

Abstract
Glycogen storage disease type-Ia (GSD-Ia) is caused by a lack of glucose-6-phosphatase-α (G6Pase-α or G6PC) activity. We have shown that gene therapy mediated by a recombinant adeno-associated virus (rAAV) vector expressing human G6Pase-α normalizes blood glucose homeostasis in the global G6pc knockout (G6pc−/−) mice for 70–90 weeks. The treated G6pc−/− mice expressing 3–63% of normal hepatic G6Pase-α activity (AAV mice) produce endogenous hepatic glucose levels 61–68% of wild-type littermates, have a leaner phenotype and exhibit fasting blood insulin levels more typical of young adult mice. We now show that unlike wild-type mice, the lean AAV mice have increased caloric intake and do not develop age-related obesity or insulin resistance. Pathway analysis shows that signaling by hepatic carbohydrate response element binding protein that improves glucose tolerance and insulin signaling is activated in AAV mice. In addition, several longevity factors in the calorie restriction pathway, including the NADH shuttle systems, NAD+ concentrations and the AMP-activated protein kinase/sirtuin 1/peroxisome proliferator-activated receptor-γ coactivator 1α pathway are upregulated in the livers of AAV mice. The finding that partial restoration of hepatic G6Pase-α activity in GSD-Ia mice not only attenuates the phenotype of hepatic G6Pase-α deficiency but also prevents the development of age-related obesity and insulin resistance seen in wild-type mice may suggest relevance of the G6Pase-α enzyme to obesity and diabetes.
Introduction
Glycogen storage disease type-Ia (GSD-Ia, MIM232200) is characterized by impaired glucose homeostasis caused by deficiencies in glucose-6-phosphatase-α (G6Pase-α or G6PC). G6Pase-α is a key enzyme for endogenous glucose production in the liver, kidney and intestine, catalyzing the hydrolysis of G6P to glucose in the terminal, rating limiting step of gluconeogenesis and glycogenolysis (1). The metabolic disruption present in GSD-Ia patients can be adequately managed with dietary therapies, if there is strong compliance. Infants typically receive nocturnal nasogastric infusion of glucose to avoid hypoglycemia (2). Patients 3 years or older are prescribed uncooked cornstarch, a slow release carbohydrate, to prolong the length of euglycemia between meals (3). These dietary therapies enable patients to maintain normoglycemia, but the underlying pathological processes remain uncorrected and hepatocellular adenoma (HCA), which may undergo transformation to hepatocellular carcinoma (HCC), is common (1). We had previously generated G6Pase-α-deficient (G6pc−/−) mice that manifest all of the symptoms of human GSD-Ia (4). However, in these mice, dietary therapy is difficult and not very effective, and the few mice that can survive weaning typically live not more than 3 months (4). Using G6pc−/− mice, we have shown that systemic administration of rAAV-GPE, a recombinant adeno-associated virus (rAAV) pseudotype 2/8 vector expressing human G6Pase-α directed by the human G6PC promoter/enhancer (GPE), delivers G6Pase-α to the liver and corrects blood glucose homeostasis (5). In a long-term study, we have shown that 70–90-week-old rAAV-GPE-treated G6pc−/− mice, expressing 3–63% of wild-type hepatic G6Pase-α activity (AAV mice), maintain glucose homeostasis and show no evidence of HCA/HCC (6). Interestingly, as they age, these mice have a leaner phenotype and exhibit fasting blood insulin levels closer to those of young adult wild-type mice than their similarly aged controls (6).
Several mechanisms might contribute to this observation. First, the old AAV mice exhibit elevated concentrations of hepatic glucose-6-phosphate (G6P) (6) that activates the carbohydrate response element binding protein (ChREBP), a lipogenic transcription factor (7). Studies have shown that wild-type mice engineered to overexpress hepatic ChREBP and maintained on a high-fat diet exhibit improved glucose tolerance compared with controls (8). So activation of ChREBP signaling may be one explanation. Secondly, hepatic endogenous glucose produced in the old AAV mice averaged 61–68% of those of control littermates (6), suggesting that AAV mice live with a chronic reduction of hepatic glucose production. This suggests that the calorie restriction (CR) pathway may also contribute to more healthy aging of these mice (9,10). Within the CR pathway, the mitochondrial (m) enzymes, malate dehydrogenase (mMDH) and aspartate aminotransferase (mAAT) of the malate–aspartate (MA) shuttle system (11 ) and glycerol-3-phosphate (GP) dehydrogenase (mGPDH) of the GP shuttle system (11,12), are metabolic longevity regulators (11). In older wild-type mice, CR has been shown to reverse the decline in activities of mMDH, mAAT, cytosolic (c) MDH and cAAT of the MA shuttle system (13). Moreover, Lou/C rats that display improved insulin sensitivity and resistance to age- or diet-induced obesity (14,15) exhibit elevated hepatic mGPDH levels. The NADH shuttle systems regulate intracellular concentrations of the primary redox carriers, NAD+ and NADH, by re-oxidizing cytosolic NADH produced by glycolysis and transferring electrons into the mitochondrial respiratory chain, connecting glycolysis with mitochondrial oxidative phosphorylation (11,12). Notably, intracellular NAD+ concentrations decrease in mice with age and/or obesity, but increase under CR (16–18).
Aging is also associated with mitochondrial dysfunction; so protection of mitochondria may also contribute to the observed phenotype (9,10). The peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) is a master regulator of energy metabolism and mitochondrial biogenesis, playing key roles in the maintenance of mitochondrial integrity, biogenesis and function (9,10). In wild-type mice, a moderate reduction in hepatic PGC-1α expression is associated with reduced hepatic insulin sensitivity (19), whereas overexpression of hepatic PGC-1α stimulates mitochondrial function (20). PGC-1α can be activated either via phosphorylation by AMP-activated protein kinase (AMPK) that regulates energy homeostasis (21) or via deacetylation by sirtuin 1 (SIRT1), an NAD+-dependent deacetylase that plays key roles in longevity mediated by CR (18).
