

Abstract
Nebulin is a giant filamentous protein that is coextensive with the actin filaments of the skeletal muscle sarcomere. Nebulin mutations are the main cause of nemaline myopathy (NEM), with typical adult patients having low expression of nebulin, yet the roles of nebulin in adult muscle remain poorly understood. To establish nebulin's functional roles in adult muscle, we studied a novel conditional nebulin KO (Neb cKO) mouse model in which nebulin deletion was driven by the muscle creatine kinase (MCK) promotor. Neb cKO mice are born with high nebulin levels in their skeletal muscles, but within weeks after birth nebulin expression rapidly falls to barely detectable levels Surprisingly, a large fraction of the mice survive to adulthood with low nebulin levels (<5% of control), contain nemaline rods and undergo fiber-type switching toward oxidative types. Nebulin deficiency causes a large deficit in specific force, and mechanistic studies provide evidence that a reduced fraction of force-generating cross-bridges and shortened thin filaments contribute to the force deficit. Muscles rich in glycolytic fibers upregulate proteolysis pathways (MuRF-1, Fbxo30/MUSA1, Gadd45a) and undergo hypotrophy with smaller cross-sectional areas (CSAs), worsening their force deficit. Muscles rich in oxidative fibers do not have smaller weights and can even have hypertrophy, offsetting their specific-force deficit. These studies reveal nebulin as critically important for force development and trophicity in adult muscle. The Neb cKO phenocopies important aspects of NEM (muscle weakness, oxidative fiber-type predominance, variable trophicity effects, nemaline rods) and will be highly useful to test therapeutic approaches to ameliorate muscle weakness.
Introduction
Nebulin is a large sarcomeric protein in skeletal muscle, located along the length of the thin filament, with its C-terminus anchored in the Z-disk and its N-terminus positioned near the thin filament pointed end (1–4). The NEB gene has 183 exons that are predicted to encode a protein of maximally ∼900 kDa (5). (Note that a list of abbreviations is provided at the end of article.) The majority of nebulin is composed of ∼35-residue domains that bind actin with high affinity, are organized into super-repeats that match the repeat of F-actin (6,7), and that might determine the minimal length of the thin filament (1,8). The pivotal role of nebulin in skeletal muscle function is illustrated by the many NEB mutations that account for ∼50% of nemaline myopathy (NEM), the most common of the non-dystrophic congenital myopathies (9,10), and characterized by muscle weakness and muscle atrophy (11). Several studies on skeletal muscles of patients with nebulin-based NEM have revealed low levels of nebulin (12–14), but how universal this finding is and, importantly, the detailed mechanisms by which reduced nebulin levels cause NEM remain to be established.
A conventional mouse model (Neb KO) (15,16) and a model in which NEB exon 55 is deleted (NebΔex55) (17) exist, but mice die within days after birth due to complex developmental delays and the rapid onset of marked abnormalities associated with NEM. The typical NEM patients with NEB mutations survive to adulthood, express low (but detectable) levels of nebulin and display a considerably milder phenotype compared with the Neb KO and NebΔex55 mouse models (12). Thus, early postnatal death of existing models severely limits their usefulness for understanding the roles of nebulin in adult muscle and how nebulin deficiency gives rise to typical forms of NEM. To study the effect of nebulin deficiency in adult muscle, we made a conditional nebulin KO mouse model (Neb cKO). Mice survive to adult age and akin to NEM patients, skeletal muscles in adult Neb cKO mice had much reduced levels of nebulin and contained nemaline rod bodies. Importantly, relative to control (Ctrl) mice, the Neb cKO mice had severe muscle weakness (reduction in specific force). Structural and functional studies revealed that its mechanistic basis includes sarcomere defects. Additionally, hypotrophy in muscles rich in glycolytic fibers reduced their CSA and this added to their force deficit. The implications of these novel findings for the roles of nebulin in normal muscle function as well as in the pathophysiology of nebulin-based NEM are discussed.
