

Abstract
Berberine interfering with cancer reprogramming metabolism was confirmed in our previous study. Lipid metabolism and mitochondrial function were also the core parts in reprogramming metabolism. In the presence of some energy-related inhibitors, including C75, compound C, and TOFA, the discrete roles of berberine in lipid metabolism and mitochondrial function were elucidated. An altered lipid metabolism induced by berberine was observed under the inhibition of FASN, AMPK, and ACC in breast cancer cell MCF-7. And the reversion of berberine-induced lipid suppression indicated that ACC inhibition might be involved in that process instead of FASN inhibition. A robust apoptosis induced by berberine even under the inhibition of AMPK and lipid synthesis was also indicated. Finally, mitochondrial function regulation under the inhibition of AMPK and ACC might be in an ACL-independent manner. Undoubtedly, the detailed mechanisms of berberine interfering with lipid metabolism and mitochondrial function combined with energy-related inhibitors need further investigation, including the potential compensatory mechanisms for ATP production and the upregulation of ACL.
1. Introduction
Lipid metabolism plays discrete roles in cancer reprogramming metabolism, including the conventional roles, namely, membrane structure generation, providing signaling molecules and posttranslational modification of proteins, and the novel roles, namely, participating in autophagy and metastasis, involved in microenvironment and angiogenesis. It was systematically expounded in Baenke's review [1]. Even lipolysis also contributes to cancer pathogenesis [2]. Collectively, lipid metabolism in cancer cells was context dependent and would generate intricate connections in cancer reprogramming metabolism. Mitochondrial function was another core part of cancer metabolism [3–6] and was also implied in many aspects, including therapeutic resistance [7–17], autophagy [18–21], drug sensitivity [22–24], apoptosis [25–38], metastasis [39–42], and angiogenesis [43]. Mitochondrial function was regarded as a metabolic symbiosis between tumor stromal cells and epithelial cancer cells in human breast cancer [5]. The final exhibited mitochondrial function might be a result of concerted action of the dysfunction of stromal cells and the biogenesis of epithelial cancer cells. Our study focused on the effect of berberine on lipid metabolism and mitochondrial function in presence of C75, compound C, and TOFA in cancer cells, which is relatively rare. We tested the induced apoptosis, lipid droplets content, and mitochondrial function alteration in presence of these inhibitors, in order to explore the underlying mechanisms of berberine regulating reprogramming metabolism of cancer cells when combined with conventional inhibitors.
C75 (4-methylene-2-octyl-5-oxotetra-hydrofuran-3-carboxylic acid) is an analogue of cerulenin [44] and is a stable inhibitor of fatty acid synthase (FASN). In pancreatic ductal adenocarcinoma cells Colo357, C75 induced increased apoptosis in a dose-dependent manner in vitro and 50% of reduction of primary pancreatic tumor weight in mouse orthotopic tumor in vivo, accompanied by an antimetastasis effect [45]; similar results of growth suppression were also observed in LNCaP prostate cancer cell in vitro and in LNCaP/tk-luc bearing animal model in vivo [46]. And, in breast cancer cell lines, C75 inhibited FASN and stimulated beta-oxidation through carnitine palmitoyltransferase-1 (CPT-1) regulation [47, 48] or directly suppressing HER2 and FASN phosphorylation [49]. And this similar efficacy was also observed in lung cancer cells A549 [50]. Compound C (also called dorsomorphin, 6-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-3-pyridin-4-yl-pyrazolo [1,5-a] pyrimidine) is a reversible ATP-competitive inhibitor of adenosine 5′-monophosphate- (AMP-) activated protein kinase (AMPK) [51]. It induced apoptosis in A549, SMMC-7721, and HeLa cells and exerted transcriptional repression through increasing p53 expression and decreasing the expression of Bcl-2 and Bcl-xl and the phosphorylation of eIF2α on Ser51 [52]. Compound C also induced autophagy in cancer cells, including human glioma cells U251, rat glioma cells C6, mouse fibrosarcoma L929, and mouse melanoma cells B16. Notably, the routine AMPK-dependent manner was challenged and discussed in some studies. An induced protective autophagy through AMPK inhibition-independent blockade of Akt/mTOR pathway was presented [51]. And this AMPK-independent manner was also illustrated in the induced necroptosis and autophagy of glioma cells [53], in the sensitization of TRAIL-induced apoptosis in Caki human renal cancer cells [54], and in the inhibition of transcriptional activation of UPR-targeted genes [55]. And in human colorectal cancer cells, including HCT116, DLD-1, SW480, and KM12C, an induced apoptotic or autophagic death was observed simultaneously [56]. The concurrent effect of apoptosis and autophagy by compound C was also found in skin cancer cells and was demonstrated by means of p53 status [57]. TOFA (5-tetradecyloxy-2-furoic acid) is an allosteric inhibitor of acetyl-CoA carboxylase (ACC), also reducing endogenous fatty acid for phospholipid composition of cell membrane [58]. Initially, TOFA showed an inhibition on fatty acid synthesis in hepatocytes, rat liver homogenates, or male rat liver [59–62], and now it showed a wider range of proliferation inhibition on cancer cells, including ovarian cancer cells COC1 and COC1/DDP [63], human prostatic carcinoma cells LNCaP [58], lung cancer cells NCI-H460, and colon carcinoma cells HCT-8 and HCT-15 [64].
Referring to the interactions between berberine and C75, compound C and TOFA in cancer cells, a definitive conclusion warranted more caution because of the insufficient researches. In liver cells HL-7702, the induced Raf-1 signaling stimulation by berberine was blocked by compound C [26]. And in human hepatoma cell line HepG2, compound C suppressed apoptosis and autophagy induced by berberine [65] but showed no influence on the stimulated glucose utilization and lactate production induced by berberine [66]. Our study emphasized the regulation of berberine on lipid metabolism and mitochondrial function in presence of energy-related inhibitors in breast cancer cells MCF-7, in order to elucidate the underlying mechanisms of berberine interfering with cancer reprogramming metabolism.