We now show that the underlying mechanisms responsible for the beneficial metabolic phenotype of AAV mice correlate with activation of ChREBP signaling, increases in the expression of NADH shuttle systems, increases in NAD+ concentrations and activation of the AMPK/SIRT1/PGC-1α pathway in the livers of AAV mice.
Results
AAV mice do not develop age-related obesity or insulin resistance
We have previously shown that rAAV-GPE-treated G6pc−/− mice expressing 3–9% (AAV-L mice) and 22–63% (AAV-M mice) of wild-type hepatic G6Pase-α activity maintain blood glucose homeostasis for 70–90 weeks, show no evidence of HCA and tolerate a 24 h fast (6). Moreover, as they age, AAV-L and AAV-M mice have a leaner phenotype and exhibit fasting blood insulin levels more typical of young adult mice (6). As mice expressing 3–63% of wild-type hepatic G6Pase-α activity exhibit similar metabolic profiles (6), we now refer to mice expressing G6Pase-α activity in this range collectively as ‘AAV mice’. To examine the mechanisms underlying the leaner phenotype and greater insulin sensitivity, we generated more AAV mice by infusing 2-week-old G6pc−/− mice with rAAV-GPE ranging from 5 × 1012 to 1013 vp/kg and their metabolic profiles were monitored up to the age of 90 weeks.
Hepatic G6Pase-α activity in old (75–90-week-old) wild-type mice averaged 171.8 ± 7.1 nmol/min/mg (Fig. 1A). The treated mice expressing G6Pase-α activity ranging from 11.1 to 65.8 nmol/min/mg, equivalent to 7–38% of wild-type hepatic G6Pase-α activity, were classified as AAV mice with low-to-medium G6Pase-α activity (Fig. 1A). The 75–90-week-old AAV mice could sustain a 24 h fast (Fig. 1B) and their 24 h fasted blood glucose levels were 88.8 ± 6.4 mg/dl, indistinguishable from the results observed in 70–90-week-old AAV-L/AAV-M mice reported in our earlier study (6).
Figure 1.

Biochemical and phenotypic analyses of wild-type and AAV mice. (A) Hepatic microsomal G6Pase-α activity in the AAV mice is shown at the indicated ages in weeks (W). Hepatic microsomal G6Pase-α activity in 75–90-week-old wild-type mice (n = 18) averaged 171.8 ± 7.1 nmol/min/mg (100%). (B) Blood glucose levels following a 24 h fast in 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (C) Hepatic microsomal G6P uptake activity of 75–90-week-old wild-type (n = 10), AAV (n = 8) or 6-week-old G6pc−/− (n = 6) mice. (D) Hepatic G6PT mRNA expression in 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (E) Serum metabolite profiles in 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (F) BWs of 12-week-old wild-type (n = 8) and AAV mice (n = 8) and 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (+/+), wild-type mice; (−/−), G6pc−/− mice. Data are mean ± SEM. *P < 0.05 and **P < 0.005.
Hepatic endogenous glucose production is controlled by functional coupling of G6Pase-α to the G6P transporter (G6PT), which is the rate-limiting component of the G6Pase-α/G6PT complex (1). As expected, the G6pc−/− hepatic microsomes containing an intact G6PT exhibited no significant G6P uptake activity (1,6) (Fig. 1C), whereas microsomes from AAV livers showed uptake activity 50% of wild-type activity (Fig. 1C). Confirming our earlier study (6), restoration of 7–38% of wild-type G6Pase-α activity in AAV mice led to 1.9-fold increases in hepatic G6PT mRNA expression over that in wild-type mice (Fig. 1D). Overall, the metabolic profiles of the old AAV mice (Fig. 1E) were indistinguishable from the AAV-L/AAV-M mice reported in our earlier study (6).
At 12 weeks, the average body weights (BW) of wild-type and AAV mice were 24.2 ± 1 and 21.3 ± 0.8 g, respectively (Fig. 1F), whereas at 75–90 weeks, they were 36.7 ± 1.2 and 21.3 ± 1.2 g, respectively (Fig. 1F). This showed that while wild-type mice increased BW with age, the treated mice kept a constant BW. Histological analysis of liver biopsy samples revealed no hepatic nodules or histological abnormalities in any of the treated or wild-type mice. There was a 2.5-fold increase in glycogen accumulation in the AAV mice (Fig. 2A), but hepatic triglyceride contents were similar to wild-type mice (Fig. 2A).
Figure 2.

Phenotype and insulin tolerance profiles of wild-type and AAV mice. (A) Hepatic glycogen and triglyceride contents in 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (B) Hepatic levels of pyruvate and lactate in 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (C) Twenty-four hour fasted blood insulin levels in 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (D) Insulin tolerance profiles of 12-week-old wild-type (triangle, n = 8) and AAV mice (filled triangle, n = 8), and 75–90-week-old wild-type mice (circle, n = 10) and AAV mice expressing 7–8% (square, n = 2) or 16–38% (filled square, n = 6) mice treated with 0.25 IU/kg of insulin. Values are reported as a percent of respective level of each group at zero time. (E) Body fat contents in 12-week-old wild-type (n = 8) and AAV mice (n = 8) and 75–90-week-old wild-type (n = 10) and AAV (n = 8) mice. (+/+), wild-type mice. Data are mean ± SEM. *P < 0.05 and **P < 0.005.
Under aerobic conditions, pyruvate is converted to acetyl-CoA, whereas under anaerobic conditions, pyruvate is converted to lactate. Hepatic concentrations of pyruvate and lactate were similar between the old AAV and control littermates (Fig. 2B), suggesting that glucose flux via glycolysis and gluconeogenesis was maintained in the old AAV mice.