Results
Generation of NEB cKO mice that survive to adulthood with low nebulin levels
A conditional mouse model was made in which the translational start codon of the Neb gene was floxed and its deletion directed by a MCK-Cre transgene (see Materials and Methods for details). Neb cKO mice appear normal at birth but about half die within 3 months (median survival 83 days), with most of the remaining mice surviving into adulthood (Fig. 1A). Adult Neb cKO mice have relative to Ctrl mice a ∼40% lower body weight (BW) (Fig. 1B, Supplementary Material, Table S1). Western blot studies revealed a severe reduction in nebulin levels (Fig. 1C). At the earliest characterized age of 3 weeks, the average nebulin level was 48% of Ctrl levels, at 5 weeks 4.8% and at 6 months 2.2% (Fig. 1C, inset). Having mice that survive into adulthood with very low levels of nebulin provides unique opportunities to study the roles of nebulin in adult muscles and improve our understanding of NEM.
Figure 1.

Neb cKO mice survive to adulthood while expressing low nebulin levels. (A) Left, survival curves show that the median survival time of cKO mice is ∼3 months and about half of the mice survive at least 6 months. (B) Body weight (BW) is smaller in cKO mice. (C) Top, example of WB with anti-nebulin antibody and MHC of corresponding Ponceau S-stained membrane in muscles types of Neb cKO mice (5 weeks). Bottom, mean nebulin expression in cKO (relative to MHC expression and normalized to Ctrl) as a function of age. Inset shows mean values in Neb cKO as % of Ctrl levels. (For statistical details including group sizes, see Materials and Methods, section ‘Statistics’. Also note that a list of abbreviations has been provided at the end of the article.)
Effects of nebulin deficiency on muscle trophicity
Multiple muscle types were studied using 3-week-, 5-week- and 6-month-old Neb cKO and Ctrl mice. Supplementary Material, Table S1 shows absolute muscle weights as well as BW and tibia length (TL) normalized weights, because TL normalization lessens effects of body size differences among genotypes and age groups, TL normalized data are emphasized below. Analysis revealed that different muscles respond differently to nebulin deficiency. The gastrocnemius (Gast) and quadriceps (Quad) had, at all tested ages, weights that were 60–70% smaller than those of the Ctrl muscles (Fig. 2). The tibialis cranialis (TC) and extensor digitorum longus (EDL) had weights that were much smaller at 5 weeks but less at 6 months (Fig. 2). Finally, the diaphragm (Diaph) and soleus (Sol) had minimal weight changes except at 6 months when the Sol weight was increased by ∼50% (Fig. 2). Thus, different muscle types respond differently to nebulin deficiency, relative to Ctrl mice some muscles have weights that are much smaller and others are hypertrophied.
Figure 2.
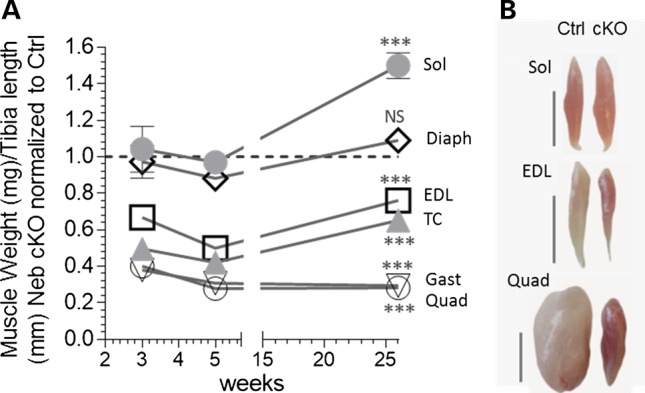
Effect of nebulin deficiency on muscle weight varies with muscle type. (A) Muscle weights as function of age (3 and 5 weeks, 6 months) in Neb cKO mice (results plotted relative to Ctrl). The weights of Gast and Quad muscles are much smaller at all ages. Weights of TC and EDL muscles are smallest at 5 weeks and less at 3 weeks and 6 months. The Diaph and Sol are unchanged except at 6 months when the Sol muscle is hypertrophied. (B) Examples of Sol, EDL and Quad muscles (top to bottom) in 6-month-old mice. Bar: 5 mm. Note smaller Quad and EDL muscles but larger Sol muscle in Neb cKO mice, and the redder muscle color of muscles of Neb cKO mice.
