

Abstract
Fanconi anemia (FA) is a recessive X-linked and autosomal genetic disease associated with bone marrow failure and increased cancer, as well as severe germline defects such as hypogonadism and germ cell depletion. Although deficiencies in FA factors are commonly associated with germ cell defects, it remains unknown whether the FA pathway is involved in unique epigenetic events in germ cells. In this study, we generated Fancb mutant mice, the first mouse model of X-linked FA, and identified a novel function of the FA pathway in epigenetic regulation during mammalian gametogenesis. Fancb mutant mice were infertile and exhibited primordial germ cell (PGC) defects during embryogenesis. Further, Fancb mutation resulted in the reduction of undifferentiated spermatogonia in spermatogenesis, suggesting that FANCB regulates the maintenance of undifferentiated spermatogonia. Additionally, based on functional studies, we dissected the pathway in which FANCB functions during meiosis. The localization of FANCB on sex chromosomes is dependent on MDC1, a binding partner of H2AX phosphorylated at serine 139 (γH2AX), which initiates chromosome-wide silencing. Also, FANCB is required for FANCD2 localization during meiosis, suggesting that the role of FANCB in the activation of the FA pathway is common to both meiosis and somatic DNA damage responses. H3K9me2, a silent epigenetic mark, was decreased on sex chromosomes, whereas H3K9me3 was increased on sex chromosomes in Fancb mutant spermatocytes. Taken together, these results indicate that FANCB functions at critical stages of germ cell development and reveal a novel function of the FA pathway in the regulation of H3K9 methylation in the germline.
Introduction
The germline is responsible for the continuity of life. Therefore, the integrity of the genome must be precisely maintained in the germline. Indeed, the mutation rate of the germline is significantly lower than in somatic cells (1,2). A potential DNA repair pathway that ensures the fidelity of the germline is the Fanconi anemia (FA)-BRCA pathway. FA is a rare genetic disorder caused by mutation of an X-linked gene or biallelic mutation of autosomal genes. FA is associated with bone marrow failure, early onset of cancer, developmental defects and also germline defects such as reduced fertility, hypogonadism and testicular failure (3). These phenotypes are typically attributed to defective DNA repair and resultant genomic instability (4). At least 17 FA genes have been identified. Germline defects are also common in mouse models that harbor deficiencies for FA genes, including Fanca, Fancc, Brca2 (Fancd1), Fancd2, Fancf, Fancg, Fancl, Fancm and Slx4 (Fancp, also known as Btbd12) (5–14).
Eight of the FA proteins, FANCA/B/C/E/F/G/L/M, form the FA core complex (15). In part, because each component of the FA core complex is required for its function, and in part, because FA core complex components have some distinct functions, other FA proteins do not compensate for the loss of a specific FA core complex component (16–18). Once the DNA damage response is activated in response to DNA interstrand crosslinks (ICLs) or replication stress, FANCM recognizes the DNA damage sites and recruits other FA core complex components (19,20). Following this, FANCL, an E3 ligase in the FA core complex, monoubiquitinates the FANCD2-FANCI heterodimer (21–23). Monoubiquitinated FANCD2 and FANCI then localize to chromatin (21,23–25). FANCD2-I then recruits downstream repair factors: BRCA2 (FANCD1) (25), which is in a complex with BRIP1 (FANCJ, also known as BACH1) (26), PALB2 (FANCN) (27) and RAD51C (FANCO) (28), and the SLX4 (FANCP) nuclease (29).
At the cellular level, FA is characterized by chromosomal instability and hypersensitivity to DNA crosslinking agents, such as mitomycin C (MMC). Monoubiquitination of FANCD2 and FANCI by the FA pathway specifically mediates resistance to MMC (23,30). However, despite evidence for a role in somatic DNA repair and homologous recombination (HR) (31), the function of the FA pathway in the germline is largely unknown.
DNA damage response (DDR) pathways have critical functions in the regulation of the sex chromosomes during meiosis in the male germline. Many genes on the sex chromosomes are silenced during meiosis in response to a state of unsynapsis in a process known as meiotic sex chromosome inactivation (MSCI). MSCI is an essential step in germ cell development (32,33), during which DDR proteins such as BRCA1 and ATR accumulate on the axes of sex chromosomes and regulate DDR pathways on the unsynapsed axes (34–36). MDC1, a binding partner of H2AX phosphorylated at serine 139 (γH2AX), amplifies γH2AX from axes onto the chromosome-wide domain and initiates MSCI (37). Because the FA pathway proteins FANCA and FANCD2 associate with BRCA1 (30,38), and because ATR regulates monoubiquitination of FANCD2 (39), we reasoned that the FA pathway is involved in the regulation of the sex chromosomes during meiosis. Indeed, FANCD2 accumulates on meiotic sex chromosomes and colocalizes with BRCA1 (30).
To reveal the function of the FA pathway in the germline, we focused on the FANCB protein based upon its localization to meiotic sex chromosomes (described below). Human FANCB is crucial for FANCL stability and FANCD2 ubiquitination (17,40), and is considered to be a critical factor in the FA pathway. Interestingly, FANCB is the only identified X-linked FA factor. For this study, we generated Fancb mutant mice, which represent the first mouse model of X-linked FA. Fancb mutant mice are infertile, and FANCB regulates three critical stages in the male germline: PGCs in embryos, undifferentiated spermatogonia and meiosis in adult testes. Our study provides a novel link between the FA DNA repair pathway and regulation of H3K9 methylation in the germline.
Results
FANCB localizes on the sex chromosomes during meiosis
To determine whether FANCB, an FA pathway protein, accumulates on the sex chromosomes during meiosis, we performed immunofluorescence microscopy on meiotic chromosome spreads from wild-type mice. We found that signals detected by anti-human FANCB antibody localized on the mouse sex chromosomes during meiosis (Supplementary Material, Fig. S1). To confirm the specificity of the signals, we generated two independent antibodies raised against amino acids 140–320 of mouse FANCB that recognizes a region distinct from that of the anti-human FANCB antibody. These new antibodies confirmed the localization of FANCB on the sex chromosomes in wild-type meiosis (Fig. 1A and Supplementary Material, Fig. S1). FANCB was not detected on the autosome regions in the leptotene, zygotene, pachytene or diplotene stages. However, FANCB localized around the unsynapsed axes of the sex chromosomes beginning at the early pachytene stage. FANCB signals progressively increased from axes to the entire domain of the sex chromosomes by the early diplotene stage, but disappeared from the sex chromosomes in the late diplotene stage. These results raise the possibility that FANCB is involved in the regulation of the sex chromosomes during meiosis.
Figure 1.

FANCB localization on meiotic sex chromosomes and generation of Fancb mutant mice. (A) Immunostaining of meiotic chromosome spreads using an anti-mouse FANCB antibody. SYCP3 is a marker for meiotic chromosome axes. Dotted squares represent the areas surrounding sex chromosomes, and the areas are magnified in the right panels. Scale bars: 10 µm. (B) Structure of Fancb gene and FANCB protein. The site of ZFN-induced mutation of line #84 mutant mice is shown. Asterisk indicates targeting site. White boxes represent non-coding regions, and gray boxes represent coding regions. The mutation leads to a frame change (shown with a black box) and a premature stop codon. (C) Immunoprecipitation and western blot analysis showing the absence of full-length FANCB protein in Fancb mutant testes. (D) Body weights compared for wild-type and Fancb mutant mice at 4 and 10 weeks after birth. Mean and SEM for independent mice are shown. Numbers of mice analyzed at 4 and 10 weeks, respectively: seven and six wild-type, and seven and six Fancb mutant mice. *P < 0.05. Unpaired t-test. (E) Fertility test: mean numbers of litters and SEM for independent mice are shown. Numbers of mice tested: seven wild-type and seven Fancb mutant mice. P < 0.05. Unpaired t-test. (F) Testis weights/body weight (mg/g) of wild-type and Fancb mutant mice at 4 and 10 weeks. Mean and SEM for independent mice are shown. Numbers of mice analyzed at 4 and 10 weeks, respectively: seven and six wild-type, and seven and six Fancb mutant mice. *P < 0.05. Unpaired t-test. (G) Representative photo of the testes of wild-type and Fancb mutant mice. (H) Immunostaining of testicular sections for H4K16 acetylation and protamine 1 (PRM1) from 10-week-old wild-type and Fancb mutant mice. Scale bars: 100 µm. The areas shown with white dotted squares are magnified in the right panels.