Fasting blood insulin levels in old wild-type and AAV mice were 1.33 ± 0.19 and 0.63 ± 0.11 ng/ml, respectively (Fig. 2C). All were within the normal range, although insulin levels in the old AAV mice were closer to those in 10–20-week-old young adult mice than those in the old wild-type mice (22). Because the AAV mice exhibit increased insulin sensitivity, a reduced insulin dose of 0.25 IU/kg was chosen to monitor blood insulin tolerance profiles. Following an intraperitoneal insulin injection, blood glucose levels in 12-week-old wild-type and AAV mice decreased, in parallel, with time (Fig. 2D). Blood glucose levels in the 75–90-week-old wild-type mice failed to decrease following insulin injection (Fig. 2D), reflecting age-related decrease in insulin sensitivity (23). In contrast, the 75–90-week-old AAV mice exhibiting either 7–8% (AAV-L mice) or 16–38% (AAV-M mice) of wild-type hepatic G6Pase-α activity exhibited similar insulin tolerance profiles that are closer to those of young adult mice (Fig. 2D), demonstrating that restoring 7–38% of wild-type hepatic G6Pase-α activity protected the mice against an age-related insulin resistance.
Whole-body energy metabolism in old AAV mice
The old AAV mice were also protected against age-related obesity with an overall reduction in total body fat contents (Fig. 2E). Indirect calorimetry analysis showed that the old AAV mice consumed significantly greater O2 and produced more CO2 than their control littermates (Fig. 3A). Both daily food intake/BW (Fig. 3B) and energy expenditure (Fig. 3C) were also elevated in the old AAV mice compared with the controls, whereas respiratory quotient (RQ) values (Fig. 3D) and activity (data not shown) were similar. In summary, the old AAV mice have higher catabolic rates that confer resistance to age-related obesity.
Figure 3.

Indirect calorimetry analysis of 75–90-week-old wild-type and AAV mice using the CLAMS. (A) O2 consumption and CO2 production in wild-type (circle) and AAV (filled circle) mice. (B) Daily food intake. (C) Energy expenditure. (D) The RQ values. (+/+), wild-type mice (n = 10) and AAV mice (n = 8). *P < 0.05 and **P < 0.005.
Activation of hepatic ChREBP signaling in old AAV mice
To obtain sufficient tissue to examine the mechanisms underlying the beneficial metabolic phenotype of the old AAV mice, we augmented samples collected in this study with frozen liver samples from a similar earlier study of 70–90-week-old AAV-L/AAV-M mice expressing 3–63% of wild-type hepatic G6Pase-α activity (6). Quantitative real-time reverse transcriptase–polymerase chain reaction (RT–PCR) analysis showed that levels of hepatic transcripts of genes involved in ChREBP signaling, NADH shuttle systems and the AMPK/SIRT1/PGC-1α pathway reported later in this study were similar between AAV-L and AAV-M mice and were combined together as the AAV mice. The results described in the following sections were obtained from a combined total of 23 AAV mice expressing 3–63% of wild-type hepatic G6Pase-α activity.
Hepatic G6P levels were 1.6-fold higher in the old AAV mice, compared with the controls (Fig. 4A). Hepatic ChREBP transcripts were increased 1.5-fold in the old AAV mice, but the increase in hepatic ChREBP proteins was not statistically significant (Fig. 4B). G6P activates ChREBP signaling by promoting ChREBP nuclear translocation (7), and consistent with this, the percentage of hepatic nuclear ChREBP protein contents increased markedly in the old AAV mice, compared with control littermates (Fig. 4C). Consistently, the expression of many ChREBP-regulated hepatic genes (7,8) was induced, including the liver isoform of pyruvate kinase (L-PK) in glycolysis and acetyl-CoA carboxylase isoform-1 (ACC1), elongation of very long chain fatty acids protein-6 (ELOVL6), fatty acid synthase (FAS) and stearoyl-CoA desaturase 1 (SCD1) in lipogenesis (Fig. 4D).
Figure 4.

Analysis of hepatic ChREBP signaling in 70–90-week-old wild-type and AAV mice. (A) Hepatic G6P levels from wild-type (n = 26) and AAV (n = 20) mice. (B) Quantification of ChREBP mRNA by real-time RT–PCR, ChREBP protein levels by densitometric and western-blot analysis of ChREBP or β-actin. For quantitative RT–PCR, data represent mean ± SEM for wild-type (n = 24) and AAV (n = 22) mice, and for densitometric analysis, data represent mean ± SEM of four separate replicas of western blots. (C) Immunohistochemical analysis of hepatic ChREBP nuclear localization and quantification of nuclear ChREBP-translocated cells. Plates shown, from left to right, are at magnifications of ×100, ×400 and ×400. Data represent mean ± SEM for wild-type (n = 5) and AAV (n = 11) mice. (D) Quantification of mRNA for L-PK, ACC1, ElOVL6, FAS and SCD1 by real-time RT–PCR. Data represent mean ± SEM for wild-type (n = 24) and AAV (n = 22) mice. *P < 0.05 and **P < 0.005.
Despite the induction of lipogenic genes, hepatic triglyceride levels were similar between old AAV and wild-type littermates (Fig. 2A). Energy metabolism is also regulated by factors that modulate mitochondria β-oxidation, including peroxisome proliferator-activated receptor α (PPARα), carnitine palmitoyl transferase 1α (CPT1α) and fibroblast growth factor 21 (FGF21) (24). Compared with wild-type littermates, hepatic mRNAs for PPARα in AAV mice were decreased and hepatic mRNAs for CPT1α were unchanged (Fig. 5A). However, hepatic mRNAs for FGF21 (24) were elevated 1.8-fold over wild-type mice (Fig. 5A).
Figure 5.