2. Materials and Methods
2.1. Cell Culture and Reagents
Human breast cancer cell line MCF-7 was purchased from American Type Culture Collection (America) and was cultured with RPMI1640 medium containing fetal bovine serum (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL), at 37°C in a humidified atmosphere of 5% CO2 in air.
2.2. Cell Viability Assay
MTT assay was conducted to determine cell growth and viability. MCF-7 cells were seeded in 96-well plate at density of 1 × 105/mL. After cell adhesion, a 24-hour serum starvation was carried out for synchronization before drug treatment. Pretreatments of C75, CC, and TOFA for 3 hours were conducted; the concentrations of these stocks were 5 mg/mL, 4 mg/mL, and 5 mg/mL, respectively, which were soluble in DMSO. And the dilutions were 1 : 1000, 1 : 1000, and 1 : 500, respectively, for C75, CC, and TOFA. Berberine treatment duration was 24 hours, and concentrations were 10 μM or 25 μM as required. Cell viability was determined under 570 nm and expressed as percentage of control.
2.3. Mitochondrial Membrane Potential Assay
MCF-7 cells were seeded in 96-well plate at density of 1 × 105/mL. After cell adhesion, a 24-hour serum starvation was carried out for synchronization. Drug treatment was according to the mentioned above. Cells were incubated with JC-1 probe (working concentration is 2.5 μg/mL) at 37°C for 1 hour. Determination was conducted under Ex/Em 550 nm/600 nm for red fluorescence and Ex/Em 485 nm/535 nm for green fluorescence, and the ratio of red and green was calculated.
2.4. ROS Level Assay
MCF-7 cells were seeded in 96-well plate at density of 1 × 105/mL. After cell adhesion, drug treatment was according to the mentioned above. Cells were incubated with H2DCF-DA probe (stock concentration is 1 mM and working concentration is 10 μM) for 1 hour and determined by multifunctional microplate reader SpectraMax M5 under Ex/Em 485 nm/530 nm. Then the diluted Hoechst 33342 was determined by M5 under Ex/Em 355 nm/465 nm.
2.5. Lipid Droplets Assay
MCF-7 cells were seeded in 96-well plate at density of 1 × 105/mL. After cell adhesion, drug treatment was according to the mentioned above. Cells were fixed with formalin (10%) and stained in Oil-Red-O (sigma, O0625) solution (stock solution is 5 g/L, dissolved in 2-isopropanol; ratio of the working solution is 6 : 4 [Oil-Red-O stock: MilliQ]) for 1 hour. Cells were rinsed with 60% propanol briefly, then with dH2O exhaustively. Isopropanol was added to determine lipid droplets content in cells. The extracted dye was transferred into a new 96-well plate for determination under 490 nm by M5, and the values were presented as the percentage of control.
2.6. Mitochondrial Function Assay
MCF-7 cells were seeded in 96-well plate at density of 1 × 105/mL. After cell adhesion, a 24-hour serum starvation was carried out for synchronization. Drug treatment was according to the mentioned above. Mitochondrial deep red probe solution was incubated and protected from light for 1 hour (working concentration is 1 μM in PBS). Then, cells were fixed with paraformaldehyde (3.7%) for 45 mins and were incubated with triton X-100 (0.2%) under room temperature for 30 mins. Function determination was conducted under Ex/Em 644 nm/665 nm with multifunctional microplate reader SpectraMax M5 and DAPI staining determination under Ex/Em 355 nm/460 nm.
2.7. ATP Production Assay
MCF-7 cells were seeded in Petri dish at density of 2 × 106/mL. After cell adhesion, drug treatment was according to the mentioned above. According to the protocol of ATP colorimetric assay kit, determination was conducted under absorbance wavelength of 570 nm and values were presented as the percentage of control.
2.8. Western Blotting
MCF-7 cells were seeded in 45 mm flask at density of 1 × 106/mL. After cell adhesion, drug treatment was according to the mentioned above. Cell lysate was collected and spun for 5 mins under 4°C, and the total protein content was determined according to BCA kit protocol. The equal weight of total protein was subjected to SDS-PAGE (6%) gels and transferred to methanol-treated PVDF membrane. One-hour blocking in nonfat milk (5%), 2-hour incubation for the first antibodies (cell signaling, 1 : 1000 dilution), and 1-hour incubation for the secondary antibodies were performed. ECL advanced western blotting detection kit (Amersham) was used for visualization, and the band density was normalized by the density of beta-actin.
3. Results and Discussion
3.1. Results
3.1.1. Cell Viability of Human Breast Cancer Cells MCF-7 Was Not Inhibited by Pretreatments of C75, Compound C, and TOFA, Even in the Presence of Berberine
FASN inhibitor C75, AMPK inhibitor compound C (CC), and ACC inhibitor TOFA were conducted to test cell viability influence in human breast cancer cell line MCF-7. After 3-hour pretreatments of C75, CC, and TOFA, 24-hour cell viability was determined by MTT assay with no significance. And, in the presence of berberine (25 μM for 24 hours), these pretreatments also showed no significant variance, which was shown in Figure 1.
Figure 1.
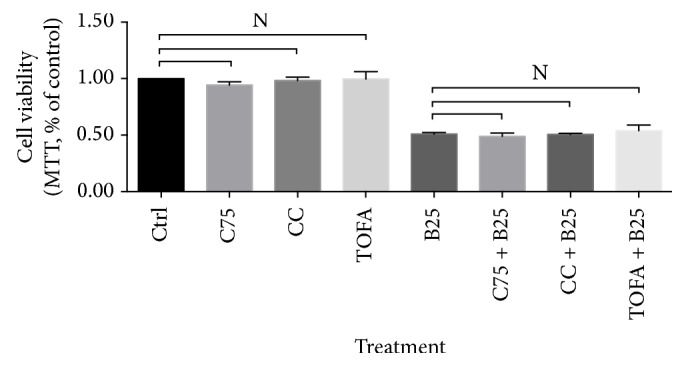
Cell viability of human breast cancer cell line MCF-7 in presence of C75, CC, and TOFA. Cell viability was obtained by MTT assay after pretreatment of C75 (5 μg/mL), CC (4 μg/mL), and TOFA (10 μg/mL) for 3 hours and treatment of berberine (25 μM) for 24 hours. Cell viability was presented as percentage of control, and significance difference was calculated by Student's test, p value ∗ < 0.05.
