-002-0859-9 [DOI] [PubMed] [Google Scholar]
- 7. Keats EC, Khan ZA. Vascular stem cells in diabetic complications: evidence for a role in the pathogenesis and the therapeutic promise. Cardiovasc Diabetol 2012; 11:37; PMID:22524626; http://dx.doi.org/ 10.1186/1475-2840-11-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan ZA, Chakrabarti S. Therapeutic targeting of endothelial dysfunction in chronic diabetic complications. Recent Pat Cardiovasc Drug Discov 2006; 1:167-75; PMID:18221084; http://dx.doi.org/ 10.2174/157489006777442531 [DOI] [PubMed] [Google Scholar]
- 9. Khan ZA, Farhangkhoee H, Chakrabarti S. Towards newer molecular targets for chronic diabetic complications. Curr Vasc Pharmacol 2006; 4:45-57; PMID:16472176; http://dx.doi.org/ 10.2174/157016106775203081 [DOI] [PubMed] [Google Scholar]
- 10. Howangyin KY, Silvestre JS. Diabetes mellitus and ischemic diseases: molecular mechanisms of vascular repair dysfunction. Arterioscler Thromb Vasc Biol 2014; 34:1126-35; PMID:24675660; http://dx.doi.org/ 10.1161/ATVBAHA.114.303090 [DOI] [PubMed] [Google Scholar]
- 11. Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, Stefanadis C. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol 2013; 62:667-76; PMID:23948511; http://dx.doi.org/ 10.1016/j.jacc.2013.03.089 [DOI] [PubMed] [Google Scholar]
- 12. Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res 2008; 78:413-21; PMID:18349136; http://dx.doi.org/ 10.1093/cvr/cvn081 [DOI] [PubMed] [Google Scholar]
- 13. Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002; 106:2781-6; PMID:12451003; http://dx.doi.org/ 10.1161/01.CIR.0000039526.42991.93 [DOI] [PubMed] [Google Scholar]
- 14. Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, Menegolo M, de Kreutzenberg SV, Tiengo A, Agostini C, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol 2005; 45:1449-57; PMID:15862417; http://dx.doi.org/ 10.1016/j.jacc.2004.11.067 [DOI] [PubMed] [Google Scholar]
- 15. Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, Grego F, Vigili de Kreutzenberg S, Tiengo A, Agostini C, et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol 2006; 26:2140-6; PMID:16857948; http://dx.doi.org/ 10.1161/01.ATV.0000237750.44469.88 [DOI] [PubMed] [Google Scholar]
- 16. Botolin S, Faugere M-C, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-γ2 expression in type I diabetic mice. Endocrinology 2005; 146:3622-31; PMID:15905321; http://dx.doi.org/ 10.1210/en.2004-1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology 2007; 148:198-205; PMID:17053023; http://dx.doi.org/ 10.1210/en.2006-1006 [DOI] [PubMed] [Google Scholar]
- 18. Leidig-Bruckner G, Ziegler R. Diabetes mellitus a risk for osteoporosis? Exp Clin Endocrinol Diabetes 2001; 109(Suppl 2):S493-514; PMID:11460594; http://dx.doi.org/ 10.1055/s-2001-18605 [DOI] [PubMed] [Google Scholar]
- 19. Tuominen JT, Impivaara O, Puukka P, Rönnemaa T. Bone mineral density in patients with type 1 and type 2 diabetes. Diabetes Care 1999; 22:1196-200; PMID:10388989; http://dx.doi.org/ 10.2337/diacare.22.7.1196 [DOI] [PubMed] [Google Scholar]
- 20. Wang A, Midura RJ, Vasanji A, Wang AJ, Hascall VC. Hyperglycemia diverts dividing osteoblastic precursor cells to an adipogenic pathway and induces synthesis of a hyaluronan matrix that is adhesive for monocytes. J Biol Chem 2014; 289:11410-20; PMID:24569987; http://dx.doi.org/ 10.1074/jbc.M113.541458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dhaliwal R, Cibula D, Ghosh C, Weinstock RS, Moses AM. Bone quality assessment in type 2 diabetes mellitus. Osteoporos Int 2014; 25:1969-73; PMID:24718377; http://dx.doi.org/ 10.1007/s00198-014-2704-7 [DOI] [PubMed] [Google Scholar]
- 22. de Liefde II, van der Klift M, de Laet CE, van Daele PL, Hofman A, Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos Int 2005; 16:1713-20; PMID:15940395; http://dx.doi.org/ 10.1007/s00198-005-1909-1 [DOI] [PubMed] [Google Scholar]
- 23. Janghorbani M, Feskanich D, Willett WC, Hu F. Prospective study of diabetes and risk of hip fracture: the Nurses’ Health Study. Diabetes Care 2006; 29:1573-8; PMID:16801581; http://dx.doi.org/ 10.2337/dc06-0440 [DOI] [PubMed] [Google Scholar]
- 24. Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol 2007; 166:495-505; PMID:17575306; http://dx.doi.org/ 10.1093/aje/kwm106 [DOI] [PubMed] [Google Scholar]
- 25. Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ 2009; 180:32-9; PMID:19073651; http://dx.doi.org/ 10.1503/cmaj.080486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McDonough AK, Rosenthal RS, Cao X, Saag KG. The effect of thiazolidinediones on BMD and osteoporosis. Nat Clin Pract Endocrinol Metab 2008; 4:507-13; PMID:18695700; http://dx.doi.org/ 10.1038/ncpendmet0920 [DOI] [PubMed] [Google Scholar]