Analysis of hepatic expression of β-oxidation modulating genes, Akt and NADH shuttle systems in 70–90-week-old wild-type and AAV mice and hepatic concentrations of NAD+ and NADH. For quantitative RT–PCR, the data represent the mean ± SEM for 70–90-week-old wild-type (n = 22) and AAV (n = 22) mice. (A) Quantification of mRNA for hepatic PPARα, CPT1α, FGF21 and Akt by real-time RT–PCR and quantification of protein levels by densitometric analysis. The data for densitometric analysis represent the mean ± SEM of four separate replicas of western blots. (B) Western-blot analysis of Akt, p-Akt-S473, p-Akt-T308 or β-actin. (C) Quantification of mRNA for members of hepatic MA and GP shuttle systems by real-time RT–PCR. (D) Western-blot analysis of mMDH, mAAT, cMDH, cAAT, mGPDH, cGPDH or β-actin and quantification of protein levels by densitometry of seven separate replicas of western blots. (E) Hepatic levels of NAD+ and NADH in 12-week-old wild-type (n = 12) and AAV (n = 12) mice and 70–90 week-old wild-type (n = 10) and AAV (n = 13) mice. *P < 0.05 and **P < 0.005.
SCD1 catalyzes the conversion of saturated fatty acids into the beneficial monounsaturated fatty acids (25). Studies have shown that mice overexpressing hepatic ChREBP along with increased SCD1 exhibit improved insulin signaling that correlates with phosphorylation and activation of protein kinase B/Akt (8). Consistent with that, hepatic Akt mRNA (Fig. 5A) and total Akt protein (Fig. 5A and B) were similar between old AAV and wild-type mice, but levels of the active, phosphorylated forms of Akt (26), p-Akt-S473 and p-Akt-T308, were increased 1.8- and 2.0-fold, respectively, in the old AAV mice (Fig. 5A and B).
Increase in NADH shuttle system expression and NAD+ concentrations in old AAV mice
The mitochondrial members of the NADH shuttle systems, mMDH, mAAT and mGPDH, are longevity regulators in yeast (11). Although hepatic mAAT transcripts were similar between old wild-type and AAV mice, hepatic mRNAs were elevated relative to wild-type mice for cAAT (3.7-fold), mMDH (1.3-fold) and cMDH (1.2-fold) of the MA shuttle and cGPDH (2.2-fold) and mGPDH (1.8-fold) of the GP shuttle in the old AAV mice (Fig. 5C). Western-blot analysis showed corresponding increases in protein levels of mMDH, cMDH, cGPDH and mGPDH in the old AAV mice (Fig. 5D), although protein levels for mAAT and cAAT were similar to controls (Fig. 5D).
Intracellular NAD+ concentrations decrease with both age and obesity (16,17) but increase under CR (18). At 12 weeks, hepatic NAD+ levels were similar between wild-type and AAV mice (Fig. 5E). In wild-type mice, hepatic NAD+ concentrations decreased with age, averaging 74% of that of young wild-type mice by 75–90 weeks (Fig. 5E). In contrast, hepatic NAD+ concentrations in AAV mice remained constant. Consequently, NAD+ concentrations in the old AAV mice were on average 1.6-fold higher those of the old wild-type littermates (Fig. 5E). With increased age, hepatic levels of NADH in wild-type mice also decreased, although NADH levels in the old AAV mice were unchanged (Fig. 5E).
Activation of AMPK/SIRT1/PGC-1α pathway in old AAV mice
CR enhances mitochondrial function through activation of the AMPK/SIRT1/PGC-1α pathway (9,10). Although hepatic AMPK-α1 mRNA levels were lower in the old AAV mice compared with controls, hepatic AMPK-α2 mRNA levels were similar between old wild-type and AAV mice (Fig. 6A). Studies have shown that CR upregulates AMPK-α2 by increasing protein levels of total AMPK-α2 and p-AMPK-α2 via protein stabilization without affecting AMPK-α2 mRNA levels (27). Likewise, we showed that hepatic levels of total AMPK and the active p-AMPK-T172 in the old AAV mice were increased 1.9- and 1.6-fold, respectively, over that in the old control mice (Fig. 6B). SIRT1 is an NAD+-dependent deacetylase that is activated in response to CR or an increase in cellular NAD+ levels (18). Although hepatic SIRT1 mRNA levels were decreased in old AAV mice (Fig. 6A), hepatic SIRT1 protein levels were similar between old wild-type and AAV mice (Fig. 6B). The marked increase in hepatic NAD+ concentrations in the old AAV mice (Fig. 5E) suggests that hepatic SIRT1 activity is activated in these mice.
Figure 6.

Analysis of hepatic AMPK/SIRT1/PGC-1α signaling pathway in wild-type and AAV mice. (A) Quantification of hepatic mRNA for AMPK-α1, AMPK-α2, SIRT1 and PGC-1α by real-time RT–PCR. The data represent the mean ± SEM for 12-week-old wild-type (n = 12) and AAV (n = 11) mice and for 70–90-week-old wild-type (n = 22) and AAV (n = 22) mice. (B) Western-blot analysis of hepatic AMPK, p-AMPK-T172, SIRT1, PGC-1α or β-actin in wild-type and AAV mice and quantification of protein levels by densitometric analysis. The data represent the mean ± SEM of seven separate replicas of western blots. (C) Western-blot analysis of hepatic mitochondrial ETC chain in 70–90-week-old wild-type and AAV mice and quantification of protein levels by densitometric analysis. The data represent the mean ± SEM of seven separate replicas of western blots. *P < 0.05 and **P < 0.005.
PGC-1α, which decreases during aging (10), is activated both via phosphorylation by AMPK and via deacetylation by SIRT1 (9,10). In both wild-type and AAV mice, hepatic levels of PGC-1α mRNA (Fig. 6A) decreased in aging. However, hepatic PGC-1α transcripts in 12-week and 70–90-week-old AAV mice were approximately 2.0-fold higher than those in the respective wild-type littermates (Fig. 6A). Supporting this, PGC-1α protein levels in 12-week and 70–90-week-old AAV mice were 1.3- and 1.7-fold higher than those in the respective wild-type controls (Fig. 6B).