3.1.2. Berberine Decreased the Elevated Mitochondrial Membrane Potential Induced by Compound C or TOFA
Berberine significantly decreased mitochondrial membrane potential in human breast cancer cells MCF-7. The pretreatments of CC and TOFA dramatically increased mitochondrial membrane potential. But the subsequent treatment of berberine at dosage of 25 μM for 24 hours reversed this increase with significance, as shown in Figure 2.
Figure 2.
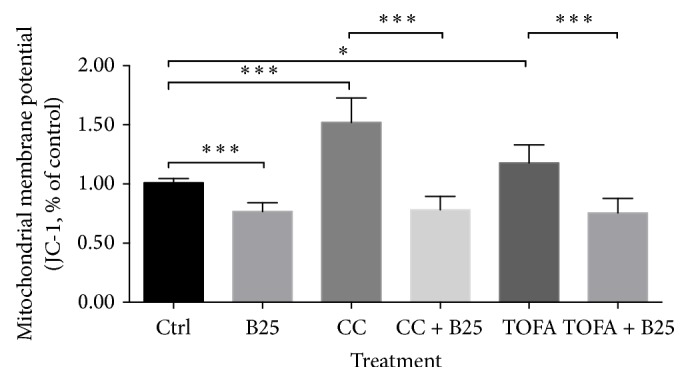
Mitochondrial membrane potential alteration of MCF-7 cells in presence of CC and TOFA. Mitochondrial membrane potential of MCF-7 cells was assayed by JC-1 probe after pretreatment of CC (4 μg/mL) and TOFA (10 μg/mL) for 3 hours and treatment of berberine (25 μM) for 24 hours. Mitochondrial membrane potential alterations were presented as percentage of control, and all significant difference was calculated by Student's test and presented as ∗ and ∗∗∗, p value <0.05 and <0.005.
3.1.3. Berberine Interfered with ROS Generation after CC or TOFA Pretreatment
Although berberine increased ROS level in MCF-7 cells, it remarkably reduced ROS level induced by TOFA, as shown in Figure 3. While in CC pretreatment groups, ROS content was not disturbed by CC pretreatment and also not disturbed by subsequent treatment of berberine at dosage of 25 μM for 24 hours.
Figure 3.
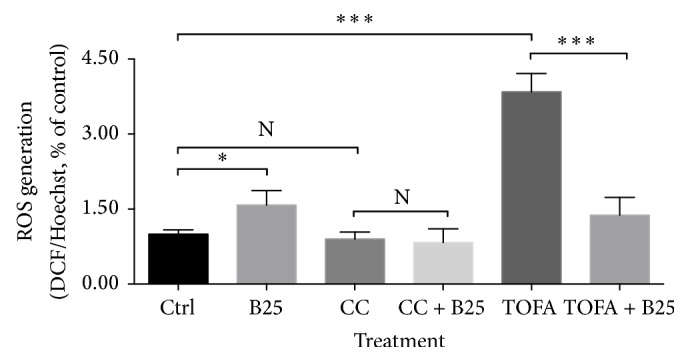
ROS level of MCF-7 cells in presence of CC and TOFA. ROS level of MCF-7 cells was assayed by H2DCF-DA/Hoechst 33342 after pretreatment of CC (4 μg/mL) and TOFA (10 μg/mL) for 3 hours and treatment of berberine (25 μM) for 24 hours. ROS generation was presented as percentage of control, and all significant difference was calculated by Student's test and presented as ∗ and ∗∗∗, p value <0.05 and <0.005.
3.1.4. TOFA and C75 Influenced the Efficacy of Berberine on Lipid Droplets
No significant variations were found in lipid droplets content after the treatment of C75, TOFA, and the combination of both in MCF-7 cells, as shown in Figures 4(a), 4(b), and 4(c). Berberine treatment inhibited the content of lipid droplets significantly in MCF-7 cells, and the less lipid droplets were also observed in MDA-MB-231 cells in our previous study. But, in the presence of C75, TOFA, or the combination of both, the decreased content of lipid droplets induced by berberine changed. Briefly, berberine increased the content of lipid droplets after TOFA pretreatment and decreased the content of lipid droplets after C75 pretreatment. And, after the pretreatment of the combination of C75 and TOFA, berberine showed no significance on lipid droplets alteration.
Figure 4.

Lipid droplets alteration of MCF-7 cells in presence of C75, TOFA, and the combination of both. Lipid droplets alteration of MCF-7 cells was assayed by Oil-Red-O staining after pretreatment of C75 (5 μg/mL) (a), TOFA (10 μg/mL) (b), and the combination of both (c) for 3 hours and treatment of berberine (10 μM) for 24 hours. Lipid droplets content was presented as percentage of control, and all significant difference was calculated by Student's test and presented as ∗, p value <0.05.
3.1.5. In the Presence of C75, CC, and TOFA, the Berberine-Induced Mitochondrial Function Alterations Changed
In the absence of C75, CC, and TOFA, the berberine-induced mitochondrial function alterations changed in MCF-7 cells using a real-time determination, as shown in Figure 5. However, in the presence of C75, mitochondrial function elevated moderately and the subsequent berberine treatment suppressed this elevation slightly in Figure 5(a). And, in the presence of CC, the elevated mitochondrial function was suppressed dramatically by berberine treatment in Figure 5(b). Conversely, in the presence of TOFA, mitochondrial function was inhibited remarkably by pretreatment and the subsequent berberine treatment reversed this inhibition substantially in Figure 5(c).
Figure 5.