- 27. Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology 2004; 145:401-6; PMID:14500573; http://dx.doi.org/ 10.1210/en.2003-0746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, et al. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab 2006; 91:3349-54; PMID:16608888; http://dx.doi.org/ 10.1210/jc.2005-2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yaturu S, Bryant B, Jain SK. Thiazolidinedione treatment decreases bone mineral density in type 2 diabetic men. Diabetes Care 2007; 30:1574-6; PMID:17363747; http://dx.doi.org/ 10.2337/dc06-2606 [DOI] [PubMed] [Google Scholar]
- 30. Oikawa A, Siragusa M, Quaini F, Mangialardi G, Katare RG, Caporali A, van Buul JD, van Alphen FP, Graiani G, Spinetti G, et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler Thromb Vasc Biol 2010; 30:498-508; PMID:20042708; http://dx.doi.org/ 10.1161/ATVBAHA.109.200154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spinetti G, Cordella D, Fortunato O, Sangalli E, Losa S, Gotti A, Carnelli F, Rosa F, Riboldi S, Sessa F, et al. Global remodeling of the vascular stem cell niche in bone marrow of diabetic patients: implication of the microRNA-155/FOXO3a signaling pathway. Circ Res 2013; 112:510-22; PMID:23250986; http://dx.doi.org/ 10.1161/CIRCRESAHA.112.300598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fadini GP, Agostini C, Avogaro A. Characterization of endothelial progenitor cells. Biochem Biophys Res Commun 2005; 336:1-2; PMID:16084490; http://dx.doi.org/ 10.1016/j.bbrc.2005.07.119 [DOI] [PubMed] [Google Scholar]
- 33. Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 2004; 53:195-9; PMID:14693715; http://dx.doi.org/ 10.2337/diabetes.53.1.195 [DOI] [PubMed] [Google Scholar]
- 34. Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res 2001; 89:E1-7; PMID:11440984; http://dx.doi.org/ 10.1161/hh1301.093953 [DOI] [PubMed] [Google Scholar]
- 35. Yue WS, Lau KK, Siu CW, Wang M, Yan GH, Yiu KH, Tse HF. Impact of glycemic control on circulating endothelial progenitor cells and arterial stiffness in patients with type 2 diabetes mellitus. Cardiovasc Diabetol 2011; 10:113; PMID:22185563; http://dx.doi.org/ 10.1186/1475-2840-10-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keats EC, Dominguez JM, 2nd, Grant MB, Khan ZA. Switch from canonical to noncanonical Wnt signaling mediates high glucose-induced adipogenesis. Stem Cells 2014; 32:1649-60; PMID:24496952; http://dx.doi.org/ 10.1002/stem.1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keats E, Khan ZA. Unique responses of stem cell-derived vascular endothelial and mesenchymal cells to high levels of glucose. PLoS One 2012; 7:e38752; PMID:22701703; http://dx.doi.org/ 10.1371/journal.pone.0038752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Albiero M, Poncina N, Tjwa M, Ciciliot S, Menegazzo L, Ceolotto G, Vigili de Kreutzenberg S, Moura R, Giorgio M, Pelicci P, et al. Diabetes causes bone marrow autonomic neuropathy and impairs stem cell mobilization via dysregulated p66Shc and Sirt1. Diabetes 2014; 63:1353-65; PMID:24270983; http://dx.doi.org/ 10.2337/db13-0894 [DOI] [PubMed] [Google Scholar]
- 39. Barthelmes D, Irhimeh MR, Gillies MC, Karimipour M, Zhou M, Zhu L, Shen WY. Diabetes impairs mobilization of mouse bone marrow-derived Lin(-)/VEGF-R2(+) progenitor cells. Blood Cells Mol Dis 2013; 51:163-73; PMID:23714230; http://dx.doi.org/ 10.1016/j.bcmd.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 40. Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell 1974; 3:127-33; PMID:4426090; http://dx.doi.org/ 10.1016/0092-8674(74)90116-0 [DOI] [PubMed] [Google Scholar]
- 41. Novakofski J. Adipogenesis: usefulness of in vitro and in vivo experimental models. J Anim Sci 2004; 82:905-15; PMID:15032449 [DOI] [PubMed] [Google Scholar]
- 42. Nakamura T, Shiojima S, Hirai Y, Iwama T, Tsuruzoe N, Hirasawa A, Katsuma S, Tsujimoto G. Temporal gene expression changes during adipogenesis in human mesenchymal stem cells. Biochem Biophys Res Commun 2003; 303:306-12; PMID:12646203; http://dx.doi.org/ 10.1016/S0006-291X(03)00325-5 [DOI] [PubMed] [Google Scholar]
- 43. Camp HS, Ren D, Leff T. Adipogenesis and fat-cell function in obesity and diabetes. Trends Mol Med 2002; 8:442-7; PMID:12223316; http://dx.doi.org/ 10.1016/S1471-4914(02)02396-1 [DOI] [PubMed] [Google Scholar]
- 44. Yu WH, Li FG, Chen XY, Li JT, Wu YH, Huang LH, Wang Z, Li P, Wang T, Lahn BT, et al. PPARγ suppression inhibits adipogenesis but does not promote osteogenesis of human mesenchymal stem cells. Int J Biochem Cell Biol 2012; 44:377-84; PMID:22120652; http://dx.doi.org/ 10.1016/j.biocel.2011.11.013 [DOI] [PubMed] [Google Scholar]
- 45. Fajas L, Auboeuf D, Raspé E, Schoonjans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruchart JC, Deeb S, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem 1997; 272:18779-89; PMID:9228052; http://dx.doi.org/ 10.1074/jbc.272.30.18779 [DOI] [PubMed] [Google Scholar]