PGC-1α is a master regulator of energy metabolism and mitochondrial biogenesis (9,10). Mitochondria are responsible for the production of cellular energy through the electron transport chain (ETC) that consists of five protein complexes I–V (28). Complexes I–IV transfer high-energy electrons and complex V is an ATP synthase. The activities of the mitochondrial ETC protein complexes decline with age (28). The increase in PGC-1α expression in the old AAV mice correlated with the increases in protein levels of all five ETC complexes, I, II, III, IV and V (Fig. 6C). Taken together, the results suggest that the AMPK/SIRT1/PGC-1α signaling pathway is activated in the AAV mice, leading to stimulation of mitochondrial energy metabolism, providing another mechanism that protects the mice against age-related obesity and insulin resistance.
Discussion
Previous long-term studies have shown that rAAV-GPE-mediated gene transfer in G6pc−/− mice, which restores >3% of normal hepatic G6Pase-α activity, corrects the hallmark metabolic defects of GSD-Ia and prevents the chronic formation of HCA/HCC (6). It is known that patients with type 2 diabetes exhibit increased hepatic G6Pase-α expression, which has led to the suggestion that G6Pase-α constitutes a potential pharmaceutical target for treating type 2 diabetes (29,30). Therefore, it was interesting to discover that the AAV mice, expressing 3–63% of wild-type hepatic G6Pase-α activity, exhibit better metabolic controls as they age than their control littermates (6). This finding also starts to inform the potential safety margin that might exist in any diabetes therapy that seeks to downregulate G6Pase-α. By 70–90 weeks, the AAV mice have fasting blood insulin levels closer to those of young adult mice (22) along with a significantly leaner phenotype. We now show that although wild-type mice gain both fat and insulin resistance with age, AAV mice do not develop age-related insulin resistance or obesity. We further show that at least some of the molecular mechanisms underlying the improved metabolic phenotype are common to the pathways seen in animals subject to CR, suggesting that the reduced hepatic glucose output in AAV mice mimics hepatic CR. The improved metabolic phenotype arises from activation of ChREBP signaling, increases in the expression of NADH shuttle systems, increases in NAD+ concentrations and activation of the AMPK/SIRT1/PGC-1α signaling pathway in the livers of the old AAV mice (Fig. 7).
Figure 7.
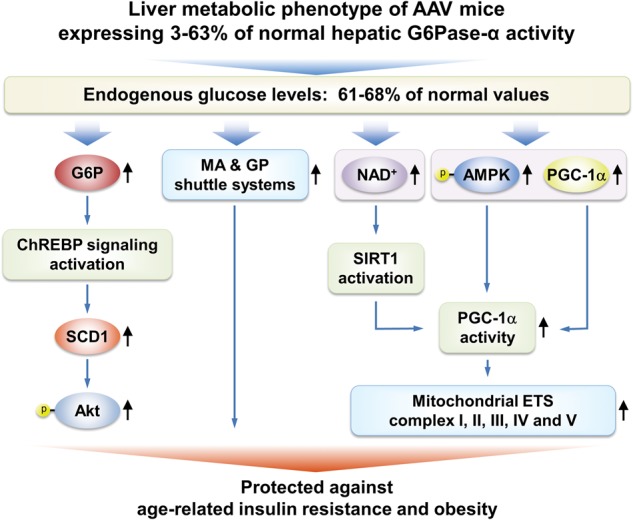
Mechanisms that underlie protection of AAV mice against age-related obesity and insulin resistance. Several signaling pathways in the livers of AAV mice contribute to the beneficial metabolic phenotype. The ChREBP-SCD1-Akt signaling pathway, activated by elevated hepatic G6P levels, leads to improved insulin signaling and glucose tolerance. The increases in the expression of the MA and GP shuttle systems that couple mitochondrial ATP synthesis to the regeneration of NAD+ confer insulin sensitivity and resistance to age-related obesity. The activation of the AMPK/SIRT1/PGC-1α pathway that leads to increased expression of mitochondrial ETC complexes and protect against age-related mitochondrial dysfunction has three components: (i) the increased expression of PGC-1α, a master regulator of mitochondrial biogenesis; (ii) PGC-1α activation via deacetylation by SIRT1 which is activated by elevated cellular concentrations of NAD+ and (iii) PGC-1α activation via phosphorylation by p-AMPK.
Interprandial blood glucose homeostasis is maintained by G6Pase-α-mediated hydrolysis of intracellular G6P to glucose in the terminal step of gluconeogenesis and glycogenolysis (1). As G6Pase-α expression is tissue-restricted, the primary contributor to blood glucose homeostasis is the liver, which accounts for ∼80% of the blood glucose, supported by the kidney, which accounts for ∼20%, with a further minor contribution from the intestine (1). Our previous studies have shown that restoration of just 3% of wild-type hepatic G6Pase-α activity in the global G6Pase-α knockout mouse is sufficient for viability, and here we show that this also confers resistance to the development of age-related insulin resistance that can lead to diabetes. These findings align well with the observation that the conditional liver-specific knockout (L-G6pc−/−) mice, who still have renal and intestinal glucose outputs, are viable, have a normal survival rate, exhibit normal glucose profiles and do not develop high-fat/sucrose diet-induced diabetes or obesity (31,32). Consistent with this, peripheral glucose metabolism and thermogenesis are activated in white adipose tissue, brown adipose tissue and skeletal muscle of L-G6pc−/− mice (32).
The rAAV-GPE-mediated G6Pase-α transgene expression primarily targeted the liver, and very little transgene expression was observed in the kidney and intestine (6,33). Consequently, kidney and intestine of AAV mice remained G6pc-null and incapable of endogenous glucose production. In the absence of endogenous glucose production from the kidney and intestine, the AAV mice expressing 3–63% of wild-type hepatic G6Pase-α activity, which produce reduced levels of hepatic endogenous glucose averaging 61–68% of those of control littermates (6), are living under chronic reduction of hepatic glucose production, similar to animals living under CR.