Effects of nebulin deficiency on fiber-type composition
The muscles from cKO mice were redder in color, suggesting changes in fiber type toward more myoglobin-rich type I and IIA fibers (18). We therefore thoroughly examined the effects of nebulin deficiency on the fiber-type composition and fiber CSA using immunofluorescence on cross-sections of muscles as well as a myosin heavy chain (MHC) gel analysis on a wide range of muscle types. For the CSA analysis, muscles were cut at their mid-belly and stained with antibodies specific to myosin type I, IIA or IIB and co-stained with anti-laminin antibodies to trace the cell boundary and measure CSA (Fig. 3 left shows examples of Sol (top) and EDL (bottom) muscles). In the Sol muscle, all three fiber types were smaller in the cKO than in the Ctrl (Fig. 3A), the total number of fibers was increased (Fig. 3B) and the fraction of fibers that were type I was increased at the expense of type IIA and type IIB (Fig 3C and Supplementary Material, Table S2). Figure 3D highlights that the Sol muscle CSA is ∼2/3 type I and 1/3 IIA in Ctrl mice, and that in cKO mice the Sol muscle is nearly exclusively type I.
Figure 3.

Nebulin deficiency alters fiber-type composition and CSA. Mid-belly muscle cross-sections (6 months) were stained with MHC isotype I, IIA or IIB. Examples are shown at the left with Sol at top and EDL at bottom. (A–D) Sol data. (A) Fiber-type CSA; (B) total number of fibers per Sol muscle; (C) fiber-type composition; (D) CSA of Sol muscle taken up by the different fiber types. (E–H) EDL data. (E) fiber-type CSA; (F) total number of fibers per EDL muscle; (G) fiber-type composition; and (H) muscle CSA.
In the EDL, nebulin deficiency resulted in larger type I and type IIA fibers, but ∼80% smaller type IIB fibers (Fig. 3E). The total number of fibers was unaltered (Fig. 3F) whereas an increased proportion of all fibers was type I and IIA and a much reduced fraction was type IIB (Fig. 3G). The muscle CSA was converted toward oxidative fiber types (Fig. 3H). Furthermore, type I and IIA fibers hypertrophy and IIB fibers atrophy (Supplementary Material, Table S2). We also determined the minimum feret distance (MinFeret) as an alternative measure of cross-sectional fiber size. (MinFeret is relatively insensitive to section angle variation.) Supplementary Material, Table S2 shows that MinFeret findings were similar to those obtained by measuring CSA. Finally, to determine whether our findings extrapolate to other muscle types, we studied the Quad. A severe reduction was present in type IIB fiber size and number (Supplementary Material, Fig. S2). Thus, a consistent fiber-type switch away from type IIB fibers was found with hypertrophy of type I and IIA fiber types and severe atrophy of remaining type IIB fibers.
MHC isoform expression was also studied using gel electrophoresis as a high-throughput technique. Examples of MHC expression patterns are shown in Figure 4A. MHC type I and IIB are well separated on gels (bottom and middle bands, respectively), whereas the top band is likely to be majority IIA that overlaps with a small amount of IIX that exists in mouse skeletal muscle (19). We attempted to separate IIA and IIX bands, but were not successful and instead refer to the top band as IIA(X). The obtained MHC composition in Ctrl and Neb cKO samples at three ages is shown in Figure 4B and C and in Supplementary Material, Table S3. Interestingly, only minimal MHC type changes occurred in the Diaph but in all other muscle types both type I and IIA(X) were increased (Fig. 4B). In the Sol muscle, MHC type I became more prominent at the expense of type IIA(X) and in all other muscle types both type I and IIA(X) were increased at the expense of type IIB MHC (Fig. 4C, Supplementary Material, Table S3). Because we noted a correlation between MHC IIB content in Ctrl muscle and the degree of atrophy in cKO mice, we studied MHC composition and atrophy in an additional four forelimb muscle (Biceps, B; Triceps medialis, Tm; Triceps lateralis, TLa and Triceps longus, Tlo). Linear regression analysis revealed a significant positive correlation (Fig. 4D). Thus, a greater MHC IIB content of muscle (in Ctrl mice) predicts more severe atrophy in nebulin-deficient mice.