Mitochondrial function alteration of MCF-7 cells in presence of C75, CC, and TOFA. Mitochondrial function alteration of MCF-7 cells was assayed by mitochondrial deep red real-time determination after pretreatment of C75 (5 μg/mL) (a), CC (4 μg/mL), (b) and TOFA (10 μg/mL) (c) for 3 hours and treatment of berberine (25 μM) for 12 hours. During 12-hour duration, a real-time alteration of mitochondrial function was detected and the curves were presented as percentage of responding value on the initial time point.
3.1.6. CC and TOFA Influenced the Efficacy of Berberine on ATP Production
Berberine significantly increased ATP level in MCF-7 cells in our previous study. And CC or TOFA pretreatment could also increase ATP level in MCF-7 cells. But, in the presence of CC, this elevation induced by berberine treatment was not observed as shown in Figure 6. And, in the presence of TOFA, this elevation was reversed significantly by berberine treatment.
Figure 6.
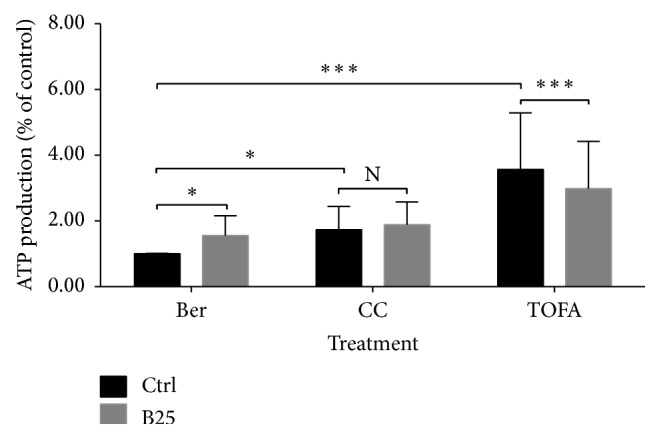
ATP production of MCF-7 cells in presence of CC and TOFA. ATP production of MCF-7 cells was assayed by ATP assay kit after pretreatment of CC (4 μg/mL) and TOFA (10 μg/mL) for 3 hours and treatment of berberine (25 μM) for 24 hours. ATP production was presented as percentage of control, and all significant difference was calculated by Student's test and presented as ∗ and ∗∗∗, p value <0.05 and <0.005.
3.1.7. Berberine Increased Phosphor ACL despite the Presence of CC or TOFA
Berberine increased phosphor ACL in MCF-7 cells. Even after CC inhibited phosphor AMPK significantly, the elevated expression of phosphor ACL induced by berberine was still observed in Figure 7(b). Similar situation was also observed in TOFA pretreatment. After TOFA inhibited total ACC significantly, berberine treatment increased the expression of phosphor ACL, the same as the increase in the absence of TOFA in Figure 7(c).
Figure 7.

Phosphor ACL alteration of MCF-7 cells in presence of CC and TOFA. Phosphor ACL alteration of MCF-7 cells was assayed by western blotting after pretreatment of CC (4 μg/mL) and TOFA (10 μg/mL) for 3 hours and treatment of berberine (25 μM) for 24 hours. The expression alterations of the corresponding proteins were visualized by band densitometry (a) and were presented as percentage of control (b). All significant difference was calculated by Student's test and presented as ∗, ∗∗, and ∗∗∗∗, p value <0.05, <0.01, and <0.001.
3.2. Discussion
3.2.1. Berberine Induced Apoptosis after C75, CC, and TOFA Pretreatments
No significances were observed in cell viability after pretreatments of C75, CC, and TOFA despite the absence or presence of berberine. The MTT assay for cell viability excluded the potential impact of inhibitors on cell growth, which facilitated the unilateral mechanism study of berberine on cell lipid metabolism. In the absence of CC or TOFA, berberine induced a significant reduction of mitochondrial membrane potential in MCF-7 cells. However, in the presence of CC or TOFA, this substantial reduction induced by berberine was still observed with significance, even with more significant differences. In other words, the suppression induced by berberine still lingered in independent pattern with the existence of CC or TOFA. Given that CC is an inhibitor of AMPK and TOFA is an inhibitor of ACC, the decreased mitochondrial membrane potential independent of CC or TOFA indicated a slight relationship between mitochondrial depolarization and AMPK inhibition or ACC inhibition. Actually, AMPK regulation or ACC inhibition by berberine was also found in others' studies [67–69]. And, in our study, this independent manner indicated that berberine efficacy on mitochondrial membrane potential was not associated with AMPK or ACC inhibition. Mitochondrial membrane potential assay by JC-1 probe also determined the proportion of nonapoptosis cells. This robust suppression of berberine indicated the induced apoptosis even under AMPK inhibition and lipid synthesis inhibition. ROS generation was another hallmark event of apoptosis, and ROS level also elevated after berberine treatment. But, in the presence of CC, the elevation induced by berberine was abolished, which indicated a blockage of berberine-induced apoptosis. However, in the presence of TOFA, the elevation induced by berberine was reversed significantly. The substantial increase of ROS level induced by TOFA was inhibited dramatically by berberine treatment. The efficacy of stimulating ROS level by berberine in the absence of TOFA changed into suppressing ROS level in the presence of TOFA. This might suggest a palliation of berberine on TOFA-induced ROS generation, also indicating that berberine might attenuate the TOFA-induced apoptosis.