- 46. Saladin R, Fajas L, Dana S, Halvorsen YD, Auwerx J, Briggs M. Differential regulation of peroxisome proliferator activated receptor gamma1 (PPARgamma1) and PPARgamma2 messenger RNA expression in the early stages of adipogenesis. Cell Growth Differ 1999; 10:43-8; PMID:9950217 [PubMed] [Google Scholar]
- 47. Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 2000; 16:145-71; PMID:11031233; http://dx.doi.org/ 10.1146/annurev.cellbio.16.1.145 [DOI] [PubMed] [Google Scholar]
- 48. Brun RP, Kim JB, Hu E, Altiok S, Spiegelman BM. Adipocyte differentiation: a transcriptional regulatory cascade. Curr Opin Cell Biol 1996; 8:826-32; PMID:8939673; http://dx.doi.org/ 10.1016/S0955-0674(96)80084-6 [DOI] [PubMed] [Google Scholar]
- 49. Garten A, Schuster S, Kiess W. The insulin-like growth factors in adipogenesis and obesity. [v-vi.]. Endocrinol Metab Clin North Am 2012; 41:283-95, v-vi; PMID:22682631; http://dx.doi.org/ 10.1016/j.ecl.2012.04.011 [DOI] [PubMed] [Google Scholar]
- 50. Rodríguez Fernández JL, Ben-Ze’ev A. Regulation of fibronectin, integrin and cytoskeleton expression in differentiating adipocytes: inhibition by extracellular matrix and polylysine. Differentiation 1989; 42:65-74; PMID:2633939; http://dx.doi.org/ 10.1111/j.1432-0436.1989.tb00608.x [DOI] [PubMed] [Google Scholar]
- 51. Bortell R, Owen TA, Ignotz R, Stein GS, Stein JL. TGF β 1 prevents the down-regulation of type I procollagen, fibronectin, and TGF β 1 gene expression associated with 3T3-L1 pre-adipocyte differentiation. J Cell Biochem 1994; 54:256-63; PMID:8175900; http://dx.doi.org/ 10.1002/jcb.240540214 [DOI] [PubMed] [Google Scholar]
- 52. Cawthorn WP, Heyd F, Hegyi K, Sethi JK. Tumour necrosis factor-α inhibits adipogenesis via a β-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ 2007; 14:1361-73; PMID:17464333; http://dx.doi.org/ 10.1038/sj.cdd.4402127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Petruschke T, Hauner H. Tumor necrosis factor-alpha prevents the differentiation of human adipocyte precursor cells and causes delipidation of newly developed fat cells. J Clin Endocrinol Metab 1993; 76:742-7; PMID:8445033 [DOI] [PubMed] [Google Scholar]
- 54. Spinella-Jaegle S, Rawadi G, Kawai S, Gallea S, Faucheu C, Mollat P, Courtois B, Bergaud B, Ramez V, Blanchet AM, et al. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J Cell Sci 2001; 114:2085-94; PMID:11493644 [DOI] [PubMed] [Google Scholar]
- 55. Beresford JN, Bennett JH, Devlin C, Leboy PS, Owen ME. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci 1992; 102:341-51; PMID:1400636 [DOI] [PubMed] [Google Scholar]
- 56. Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J. Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J Bone Miner Res 2012; 27:2344-58; PMID:22729939; http://dx.doi.org/ 10.1002/jbmr.1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Muruganandan S, Roman AA, Sinal CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol Life Sci 2009; 66:236-53; PMID:18854943; http://dx.doi.org/ 10.1007/s00018-008-8429-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. James AW, Leucht P, Levi B, Carre AL, Xu Y, Helms JA, Longaker MT. Sonic Hedgehog influences the balance of osteogenesis and adipogenesis in mouse adipose-derived stromal cells. Tissue Eng Part A 2010; 16:2605-16; PMID:20367246; http://dx.doi.org/ 10.1089/ten.tea.2010.0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kang S, Bennett CN, Gerin I, Rapp LA, Hankenson KD, Macdougald OA. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor γ. J Biol Chem 2007; 282:14515-24; PMID:17351296; http://dx.doi.org/ 10.1074/jbc.M700030200 [DOI] [PubMed] [Google Scholar]
- 60. Li Y-M, Schilling T, Benisch P, Zeck S, Meissner-Weigl J, Schneider D, Limbert C, Seufert J, Kassem M, Schütze N, et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem Biophys Res Commun 2007; 363:209-15; PMID:17868648; http://dx.doi.org/ 10.1016/j.bbrc.2007.08.161 [DOI] [PubMed] [Google Scholar]
- 61. Shilpa K, Dinesh T, Lakshmi BS. An In Vitro Model to Probe the Regulation of Adipocyte Differentiation under Hyperglycemia. Diabetes Metab J 2013; 37:176-80; PMID:23807920; http://dx.doi.org/ 10.4093/dmj.2013.37.3.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS, Attie AD. The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci U S A 2000; 97:11371-6; PMID:11027337; http://dx.doi.org/ 10.1073/pnas.97.21.11371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP, Rajala MW, Du X, Rollman B, Li W, et al. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem 2005; 280:4617-26; PMID:15536073; http://dx.doi.org/ 10.1074/jbc.M411863200 [DOI] [PubMed] [Google Scholar]
- 64. Chuang CC, Yang RS, Tsai KS, Ho FM, Liu SH. Hyperglycemia enhances adipogenic induction of lipid accumulation: involvement of extracellular signal-regulated protein kinase 1/2, phosphoinositide 3-kinase/Akt, and peroxisome proliferator-activated receptor γ signaling. Endocrinology 2007; 148:4267-75; PMID:17540722; http://dx.doi.org/ 10.1210/en.2007-0179 [DOI] [PubMed] [Google Scholar]