FANCB is required for male fertility
To address the function of FANCB, we generated Fancb mutant mice harboring a mutation in exon 2 of the X-linked Fancb gene using zinc-finger nuclease (ZFN) technology (Fig. 1B). After the injection of the ZFN into pronuclei of 1-cell stage embryos, we obtained four independent mouse lines harboring mutations in the Fancb gene. Among them, three were males and all of them were infertile, suggesting that FANCB is essential in the male germline (Supplementary Material, Fig. S2). Owing to the X-linkage of the Fancb gene and male infertility in the Fancb mutants, we were unable to obtain female homozygous Fancb mutants; instead, we used a female line (#84) as a founder to generate male Fancb mutants for further analyses. The Fancb mutant (#84) harbors a five base pair deletion that generates a truncated protein owing to a premature stop codon (Fig. 1B). Although the truncated protein was expressed and localized normally on sex chromosomes during meiosis (Supplementary Material, Fig. S1), FANCB protein was not detected by western blotting using the antibody against the C-terminus of FANCB in Fancb mutants (Fig. 1C), suggesting that the C-terminus of FANCB is truncated. In this experiment, owing to the low protein expression of FANCB because of low RNA transcription in germ cells (Supplementary Material, Table S1), western blotting was performed after immunoprecipitation (40). In somatic cells, protein expression of FANCB is also low in spite of its critical function, and immunoprecipitation is also necessary to detect FANCB protein by western blotting (40).
Fancb mutants were viable but consistently smaller than their wild-type littermates (Fig. 1D), suggesting that growth retardation was caused by the Fancb mutation. Consistent with other independent ZFN male mutants (Supplementary Material, Fig. S2), the Fancb mutants were sterile (Fig. 1E), and mutant testes at 4 and 10 weeks were significantly smaller than those of littermate controls (Fig. 1F and G). We compared the development of testicular seminiferous tubules from the Fancb mutants with tubules from littermate controls by histological analysis. The majority of the Fancb mutants' seminiferous tubules were markedly atrophic and largely devoid of spermatogenic cells (Fig. 1H). Although the other tubules contained germ cells up to elongating spermatids, as detected by H4K16 acetylation, these tubules were devoid of the final step of elongated spermatids that undergo histone-to-protamine exchange (Fig. 1H, magnified panels). These data indicate that FANCB is essential in the male germline.
Primordial germ cells are substantially reduced in Fancb mutant mice
As the Fancb mutant mice exhibited a small testis phenotype and because most mutant tubules were devoid of germ cells in adults, we first investigated whether germ cell defects occurred in PGCs during embryonic development. We performed immunostaining of PGCs from Fancb mutant embryos at E9.5, E11.5 and E13.5 using the germ cell marker SOX2. At E9.5, the number of PGCs detected by the presence of SOX2 in the Fancb mutants was already less than in wild-type littermates (Fig. 2A and D). The number of PGCs was substantially decreased compared with wild-type littermates at E11.5 and E13.5 (Fig. 2B, C and E), whereas the distribution of GATA4-positive Sertoli cells remained comparable between controls and Fancb mutants at E13.5 (Fig. 2C). However, there was no apparent change in the location of PGCs between Fancb mutants and wild-type littermates, and PGCs were not detected outside the genital ridge/gonad, suggesting that mutation of Fancb did not perturb PGC migration to gonads. These results suggest that FANCB is important for PGC proliferation and/or survival. Previously, PGC defects were also observed in other FA mutants (6,41,42). Together, the common PGC defect in FA mutant mice suggests that the FA pathway is collectively involved in PGC proliferation.
Figure 2.

Reduction of PGCs in Fancb mutant mice. (A–C) Immunostaining of mouse whole embryos at E9.5, E11.5 and E13.5. SOX2 is a marker for PGCs, and GATA4 is a marker for gonadal somatic cells. Scale bars: 100 µm. (D) Total numbers of the SOX2-positive PGCs per embryo at E9.5. (E) Total numbers of PGCs in each genital ridge (cells/genital ridge) at E11.5. Mean and SEM for independent mice are shown. Numbers of mice analyzed at E9.5 and E11.5, respectively: five wild-type and four Fancb mutant mice. *P < 0.05. Unpaired t-test.
FANCB regulates maintenance of undifferentiated spermatogonia
To determine whether FANCB has a function during spermatogenesis after birth, we examined Fancb mutant testes in detail. In accord with the testicular defects, the diameter of the seminiferous tubules in Fancb mutants was significantly smaller than tubules in wild-type mice at both 4 weeks and 10 weeks (Fig. 3A and B). In Fancb mutant testes, Sertoli cells (detected by the presence of WT1, a Sertoli cell marker) were enriched on the periphery of the tubules (Fig. 3A), suggesting the significant depletion of germ cells and relative enrichment of Sertoli cells. Consistent with germ cell loss during spermatogenesis in Fancb mutants, the number of apoptotic cells detected by TUNEL assay was continuously high in Fancb mutant testes (Fig. 3C and D). Apoptosis was observed at the periphery of tubules and in differentiated spermatids (Supplementary Material, Fig. S3). Next, we examined whether depletion of germ cells occurs progressively during spermatogenesis, in addition to the reduction of progenitor cells resulting from PGC loss. Because the maintenance of undifferentiated spermatogonia is required for continuous sperm production during spermatogenesis, we examined whether the number of PLZF-positive undifferentiated spermatogonia, which includes spermatogonial stem cells, is altered in Fancb mutants. Although the majority of tubules already lack germ cells and contain Sertoli cells only at 4 weeks, the number of PLZF-positive cell-containing tubules (PLZF-positive tubules) progressively decreased in tubules from 4 weeks up to 6 months (Fig. 3E–G). Further, among the germ cell-containing tubules, the number of PLZF-positive cells gradually decreased (Fig. 3H). These results suggest that FANCB regulates maintenance of undifferentiated spermatogonia.
Figure 3.

FANCB regulates the maintenance of undifferentiated spermatogonia. (A) Immunostaining of testicular sections at 10 weeks old. WT1 is a marker for Sertoli cells. Dotted circles are the areas of representative tubules. (B) Quantification of seminiferous tubule areas (103 μm2) at 4 and 10 weeks after birth. Mean and SEM for independent mice are shown. A total of 100 tubules from three independent mice were analyzed at 4 and 10 weeks, for both wild-type and Fancb mutant mice. (C) TUNEL assays of testicular sections at 10 weeks old. (D) Scoring of the number of TUNEL-positive cells per germ cell-containing tubule. Mean and SEM for independent mice are shown. Total numbers of tubules analyzed at 4 and 10 weeks, respectively: 303 and 430 wild-type, and 394 and 138 Fancb mutant mice. Three independent mice were analyzed for each dataset. (E and F) Immunostaining of testicular sections at 10 weeks old for panels in E and at 6-months old for panels in F. PLZF is a marker for undifferentiated spermatogonia. Arrowheads: PLZF-positive cells. (G) Scoring of the percentage of PLZF-positive tubules per total tubule. One hundred tubules from three independent mice were analyzed at 4 weeks, 10 weeks and 6 months, in both wild-type and Fancb mutant mice. Mean and SEM for independent mice are shown. (H) Scoring of the number of PLZF-positive cells per PLZF-positive tubule. One hundred tubules from three independent mice were analyzed at 4 weeks, 10 weeks and 6 months, both for wild-type and Fancb mutant mice. Mean and SEM for independent mice are shown. (I and J) Immunostaining of testicular sections at 10 weeks old for panels in I, and at 6 months old for panels in J. (K) Scoring of the percentage of γH2AX-positive tubules per total tubule. Mean and SEM for independent mice are shown. One hundred tubules from three independent mice were analyzed at 4 and 10 weeks, in both wild-type and Fancb mutant mice. One hundred tubules (two independent mice) were analyzed at 6 months, in both wild-type and Fancb mutant mice. Scale bars: 100 µm. *P < 0.05. Unpaired t-test.