3.2.2. Berberine Regulated Lipid Metabolism in the Presence of C75 and TOFA
Lipid metabolism was found closely related to membranes synthesis, energy homeostasis, and signaling function for cancer cells [70, 71]. Lipid droplets content was also higher than that of normal tissue. In colorectal cancer, the high level of lipid droplets was regarded as a distinctive mark of cancer stem cells [72]. We chose breast cancer cells MDA-MB-231 to conduct Oil-Red-O staining, because it contained much more lipid droplets and a clearer observation could be obtained. Less lipid droplets were clearly visible after berberine treatment in MDA-MB-231 cells (data were not shown). Then, the lipid droplets content in MCF-7 cells assayed by spectrometer M5 also confirmed the reduced quantity induced by berberine. Despite ACC inhibition by TOFA or FASN inhibition by C75, the lipid droplets content in MCF-7 cells showed no significant alteration. This indicated that the ACC inhibition or FASN inhibition would not directly alter the ultimate lipid droplets content. But, in the presence of TOFA or C75, we found that TOFA reversed the efficacy of berberine on lipid metabolism but not C75. And in the combination treatment of TOFA and C75, because of the existence of TOFA, the abolished efficacy was obtained, as shown in Figure 4 with no significance. Collectively, ACC inhibition by TOFA reversed the suppression of berberine on lipid metabolism, while FASN inhibition was not powerful as ACC inhibition. Actually, in our previous study, berberine was also found to inhibit ACC expression in a dose and time dependent manner in MCF-7 cells [73]. Since berberine and TOFA both inhibited ACC expression, the content of lipid droplets in presence of TOFA needed further investigations.
3.2.3. Berberine Regulated Mitochondrial Function in the Presence of C75, CC, and TOFA
Under the different backgrounds, the efficacy of berberine on mitochondrial function in real time showed different characteristics. Briefly, in the presence of C75 and CC, berberine exhibited suppression on mitochondrial function in varying levels. The former was slight and the latter was conspicuous, but both were suppression. On the contrary, in the presence of TOFA, berberine exhibited stimulation on mitochondrial function with a substantial level. And, in our previous study, the efficacy of berberine on mitochondrial function was prone to be stimulation in a short-term manner [73]. In other words, after AMPK inhibition, the efficacy of berberine transformed into slight suppression on mitochondrial function; after FASN inhibition, the efficacy transformed into suppression with a greater degree; and, after ACC inhibition, the stimulation by berberine was amplified and observed significantly. Therefore, the efficacy of berberine on mitochondrial function was closely related to ACC inhibition and could be blocked by AMPK or FASN inhibition. Since berberine was found to inhibit ACC through phosphorylation in our previous study, TOFA took over the ACC inhibition and amplified stimulation of berberine substantially. In ATP level assay, CC abolished the elevated ATP content induced by berberine and TOFA reversed the elevated ATP significantly. Collectively, the slight suppression on mitochondrial function by berberine in presence of CC was companied with the abolishment of the elevated ATP content; and the remarkable stimulation on mitochondrial function by berberine in presence of TOFA was companied with the reversal of the elevated ATP content. Therefore, the latter, the remarkable stimulation of mitochondrial function accompanied by the reduced ATP content, should deserve more attention than the former. This might be due to the fact that mitochondrial function determination was a real-time assay while ATP level was an end-point assay. ATP level indicated the final quantity in this process compared with the control group, while function assay showed a dynamic change of this process. Although the dynamic process was stimulated, the net output of this process might be reduced finally. In this process, many other compensatory mechanisms might be involved and lead to the final reduction of ATP content in MCF-7 cells. Besides, the upregulated ACL was regarded as a key player in lipid metabolism in cancer cells [74–76], but the berberine-induced ACL upregulation still needed further investigation. The robust upregulation of ACL induced by berberine in the absence or presence of CC and TOFA was observed with significance. TOFA pretreatment blocked the conversion of citrate to malonyl-CoA and the conversion of malonyl-CoA to acetyl-CoA, both of which were catalyzed by ACC. Citrate and acetyl-CoA had no other way out but the acetyl-CoA producing pathway. Finally, the upregulated ACL by berberine leads to an accumulation of acetyl-CoA in cytosol. The CC-induced AMPK inhibition also did not regulate this acetyl-CoA accumulation process.
4. Conclusion
Berberine regulated lipid metabolism under the inhibitions of AMPK, FASN, and TOFA in breast cancer cells MCF-7. The robust suppression of mitochondrial membrane potential still lingered in independent pattern with the existence of CC or TOFA, indicating a berberine-induced apoptosis even under AMPK inhibition and lipid synthesis inhibition. A palliation of berberine on TOFA-induced ROS increase also suggested the attenuation on the TOFA-induced apoptosis. Meanwhile, ACC inhibition by TOFA reversed the berberine-induced suppression on lipid metabolism, while FASN inhibition was not as powerful as ACC inhibition on reversing the suppressed lipid metabolism. Besides, the altered mitochondrial function in the presence of CC and TOFA and the robust upregulation of ACL in the absence or presence of CC and TOFA with significance also suggested the interference of berberine with cancer reprogramming metabolism. Undoubtedly, the detailed mechanisms of berberine interfering with lipid metabolism under the condition of energy-related inhibitors needed further investigation, including the potential compensatory mechanisms for ATP production and the upregulation of ACL.
Acknowledgments
This study was supported by Fundamental Research Funds for the Central Universities (lzujbky-2014-148) and University of Macau Research Fund (UL016A/09-Y2/CMS/WYT01/ICMS).
Conflict of Interests
The authors declared that they have no competing interests.
Authors' Contribution
Wen Tan and Zhangfeng Zhong contributed equally to the work.