- 65. Downward J. Lipid-regulated kinases: some common themes at last. Science 1998; 279:673-4; PMID:9471728; http://dx.doi.org/ 10.1126/science.279.5351.673 [DOI] [PubMed] [Google Scholar]
- 66. Xu J, Liao K. Protein kinase B/AKT 1 plays a pivotal role in insulin-like growth factor-1 receptor signaling induced 3T3-L1 adipocyte differentiation. J Biol Chem 2004; 279:35914-22; PMID:15192111; http://dx.doi.org/ 10.1074/jbc.M402297200 [DOI] [PubMed] [Google Scholar]
- 67. Cho HJ, Park J, Lee HW, Lee YS, Kim JB. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem Biophys Res Commun 2004; 321:942-8; PMID:15358118; http://dx.doi.org/ 10.1016/j.bbrc.2004.07.050 [DOI] [PubMed] [Google Scholar]
- 68. Guillet-Deniau I, Pichard A-L, Koné A, Esnous C, Nieruchalski M, Girard J, Prip-Buus C. Glucose induces de novo lipogenesis in rat muscle satellite cells through a sterol-regulatory-element-binding-protein-1c-dependent pathway. J Cell Sci 2004; 117:1937-44; PMID:15039461; http://dx.doi.org/ 10.1242/jcs.01069 [DOI] [PubMed] [Google Scholar]
- 69. Yue T, Yin J, Li F, Li D, Du M. High glucose induces differentiation and adipogenesis in porcine muscle satellite cells via mTOR. BMB Rep 2010; 43:140-5; PMID:20193134; http://dx.doi.org/ 10.5483/BMBRep.2010.43.2.140 [DOI] [PubMed] [Google Scholar]
- 70. Aguiari P, Leo S, Zavan B, Vindigni V, Rimessi A, Bianchi K, Franzin C, Cortivo R, Rossato M, Vettor R, et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc Natl Acad Sci U S A 2008; 105:1226-31; PMID:18212116; http://dx.doi.org/ 10.1073/pnas.0711402105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996; 19:257-67; PMID:8742574; http://dx.doi.org/ 10.2337/diacare.19.3.257 [DOI] [PubMed] [Google Scholar]
- 72. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000; 49:1939-45; PMID:11078463; http://dx.doi.org/ 10.2337/diabetes.49.11.1939 [DOI] [PubMed] [Google Scholar]
- 73. Sano T, Umeda F, Hashimoto T, Nawata H, Utsumi H. Oxidative stress measurement by in vivo electron spin resonance spectroscopy in rats with streptozotocin-induced diabetes. Diabetologia 1998; 41:1355-60; PMID:9833944; http://dx.doi.org/ 10.1007/s001250051076 [DOI] [PubMed] [Google Scholar]
- 74. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005; 54:1615-25; PMID:15919781; http://dx.doi.org/ 10.2337/diabetes.54.6.1615 [DOI] [PubMed] [Google Scholar]
- 75. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404:787-90; PMID:10783895; http://dx.doi.org/ 10.1038/35008121 [DOI] [PubMed] [Google Scholar]
- 76. Schwartz AV. Diabetes Mellitus: Does it Affect Bone? Calcif Tissue Int 2003; 73:515-9; PMID:14517715; http://dx.doi.org/ 10.1007/s00223-003-0023-7 [DOI] [PubMed] [Google Scholar]
- 77. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006; 114:597-605; PMID:16894049; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.106.621854 [DOI] [PubMed] [Google Scholar]
- 78. Jakuš V, Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol Res 2004; 53:131-42; PMID:15046548 [PubMed] [Google Scholar]
- 79. Franke S, Siggelkow H, Wolf G, Hein G. Advanced glycation endproducts influence the mRNA expression of RAGE, RANKL and various osteoblastic genes in human osteoblasts. Arch Physiol Biochem 2007; 113:154-61; PMID:17922311; http://dx.doi.org/ 10.1080/13813450701602523 [DOI] [PubMed] [Google Scholar]
- 80. Katayama Y, Akatsu T, Yamamoto M, Kugai N, Nagata N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J Bone Miner Res 1996; 11:931-7; PMID:8797113; http://dx.doi.org/ 10.1002/jbmr.5650110709 [DOI] [PubMed] [Google Scholar]
- 81. Santana RB, Xu L, Chase HB, Amar S, Graves DT, Trackman PC. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes 2003; 52:1502-10; PMID:12765963; http://dx.doi.org/ 10.2337/diabetes.52.6.1502 [DOI] [PubMed] [Google Scholar]
- 82. Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L, Graves DT. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 2007; 40:345-53; PMID:17064973; http://dx.doi.org/ 10.1016/j.bone.2006.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kume S, Kato S, Yamagishi S, Inagaki Y, Ueda S, Arima N, Okawa T, Kojiro M, Nagata K. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J Bone Miner Res 2005; 20:1647-58; PMID:16059636; http://dx.doi.org/ 10.1359/JBMR.050514 [DOI] [PubMed] [Google Scholar]
- 84. Zhang Y, Yang JH. Activation of the PI3K/Akt pathway by oxidative stress mediates high glucose-induced increase of adipogenic differentiation in primary rat osteoblasts. J Cell Biochem 2013; 114:2595-602; PMID:23757055; http://dx.doi.org/ 10.1002/jcb.24607 [DOI] [PubMed] [Google Scholar]
- 85. Christodoulides C, Lagathu C, Sethi JK, Vidal-Puig A. Adipogenesis and WNT signalling. Trends Endocrinol Metab 2009; 20:16-24; PMID:19008118; http://dx.doi.org/ 10.1016/j.tem.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 1997; 11:3286-305; PMID:9407023; http://dx.doi.org/ 10.1101/gad.11.24.3286 [DOI] [PubMed] [Google Scholar]