Figure 4.
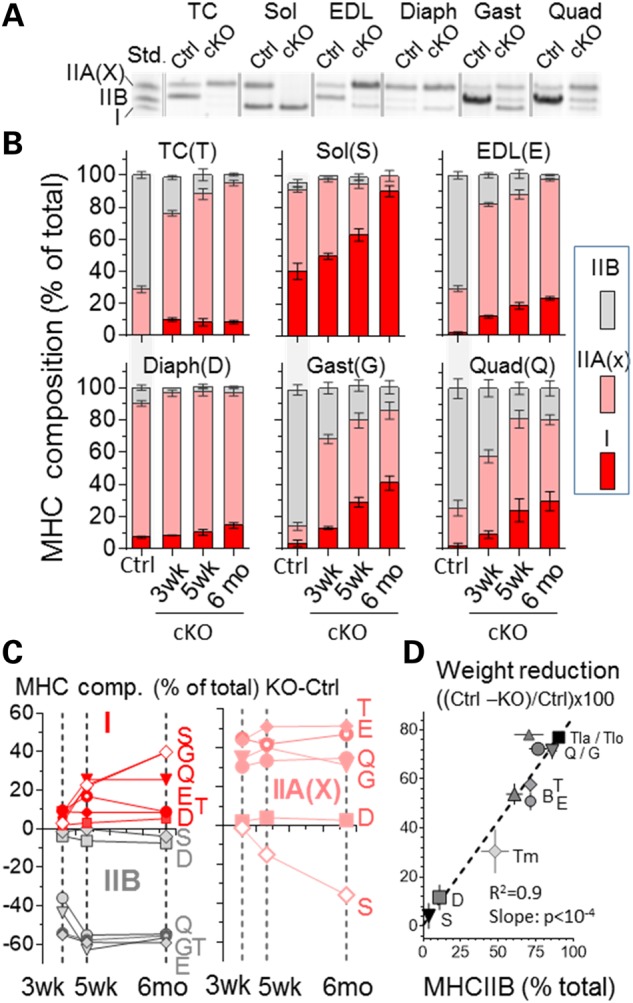
Nebulin deficiency shifts myosin expression from glycolytic (IIB) to oxidative (I/IIA) types. (A) Example of typical electrophoretic pattern of whole muscle lysate from 6-month-old WT and Neb cKO mice. (Left lane is a MHC type standard, Std.) (B) MHC type composition as function of age. For simplicity sake, MHC composition of Ctrl is only shown for 5-week-old mice. (C) Change in MHC composition as function of age. In all muscle types, nebulin deficiency increases MHC type I (left top), decreases type IIB (left bottom) and increases MHC IIA(X), except for the Sol where IIA(X) goes down. (D) MHC IIB content in control muscle correlates with muscle weight in nebulin-deficient muscle (5 weeks). (See B for muscle abbreviations. Additionally: Biceps, B; Triceps medialis, Tm; Triceps lateralis, Tla; and Triceps longus,Tlo).
Gene expression analysis
A microarray analysis was performed on muscles from 5-week-old mice (an early age at which nebulin levels are near zero). We studied the Quad (a muscle type that has a much smaller weight in cKO than in Ctrl) and the Sol (which has a normal weight at this age). Differential gene expression for the cKO versus Ctrl revealed up-regulation of 568 genes in the Quad and 434 genes in the Sol (overlap 19.4%) and downregulation of 295 genes in the Quad and 241 genes in the Sol (overlap 15.3%), see Supplementary Material, Table S4A–E. Notably, no genes were found that were up in the Sol and down in the Quad and neither vice versa. A list of top hits is shown in Supplementary Material, Table S4F with results in common with the conventional Neb KO (16) highlighted in bold. Upregulation of Ankrd1 (CARP), Dsp (desmoplakin) and Sln (sarcolipin) is common among the cKO Quad and Sol muscles and the Neb KO, indicating that these changes are closely linked to nebulin deficiency. We also listed the expression levels of nebulin-binding proteins (Supplementary Material, Table S4G) and except for changes that reflect fiber-type switches, few significant changes occur with the exception of the recently discovered nebulin-binding protein KLHL40 (20) that is greatly increased in the Sol. Among the genes that are up in the Sol but not in the Quad, there is significant over-representation of ribosomal proteins (P-value = 1.05 × 10−21), suggesting increased protein synthesis in the Sol. Candidates that are up in the Quad but not the Sol include Fbxo30/MUSA1 [ubiquitin ligase downstream of BMP signaling that is necessary for muscle atrophy (21)] and Gadd45a (see Discussion). Although microarray analysis did not reveal differences in transcript levels for the E3 ubiquitin ligases atrogin-1 and MuRF-1, two atrogenes that are upregulated in multiple atrophy models (22), at the protein level atrogin-1 was equally expressed, but MuRF-1 was significantly upregulated in the cKO with a significant increase in the Quad (Supplementary Material, Fig. S3).