References
- 1.Baenke F., Peck B., Miess H., Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Disease Models & Mechanisms. 2013;6(6):1353–1363. doi: 10.1242/dmm.011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hung Y. U.-H., Chan Y. I.-S., Chang Y.-S., et al. Fatty acid metabolic enzyme acyl-CoA thioesterase 8 promotes the development of hepatocellular carcinoma. Oncology Reports. 2014;31(6):2797–2803. doi: 10.3892/or.2014.3155. [DOI] [PubMed] [Google Scholar]
- 3.Chiaradonna F., Moresco R. M., Airoldi C., et al. From cancer metabolism to new biomarkers and drug targets. Biotechnology Advances. 2012;30(1):30–51. doi: 10.1016/j.biotechadv.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Mayevsky A. Mitochondrial function and energy metabolism in cancer cells: past overview and future perspectives. Mitochondrion. 2009;9(3):165–179. doi: 10.1016/j.mito.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Sotgia F., Whitaker-Menezes D., Martinez-Outschoorn U. E., et al. Mitochondria ‘fuel’ breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. 2012;11(23):4390–4401. doi: 10.4161/cc.22777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Outschoorn U., Sotgia F., Lisanti M. P. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Seminars in Oncology. 2014;41(2):195–216. doi: 10.1053/j.seminoncol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Griguer C. E., Oliva C. R. Bioenergetics pathways and therapeutic resistance in gliomas: emerging role of mitochondria. Current Pharmaceutical Design. 2011;17(23):2421–2427. doi: 10.2174/138161211797249251. [DOI] [PubMed] [Google Scholar]
- 8.Cho K., Shin H.-W., Kim Y.-I., et al. Mad1 mediates hypoxia-induced doxorubicin resistance in colon cancer cells by inhibiting mitochondrial function. Free Radical Biology & Medicine. 2013;60:201–210. doi: 10.1016/j.freeradbiomed.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 9.Thomas K. J., Jacobson M. R. Defects in mitochondrial fission protein dynamin-related protein 1 are linked to apoptotic resistance and autophagy in a lung cancer model. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0045319.e45319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Outschoorn U. E., Goldberg A., Lin Z., et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biology & Therapy. 2011;12(10):924–938. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Outschoorn U. E., Lin Z., Ko Y.-H., et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10(15):2521–2528. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Y., Berners-Price S. J., Thorburn D. R., et al. Serine protease inhibition and mitochondrial dysfunction associated with cisplatin resistance in human tumor cell lines: targets for therapy. Biochemical Pharmacology. 1997;53(11):1673–1682. doi: 10.1016/s0006-2952(97)00015-4. [DOI] [PubMed] [Google Scholar]
- 13.Landriscina M., Amoroso M. R., Piscazzi A., Esposito F. Heat shock proteins, cell survival and drug resistance: the mitochondrial chaperone TRAP1, a potential novel target for ovarian cancer therapy. Gynecologic Oncology. 2010;117(2):177–182. doi: 10.1016/j.ygyno.2009.10.078. [DOI] [PubMed] [Google Scholar]
- 14.Ling X., Wen L., Zhou Y. Role of mitochondrial translocation of telomerase in hepatocellular carcinoma cells with multidrug resistance. International Journal of Medical Sciences. 2012;9(7):545–554. doi: 10.7150/ijms.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bustamante J., Caldas Lopes E., Garcia M., Di Libero E., Alvarez E., Hajos S. E. Disruption of mitochondrial membrane potential during apoptosis induced by PSC 833 and CsA in multidrug-resistant lymphoid leukemia. Toxicology and Applied Pharmacology. 2004;199(1):44–51. doi: 10.1016/j.taap.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 16.Chen X., Wei S., Ma Y., et al. Quantitative proteomics analysis identifies mitochondria as therapeutic targets of multidrug-resistance in ovarian cancer. Theranostics. 2014;4(12):1164–1175. doi: 10.7150/thno.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song I. S., Kim H. K., Lee S. R., et al. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. International Journal of Cancer. 2013;133(6):1357–1367. doi: 10.1002/ijc.28149. [DOI] [PubMed] [Google Scholar]
- 18.Salem A. F., Whitaker-Menezes D., Lin Z., et al. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012;11(13):2545–2556. doi: 10.4161/cc.20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo J. Y., White E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy. 2013;9(10):1636–1638. doi: 10.4161/auto.26123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strohecker A. M., Guo J. Y., Karsli-Uzunbas G., et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discovery. 2013;3(11):1272–1285. doi: 10.1158/2159-8290.cd-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo J. Y., Karsli-Uzunbas G., Mathew R., et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes & Development. 2013;27(13):1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim H. K., Song I.-S., Lee S. Y., et al. B7-H4 downregulation induces mitochondrial dysfunction and enhances doxorubicin sensitivity via the cAMP/CREB/PGC1-α signaling pathway in HeLa cells. Pflügers Archiv: European Journal of Physiology. 2014;466(12):2323–2338. doi: 10.1007/s00424-014-1493-3. [DOI] [PubMed] [Google Scholar]
- 23.Abdi J., Garssen J., Faber J., Redegeld F. A. Omega-3 fatty acids, EPA and DHA induce apoptosis and enhance drug sensitivity in multiple myeloma cells but not in normal peripheral mononuclear cells. The Journal of Nutritional Biochemistry. 2014;25(12):1254–1262. doi: 10.1016/j.jnutbio.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 24.Elliott R. L., Jiang X. P., Head J. F. Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Research and Treatment. 2012;136(2):347–354. doi: 10.1007/s10549-012-2283-2. [DOI] [PubMed] [Google Scholar]