- 87. Miller JR. The Wnts. Genome Biol 2002; 3:S3001; PMID:11806834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab 2006; 4:263-73; PMID:17011499; http://dx.doi.org/ 10.1016/j.cmet.2006.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer SR. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J 2003; 376:607-13; PMID:12954078; http://dx.doi.org/ 10.1042/BJ20030426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor γ and β-catenin signaling during adipogenesis. A glycogen synthase kinase 3β phosphorylation-defective mutant of β-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem 2004; 279:45020-7; PMID:15308623; http://dx.doi.org/ 10.1074/jbc.M407050200 [DOI] [PubMed] [Google Scholar]
- 91. Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor γ and β-catenin. Mol Cell Biol 2006; 26:5827-37; PMID:16847334; http://dx.doi.org/ 10.1128/MCB.00441-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond) 2005; 29(Suppl 1):S13-6; PMID:15711576; http://dx.doi.org/ 10.1038/sj.ijo.0802907 [DOI] [PubMed] [Google Scholar]
- 93. Semënov MV, Habas R, MacDonald BT, He X. SnapShot: noncanonical Wnt signaling pathways. Cell 2007; 131:1378. e1-. e2. [DOI] [PubMed] [Google Scholar]
- 94. Takada I, Mihara M, Suzawa M, Ohtake F, Kobayashi S, Igarashi M, Youn MY, Takeyama K, Nakamura T, Mezaki Y, et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat Cell Biol 2007; 9:1273-85; PMID:17952062; http://dx.doi.org/ 10.1038/ncb1647 [DOI] [PubMed] [Google Scholar]
- 95. Fleming I, MacKenzie SJ, Vernon RG, Anderson NG, Houslay MD, Kilgour E. Protein kinase C isoforms play differential roles in the regulation of adipocyte differentiation. Biochem J 1998; 333:719-27; PMID:9677333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhou Y, Wang D, Li F, Shi J, Song J. Different roles of protein kinase C-betaI and -delta in the regulation of adipocyte differentiation. Int J Biochem Cell Biol 2006; 38:2151-63; PMID:16950644; http://dx.doi.org/ 10.1016/j.biocel.2006.06.009 [DOI] [PubMed] [Google Scholar]
- 97. Frevert EU, Kahn BB. Protein kinase C isoforms epsilon, eta, delta and zeta in murine adipocytes: expression, subcellular localization and tissue-specific regulation in insulin-resistant states. Biochem J 1996; 316:865-71; PMID:8670164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Webb PR, Doyle C, Anderson NG. Protein kinase C-epsilon promotes adipogenic commitment and is essential for terminal differentiation of 3T3-F442A preadipocytes. Cell Mol Life Sci 2003; 60:1504-12; PMID:12943236; http://dx.doi.org/ 10.1007/s00018-003-2337-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Accili D, Taylor SI. Targeted inactivation of the insulin receptor gene in mouse 3T3-L1 fibroblasts via homologous recombination. Proc Natl Acad Sci U S A 1991; 88:4708-12; PMID:2052553; http://dx.doi.org/ 10.1073/pnas.88.11.4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cinti S, Eberbach S, Castellucci M, Accili D. Lack of insulin receptors affects the formation of white adipose tissue in mice. A morphometric and ultrastructural analysis. Diabetologia 1998; 41:171-7; PMID:9498650; http://dx.doi.org/ 10.1007/s001250050886 [DOI] [PubMed] [Google Scholar]
- 101. Irwin R, Lin HV, Motyl KJ, McCabe LR. Normal bone density obtained in the absence of insulin receptor expression in bone. Endocrinology 2006; 147:5760-7; PMID:16973725; http://dx.doi.org/ 10.1210/en.2006-0700 [DOI] [PubMed] [Google Scholar]
- 102. Haffner SM, Stern MP, Hazuda HP, Pugh JA, Patterson JK. Hyperinsulinemia in a population at high risk for non-insulin-dependent diabetes mellitus. N Engl J Med 1986; 315:220-4; PMID:3523246; http://dx.doi.org/ 10.1056/NEJM198607243150403 [DOI] [PubMed] [Google Scholar]
- 103. Li C, Ford ES, Zhao G, Mokdad AH. Prevalence of pre-diabetes and its association with clustering of cardiometabolic risk factors and hyperinsulinemia among U.S. adolescents: National Health and Nutrition Examination Survey 2005-2006. Diabetes Care 2009; 32:342-7; PMID:18957533; http://dx.doi.org/ 10.2337/dc08-1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhang HH, Huang J, Düvel K, Boback B, Wu S, Squillace RM, Wu CL, Manning BD. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One 2009; 4:e6189; PMID:19593385; http://dx.doi.org/ 10.1371/journal.pone.0006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem 2007; 102:1343-57; PMID:17975793; http://dx.doi.org/ 10.1002/jcb.21573 [DOI] [PubMed] [Google Scholar]
- 106. Kershnar AK, Daniels SR, Imperatore G, Palla SL, Petitti DB, Pettitt DJ, Marcovina S, Dolan LM, Hamman RF, Liese AD, et al. Lipid abnormalities are prevalent in youth with type 1 and type 2 diabetes: the SEARCH for Diabetes in Youth Study. J Pediatr 2006; 149:314-9; PMID:16939739; http://dx.doi.org/ 10.1016/j.jpeds.2006.04.065 [DOI] [PubMed] [Google Scholar]