Next, to examine whether germ cell loss occurs during differentiation in spermatogenesis, we scored the number of tubules containing differentiated spermatocytes after immunostaining with γH2AX (a marker for nuclei at the leptotene and zygotene stages, and of the XY body in the pachytene stage) and H1T (a marker that is enriched in nuclei from the mid pachytene stage onward; Fig. 3I–K). Although more than half of the testicular tubules are devoid of germ cells in Fancb mutants, remaining germ cells underwent meiotic progression detected with γH2AX and H1T (Fig. 3I), reached the round spermatid stage as detected with H4K16 acetylation, and proceeded up to the elongating spermatid stage (Fig. 1H). The population of these γH2AX-positive (germ-cell-containing) tubules in Fancb mutants was decreased from 4 weeks up to 6 months (Fig. 3K). Notably, the decrease of PLZF-positive and γH2AX-positive tubules was comparable from 4 weeks to 6 months in Fancb mutants (Fig. 3G and K), suggesting that the main cause of germ cell loss could be attributed to defects in the maintenance of undifferentiated spermatogonia rather than defects in differentiation.
FANCB is not essential for meiotic recombination but is required for FANCD2 foci formation on autosomes
During male meiosis, two essential events involve the DDR: one is meiotic recombination, which takes place during the early and middle stages of prophase I: leptotene, zygotene and pachytene; the other is MSCI, which takes place during the middle and late stages of prophase I: pachytene and diplotene (Fig. 4A). Given the role of FANCB in the somatic DDR (40,43), it is possible that FANCB is involved in these two steps at different times during meiosis. Therefore, we investigated whether FANCB regulates meiotic stage progression. The population of each stage of meiotic prophase I was not altered in Fancb mutants (Fig. 4B). This result suggests that FANCB is not involved in meiotic prophase I stage progression.
Figure 4.

Roles of FANCB in meiotic recombination. (A) Schematic of meiotic prophase and the involvement of two DDR events. (B) The percentage of each stage of meiotic prophase as judged by SYCP3 immunostaining. Total numbers of cells analyzed: 263 from wild-type, and 230 from Fancb mutant mice. Mean and SEM for independent mice are shown. Three independent mice were analyzed for each dataset. (C–I) Immunostaining of chromosome spreads. Scale bars: 10 µm. (C–E) RAD51 foci in zygotene and pachytene spermatocytes. In the right panels: scoring of the number of RAD51 foci per cell at the indicated stages. Distribution of data is shown as dots; mean ± SEM for independent mice is shown as bars. Total numbers of analyzed nuclei are indicated in the panels. For each panel, four independent mice were analyzed for both wild-type and Fancb mutant mice. (F) SYCP1 localization in pachytene spermatocytes. (G and H) FANCD2 foci in zygotene and pachytene spermatocytes. (I) MLH1 foci in pachytene spermatocytes. In the right panel: scoring of the number of MLH1 foci per cell at the mid pachytene stage. Distribution of data is shown as dots; mean ± SEM for independent mice is shown as bars. Total numbers of analyzed nuclei are indicated in the panel. Five independent mice were analyzed for both wild-type and Fancb mutant mice. Arrows: sex chromosomes; arrowheads: RAD51 foci in panel (E) and FANCD2 foci in panel (H). *P < 0.05. ns: not significant. Unpaired t-test.
To examine the possible role of FANCB in meiotic recombination, we examined the localization of RAD51, which is a marker of sites of DNA double-strand breaks (DSBs) during meiosis. Between wild-type and Fancb mutants, no significant difference was observed in RAD51 foci formation and disappearance; RAD51 signals accumulated at the sites of DSBs in the early zygotene stage and were decreased in the late zygote stage of mice in each genotype (Fig. 4C and D). These signals largely disappeared by the mid pachytene stage, except from the axes of sex chromosomes and some autosomal regions, regardless of the genotype (arrows and arrowheads, respectively, in Fig. 4E). At this stage, the number of autosomal RAD51 foci was slightly increased in the Fancb mutants (Fig. 4E), suggesting an increased number of persistent DSBs owing to a defect in DSB repair on autosomes in Fancb mutants. However, consistent with normal RAD51 foci formation in the zygote stage, we detected normal chromosome synapsis in Fancb mutants (Fig. 4F), as detected by double staining for two components of the synaptonemal complex that are exclusively present on synapsed axes, SYCP3 and SYCP1. Concomitant with meiotic recombination, FANCD2 foci accumulated on the synapsed axes of autosomes during the late zygotene to early pachytene stages in wild-type control littermates (Fig. 4G and H) (30). However, in Fancb mutants, FANCD2 foci on synapsed regions were abolished (Fig. 4G and H), indicating that FANCB is required for FANCD2 foci formation on synapsed axes at these stages. This result reveals a parallel between somatic cells and meiotic cells in that the FA core complex is required for FANCD2 foci formation, and suggests that FANCB activates the FA pathway during meiotic recombination.
Next, we examined whether defective FANCD2 foci in Fancb mutants were associated with an altered outcome of meiotic recombination. To this end, we examined the localization of MLH1, a mismatch repair factor that localizes on the sites of crossover in the mid pachytene stage. The numbers of MLH1 foci in mid pachytene spermatocytes were comparable between the Fancb mutants and their control littermates (Fig. 4I). These results indicate that neither FANCB nor FANCD2 foci are required for crossover formation.
FANCB is required for FANCD2 foci on the sex chromosomes during meiosis
Next, we tested whether FANCB regulates other DDR events that occur on the sex chromosomes during meiosis. In normal meiosis, BRCA1 accumulated on unsynapsed axes of the sex chromosomes (Fig. 5A); BRCA1 is required for ATR loading (34) and amplification of DDR signals along the axes (36). Together with its activator TOPBP1, ATR accumulated along the unsynapsed axes of the chromosomes and spread through the chromosome-wide domain (Fig. 5B and C), as did γH2AX (Fig. 5D) (35). MDC1, the binding partner of γH2AX, localized on the sex chromosomes (Fig. 5E). Furthermore, MDC1 is required for spreading the TOPBP1–ATR complex and γH2AX to the chromosome-wide domain, presumably based on the interaction between MDC1 and TOPBP1 (37). In Fancb mutants, localization of these factors was unchanged in 100% of observed Fancb mutant cells (four independent mice; total number of cells analyzed: n = 100 for BRCA1, n = 100 for ATR, n = 116 for γH2AX; Fig. 5A–E). Therefore, FANCB is not required for DDR events that occur at the onset of MSCI.
Figure 5.

Regulation of DDR factors on the sex chromosomes in Fancb mutant mice. (A–G and I) Immunostaining of chromosome spreads of pachytene spermatocytes. In panel G, dotted squares represent areas surrounding sex chromosomes, and the areas are magnified in the bottom panels. Arrows: sex chromosomes. Scale bars: 10 µm unless otherwise indicated. (H) Scoring of the number of RAD51 foci per X axis, Y axis, or pseudo-autosomal region (PAR) in mid-pachytene spermatocytes. Distribution of data is shown as dots; mean ± SEM for independent mice is shown as bars. Total numbers of analyzed nuclei are indicated in the panel. Four independent mice were analyzed for both wild-type and Fancb mutant mice. *P < 0.05. Unpaired t-test. (J) Model of the role of FANCB in DDR pathways on the sex chromosomes during meiosis.
Next, we examined DDR factors that act downstream of the TOPBP1–ATR–MDC1–γH2AX network on the sex chromosomes. In the early pachytene through early diplotene stages, FANCD2 foci localized on unsynapsed axes of the sex chromosomes (Fig. 5F) (30). Similar to FANCD2 foci on synapsed axes at earlier stages (Fig. 4E), FANCD2 foci on the sex chromosomes were abolished in Fancb mutants (Fig. 5F). This result suggests that the FA pathway is activated on the sex chromosomes and that FANCB is an essential factor for this activation. Next, we investigated how the FA pathway regulates other DDR/repair factors that localize on the sex chromosomes. In wild-type, RAD51 foci are amplified along the unsynapsed axes of the sex chromosome in a BRCA1-dependent manner during the early-to-mid pachytene stages (36). In Fancb mutants, RAD51 foci were decreased on X chromosome axes, but were not affected on Y chromosome axes or pseudo-autosomal regions in Fancb mutants (Fig. 5G and H). This result suggests that FANCB is involved in the amplification of RAD51 foci along the X-axes. On the other hand, SLX4 (FANCP), another DNA repair factor that localizes on the on the sex chromosomes during meiosis (44), accumulated normally in Fancb mutants (Fig. 5I). Although activation of the FA pathway, as shown by FANCD2 foci, and amplification of RAD51 foci are FANCB-dependent, SLX4 was normally regulated in Fancb mutants. Thus, FANCB is required for FANCD2 foci on sex chromosomes and amplification of RAD51 foci on the X chromosome axes (Fig. 5J).