- 25.Ralph S. J., Rodríguez-Enríquez S., Neuzil J., Moreno-Sánchez R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Molecular Aspects of Medicine. 2010;31(1):29–59. doi: 10.1016/j.mam.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Sun S., Han Y., Liu J., et al. Trichostatin a targets the mitochondrial respiratory chain, increasing mitochondrial reactive oxygen species production to trigger apoptosis in human breast cancer cells. PLoS ONE. 2014;9(3) doi: 10.1371/journal.pone.0091610.e91610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das S., Das J., Samadder A., Boujedaini N., Khuda-Bukhsh A. R. Apigenin-induced apoptosis in A375 and A549 cells through selective action and dysfunction of mitochondria. Experimental Biology and Medicine. 2012;237(12):1433–1448. doi: 10.1258/ebm.2012.012148. [DOI] [PubMed] [Google Scholar]
- 28.Singhapol C., Pal D., Czapiewski R., Porika M., Nelson G., Saretzki G. C. Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0052989.e52989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K.-Y., Cho H.-J., Yu S.-N., et al. Interplay of reactive oxygen species, intracellular Ca2+ and mitochondrial homeostasis in the apoptosis of prostate cancer cells by deoxypodophyllotoxin. Journal of Cellular Biochemistry. 2013;114(5):1124–1134. doi: 10.1002/jcb.24455. [DOI] [PubMed] [Google Scholar]
- 30.Lin X., Wu G., Huo W.-Q., Zhang Y., Jin F.-S. Resveratrol induces apoptosis associated with mitochondrial dysfunction in bladder carcinoma cells. International Journal of Urology. 2012;19(8):757–764. doi: 10.1111/j.1442-2042.2012.03024.x. [DOI] [PubMed] [Google Scholar]
- 31.Kil I. S., Jung K. H., Nam W. S., Park J.-W. Attenuated mitochondrial NADP+-dependent isocitrate dehydrogenase activity enhances EGCG-induced apoptosis. Biochimie. 2011;93(10):1808–1815. doi: 10.1016/j.biochi.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 32.Ballot C., Kluza J., Lancel S., et al. Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis. 2010;15(7):769–781. doi: 10.1007/s10495-010-0471-2. [DOI] [PubMed] [Google Scholar]
- 33.Kil I. S., Chung K. H., Park J.-W. Silencing of mitochondrial NADP+-dependent isocitrate dehydrogenase gene enhances selenite-induced apoptosis. Free Radical Research. 2010;44(3):332–339. doi: 10.3109/10715760903494184. [DOI] [PubMed] [Google Scholar]
- 34.Hail N., Jr., Lotan R. Cancer chemoprevention and mitochondria: targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Molecular Nutrition & Food Research. 2009;53(1):49–67. doi: 10.1002/mnfr.200700527. [DOI] [PubMed] [Google Scholar]
- 35.Heerdt B. G., Houston M. A., Augenlicht L. H. Short-chain fatty acid-initiated cell cycle arrest and apoptosis of colonic epithelial cells is linked to mitochondrial function. Cell Growth & Differentiation. 1997;8(5):523–532. [PubMed] [Google Scholar]
- 36.Dey R., Moraes C. T. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. The Journal of Biological Chemistry. 2000;275(10):7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- 37.Hail N., Jr., Youssef E. M., Lotan R. Evidence supporting a role for mitochondrial respiration in apoptosis induction by the synthetic retinoid CD437. Cancer Research. 2001;61(18):6698–6702. [PubMed] [Google Scholar]
- 38.Lebedeva I. V., Su Z.-Z., Sarkar D., et al. Melanoma differentiation associated gene-7, mda-7/interleukin-24, induces apoptosis in prostate cancer cells by promoting mitochondrial dysfunction and inducing reactive oxygen species. Cancer Research. 2003;63(23):8138–8144. [PubMed] [Google Scholar]
- 39.Lu J., Tan M., Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Letters. 2015;356(2):156–164. doi: 10.1016/j.canlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ladiges W., Wanagat J., Preston B., Loeb L., Rabinovitch P. A Mitochondrial view of aging, reactive oxygen species and metastatic cancer. Aging Cell. 2010;9(4):462–465. doi: 10.1111/j.1474-9726.2010.00579.x. [DOI] [PubMed] [Google Scholar]
- 41.Lee G.-H., Yan C., Shin S.-J., et al. BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene. 2010;29(14):2130–2141. doi: 10.1038/onc.2009.491. [DOI] [PubMed] [Google Scholar]
- 42.Lee G.-H., Chae H.-J., Kim H.-R. Monoamine carboxylate transporters are involved in BI-1-associated cancer metastasis in HT1080 colon fibrosarcoma cells. International Journal of Oncology. 2011;39(1):209–216. doi: 10.3892/ijo.2011.1024. [DOI] [PubMed] [Google Scholar]
- 43.Sutendra G., Dromparis P., Kinnaird A., et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2013;32(13):1638–1650. doi: 10.1038/onc.2012.198. [DOI] [PubMed] [Google Scholar]
- 44.Chen C., Han X., Zou X., et al. 4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75), an inhibitor of fatty-acid synthase, suppresses the mitochondrial fatty acid synthesis pathway and impairs mitochondrial function. The Journal of Biological Chemistry. 2014;289(24):17184–17194. doi: 10.1074/jbc.m114.550806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruggeri B. A., Wang H. Q., Altomare D. A., Jones-Bolin S., Zhou H., Testa J. R. Anti-tumor effects of fatty acid synthase inhibitor C75 on orthotopically-implanted human pancreatic ductal adenocarcinoma in nude mice. Proceedings of the 95th Annual AACR Meeting; March 2004; Orlando, Fla, USA. American Association for Cancer Research; [Google Scholar]
- 46.Chen H.-W., Chang Y.-F., Chuang H.-Y., Tai W.-T., Hwang J.-J. Targeted therapy with fatty acid synthase inhibitors in a human prostate carcinoma LNCaP/tk-luc-bearing animal model. Prostate Cancer and Prostatic Diseases. 2012;15(3):260–264. doi: 10.1038/pcan.2012.15. [DOI] [PubMed] [Google Scholar]
- 47.Thupari J. N., Landree L. E., Ronnett G. V., Kuhajda F. P. C75 increases peripheral energy utilization and fatty acid oxidation in diet-induced obesity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(14):9498–9502. doi: 10.1073/pnas.132128899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puig T., Vázquez-Martín A., Relat J., et al. Fatty acid metabolism in breast cancer cells: differential inhibitory effects of epigallocatechin gallate (EGCG) and C75. Breast Cancer Research and Treatment. 2008;109(3):471–479. doi: 10.1007/s10549-007-9678-5. [DOI] [PubMed] [Google Scholar]