Studies have shown that the absence of hepatic glucose production in L-G6pc−/− mice stimulates energy expenditure through increased expression of hepatic FGF21 (32), a target gene for ChREBP (34) and a major regulator of energy metabolism that stimulates energy expenditure, fat utilization and lipid excretion (35). Moreover, a recent study (8) showed that mice overexpressing hepatic ChREBP exhibit improved glucose and lipid metabolism resulting from Akt activation and increases in the expression of SCD1, which converts saturated fatty acids into the beneficial monounsaturated fatty acids (25). In the old AAV mice, hepatic ChREBP signaling is stimulated along with elevated expression of FGF21 and SCD1, and the active p-Akt-S473 and p-Akt-T308 provide one reason why these mice are protected against age-related insulin resistance and obesity.
The available evidence has suggested that expression of the NADH shuttle systems, which play important roles in activating targets of the CR pathway (11,12), is likely upregulated in the AAV mice. First, Lou/C rats exhibiting improved insulin sensitivity and resistance to age- or diet-induced obesity (14,15) exhibit elevated levels of hepatic mGPDH. Secondly, the mitochondrial components of the MA and GP shuttle systems are novel longevity factors in the CR pathway in yeast (11). Thirdly, cGDPH and cMDH are genes upregulated by ChREBP (36). We show that mRNAs and proteins for mMDH, cMDH of the MA shuttle and mGPDH and cGPDH of the GP shuttle are increased in the old AAV mice. Studies have shown that functional activity of the GP shuttle requires equal molar proportions of mGPDH and cGPDH (12). Consistent with this, we show that hepatic cGPDH and mGPDH proteins are equally elevated 1.9-fold in the old AAV mice, compared with wild-type controls. The mitochondrial inner membrane is impermeable to NAD+ and NADH, and the NADH shuttle systems are required to couple mitochondrial ATP synthesis to the regeneration of NAD+ (12). The concentrations of NAD+, which play a critical role in mitochondrial aging and disease (18), are increased under CR and decreased in obesity and the aged (16–18). At 12 weeks, hepatic NAD+ concentrations were similar between wild-type and AAV mice. Although hepatic NAD+ concentrations decreased in wild-type mice during aging, hepatic NAD+ concentrations in the old AAV mice remained high, similar to the levels of young adult mice, reflecting the benefits of CR along with elevated NADH shuttle system expression.
A hallmark of aging is mitochondrial dysfunction (9,10). We show that the AMPK/SIRT1/PGC-1α pathway that regulates mitochondrial function and aging is activated in the old AAV mice. AMPK and SIRT1 are stress and bioenergetic sensors that stimulate mitochondrial gene expression selectively in response to energy levels (9,10,18,21). AMPK also plays a role in insulin sensitivity during CR (27). The old AAV mice exhibit increased protein levels of total AMPK and the active form, p-AMPK-T172. SIRT1 can be activated by increased NAD+ levels without changing SIRT1 protein levels (9,18), suggesting that elevated concentrations of hepatic NAD+ in the old AAV mice activate SIRT1 activity. Supporting this, levels of both mRNA and protein for PGC-1α, the master of mitochondrial biogenesis (9,10), which can be activated both by AMPK-mediated phosphorylation and by SIRT1-mediated deacetylation (9,10), are upregulated in the old AAV mice. Variation of PGC-1α levels exhibits profound effects on lipid and glucose metabolism. Mice overexpressing hepatic PGC-1α exhibit improved mitochondrial function and increased fatty acid oxidation (20). In contrast, PGC-1α heterozygous mice display decreased expression of genes involved in mitochondrial beta-oxidation and reduced hepatic insulin sensitivity (19). We show that protein levels of complexes I–V, components of the mitochondrial ETC (28), are upregulated in the old AAV mice. Taken together, one key mechanism responsible for protection of the AAV mice against age-related insulin resistance and obesity is activation of the AMPK/SIRT1/PGC-1α pathway.
In summary, we have demonstrated that AAV mice titrated to express 3–63% of normal hepatic G6Pase-α activity but lacking renal and intestinal endogenous glucose production not only maintain glucose homeostasis in the absence of HCA/HCC but are also protected against age-related insulin resistance and obesity. We further show that some of the underlying mechanisms responsible for the beneficial metabolic phenotype of the old AAV mice arise from activation of ChREBP signaling, increase in the expression of NADH shuttle systems, increase in cellular NAD+ concentrations and activation of the AMPK/SIRT1/PGC-1α pathway in the liver of the old AAV mice. Our study suggests that full restoration of normal G6Pase-α activity will not be required to confer significant therapeutic benefits in GSD-Ia by gene therapy. The finding that a moderate reduction of hepatic G6Pase-α activity in mice may promote a leaner phenotype and prevent the development of age-related decrease in insulin sensitivity lends weight to the suggestion that G6Pase-α constitutes a potential pharmaceutical target for treating type 2 diabetes and provides insight into the safety margin in such activity-reducing therapies.
Materials and Methods
Infusion of G6pc−/− mice with rAAV-GPE
All animal studies were conducted under an animal protocol approved by the NICHD Animal Care and Use Committee. The rAAV-GPE vector was infused into 2-week-old G6pc−/− mice (4), as described previously (5,6). Age-matched G6pc+/+/G6pc+/− and 6-week-old G6pc−/− mice were used as controls. Liver samples from wild-type and rAAV-GPE-treated mice were collected at sacrifice following a 24 h fast.
Phosphohydrolase and microsomal G6P uptake assays
Microsome isolation, phosphohydrolase and G6P uptake assays were determined essentially, as described previously (4,6). In phosphohydrolase assays, reaction mixtures (100 μl) containing 50 mm cacodylate buffer, pH 6.5, 10 mm G6P and appropriate amounts of microsomal preparations were incubated at 37°C for 10 min. Disrupted microsomal membranes were prepared by incubating intact membranes in 0.2% deoxycholate for 20 min at 0°C. Non-specific phosphatase activity was estimated by pre-incubating disrupted microsomal preparations at pH 5 for 10 min at 37°C to inactivate the acid-labile G6Pase-α.