MDC1 is required for FANCB localization
The results above revealed the role of FANCB in DDR-related pathways on the sex chromosomes. To further clarify this role, we tested the localization of FANCB in mutant backgrounds for several different DDR factors. In Mdc1 knockout mice, FANCB localization on sex chromosomes was abolished (Fig. 6A). Thus, FANCB localization is MDC1-dependent. However, we found that FANCB was present on the sex chromosomes in the testes of two types of Brca1 mutants (Fig. 6B and C): Brca1 exon 11 conditional deletion mice using germline-specific Ddx4-Cre, and double mutants of Brca1 exon 11 deletion and 53bp1; deletion of 53bp1 rescues the somatic phenotype of Brca1 deletion but not the meiotic phenotype (36). Because MDC1 operates in the Brca1 mutants and forms aberrant γH2AX domains (36), FANCB may be recruited by these aberrant γH2AX domains in the Brca1 mutants.
Figure 6.
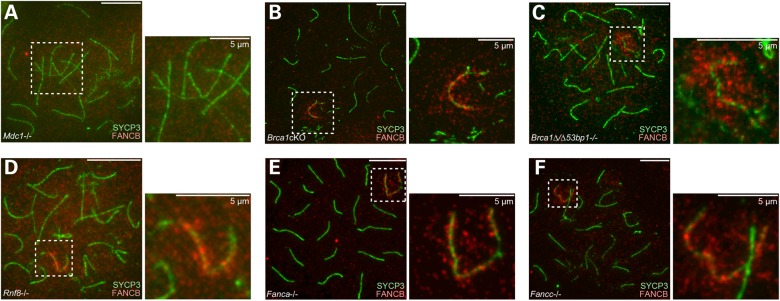
MDC1 is required for FANCB localization on the sex chromosomes. (A–F) Immunostaining of chromosome spreads of pachytene spermatocytes from mice that are mutant for different DDR factors as indicated. Dotted squares represent areas surrounding sex chromosomes, and the areas are magnified in the right panels. Scale bars: 10 µm unless otherwise indicated.
RNF8 is an MDC1-interacting protein and recruits FA core components to sites of DNA ICLs in somatic cells (45). During meiosis, MDC1 and RNF8 have distinct functions in, respectively, sex chromosome inactivation and the establishment of active epigenetic modifications for gene activation in round spermatids (37,46). In accordance with this, FANCB localization on sex chromosomes is dependent on MDC1, but it is unchanged in Rnf8 knockout mice (Fig. 6D). Furthermore, FANCB localization to the sex chromosomes was also unaltered in knockouts of two other subunits of the FA core complex, Fanca and Fancc (Fig. 6E and F). This is interesting given recent reports that, in contrast to meiosis, FANCA and FANCC are required for the FA core complex to bind at DNA damage sites in somatic cells (17,47). Together, these results suggest that FANCB recruitment is not regulated by RNF8 and that it is independent of other FA core subunits.
FANCB regulates H3K9 methylation on meiotic sex chromosomes
On the sex chromosomes, the initiation of chromosome-wide silencing is mediated by the action of γH2AX-MDC1 signaling in the early pachytene stage and is followed by establishment of a cascade of both inactive and active epigenetic modifications after the late pachytene and diplotene stages (33). DDR factors can regulate downstream epigenetic modifications on the sex chromosomes. For example, RNF8 is required for the establishment of a cascade of active epigenetic modifications and subsequent activation of reproduction genes in round spermatids (46). Therefore, we reasoned that the FA pathway, activated by FANCB, could potentially regulate downstream epigenetic modifications; thus, we examined whether FANCB is involved in such regulation of the sex chromosomes.
Because RNF8 is one of the factors upstream of the FA pathway in the repair of ICLs in somatic cells (45), we first investigated the possibility that FANCB regulates active epigenetic modifications mediated by RNF8. In the early pachytene stage, RNF8 mediates polyubiquitination of an unknown target (detected with both FK2 and anti-ubiquitinated H2A E6C5 antibodies) and H4K20me1 on the sex chromosomes (46,48). Further, from the diplotene stage onward, H3K4me2, a marker for transcriptional activation also accumulates on sex chromosomes in an RNF8-dependent manner (46). However, in Fancb mutant spermatocytes, accumulation of ubiquitination, H4K20me1 and H3K4me2 was not affected on sex chromosomes (Fig. 7A–D).
Figure 7.

FANCB regulates H3K9 methylation on sex chromosomes. (A–F, H, I, K and L) Immunostaining of chromosome spreads. Arrows: sex chromosomes. Scale bars: 10 µm. (G and J) Quantification of H3K9me2/3 signal intensity on sex chromosomes and autosome regions. Relative Mean Fluorescence Intensity (RMFI) was calculated as described in the Materials and Methods section. Mean and SEM for independent mice are shown. Numbers of analyzed nuclei are indicated in the panels. Three independent mice were analyzed for both wild-type and Fancb mutant mice. *P < 0.05 by one-way ANOVA followed by Tukey's range test.
Next, we investigated an alternative possibility that FANCB regulates silent modifications on the sex chromosomes. H3K9me2, an epigenetic modification for gene silencing, starts to accumulate on sex chromosomes during the transition between the pachytene-to-diplotene stages (49,50). In Fancb mutants, accumulation of H3K9me2 on sex chromosomes was largely diminished during the late pachytene-to-diplotene transition (Fig. 7E and F). To further corroborate this observation, we quantified the signal intensity of H3K9me2, both on the XY body and autosomes regions, and confirmed that accumulation of H3K9me2 on the XY body was significantly decreased during the late pachytene and diplotene transition in Fancb mutants (Fig. 7G). While FANCB positively regulates H3K9me2 accumulation, we found that FANCB negatively regulates another epigenetic modification associated with gene silencing, H3K9me3. In the wild-type, H3K9me3 initially accumulates on the sex chromosome in the early pachytene stage and disappears in the mid pachytene stage owing to histone H3 replacement, but then reaccumulates on sex chromosomes in the diplotene stage (51). In Fancb mutants, H3K9me3 intensity was increased on the sex chromosomes in comparison with wild-type sex chromosomes, in both the early pachytene and diplotene stages (Fig. 7H–J). Therefore, H3K9me2 and H3K9me3 are distinctly regulated on the sex chromosomes in Fancb mutants.
To test whether these changes in histone modifications are associated with transcriptional changes of the sex chromosomes, we performed immunostaining of RNA polymerase II. On the sex chromosomes, RNA polymerase II was largely excluded, both in the wild-type and in Fancb mutant mice (Fig. 7K). Consistent with the normal accumulation of γH2AX and MDC1 on the sex chromosomes in Fancb mutants, this result suggests that initiation of MSCI is not perturbed in Fancb mutants. However, we were unable to perform detailed gene expression analysis using purified pachytene spermatocytes because of the extensive germ cell loss in in Fancb mutants. We further investigated whether FANCB is required for the localization of SCML2, a recently identified germline-specific polycomb protein that accumulates on the sex chromosome (48,52). The localization of SCML2 was not changed in Fancb mutants (Fig. 7L). SCML2 suppresses excessive accumulation of H3K9me1 on the sex chromosomes (48), and we observed no difference in H3K9me1 accumulation patterns in Fancb mutants versus wild-type (Supplementary Material, Fig. S4). Thus, regulation of H3K9me1 is independent of FANCB (Fig. 8). Together with the report described above, our data suggest that H3K9 methylation is distinctly regulated on the sex chromosomes during meiosis at the levels of mono-, di- and trimethylation.
Figure 8.

Model of a FANCB-regulated epigenetic pathway on the sex chromosomes. Summary of the FANCB-regulated pathway, and SCML2- and RNF8-dependent pathways, on the sex chromosomes during meiosis. The SCML2- and RNF8-dependent pathways were identified previously.