Structural changes in NEB cKO mice
Electron microscopy (EM) studies revealed overall normal looking sarcomeres in Neb cKO muscles. However, compared with Ctrl sarcomeres, Z-disks of cKO sarcomere were often irregular and wavy (Fig. 5A–F). EM showed in Ctrl muscle well-defined H-zones (that demarcate the pointed ends of the thin filaments) (Fig. 5E). In cKO sarcomeres, H-zones were absent and A-band density gradually decreased toward the M-band (Fig. 5F), suggesting that nebulin deficiency results in thin filaments that are of variable length. Additionally the micrographs revealed local areas with nemaline rods, a hallmark of NEM (Fig. 5G and H). These dense structures, packed with Z-disk and thin filament proteins, might affect force production. We measured the area of individual rod bodies and found that the rod bodies are smaller in EDL than in Sol cKO (Fig. 5I left). However, due to a difference in the number of rod bodies (∼15 per 100 μm2 fiber in EDL and ∼5 per 100 μm2 in Sol), no difference in the total area that is composed of rod bodies was found; in both Sol and EDL muscle rod bodies made up ∼3% of the fiber area (Fig. 5I, right).
Figure 5.

Ultrastructural studies of EDL and Sol muscle of Ctrl and Neb cKO mice (6 months). (A) Ctrl EDL and (C) Ctrl Sol muscles show normal sarcomere structure. (B) cKO EDL and (D) cKO Sol sarcomeres have an overall normal structure, except that Z-disks tend to be wavy. (E) Densitometry of Ctrl EDL sarcomere and (F) cKO EDL sarcomere. Analyzed regions indicated by the box reveal a clear H-zone in Ctrl (see also arrow heads) but not in Neb cKO. (G–I) Rod body analysis. (G and H) Selected areas rich in rod bodies (dark elongated structures). (I) The area per rod body (left) was less in EDL than in Sol cKO muscles but the combined rod body area (expressed as fraction of fiber area) was not different (Calibration bars: 1.0 µm.).
Effects of nebulin deficiency on muscle force
We tested the functional performance of Neb cKO mice using whole animal experiments by providing mice (6 months old) access to a free-running wheel and measuring the average 24 h running duration, speed and distance (see Materials and Methods). Neb cKO mice did exercise, but, compared with Ctrl mice, with a much reduced duration (1.2 versus 5.4 h), speed (0.4 versus 1.1 km/h) and distance ran (0.5 versus 6.6 km), see Figure 6A. This reduced exercise performance suggests compromised muscle function. In vitro studies were therefore performed on intact EDL and Sol muscle, selected for their suitability for this type of work and their glycolytic (type IIB) and oxidative (type I and IIA) fiber-type predominance, respectively. Muscles were dissected from 5-week- and 6-month-old mice; muscles were electrically stimulated and the produced force measured (examples in Fig. 6B). The Sol of NEB cKO mice produced normal maximal tetanic forces compared with controls, whereas in EDL muscle tetanic force levels were 80–90% reduced (Fig. 6B and C). We determined the CSA of the studied muscles and used this to calculate specific force (force divided by area). Specific force levels were significantly reduced at both ages and in both muscle types (Fig. 6D) indicating that nebulin deficiency reduces the force-generating capacity of skeletal muscle.