- 49.Jin Q., Yuan L. X., Boulbes D., et al. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Research. 2010;12(6, article R96) doi: 10.1186/bcr2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Relat J., Blancafort A., Oliveras G., et al. Different fatty acid metabolism effects of (−)-Epigallocatechin-3-gallate and C75 in adenocarcinoma lung cancer. BMC Cancer. 2012;12, article 280 doi: 10.1186/1471-2407-12-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vucicevic L., Misirkic M., Janjetovic K., et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7(1):40–50. doi: 10.4161/auto.7.1.13883. [DOI] [PubMed] [Google Scholar]
- 52.Dai R. Y., Zhao X. F., Li J. J., et al. Implication of transcriptional repression in compound C-induced apoptosis in cancer cells. Cell Death & Disease. 2013;4, article e883 doi: 10.1038/cddis.2013.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu X., Chhipa R. R., Nakano I., Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Molecular Cancer Therapeutics. 2014;13(3):596–605. doi: 10.1158/1535-7163.mct-13-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jang J. H., Lee T. J., Yang E. S., et al. Compound C sensitizes Caki renal cancer cells to TRAIL-induced apoptosis through reactive oxygen species-mediated down-regulation of c-FLIPL and Mcl-1. Experimental Cell Research. 2010;316(13):2194–2203. doi: 10.1016/j.yexcr.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 55.Saito S., Furuno A., Sakurai J., Park H.-R., Shin-ya K., Tomida A. Compound C prevents the unfolded protein response during glucose deprivation through a mechanism independent of AMPK and BMP signaling. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0045845.e45845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang W.-L., Perillo W., Liou D., Marambaud P., Wang P. AMPK inhibitor compound C suppresses cell proliferation by induction of apoptosis and autophagy in human colorectal cancer cells. Journal of Surgical Oncology. 2012;106(6):680–688. doi: 10.1002/jso.23184. [DOI] [PubMed] [Google Scholar]
- 57.Huang S.-W., Wu C.-Y., Wang Y.-T., et al. P53 modulates the AMPK inhibitor compound C induced apoptosis in human skin cancer cells. Toxicology and Applied Pharmacology. 2013;267(1):113–124. doi: 10.1016/j.taap.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 58.Guseva N. V., Rokhlin O. W., Glover R. A., Cohen M. B. TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status. Cancer Biology & Therapy. 2011;12(1):80–85. doi: 10.4161/cbt.12.1.15721. [DOI] [PubMed] [Google Scholar]
- 59.Fukuda N., Ontko J. A. Interactions between fatty acid synthesis, oxidation, and esterification in the production of triglyceride-rich lipoproteins by the liver. Journal of Lipid Research. 1984;25(8):831–842. [PubMed] [Google Scholar]
- 60.McCune S. A., Harris R. A. Mechanism responsible for 5-(tetradecyloxy)-2-furoic acid inhibition of hepatic lipogenesis. The Journal of Biological Chemistry. 1979;254(20):10095–10101. [PubMed] [Google Scholar]
- 61.Panek E., Cook G. A., Cornell N. W. Inhibition by 5-(tetradecyloxy)-2-furoic acid of fatty acid and cholesterol synthesis in isolated rat hepatocytes. Lipids. 1977;12(10):814–818. doi: 10.1007/bf02533270. [DOI] [PubMed] [Google Scholar]
- 62.Kariya T., Wille L. J. Inhibition of fatty acid synthesis by RMI 14,514 (5-tetradecyloxy-2-furoic acid) Biochemical and Biophysical Research Communications. 1978;80(4):1022–1024. doi: 10.1016/0006-291X(78)91347-5. [DOI] [PubMed] [Google Scholar]
- 63.Li S., Qiu L., Wu B., et al. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Molecular Medicine Reports. 2013;8(2):373–378. doi: 10.3892/mmr.2013.1505. [DOI] [PubMed] [Google Scholar]
- 64.Wang C., Xu C., Sun M., Luo D., Liao D.-F., Cao D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochemical and Biophysical Research Communications. 2009;385(3):302–306. doi: 10.1016/j.bbrc.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu R., Zhang Z.-Q., Wang B., Jiang H.-X., Cheng L., Shen L.-M. Berberine-induced apoptotic and autophagic death of HepG2 cells requires AMPK activation. Cancer Cell International. 2014;14(1, article 49) doi: 10.1186/1475-2867-14-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu M., Xiao Y., Yin J., et al. Berberine promotes glucose consumption independently of AMP-activated protein kinase activation. PLoS ONE. 2014;9(7) doi: 10.1371/journal.pone.0103702.e103702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim H.-S., Kim M.-J., Kim E. J., Yang Y., Lee M.-S., Lim J.-S. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochemical Pharmacology. 2012;83(3):385–394. doi: 10.1016/j.bcp.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 68.Gowans G. J., Hawley S. A., Ross F. A., Hardie D. G. AMP is a true physiological regulator of amp-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metabolism. 2013;18(4):556–566. doi: 10.1016/j.cmet.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brusq J.-M., Ancellin N., Grondin P., et al. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. Journal of Lipid Research. 2006;47(6):1281–1288. doi: 10.1194/jlr.m600020-jlr200. [DOI] [PubMed] [Google Scholar]
- 70.Santos C. R., Schulze A. Lipid metabolism in cancer. The FEBS Journal. 2012;279(15):2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 71.Hirsch H. A., Iliopoulos D., Joshi A., et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 2010;17(4):348–361. doi: 10.1016/j.ccr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tirinato L., Liberale C., Di Franco S., et al. Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells. 2014;33(1):35–44. doi: 10.1002/stem.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tan W., Li N., Tan R., et al. Berberine interfered with breast cancer cells metabolism, balancing energy homeostasis. Anti-Cancer Agents in Medicinal Chemistry. 2014;15(1):66–78. doi: 10.2174/1871520614666140910120518. [DOI] [PubMed] [Google Scholar]
- 74.Zaidi N., Swinnen J. V., Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Research. 2012;72(15):3709–3714. doi: 10.1158/0008-5472.can-11-4112. [DOI] [PubMed] [Google Scholar]
- 75.Migita T., Narita T., Nomura K., et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Research. 2008;68(20):8547–8554. doi: 10.1158/0008-5472.can-08-1235. [DOI] [PubMed] [Google Scholar]
- 76.Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., Thompson C. B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]