Discussion
In this study, we show that Fancb is essential for spermatogenesis and that it regulates three aspects of spermatogenesis: maintenance of PGCs, maintenance of undifferentiated spermatogonia, and regulation of H3K9 methylation of the sex chromosomes during meiosis. Consistent with other FA-deficient mice, germline defects are a prominent phenotype of Fancb mutants. Although the molecular etiology behind the common germline defects of FA-deficient mice remains elusive, it is conceivable that the FA pathway is activated and is collectively involved in these critical stages of germ cell development. The essential step in the activation of the FA pathway in somatic cells is FANCD2 monoubiquitination. A recent in vitro reconstitution study reveals that three proteins (FANCB, FANCL and FAAP100) are the minimal subcomplex that is essential for FANCD2 monoubiquitination (47). In line with this biochemical evidence, the germ cells, and also testicular size, were severely compromised in Fancd2 mutants (8) and in Fancb mutants (this study), as compared with other mutants of FA core subunits (including FANCA, FANCC and FANCM) that display subfertility (11,13,41). Because the testis weight or seminiferous tubule phenotype of Fancb mutants were not rescued when we tried to outcross Fancb mutants to CD1 or DBA/2J strains (data not shown), these phenotypes do not appear to be dependent on strain background. Taken together, we suggest that FANCB has an essential role in germ cell development. Fancb mutants display loss of function of the FA pathway during germ cell development, as shown by a defect in FANCD2 foci on chromosomes during meiosis (Fig. 4). Despite the presence of truncated protein and its localization to the sex chromosomes in Fancb mutants (Supplementary Material, Fig. S1), these severe phenotypes suggest that FANCB is functionally null in our Fancb mutants. We observed a similar finding in the case of exon 11 deletion of Brca1, in which a severe meiotic phenotype was associated with expression and normal localization of a truncated protein (34).
Many genes involved in spermatogenesis are X-linked (53,54) and X-linkage of Fancb may reflect an essential role for the gene during germline development. Although massive germ cell loss was observed in more than half of testicular tubules in Fancb mutants owing to defects in the maintenance of PGCs and undifferentiated spermatogonia, these phenotypes appeared to be incompletely penetrant and germ cell-containing tubules were also observed in Fancb mutants (Fig. 3). However, we consistently observed the meiotic phenotypes in the rest of the germ cell-containing tubules in Fancb mutants. Our results suggest that FANCB proteins can function on the sex chromosomes after transcriptional silencing of Fancb owing to MSCI. This is the case for another X-linked protein, SCML2, which is an epigenetic regulator of the sex chromosomes despite transcriptional silencing of the Scml2 gene by MSCI (48,52).
In this study, in which we took advantage of cytological and genetic experiments, we utilized meiotic sex chromosomes as a model to dissect the molecular function of FANCB and the FA pathway. During meiosis, DDR signaling and epigenetic modifications are temporally and spatially regulated on the sex chromosomes. This feature enabled us to both dissect the pathway FANCB functions in and demonstrate that FANCB regulates H3K9 methylation. Although FANCB is not responsible for initial RAD51 foci formation at the zygotene stage, FANCB is required for the formation of FANCD2 foci both on autosomes and sex chromosomes. FANCB also has a role in suppression of RAD51 foci on autosomes and in amplifying RAD51 foci on the X chromosome axes. Other core subunits, such as FANCA, are also not involved in RAD51 foci formation during meiosis at the zygotene stage (41). Together with this study, the FA core complex is unlikely to be crucial for RAD51 loading, but the complex is likely involved in DNA repair events on the autosomes and in the regulation of DDR signaling on the X chromosome axis during meiosis.
It is conceivable that the function of the FA pathway on the sex chromosomes is independent of DSB repair and instead may serve as a signal to regulate H3K9 methylation. Many other DDR factors, besides the FA pathway, accumulate on the sex chromosomes, and their main roles could be epigenetic regulation of the sex chromosomes, as is the case with RNF8 (46). Indeed, the localization of RAD51 is confined to unsynapsed axes of the sex chromosomes, and many DDR factors localize on the chromosome-wide region of the sex chromosomes where DNA breaks may not exist. This function of FANCB could be different from its role in the repair of ICLs in somatic cells, where the FA pathway is directly involved in DNA repair. In Fancb mutant MEFs, FANCD2 foci were abrogated and RAD51 foci formation was diminished after treatment with mitomycin C (43). It would be intriguing to investigate whether H3K9 methylation is altered in an FANCB-dependent manner in somatic cells following exposure to DNA ICLs. Also, it is of note that H3K9me2 and H3K9me3 are distinctly regulated in the C. elegans germline (55). Similarly, the FA pathway could be involved in the distinct regulation of H3K9me2 and H3K9me3 in the mammalian germline. In mammals, the lysine methyltransferase Suv39h1/h2 mediates H3K9me3 on pericentric heterochromatin (56), whereas the lysine methyltransferase G9a mediates H3K9me2 on euchromatin (57). While histone lysine methyltransferases which mediate H3K9me2 and H3K9me3 on the sex chromosomes during meiosis have not been determined (58,59), it is possible that FANCB acts at the sex chromosomes by regulating Suv39h1/h2 and G9a. Despite the alteration of H3K9me2 and H3K9me3, Fancb mutants did not exhibit meiotic arrest; instead, apoptosis was observed in later stages. It is possible that the abnormal epigenetic state in Fancb mutants culminates in apoptosis in haploid spermatids.
Our study further dissects the interplay among FA proteins. Recent studies reported that FANCA and FANCC are required for the binding of components of the FA core complex to DNA damage sites in somatic cells (17,47). But during meiosis, the recruitment of FANCB to sex chromosomes is independent of FANCA and FANCC. Furthermore, we found that Fancb mutation did not affect the localization of SLX4, which is not a component of the FA core complex. SLX4 is involved in several pathways, including the FA pathway, by scaffolding various nucleases and genome stability factors (60). Therefore, the function of SLX4 could be, in part, independent of FANCB, even though it is an FA protein. This is consistent with the observation that signals for γH2AX are normal in Fancb mutant mice, whereas γH2AX is mislocalized to the autosomal region in Slx4 mutant mice (44).
Our finding that FANCB in the FA pathway regulates H3K9 methylation could raise a possible link between the FA pathway and epigenetic regulation. While beyond the scope of this study, the downstream FA pathway also has a role in regulating H3K9 methylation (K.G.A. and S.H.N. unpublished data). Although a reduction of PGCs is commonly observed in FA mutant mice, this was considered to be due to PGC proliferation defects rather due to than PGC cell death (6,42). An alternative possibility could be that the FA pathway is required for epigenetic reprogramming, a critical step in PGC development (61). Interestingly, the reduction of PGCs is already evident at E9.5 in Fancb mutants. This is earlier than the PGC defects observed in other FA mutants, with the exception of Fancl (Pog) mutants, in which the PGC defect was also evident at E9.5 (12). In line with a possible role of the FA pathway in epigenetic regulation, the base excision repair pathway is implicated in active DNA demethylation during epigenetic reprogramming in PGCs (62). It will be interesting to determine whether the FA pathway is involved in epigenetic regulation in general.
Our analyses further reveal a possible role for FANCB in the maintenance of undifferentiated spermatogonia. Progressive germ cell depletion was also observed in Fancm mutant mice (42) and other FA proteins may also be involved in the maintenance of undifferentiated spermatogonia. Based on the involvement of the FA pathway in the establishment of pluripotency, and in the maintenance of pluripotent stem cells and hematopoietic stem cells (63,64), a possible function of the FA pathway could be the maintenance of spermatogonial stem cells through the regulation of H3K9 methylation. Because the germline has a lower mutation rate than in soma, it would be intriguing to explore whether defects in PGCs and undifferentiated spermatogonia in the FA mutants are associated with defects in DNA repair that monitor the fidelity of germ cells and/or defects in epigenetic regulation. Although the extent to which defects in PGCs affect the later stage of undifferentiated spermatogonia remains unclear, there may be a cause common to different FA mutants. For example, reduced proliferation associated with compromised replication-associated DNA repair has been shown in Fancm mutants (42). Furthermore, another important question is whether the function of the FA pathway in epigenetic regulation is directly coupled to its DNA repair function or coupled with independent signaling events.