Figure 6.

Nebulin deficiency depresses running performance and lowers specific force of isolated muscles. (A) Neb cKO mice (6 months) are active on a free-running wheel but running duration, speed and distance are greatly reduced relative to age-matched Ctrl mice. (B) Examples of maximal tetanic force traces superimposed for the two genotypes (6 months) in Sol (left) and EDL muscle (right). (C) Mean tetanic force is unaltered in Sol (left) but severely reduced in EDL (right). (D) Maximal specific force (force divided by CSA) is reduced in Sol as well as EDL.
Mechanism for force reduction
Thin filament length
Because nebulin is thought to play a role in thin filament length specification, and thin filament length affects force production, we studied how nebulin deficiency in adult mice affects thin filament length. Longitudinal sections of Sol and EDL muscle were actin-stained with fluorescently labeled phalloidin, and thin filament length was measured using deconvolution microscopy (Fig. 7A shows examples). In both muscle types, nebulin deficiency results in a significant reduction in thin filament length (Fig. 7B, Table 1). To study how this affects force generation, we measured specific force as a function of sarcomere length in skinned fiber bundles activated with saturating calcium. The maximal specific force at optimal sarcomere length was much reduced in both Sol and EDL (Table 1). Due to run-down in EDL fibers when repeatedly activated, we studied the force-sarcomere length relation in only Sol and found this relation to have a descending limb that was left-shifted in the cKO by 0.1 μm per half sarcomere (Fig. 7C and Table 1). This supports the notion that nebulin deficiency reduces thin filament length, and that this contributes to the force depression in cKO muscle.
Figure 7.

Nebulin deficiency reduces thin filament length. (A) Longitudinal sections of EDL and Sol muscles fluorescently labelled with phalloidin to stain actin filaments (top), Tmod (middle) to label the pointed end of actin filaments and α-actinin (bottom) to label the Z-disks. Calibration bar: 5 µm. (B) Measurements obtained from phalloidin-labeled sarcomeres show that thin filament length is significantly reduced in both muscle types. (C) The force-sarcomere length relation (Sol muscle) has a descending limb that is shifted to the left. (Note that the force is normalized to the maximal force at optimal length; for absolute force values, see Table 1.)
Table 1.
Thin filament length and force–SL measurements in Ctrl and Neb cKO mice
| Soleus |
EDL |
|||||||
|---|---|---|---|---|---|---|---|---|
| Deconvolution microscopy | Control (n= 8) | cKO (n= 8) | t-test | Change | Control (n= 9) | cKO (n= 6) | t-test | Change |
| Sarcomere length, μm | 2.9 ± 0.01 (120) | 2.9 ± 0.02 (84) | 0 | 2.9 ± 0.01 (138) | 2.9 ± 0.03 (34) | 0 | ||
| Range, μm | (2.61–3.09) | (2.72–3.37) | NA | (2.65–3.15) | (2.69–3.18) | NA | ||
| Thin filament length (phalloidin), μm | 1.14 ± 0.01 | 0.97 ± 0.02 | *** | −15% | 1.04 ± 0.01 | 0.97 ± 0.03 | ** | −7% |
| Range, μm | (0.92–1.29) | (0.59–1.34) | NA | (0.74–1.23) | (0.69–1.28) | NA | ||
| Force–SL measurements | Control | cKO | t-test | Change, WT-KO | Control | cKO | t-test | WT-KO |
| Maximal specific force, mN/mm2 | 94.6 ± 10.4 (n= 7) | 54.8±5.5 (10) | * | 39.8 | 151.6 ± 11.9 (7) | 19.1 ± 3.9 (5) | *** | 132.4 |
| SL at half max force, μm | 3.42 ± 0.04 (n= 8) | 3.22 ± 0.04 (6) | * | 0.20 μm | 3.40 ± 0.02 (6) | ND | ND | |
| Predicted thin filament length | 1.27 ± 0.02 μm | 1.17 ± 0.03 μm | * | 0.1 μm | 1.26 ± 0.01 μm | ND | ND | |
*P < 0.05; **P < 0.01; ***P < 0.001. Change: [(KO-WT)/WT] *100.