- 107. O’Brien T, Nguyen TT, Zimmerman BR. Hyperlipidemia and diabetes mellitus. Mayo Clin Proc 1998; 73:969-76; PMID:9787748; http://dx.doi.org/ 10.4065/73.10.969 [DOI] [PubMed] [Google Scholar]
- 108. Christeff N, Homo-Delarche F, Thobie N, Durant S, Dardenne M, Nunez EA. Free fatty acid profiles in the non-obese diabetic (NOD) mouse: basal serum levels and effects of endocrine manipulation. Prostaglandins Leukot Essent Fatty Acids 1994; 51:125-31; PMID:7972266; http://dx.doi.org/ 10.1016/0952-3278(94)90088-4 [DOI] [PubMed] [Google Scholar]
- 109. Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D, Rodan GA. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol Endocrinol 1992; 6:1634-41; PMID:1333051 [DOI] [PubMed] [Google Scholar]
- 110. Bragt MCE, Popeijus HE. Peroxisome proliferator-activated receptors and the metabolic syndrome. Physiol Behav 2008; 94:187-97; PMID:18191967; http://dx.doi.org/ 10.1016/j.physbeh.2007.11.053 [DOI] [PubMed] [Google Scholar]
- 111. Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. Divergent effects of selective peroxisome proliferator-activated receptor-γ 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology 2002; 143:2376-84; PMID:12021203 [DOI] [PubMed] [Google Scholar]
- 112. Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014; 510:542-6; PMID:24847880; http://dx.doi.org/ 10.1038/nature13270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Molinuevo MS, Schurman L, McCarthy AD, Cortizo AM, Tolosa MJ, Gangoiti MV, Arnol V, Sedlinsky C. Effect of metformin on bone marrow progenitor cell differentiation: in vivo and in vitro studies. J Bone Miner Res 2010; 25:211-21; PMID:19594306; http://dx.doi.org/ 10.1359/jbmr.090732 [DOI] [PubMed] [Google Scholar]
- 114. Grossman SL, Lessem J. Mechanisms and clinical effects of thiazolidinediones. Expert Opin Investig Drugs 1997; 6:1025-40; PMID:15989661; http://dx.doi.org/ 10.1517/13543784.6.8.1025 [DOI] [PubMed] [Google Scholar]
- 115. Wagstaff AJ, Goa KL. Rosiglitazone: a review of its use in the management of type 2 diabetes mellitus. Drugs 2002; 62:1805-37; PMID:12149047; http://dx.doi.org/ 10.2165/00003495-200262120-00007 [DOI] [PubMed] [Google Scholar]
- 116. Yki-Järvinen H. The PROactive study: some answers, many questions. Lancet 2005; 366:1241-2; PMID:16214581; http://dx.doi.org/ 10.1016/S0140-6736(05)67504-6 [DOI] [PubMed] [Google Scholar]
- 117. Aubert RE, Herrera V, Chen W, Haffner SM, Pendergrass M. Rosiglitazone and pioglitazone increase fracture risk in women and men with type 2 diabetes. Diabetes Obes Metab 2010; 12:716-21; PMID:20590749; http://dx.doi.org/ 10.1111/j.1463-1326.2010.01225.x [DOI] [PubMed] [Google Scholar]
- 118. Bodmer M, Meier C, Kraenzlin ME, Meier CR. Risk of fractures with glitazones: a critical review of the evidence to date. Drug Saf 2009; 32:539-47; PMID:19530741; http://dx.doi.org/ 10.2165/00002018-200932070-00001 [DOI] [PubMed] [Google Scholar]
- 119. Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu D, Heise MA, Aftring RP, et al.; Diabetes Outcome Progression Trial (ADOPT) Study Group. Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes Outcome Progression Trial (ADOPT). Diabetes Care 2008; 31:845-51; PMID:18223031; http://dx.doi.org/ 10.2337/dc07-2270 [DOI] [PubMed] [Google Scholar]
- 120. Lecka-Czernik B. Bone loss in diabetes: use of antidiabetic thiazolidinediones and secondary osteoporosis. Curr Osteoporos Rep 2010; 8:178-84; PMID:20809203; http://dx.doi.org/ 10.1007/s11914-010-0027-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999; 402:880-3; PMID:10622252 [DOI] [PubMed] [Google Scholar]
- 122. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000; 106:473-81; PMID:10953022; http://dx.doi.org/ 10.1172/JCI10842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Benvenuti S, Cellai I, Luciani P, Deledda C, Baglioni S, Giuliani C, Saccardi R, Mazzanti B, Dal Pozzo S, Mannucci E, et al. Rosiglitazone stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells. J Endocrinol Invest 2007; 30:RC26-30; PMID:17993761; http://dx.doi.org/ 10.1007/BF03350807 [DOI] [PubMed] [Google Scholar]
- 124. Hallakou S, Doaré L, Foufelle F, Kergoat M, Guerre-Millo M, Berthault MF, Dugail I, Morin J, Auwerx J, Ferré P. Pioglitazone induces in vivo adipocyte differentiation in the obese Zucker fa/fa rat. Diabetes 1997; 46:1393-9; PMID:9287037; http://dx.doi.org/ 10.2337/diabetes.46.9.1393 [DOI] [PubMed] [Google Scholar]
- 125. Khan MA, St Peter JV, Xue JLA. A prospective, randomized comparison of the metabolic effects of pioglitazone or rosiglitazone in patients with type 2 diabetes who were previously treated with troglitazone. Diabetes Care 2002; 25:708-11; PMID:11919129; http://dx.doi.org/ 10.2337/diacare.25.4.708 [DOI] [PubMed] [Google Scholar]
- 126. Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 2002; 87:2784-91; PMID:12050251; http://dx.doi.org/ 10.1210/jcem.87.6.8567 [DOI] [PubMed] [Google Scholar]