Materials and Methods
Animals
Fancb mutant mice were produced through ZFN technology (Sigma Aldrich) (65). The mRNA of ZFNs specific to the Fancb locus was injected into fertilized eggs derived from C57BL/6J mice, and three independent Fancb-/Y males and one Fancb +/− female (lines #164, 167, 171 and 84) were obtained. The female line #84 was used as a founder and was maintained by mating with C57BL/6J male mice and genotyped with the following primer sets: for the detection of the mutant alleles in lines #164, 167, 171, Fancb-F: 5′-ACCTGATCCCTCTTGCTTACAG-3′ and Fancb-R: 5′-CAAGAATGCGGACTGGAAA-3′; for the detection of the mutant allele of line #84, Fancb-84-F: 5′-TTTCCAGGTCCCCGTTCTGA-3′ and Fancb-R: 5′-CAAGAATGCGGACTGGAAA-3′. Mdc1-KO, Rnf8-KO, Fanca-KO and Fancc-KO mice were previously described (5,66–68). Brca1 exon 11 conditional deletion mice using Ddx4-cre and the double mutants of Brca1 and 53bp1 were previously described (36).
Generation of anti-FANCB antibodies
Rabbit polyclonal antibodies against mouse FANCB were generated using MBP-tagged mouse FANCB141–320. The corresponding cDNA fragment was cloned in pMAL-c4X vector (New England Biolabs). The recombinant protein was purified using amylose resin (New England Biolabs), following the manufacturer's instructions. Polyclonal antibodies reacting with FANCB were affinity-purified with MBP-FANCB141–320 fusion protein-conjugated NHS-activated HP (GE Healthcare) after passing through MBP-conjugated NHS-activated HP. The antibodies were generated in three independent rabbits, and the results of immunostaining were consistent among them, as shown in Supplementary Material, Figure S1. One representative antibody (#61) was used for the main figures.
Histology, immunohistochemistry and TUNEL staining of testicular sections
For the preparation of testicular paraffin blocks, testes without tunica albuginea were fixed with 4% paraformaldehyde (PFA) overnight. Testes were dehydrated and embedded in paraffin. For histological analysis, paraffin sections at 6 μm thick were deparaffinized and stained with hematoxylin and eosin.
For immunohistochemistry, paraffin sections at 6 μm thick were deparaffinized and autoclaved in target retrieval solution (DAKO) at 121°C for 10 min. After incubation with blocking solution (0.15% bovine serum albumin, 0.1% Tween-20 in PBS) for 1 h at room temperature, sections were incubated at 4°C overnight with the following antibodies: mouse monoclonal anti-Protamine (Briar Patch Biosciences), 1:100; rabbit polyclonal anti-AcH4K16 (Millipore), 1:100; mouse monoclonal anti-PLZF (Calbiochem), 1:100; rabbit polyclonal anti-WT1 (Abcam), 1:100; mouse monoclonal anti-γH2AX (Millipore), 1:5000 and guinea pig polyclonal anti-H1T (gift from Mary Ann Hendel), 1:1000. The sections were washed three times for 5 min each in PBS plus 0.1% Tween 20 (PBST), and the resulting signals were detected by incubation with Alexa488- or Alexa549-conjugated secondary antibodies (Molecular Probes) and DAPI. Sections were acquired with a TiE microscope (Nikon), and images were processed with Adobe Photoshop CS6.
For TUNEL staining, sections were deparaffinized and then treated with 15 μg/ml proteinase K for 10 min at 37°C and stained with In Situ Cell Death Detection Kit (Roche) following the manufacturer's protocol. The sections were counterstained with DAPI.
Immunostaining of PGC and PCG counting
Staining of PGCs at E9.5, E11.5 and E13.5 was performed as described (69). Briefly, whole embryos or fetal gonads were fixed in 4% PFA containing 0.1% Triton X-100 at 4°C overnight. After washing in PBS containing 0.1% Triton X-100, whole embryos or fetal gonads were blocked in blocking solution (10% FBS and 3% BSA in PBS) for 60 min and incubated at 4°C overnight with the following primary antibodies: rabbit polyclonal anti-SOX2 (Seven Hills Bioreagents), 1:2000; goat polyclonal anti-GATA4 (Santa Cruz), 1:200. Whole embryos or fetal gonads were washed three times for 10 min each in 1% FBS, 3% BSA in PBS and incubated in blocking solution for 30 min. Whole embryos or fetal gonads were washed three times for 10 min each in 1% FBS, 3% BSA in PBS and thereafter incubated with secondary antibodies (Invitrogen or Jackson ImmunoResearch) and DAPI at 4°C overnight, and mounted in Vectashield (Vector Laboratories). All images of whole embryos or fetal gonads were acquired with a TE2000-E microscope (Nikon) and a CoolSNAPHQ camera (Photometrics). Image acquisition was performed using Phylum software (Improvision). Adobe Photoshop was used for composing figures. For PGC counting at E9.5 embryos and E11.5 fetal gonads, multiple focus planes (z-sections) were taken every 5 µm, starting from the base to the top of embryos or gonadal tissues to capture, respectively, whole embryos or gonadal ridges. The total numbers of SOX2 positive germ cells were counted using the merged z-sections.
Immunoprecipitation
Testes from mice were dissected and incubated in 0.5 mg/ml collagenase and 16 U/ml DNase I at 32°C for 15 min. Cells were lysed with imunnoprecipitation buffer (50 mm Tris–HCl pH 7.4, 200 mm NaCl, 0.5 mm EDTA and 1% Tritone X-100) containing complete EDTA-free medium (Roche) at 4°C for 30 min. After centrifugation, supernatants containing 3 mg of testis lysate were incubated with protein G Dynabeads (Life Technologies) coupled with 5 mg of rabbit control IgG (Millipore) and anti-human FANCB antibody (Fanconi Anemia Research Fund) at 4°C for 3 h. The samples were washed with Immunoprecipitation buffer and eluted with 2× SDS loading buffer. Proteins from 1 mg of testis lysate were fractionated by SDS–PAGE and transferred to PVDF membranes. The membrane was blocked in 5% dry milk/TBS containing 0.1% Tween 20 and analyzed using rabbit anti-human FANCB monoclonal antibody that recognizes the C-terminus of human FANCB (EPR15513: Abcam ab186729, 1:1000) and visualized using ECL western blotting substrate (Millipore).
Immunofluorescence microscopy of surface spreads of meiotic chromosomes
Analysis of sex chromosomes during meiosis was performed using hypotonic treatment as described (70). Slides were incubated in PBT (0.15% BSA, 0.1% Tween 20 in PBS) for 60 min prior to overnight incubation at room temperature with the following antibodies: mouse monoclonal FK2 (Enzo life sciences), 1:500; mouse monoclonal anti-γH2AX (Millipore), 1:5000; rabbit polyclonal anti-H3K4me2 (Millipore), 1:500; rabbit polyclonal anti-H3K9me1 (Millipore), 1:100; mouse monoclonal anti-H3K9me2 (Millipore); rabbit polyclonal anti-H3K9me3 (Millipore), 1:250; rabbit polyclonal anti-H4K20me1 (Abcam), 1:250; mouse monoclonal anti-RNA Polymerase II (Millipore), 1:100; guinea pig polyclonal anti-H1T (gift from Mary Ann Handel), 1:1000; rabbit polyclonal anti-MLH1 (Santa Cruz), 1:100; rabbit polyclonal anti-SLX4 (gift from Paula E. Cohen), 1:100; rabbit polyclonal anti-FANCB, 1:100; rabbit polyclonal anti-FANCD2 (Novus), 1:200; rabbit polyclonal anti-RAD51 (Santa Cruz), 1:50; rabbit polyclonal anti-BRCA1, 1:1500; rabbit polyclonal anti-ATR (Cell Signaling), 1:50; sheep polyclonal anti-MDC1 (AbD Serotec), 1:500; rabbit polyclonal anti-TOPBP1 (gift from Junjie Chen), 1:500; rabbit polyclonal anti-SYCP1 (Abcam), 1:1500; mouse monoclonal anti-SYCP3 (Abcam), 1:3000 and rabbit polyclonal anti-SYCP3 (Novus), 1:500. Thereafter, slides were washed three times for 5 min each in PBST, incubated with secondary antibodies (Invitrogen or Jackson ImmunoResearch) at 1:500 for 60 min in PBT, washed in PBST, and mounted in Vectashield (Vector Laboratories) with DAPI. All images of germ cells were acquired with a TiE microscope (Nikon). Adobe Photoshop CS6 was used for composing figures. Particular stages of primary spermatocytes were determined by staining for SYCP3. H3K9me2/3 signals were quantified with NIS-Elements Basic Research software. Briefly, regions of interest (ROIs) were drawn around XY bodies, denoted as XY body in Figure 7G and J, and prophase I nuclei excluding the XY body, denoted as Autosome in Figure 7G and J. The XY body and autosome ROIs were normalized to image background ROIs. For normalization of H3K9me2/3 signals, we calculated the mean of the diplotene XY body ROI signals; all ROI signals were then divided by this value (Relative Mean Fluorescence Intensity: RMFI). The independent samples were combined and statistical analyses were run through Prism 6 (GraphPad).