Cross-bridge cycling kinetics
It has been reported that nebulin regulates cross-bridge cycling kinetics and thereby the population of cross-bridges that develops force (23). We studied whether this mechanism plays a role in the muscle weakness of Neb cKO mice by measuring active tension, stiffness, ktr, and tension cost and using the two-state cross-bridge model to interpret our findings (explained in Materials and Methods ‘ktr and tension cost analysis’). Experiments were performed on single fibers from the Sol focusing on type I fibers, because they are relatively large (for the mouse) and a reasonable number type I fibers can be found in both Ctrl and cKO Sol muscle (type IIB are rare in cKO mice and IIA are small). MHC gels were used on fibers after the experiments were completed (Fig. 8A left) and only results from type I fibers were analyzed. Tension, stiffness and ktr were measured as explained in Figure 8B and C. We found that force and stiffness were similarly reduced in the Neb cKO fibers (Fig. 8B), but that ktr was significantly decreased (Fig. 8C). Tension cost was measured in fiber bundles from Sol muscle (due to the small size of mouse fibers, ATPase measurements in single fibers were unreliable, especially when using cKO fibers). As expected MHC gels revealed that Ctrl bundles contained both type I and IIA MHC while cKO bundles were nearly pure MHC I (Fig. 8A, right). Despite their much higher MHC I content [which in itself is expected to lower tension cost (24)], tension cost was significantly increased in the cKO fiber bundles (Fig. 8D).
Figure 8.

Nebulin deficiency alters cross-bridge cycling kinetics in MHC-I fibers. (A) MHC gels. Left examples of single fibers used for force, stiffness and Ktr experiments (B and C) and right examples of fiber bundles used for tension cost experiments (D). (B) Left: explanation of protocol to measure tension and stiffness. Single fibers were set to a sarcomere length of 2.4 μm and were activated with a saturating amount of calcium (pCa 4.3) and maximal specific force was measured, a 500 Hz sinusoid (peak-to-peak amplitude 0.1%) of 0.5 s duration was used to measure fiber stiffness (bottom middle) and to calculate the force-stiffness ratio (bottom right). Force and stiffness were equally reduced in Neb cKO fibers and consequently the force/stiffness ratio was unaltered. (C) Left: explanation of protocol to measure ktr. At steady-state maximal activation, a slack equivalent to 10% of the muscle length was rapidly induced resulting in unloaded shortening that was allowed to last for 15 ms followed by rapidly (∼0.5 ms) restretching the muscle fiber to its original length, after which tension redevelops. Results from a Ctrl and cKO fiber are shown superimposed. The rate constant of monoexponential tension redevelopment was determined by fitting the rise of tension (see Materials and Methods). ktr was significantly reduced in Neb cKO fibers. (D) Left, explanation of protocol to measure tension cost. Fiber bundles were activated at a range of pCa levels, and tension and ATP usage were simultaneously measured. Obtained values were plotted against each other [to determine tension cost (slope of ATPase rate versus tension)]. Tension cost is higher in cKO fiber bundles. (For additional experimental details, see Materials and Methods.)
Discussion
To gain insights in typical NEM, we studied a Neb cKO mouse model that we made. The model survives to adulthood with nebulin expression levels that are very low (<5% of control values). Analysis of Neb cKO mice revealed nemaline rods and abnormalities of fiber type, fiber number, and fiber size. Large reductions in specific force were found in the cKO mice with a mechanistic basis that includes shortening of the thin filaments and alterations in cross-bridge cycling kinetics. The force reduction is in glycolytic muscles augmented by hypotrophy that results in a much smaller muscle CSA. Below we discuss these findings in detail.