- 127. Mori Y, Murakawa Y, Okada K, Horikoshi H, Yokoyama J, Tajima N, Ikeda Y. Effect of troglitazone on body fat distribution in type 2 diabetic patients. Diabetes Care 1999; 22:908-12; PMID:10372240; http://dx.doi.org/ 10.2337/diacare.22.6.908 [DOI] [PubMed] [Google Scholar]
- 128. Beck GR, Jr., Khazai NB, Bouloux GF, Camalier CE, Lin Y, Garneys LM, Siqueira J, Peng L, Pasquel F, Umpierrez D, et al. The effects of thiazolidinediones on human bone marrow stromal cell differentiation in vitro and in thiazolidinedione-treated patients with type 2 diabetes. Transl Res 2013; 161:145-55; PMID:23022285; http://dx.doi.org/ 10.1016/j.trsl.2012.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Okazaki R, Toriumi M, Fukumoto S, Miyamoto M, Fujita T, Tanaka K, Takeuchi Y. Thiazolidinediones inhibit osteoclast-like cell formation and bone resorption in vitro. Endocrinology 1999; 140:5060-5; PMID:10537132 [DOI] [PubMed] [Google Scholar]
- 130. Bruedigam C, Eijken M, Koedam M, van de Peppel J, Drabek K, Chiba H, van Leeuwen JP. A new concept underlying stem cell lineage skewing that explains the detrimental effects of thiazolidinediones on bone. Stem Cells 2010; 28:916-27; PMID:20213769 [DOI] [PubMed] [Google Scholar]
- 131. Lecka-Czernik B, Gubrij I, Moerman EJ, Kajkenova O, Lipschitz DA, Manolagas SC, Jilka RL. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2. J Cell Biochem 1999; 74:357-71; PMID:10412038; http://dx.doi.org/ 10.1002/(SICI)1097-4644(19990901)74:3≤357::AID-JCB5≥3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 132. Jeon MJ, Kim JA, Kwon SH, Kim SW, Park KS, Park SW, Kim SY, Shin CS. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J Biol Chem 2003; 278:23270-7; PMID:12704187; http://dx.doi.org/ 10.1074/jbc.M211610200 [DOI] [PubMed] [Google Scholar]
- 133. Khan E, Abu-Amer Y. Activation of peroxisome proliferator-activated receptor-γ inhibits differentiation of preosteoblasts. J Lab Clin Med 2003; 142:29-34; PMID:12878983; http://dx.doi.org/ 10.1016/S0022-2143(03)00058-1 [DOI] [PubMed] [Google Scholar]
- 134. Okazaki R, Miura M, Toriumi M, Taguchi M, Hirota Y, Fukumoto S, Fujita T, Tanaka K, Takeuchi A. Short-term treatment with troglitazone decreases bone turnover in patients with type 2 diabetes mellitus. Endocr J 1999; 46:795-801; PMID:10724355; http://dx.doi.org/ 10.1507/endocrj.46.795 [DOI] [PubMed] [Google Scholar]
- 135. Watanabe S, Takeuchi Y, Fukumoto S, Fujita H, Nakano T, Fujita T. Decrease in serum leptin by troglitazone is associated with preventing bone loss in type 2 diabetic patients. J Bone Miner Metab 2003; 21:166-71; PMID:12720051; http://dx.doi.org/ 10.1007/s007740300026 [DOI] [PubMed] [Google Scholar]
- 136. Berberoglu Z, Gursoy A, Bayraktar N, Yazici AC, Bascil Tutuncu N, Guvener Demirag N. Rosiglitazone decreases serum bone-specific alkaline phosphatase activity in postmenopausal diabetic women. J Clin Endocrinol Metab 2007; 92:3523-30; PMID:17595249; http://dx.doi.org/ 10.1210/jc.2007-0431 [DOI] [PubMed] [Google Scholar]
- 137. Glintborg D, Andersen M, Hagen C, Heickendorff L, Hermann AP. Association of pioglitazone treatment with decreased bone mineral density in obese premenopausal patients with polycystic ovary syndrome: a randomized, placebo-controlled trial. J Clin Endocrinol Metab 2008; 93:1696-701; PMID:18285411; http://dx.doi.org/ 10.1210/jc.2007-2249 [DOI] [PubMed] [Google Scholar]
- 138. Grey A, Bolland M, Gamble G, Wattie D, Horne A, Davidson J, Reid IR. The peroxisome proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J Clin Endocrinol Metab 2007; 92:1305-10; PMID:17264176; http://dx.doi.org/ 10.1210/jc.2006-2646 [DOI] [PubMed] [Google Scholar]
- 139. Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest 2004; 113:846-55; PMID:15067317; http://dx.doi.org/ 10.1172/JCI200419900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kawaguchi H, Akune T, Yamaguchi M, Ohba S, Ogata N, Chung UI, Kubota N, Terauchi Y, Kadowaki T, Nakamura K. Distinct effects of PPARgamma insufficiency on bone marrow cells, osteoblasts, and osteoclastic cells. J Bone Miner Metab 2005; 23:275-9; PMID:15981022; http://dx.doi.org/ 10.1007/s00774-005-0599-2 [DOI] [PubMed] [Google Scholar]
- 141. Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J. Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J Bone Miner Res 2012; 27:2344-58; PMID:22729939; http://dx.doi.org/ 10.1002/jbmr.1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Botolin S, McCabe LR. Inhibition of PPARgamma prevents type I diabetic bone marrow adiposity but not bone loss. J Cell Physiol 2006; 209:967-76; PMID:16972249; http://dx.doi.org/ 10.1002/jcp.20804 [DOI] [PubMed] [Google Scholar]
- 143. Botolin S, McCabe LR. Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J Cell Biochem 2006; 99:411-24; PMID:16619259; http://dx.doi.org/ 10.1002/jcb.20842 [DOI] [PubMed] [Google Scholar]