In G6P uptake assays, microsomes were incubated at 37°C for 10 min in a reaction mixture (100 μl) containing 50 mm sodium cacodylate buffer, pH 6.5, 250 mm sucrose and 0.2 mm [U-14C]G6P (50 μCi/μmol, American Radiolabeled Chemicals, St Louis, MO, USA). The reaction was stopped by filtering immediately through a nitrocellulose membrane (BA85, Schleicher & Schuell, Keene, NH, USA). Microsomes permeabilized with 0.2% deoxycholate, to abolish G6P uptake, were used as negative controls.
Phenotype analysis
Mice were examined for hepatic nodules by ultrasound using the Vevo 2100 system (VisualSonics, Ontario, Canada). Body composition was assessed using the Bruker minispec NMR analyzer (Karlsruhe, Germany). Blood levels of glucose, cholesterol, triglyceride, lactate, urate and insulin, along with hepatic levels of glycogen, triglyceride and G6P were determined, as described previously (6). Hepatic lactate and pyruvate were measured in liver lysates deproteinized using a kit from BioVision (Mountain View, CA, USA). Lactate and pyruvate were measured using the respective assay kit from BioVision. Hepatic NAD+ and NADH were determined using the EnzyChrom NAD+/NADH assay kit (BioAssay Systems, Hayward, CA, USA).
Insulin tolerance testing of mice consisted of a 4 h fast, prior to blood sampling, followed by intraperitoneal injection of insulin at 0.25 IU/kg and repeated blood sampling via the tail vein for 1 h.
Indirect calorimetry analysis
Energy expenditure and food intake were assessed using the Comprehensive Laboratory Animal Monitoring System (CLAMS) (Columbus Instruments, Columbus, OH, USA).
Quantitative real-time RT–PCR and western-blot analysis
The mRNA expression was quantified by real-time RT–PCR in an Applied Biosystems 7300 Real-Time PCR System using Applied Biosystems TaqMan probes (Foster City, CA, USA). Data were normalized to Rpl19 RNA. Western-blot images were detected using the LI-COR Odyssey scanner and the Image studio 3.1 software (Li-Cor Biosciences, Lincoln, NE, USA). Mouse monoclonals used were β-actin (Santa Cruz Biotechnology, Dallas, TX, USA), Total OXPHOS Rodent WB Antibody Cocktail and cMDH (Abcam, Cambridge, MA, USA). Rabbit monoclonals used were p-Akt-S473, p-Akt-T308 and p-AMPK-T172 (Cell Signaling, Danvers, MA, USA). Rabbit polyclonals used were ChREBP (Novus Biologicals, Littleton, CO, USA); Akt, AMPK and mMDH (Cell Signaling); cAAT, mAAT and PGC-1α (Abcam) and SIRT1 (Millipore, Billerica, MA, USA). Goat polyclonals used were cGPDH and mGPDH (Santa Cruz Biotechnology). Protein expression was quantified by densitometry using the Multi-Gauge version 3.0 software (Fujifilm, Tokyo, Japan).
Analysis of ChREBP nuclear localization
Mouse liver paraffin sections (10 μm thickness) were treated with 0.3% hydrogen peroxide in methanol to quench endogenous peroxidases and then blocked with the Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA, USA). For ChREBP detection, liver sections were incubated serially with a rabbit antibody against ChREBP and a biotinylated anti-rabbit IgG (Vector Laboratories). The resulting complexes were detected with an ABC kit using the DAB Substrate (Vector Laboratories). Sections were counterstained with hematoxylin (Sigma-Aldrich) and visualized using a Zeiss Axioskop2 plus microscope equipped with 40×/0.50NA objectives (Carl Zeiss MicroImaging, Jena, Germany). Images were acquired using a Nikon DS-Fil digital camera and NIS-Elements F3.0 imaging software (Nikon, Tokyo, Japan). The percentage of cells in 10 randomly selected fields (number cells/field were 92.5 ± 3.1 for wild-type and 81.2 ± 2.7 for AAV mice at 400×) containing ChREBP-positive nuclei was recorded.
Statistical analysis
The unpaired t-test was performed using the GraphPad Prism Program, version 4 (San Diego, CA, USA). Statistical significance was defined as P < 0.05.
Funding
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health.
Acknowledgement
We thank Audrey Noguchi for performing indirect calorimetry analysis.
Conflict of interest statement. None declared.