Supplementary Material
Funding
This work was supported by the Lalor Foundation postdoctoral fellowship for H-S. S., the Developmental Fund at Cincinnati Children's Hospital Medical Center to S.H.N., the Research Grant (FY13-510) from the March of Dimes Foundation to S.H.N., and National Institutes of Health Grants (HL085587 to P.R.A., GM098605 to S.H.N).
Supplementary Material
Acknowledgements
We thank Tony de Falco, Takiko Daikoku and Sarah Potter for their help in embryo experiments; members of the Namekawa laboratory for discussion and helpful comments regarding the manuscript; Madeleine Carreau for the Fanca+/− mice; Manuel Buchwald for the Fancc+/− mice; Junjie Chen for providing the Mdc1-KO and Rnf8-KO mice, and for the gift of anti-TOPBP1 antibody; Mary Ann Handel for the gift of the anti-H1T antibody; Paula E. Cohen for the gift of the anti-SLX4 antibody and the Fanconi Anemia Research Fund for the gift of anti-human FANCB antibodies.
Conflict of Interest statement. None declared.
References
- 1.Kennedy S.R., Loeb L.A., Herr A.J. (2012) Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev., 133, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell C.D., Eichler E.E. (2013) Properties and rates of germline mutations in humans. Trends Genet., 29, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kee Y., D'Andrea A.D. (2012) Molecular pathogenesis and clinical management of Fanconi anemia. J. Clin. Invest., 122, 3799–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W. (2007) Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet., 8, 735–748. [DOI] [PubMed] [Google Scholar]
- 5.Cheng N.C., van de Vrugt H.J., van der Valk M.A., Oostra A.B., Krimpenfort P., de Vries Y., Joenje H., Berns A., Arwert F. (2000) Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum. Mol. Genet., 9, 1805–1811. [DOI] [PubMed] [Google Scholar]
- 6.Nadler J.J., Braun R.E. (2000) Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. Genesis, 27, 117–123. [DOI] [PubMed] [Google Scholar]
- 7.Sharan S.K., Pyle A., Coppola V., Babus J., Swaminathan S., Benedict J., Swing D., Martin B.K., Tessarollo L., Evans J.P. et al. (2004) BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development (Cambridge, England), 131, 131–142. [DOI] [PubMed] [Google Scholar]
- 8.Houghtaling S., Timmers C., Noll M., Finegold M.J., Jones S.N., Meyn M.S., Grompe M. (2003) Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev., 17, 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitney M.A., Royle G., Low M.J., Kelly M.A., Axthelm M.K., Reifsteck C., Olson S., Braun R.E., Heinrich M.C., Rathbun R.K. et al. (1996) Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood, 88, 49–58. [PubMed] [Google Scholar]
- 10.Bakker S.T., van de Vrugt H.J., Visser J.A., Delzenne-Goette E., van der Wal A., Berns M.A., van de Ven M., Oostra A.B., de Vries S., Kramer P. et al. (2012) Fancf-deficient mice are prone to develop ovarian tumours. J. Pathol., 226, 28–39. [DOI] [PubMed] [Google Scholar]
- 11.Koomen M., Cheng N.C., van de Vrugt H.J., Godthelp B.C., van der Valk M.A., Oostra A.B., Zdzienicka M.Z., Joenje H., Arwert F. (2002) Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum. Mol. Genet., 11, 273–281. [DOI] [PubMed] [Google Scholar]
- 12.Agoulnik A.I., Lu B., Zhu Q., Truong C., Ty M.T., Arango N., Chada K.K., Bishop C.E. (2002) A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum. Mol. Genet., 11, 3047–3053. [DOI] [PubMed] [Google Scholar]
- 13.Bakker S.T., van de Vrugt H.J., Rooimans M.A., Oostra A.B., Steltenpool J., Delzenne-Goette E., van der Wal A., van der Valk M., Joenje H., te Riele H. et al. (2009) Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet., 18, 3484–3495. [DOI] [PubMed] [Google Scholar]
- 14.Crossan G.P., van der Weyden L., Rosado I.V., Langevin F., Gaillard P.H., McIntyre R.E., Gallagher F., Kettunen M.I., Lewis D.Y., Brindle K. et al. (2011) Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet., 43, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mouw K.W., D'Andrea A.D. (2014) Crosstalk between the nucleotide excision repair and Fanconi anemia/BRCA pathways. DNA Repair, 19, 130–134. [DOI] [PubMed] [Google Scholar]
- 16.Chandra S., Levran O., Jurickova I., Maas C., Kapur R., Schindler D., Henry R., Milton K., Batish S.D., Cancelas J.A. et al. (2005) A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol. Ther., 12, 976–984. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y., Leung J.W., Lowery M., Matsushita N., Wang Y., Shen X., Huong D., Takata M., Chen J., Li L. (2014) Modularized functions of the Fanconi anemia core complex. Cell Rep., 7, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh T.R., Bakker S.T., Agarwal S., Jansen M., Grassman E., Godthelp B.C., Ali A.M., Du C.H., Rooimans M.A., Fan Q. et al. (2009) Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood, 114, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J.M., Kee Y., Gurtan A., D'Andrea A.D. (2008) Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood, 111, 5215–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deans A.J., West S.C. (2009) FANCM connects the genome instability disorders Bloom's syndrome and Fanconi anemia. Mol. Cell, 36, 943–953. [DOI] [PubMed] [Google Scholar]
- 21.Sims A.E., Spiteri E., Sims R.J. 3rd, Arita A.G., Lach F.P., Landers T., Wurm M., Freund M., Neveling K., Hanenberg H. et al. (2007) FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol., 14, 564–567. [DOI] [PubMed] [Google Scholar]
- 22.Meetei A.R., de Winter J.P., Medhurst A.L., Wallisch M., Waisfisz Q., van de Vrugt H.J., Oostra A.B., Yan Z., Ling C., Bishop C.E. et al. (2003) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet., 35, 165–170. [DOI] [PubMed] [Google Scholar]
- 23.Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R. 3rd, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D'Andrea A.D. et al. (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell, 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montes de Oca R., Andreassen P.R., Margossian S.P., Gregory R.C., Taniguchi T., Wang X., Houghtaling S., Grompe M., D'Andrea A.D. (2005) Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood, 105, 1003–1009. [DOI] [PubMed] [Google Scholar]
- 25.Wang X., Andreassen P.R., D'Andrea A.D. (2004) Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell Biol., 24, 5850–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumaraswamy E., Shiekhattar R. (2007) Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol. Cell. Biol., 27, 6733–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia B., Sheng Q., Nakanishi K., Ohashi A., Wu J., Christ N., Liu X., Jasin M., Couch F.J., Livingston D.M. (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell, 22, 719–729. [DOI] [PubMed] [Google Scholar]
- 28.Park J.Y., Singh T.R., Nassar N., Zhang F., Freund M., Hanenberg H., Meetei A.R., Andreassen P.R. (2014) Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene, 33, 4803–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto K.N., Kobayashi S., Tsuda M., Kurumizaka H., Takata M., Kono K., Jiricny J., Takeda S., Hirota K. (2011) Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl Acad. Sci. USA, 108, 6492–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Higuera I., Taniguchi T., Ganesan S., Meyn M.S., Timmers C., Hejna J., Grompe M., D'Andrea A.D. (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell, 7, 249–262. [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi K., Yang Y.G., Pierce A.J., Taniguchi T., Digweed M., D'Andrea A.D., Wang Z.Q., Jasin M. (2005) Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl Acad. Sci. USA, 102, 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turner J.M. (2007) Meiotic sex chromosome inactivation. Development, 134, 1823–1831. [DOI] [PubMed] [Google Scholar]
- 33.Ichijima Y., Sin H.S., Namekawa S.H. (2012) Sex chromosome inactivation in germ cells: emerging roles of DNA damage response pathways. Cell Mol. Life Sci., 69, 2559–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner J.M., Aprelikova O., Xu X., Wang R., Kim S., Chandramouli G.V., Barrett J.C., Burgoyne P.S., Deng C.X. (2004) BRCA1, histone H2AX phosphorylation, and male meiotic sex chromosome inactivation. Curr. Biol., 14, 2135–2142. [DOI] [PubMed] [Google Scholar]