- 144. He H, Liu R, Desta T, Leone C, Gerstenfeld LC, Graves DT. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology 2004; 145:447-52; PMID:14525917; http://dx.doi.org/ 10.1210/en.2003-1239 [DOI] [PubMed] [Google Scholar]
- 145. Wang W, Zhang X, Zheng J, Yang J. High glucose stimulates adipogenic and inhibits osteogenic differentiation in MG-63 cells through cAMP/protein kinase A/extracellular signal-regulated kinase pathway. Mol Cell Biochem 2010; 338:115-22; PMID:19949837; http://dx.doi.org/ 10.1007/s11010-009-0344-6 [DOI] [PubMed] [Google Scholar]
- 146. Zayzafoon M, Stell C, Irwin R, McCabe LR. Extracellular glucose influences osteoblast differentiation and c-Jun expression. J Cell Biochem 2000; 79:301-10; PMID:10967557; http://dx.doi.org/ 10.1002/1097-4644(20001101)79:2≤301::AID-JCB130≥3.0.CO;2-0 [DOI] [PubMed] [Google Scholar]
- 147. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001; 7:941-6; PMID:11479627; http://dx.doi.org/ 10.1038/90984 [DOI] [PubMed] [Google Scholar]
- 148. Yokota T, Meka CS, Medina KL, Igarashi H, Comp PC, Takahashi M, Nishida M, Oritani K, Miyagawa J, Funahashi T, et al. Paracrine regulation of fat cell formation in bone marrow cultures via adiponectin and prostaglandins. J Clin Invest 2002; 109:1303-10; PMID:12021245; http://dx.doi.org/ 10.1172/JCI0214506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Oshima K, Nampei A, Matsuda M, Iwaki M, Fukuhara A, Hashimoto J, Yoshikawa H, Shimomura I. Adiponectin increases bone mass by suppressing osteoclast and activating osteoblast. Biochem Biophys Res Commun 2005; 331:520-6; PMID:15850790; http://dx.doi.org/ 10.1016/j.bbrc.2005.03.210 [DOI] [PubMed] [Google Scholar]
- 150. Lee HW, Kim SY, Kim AY, Lee EJ, Choi J-Y, Kim JB. Adiponectin stimulates osteoblast differentiation through induction of COX2 in mesenchymal progenitor cells. Stem Cells 2009; 27:2254-62; PMID:19522015; http://dx.doi.org/ 10.1002/stem.144 [DOI] [PubMed] [Google Scholar]
- 151. Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Yamamoto M, Sugimoto T. Adiponectin and AMP kinase activator stimulate proliferation, differentiation, and mineralization of osteoblastic MC3T3-E1 cells. BMC Cell Biol 2007; 8:51; PMID:18047638; http://dx.doi.org/ 10.1186/1471-2121-8-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995; 1:1155-61; PMID:7584987; http://dx.doi.org/ 10.1038/nm1195-1155 [DOI] [PubMed] [Google Scholar]
- 153. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996; 334:292-5; PMID:8532024; http://dx.doi.org/ 10.1056/NEJM199602013340503 [DOI] [PubMed] [Google Scholar]
- 154. Kiess W, Anil M, Blum WF, Englaro P, Juul A, Attanasio A, Dötsch J, Rascher W. Serum leptin levels in children and adolescents with insulin-dependent diabetes mellitus in relation to metabolic control and body mass index. Eur J Endocrinol 1998; 138:501-9; PMID:9625360; http://dx.doi.org/ 10.1530/eje.0.1380501 [DOI] [PubMed] [Google Scholar]
- 155. Cirmanová V, Bayer M, Stárka L, Zajícková K. The effect of leptin on bone: an evolving concept of action. Physiol Res 2008; 57(Suppl 1):S143-51; PMID:18271682 [DOI] [PubMed] [Google Scholar]
- 156. Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept 2000; 92:73-8; PMID:11024568; http://dx.doi.org/ 10.1016/S0167-0115(00)00152-X [DOI] [PubMed] [Google Scholar]
- 157. Martin A, David V, Malaval L, Lafage-Proust M-H, Vico L, Thomas T. Opposite effects of leptin on bone metabolism: a dose-dependent balance related to energy intake and insulin-like growth factor-I pathway. Endocrinology 2007; 148:3419-25; PMID:17431002; http://dx.doi.org/ 10.1210/en.2006-1541 [DOI] [PubMed] [Google Scholar]
- 158. Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell 2002; 111:305-17; PMID:12419242; http://dx.doi.org/ 10.1016/S0092-8674(02)01049-8 [DOI] [PubMed] [Google Scholar]
- 159. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin Inhibits Bone Formation through a Hypothalamic Relay: A Central Control of Bone Mass. Cell 2000; 100:197-207. [DOI] [PubMed] [Google Scholar]
- 160. Motyl KJ, McCabe LR. Leptin treatment prevents type I diabetic marrow adiposity but not bone loss in mice. J Cell Physiol 2009; 218:376-84; PMID:18932203; http://dx.doi.org/ 10.1002/jcp.21608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 1995; 95:2409-15; PMID:7738205; http://dx.doi.org/ 10.1172/JCI117936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Boyce BF, Li P, Yao Z, Zhang Q, Badell IR, Schwarz EM, O’Keefe RJ, Xing L. TNF-alpha and pathologic bone resorption. Keio J Med 2005; 54:127-31; PMID:16237274; http://dx.doi.org/ 10.2302/kjm.54.127 [DOI] [PubMed] [Google Scholar]
- 163. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-α induces differentiation of and bone resorption by osteoclasts. J Biol Chem 2000; 275:4858-64; PMID:10671521; http://dx.doi.org/ 10.1074/jbc.275.7.4858 [DOI] [PubMed] [Google Scholar]