References
- 1.Chou J.Y., Jun H.S., Mansfield B.C. (2010) Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat. Rev. Endocrinol., 6, 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greene H.L., Slonim A.E., O'Neill J.A. Jr., Burr I.M. (1976) Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease. N. Engl. J. Med., 294, 423–425. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y.T., Cornblath M., Sidbury J.B. (1984) Cornstarch therapy in type I glycogen-storage disease. N. Engl. J. Med., 310, 171–175. [DOI] [PubMed] [Google Scholar]
- 4.Lei K.J., Chen H., Pan C.J., Ward J.M., Mosinger B., Lee E.J., Westphal H., Mansfield B.C., Chou J.Y. (1996) Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nat. Genet., 13, 203–209. [DOI] [PubMed] [Google Scholar]
- 5.Yiu W.H., Lee Y.M., Peng W.T., Pan C.J., Mead P.A., Mansfield B.C., Chou J.Y. (2010) Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol. Ther., 18, 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y.M., Jun H.S., Pan C.J., Lin S.R., Wilson L.H., Mansfield B.C., Chou J.Y. (2012) Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology, 56, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filhoulaud G., Guilmeau S., Dentin R., Girard J., Postic C. (2013) Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab., 24, 257–268. [DOI] [PubMed] [Google Scholar]
- 8.Benhamed F., Denechaud P.D., Lemoine M., Robichon C., Moldes M., Bertrand-Michel J., Ratziu V., Serfaty L., Housset C., Capeau J. et al. (2012) The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest., 122, 2176–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin-Montalvo A.1., de Cabo R. (2013) Mitochondrial metabolic reprogramming induced by calorie restriction. Antioxid. Redox. Signal, 19, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenz T. (2013) Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion, 13, 134–142. [DOI] [PubMed] [Google Scholar]
- 11.Easlon E., Tsang F., Skinner C., Wang C., Lin S.J. (2008) The malate–aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev., 22, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mráček T., Drahota Z., Houštěk J. (2013) The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta, 1827, 401–410. [DOI] [PubMed] [Google Scholar]
- 13.Goyary D., Sharma R. (2008) Late onset of dietary restriction reverses age-related decline of malate–aspartate shuttle enzymes in the liver and kidney of mice. Biogerontology, 9, 11–18. [DOI] [PubMed] [Google Scholar]
- 14.Taleux N., Guigas B., Dubouchaud H., Moreno M., Weitzel J.M., Goglia F., Favier R., Leverve X.M. (2009) High expression of thyroid hormone receptors and mitochondrial glycerol-3-phosphate dehydrogenase in the liver is linked to enhanced fatty acid oxidation in Lou/C, a rat strain resistant to obesity. J. Biol. Chem., 284, 4308–4316. [DOI] [PubMed] [Google Scholar]
- 15.Veyrat-Durebex C., Montet X., Vinciguerra M., Gjinovci A., Meda P., Foti M., Rohner-Jeanrenaud F. (2009) The Lou/C rat: a model of spontaneous food restriction associated with improved insulin sensitivity and decreased lipid storage in adipose tissue. Am. J. Physiol. Endocrinol. Metab., 296, E1120–E1132. [DOI] [PubMed] [Google Scholar]
- 16.Gomes A.P., Price N.L., Ling A.J., Moslehi J.J., Montgomery M.K., Rajman L., White J.P., Teodoro J.S., Wrann C.D., Hubbard B.P. et al. (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell, 155, 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kraus D., Yang Q., Kong D., Banks A.S., Zhang L., Rodgers J.T., Pirinen E., Pulinilkunnil T.C., Gong F., Wang Y.C. et al. (2014) Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature, 508, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mouchiroud L., Houtkooper R.H., Auwerx J. (2013) NAD+ metabolism: a therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol., 48, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Estall J.L., Kahn M., Cooper M.P., Fisher F.M., Wu M.K., Laznik D., Qu L., Cohen D.E., Shulman G.I., Spiegelman B.M. (2009) Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes, 58, 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris E.M., Meers G.M., Booth F.W., Fritsche K.L., Hardin C.D., Thyfault J.P., Ibdah J.A. (2012) PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol., 303, G979–G992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardie D.G. (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev., 25, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flatt P.R., Bailey C.J. (1981) Development of glucose intolerance and impaired plasma insulin response to glucose in obese hyperglycaemic (ob/ob) mice. Horm. Metab. Res., 13, 556–560. [DOI] [PubMed] [Google Scholar]
- 23.Barzilai N., Huffman D.M., Muzumdar R.H., Bartke A. (2012) The critical role of metabolic pathways in aging. Diabetes, 61, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura M.T., Yudell B.E., Loor J.J. (2014) Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res., 53, 124–144. [DOI] [PubMed] [Google Scholar]
- 25.Flowers M.T., Ntambi J.M. (2008) Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol., 19, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danielpour D., Song K. (2006) Cross-talk between IGF-I and TGF-beta signaling pathways. Cytokine Growth Factor Rev., 17, 59–74. [DOI] [PubMed] [Google Scholar]
- 27.Wang P., Zhang R.Y., Song J., Guan Y.F., Xu T.Y., Du H., Viollet B., Miao C.Y. (2012) Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes, 61, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romano A.D., Greco E., Vendemiale G., Serviddio G. (2014) Bioenergetics and mitochondrial dysfunction in aging: recent insights for a therapeutical approach. Curr. Pharm. Des., 20, 2978–2992. [DOI] [PubMed] [Google Scholar]
- 29.McCormack J.G., Westergaard N., Kristiansen M., Brand C.L., Lau J. (2001) Pharmacological approaches to inhibit endogenous glucose production as a means of anti-diabetic therapy. Curr. Pharm. Des., 7, 1451–1474. [DOI] [PubMed] [Google Scholar]
- 30.Agius L. (2007) New hepatic targets for glycaemic control in diabetes. Best Pract. Res. Clin. Endocrinol. Metab., 21, 587–605. [DOI] [PubMed] [Google Scholar]
- 31.Mutel E., Abdul-Wahed A., Ramamonjisoa N., Stefanutti A., Houberdon I., Cavassila S., Pilleul F., Beuf O., Gautier-Stein A., Penhoat A. et al. (2011) Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J. Hepatol., 54, 529–537. [DOI] [PubMed] [Google Scholar]
- 32.Abdul-Wahed A., Gautier-Stein A., Casteras S., Soty M., Roussel D., Romestaing C., Guillou H., Tourette J.A., Pleche N., Zitoun C. et al. (2014) A link between hepatic glucose production and peripheral energy metabolism via hepatokines. Mol. Metab., 3, 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee Y.M., Pan C.J., Koeberl D.D., Mansfield B.C., Chou J.Y. (2013) The upstream enhancer elements of the G6PC promoter are critical for optimal G6PC expression in murine glycogen storage disease type Ia. Mol. Genet. Metab., 110, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iizuka K., Takeda J., Horikawa Y. (2009) Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett., 583, 2882–2886. [DOI] [PubMed] [Google Scholar]
- 35.Coskun T., Bina H.A., Schneider M.A., Dunbar J.D., Hu C.C., Chen Y., Moller D.E., Kharitonenkov A. (2008) Fibroblast growth factor 21 corrects obesity in mice. Endocrinology, 149, 6018–6027. [DOI] [PubMed] [Google Scholar]
- 36.Ma L., Robinson L.N., Towle H.C. (2006) ChREBP.Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem., 281, 28721–28730. [DOI] [PubMed] [Google Scholar]