- 35.Royo H., Prosser H., Ruzankina Y., Mahadevaiah S.K., Cloutier J.M., Baumann M., Fukuda T., Hoog C., Toth A., de Rooij D.G. et al. (2013) ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes Dev., 27, 1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Broering T.J., Alavattam K.G., Sadreyev R.I., Ichijima Y., Kato Y., Hasegawa K., Camerini-Otero R.D., Lee J.T., Andreassen P.R., Namekawa S.H. (2014) BRCA1 establishes DNA damage signaling and pericentric heterochromatin of the X chromosome in male meiosis. J. Cell Biol., 205, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichijima Y., Ichijima M., Lou Z., Nussenzweig A., Camerini-Otero R.D., Chen J., Andreassen P.R., Namekawa S.H. (2011) MDC1 directs chromosome-wide silencing of the sex chromosomes in male germ cells. Genes Dev., 25, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folias A., Matkovic M., Bruun D., Reid S., Hejna J., Grompe M., D'Andrea A., Moses R. (2002) BRCA1 interacts directly with the Fanconi anemia protein FANCA. Hum. Mol. Genet., 11, 2591–2597. [DOI] [PubMed] [Google Scholar]
- 39.Andreassen P.R., D'Andrea A.D., Taniguchi T. (2004) ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev., 18, 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meetei A.R., Levitus M., Xue Y., Medhurst A.L., Zwaan M., Ling C., Rooimans M.A., Bier P., Hoatlin M., Pals G. et al. (2004) X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet., 36, 1219–1224. [DOI] [PubMed] [Google Scholar]
- 41.Wong J.C., Alon N., McKerlie C., Huang J.R., Meyn M.S., Buchwald M. (2003) Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum. Mol. Genet., 12, 2063–2076. [DOI] [PubMed] [Google Scholar]
- 42.Luo Y., Hartford S.A., Zeng R., Southard T.L., Shima N., Schimenti J.C. (2014) Hypersensitivity of primordial germ cells to compromised replication-associated DNA repair involves ATM-p53-p21 signaling. PLoS Genet., 10, e1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim T.M., Ko J.H., Choi Y.J., Hu L., Hasty P. (2011) The phenotype of FancB-mutant mouse embryonic stem cells. Mutat. Res., 712, 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holloway J.K., Mohan S., Balmus G., Sun X., Modzelewski A., Borst P.L., Freire R., Weiss R.S., Cohen P.E. (2011) Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genet., 7, e1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan Z., Guo R., Paramasivam M., Shen W., Ling C., Fox D. 3rd, Wang Y., Oostra A.B., Kuehl J., Lee D.Y. et al. (2012) A ubiquitin-binding protein, FAAP20, links RNF8-mediated ubiquitination to the Fanconi anemia DNA repair network. Mol. Cell, 47, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sin H.S., Barski A., Zhang F., Kartashov A.V., Nussenzweig A., Chen J., Andreassen P.R., Namekawa S.H. (2012) RNF8 regulates active epigenetic modifications and escape gene activation from inactive sex chromosomes in post-meiotic spermatids. Genes Dev., 26, 2737–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajendra E., Oestergaard V.H., Langevin F., Wang M., Dornan G.L., Patel K.J., Passmore L.A. (2014) The genetic and biochemical basis of FANCD2 monoubiquitination. Mol. Cell, 54, 858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hasegawa K., Sin H.S., Maezawa S., Broering T.J., Kartashov A.V., Alavattam K.G., Ichijima Y., Zhang F., Bacon W.C., Greis K.D. et al. (2015) SCML2 establishes the male germline epigenome through regulation of histone H2A ubiquitination. Dev. Cell, 32, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khalil A.M., Boyar F.Z., Driscoll D.J. (2004) Dynamic histone modifications mark sex chromosome inactivation and reactivation during mammalian spermatogenesis. Proc. Natl Acad. Sci. USA, 101, 16583–16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Namekawa S.H., Park P.J., Zhang L.F., Shima J.E., McCarrey J.R., Griswold M.D., Lee J.T. (2006) Postmeiotic sex chromatin in the male germline of mice. Curr. Biol., 16, 660–667. [DOI] [PubMed] [Google Scholar]
- 51.van der Heijden G.W., Derijck A.A., Posfai E., Giele M., Pelczar P., Ramos L., Wansink D.G., van der Vlag J., Peters A.H., de Boer P. (2007) Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nat. Genet., 39, 251–258. [DOI] [PubMed] [Google Scholar]
- 52.Luo M., Zhou J., Leu N.A., Abreu C.M., Wang J., Anguera M.C., de Rooij D.G., Jasin M., Wang P.J. (2015) Polycomb protein SCML2 associates with USP7 and counteracts histone H2A ubiquitination in the XY chromatin during male meiosis. PLoS Genet., 11, e1004954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sin H.S., Ichijima Y., Koh E., Namiki M., Namekawa S.H. (2012) Human postmeiotic sex chromatin and its impact on sex chromosome evolution. Genome Res., 22, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Y.C., Namekawa S.H. (2015) Functional significance of the sex chromosomes during spermatogenesis. Reproduction (Cambridge, England), 149, R265–r277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bessler J.B., Andersen E.C., Villeneuve A.M. (2010) Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line. PLoS Genet., 6, e1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peters A.H., Kubicek S., Mechtler K., O'Sullivan R.J., Derijck A.A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y. et al. (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell, 12, 1577–1589. [DOI] [PubMed] [Google Scholar]
- 57.Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H. et al. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev., 16, 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peters A.H., O'Carroll D., Scherthan H., Mechtler K., Sauer S., Schofer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A. et al. (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell, 107, 323–337. [DOI] [PubMed] [Google Scholar]
- 59.Tachibana M., Nozaki M., Takeda N., Shinkai Y. (2007) Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J., 26, 3346–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gibbs-Seymour I., Mailand N. (2015) SLX4: not simply a nuclease scaffold? Mol. Cell, 57, 3–5. [DOI] [PubMed] [Google Scholar]
- 61.Saitou M., Kagiwada S., Kurimoto K. (2012) Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development, 139, 15–31. [DOI] [PubMed] [Google Scholar]
- 62.Hajkova P., Jeffries S.J., Lee C., Miller N., Jackson S.P., Surani M.A. (2010) Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science (New York, NY), 329, 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haneline L.S., Gobbett T.A., Ramani R., Carreau M., Buchwald M., Yoder M.C., Clapp D.W. (1999) Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood, 94, 1–8. [PubMed] [Google Scholar]
- 64.Yung S.K., Tilgner K., Ledran M.H., Habibollah S., Neganova I., Singhapol C., Saretzki G., Stojkovic M., Armstrong L., Przyborski S. et al. (2013) Brief report: human pluripotent stem cell models of fanconi anemia deficiency reveal an important role for fanconi anemia proteins in cellular reprogramming and survival of hematopoietic progenitors. Stem Cells, 31, 1022–1029. [DOI] [PubMed] [Google Scholar]
- 65.Cui X., Ji D., Fisher D.A., Wu Y., Briner D.M., Weinstein E.J. (2011) Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat. Biotechnol., 29, 64–67. [DOI] [PubMed] [Google Scholar]
- 66.Lou Z., Minter-Dykhouse K., Franco S., Gostissa M., Rivera M.A., Celeste A., Manis J.P., van Deursen J., Nussenzweig A., Paull T.T. et al. (2006) MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell, 21, 187–200. [DOI] [PubMed] [Google Scholar]
- 67.Minter-Dykhouse K., Ward I., Huen M.S., Chen J., Lou Z. (2008) Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J. Cell Biol., 181, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen M., Tomkins D.J., Auerbach W., McKerlie C., Youssoufian H., Liu L., Gan O., Carreau M., Auerbach A., Groves T. et al. (1996) Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat. Genet., 12, 448–451. [DOI] [PubMed] [Google Scholar]
- 69.DeFalco T., Takahashi S., Capel B. (2011) Two distinct origins for Leydig cell progenitors in the fetal testis. Dev. Biol., 352, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peters A.H., Plug A.W., van Vugt M.J., de Boer P. (1997) A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res., 5, 66–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.