Survival of Neb cKO model
The Neb cKO model is the first model in which mice reach adulthood while expressing low levels of nebulin, mimicking thereby the typical form of nebulin-based NEM. For the first weeks after birth, nebulin levels are still relatively high (∼50% of control values in the Neb cKO mouse), which is likely due to the kinetics with which the MCK promotor is activated, the time required for recombination to occur, and existing wild-type nebulin transcript and protein to be degraded. The time course of nebulin reduction is not identical for all muscles; for example in 3-week-old Neb cKO mice the Sol contains nebulin at 62% of control values, whereas in the Quad this value is 26% (Fig. 1C). This difference might be due to the MCK promotor activity that varies in different muscle types (25). Additional mechanisms might be at play as well. For example, the nebulin-binding protein KLHL40 is upregulated 3.4-fold in Sol (Supplementary Material, Table S4G). KLHL40 stabilizes nebulin by inhibiting its degradation (20), and upregulation of KLHL40 in the Sol muscle of cKO mice might thereby slow down the loss of nebulin. Regardless of the underlying mechanisms, it is likely that the intermediate nebulin levels that exist in all examined muscles for several weeks after birth (Fig. 1C) is a key to the initial survival of the cKO model.
Fiber-type switching and trophicity
Nebulin deficiency caused a pronounced fiber-type switch away from the glycolytic type IIB toward oxidative type IIA and type I fibers (Figs 3 and 4). The increase in the oxidative character of the examined muscles was largely completed within 5 weeks with a more gradual increase thereafter (Fig. 4C). An increase in the number of oxidative fibers has also been shown in the Sol muscle of the Acta1(H40y) mouse model of NEM (26), albeit that its 27% increase is smaller than the increase in the number of oxidative fibers in the Sol of the Neb cKO model (∼80% increase); a change in fiber-type composition appears to be absent in Sol muscle of the TnIslow-αTmslow(Met9Arg) NEM model, although a modest switch toward oxidative fibers was present in EDL muscle (27). Our findings in the Neb cKO model are similar to those in NEM patients where a predominance of type I fibers has been reported (11,28,29) with a progressive deficiency in type II fibers as patients age (29). The molecular mechanisms that underlie the fiber-type switch in the Neb cKO model are not known and our gene expression analysis did not reveal changes in genes that are thought to be important in fiber-type switching [e.g. calcineurin/NFAT, MEF2, HDAC (18)]. Switching toward oxidative fiber types is functionally beneficial as these fibers generate ATP more efficiently than glycolytic fibers (18). Additionally, force will be produced more efficiently as well [tension cost is approximately two-fold less for IIA than IIB and approximately two less for type I than IIA (24)]. Both effects (more efficient ATP production and lower tension cost) are likely important in survival of Neb cKO mice as they will offset the greatly increased tension cost that results from altered cross-bridge cycling kinetics (see below).
Nebulin deficiency elicited a trophic response that varied greatly in different muscles (Fig. 2). For example, in 5-week-old Neb cKO mice (an early age at which nebulin levels have fallen to <5%), some muscle weights of cKO mice were much smaller than in Ctrl mice (e.g. Quad, Gast) and others were largely unchanged (Diaph and Sol). This finding may be analogous to NEM patients where different muscle groups are affected more than others (9) with the typical form of nebulin-based NEM characterized by an initially predominately proximal pattern of weakness and distal involvement usually occurring later (9). A general aspect of NEM patients is the weakness of respiratory muscle that tends to be greater than that of other muscle groups (9). It should be noted that although the weights and fiber-type composition of the Diaph of Neb cKO mice are largely unchanged, an ongoing pilot study showed that specific force of intact Diaph muscle in 6-month-old mice is reduced by 55%. Thus, it is likely that respiratory weakness is shared between the Neb cKO mouse model and NEM patients. Interestingly, our present study revealed that the MHC IIB content of Ctrl muscles predicts the muscle weight in Neb cKO mice relative to Ctrl mice; muscle types with the most type IIB fibers weigh the least (Fig. 4D). The explanation for this correlation includes the smaller size of oxidative fibers that type IIB fibers in Neb cKO switch to (∼50% CSA reduction), and, importantly, the atrophic type IIB fibers that remain in Neb cKO that have a CSA that is reduced by ∼80%.
The mechanism by which IIB fibers undergo atrophy is likely to include proteolysis as the ubiquitin ligase MuRF-1 and Fbxo30/MUSA1 [an ubiquitin ligase that is necessary for muscle atrophy (21)] were upregulated in the Quad. Increased protein degradation is also suggested by the upregulation of Gadd45a, the only factor commonly increased in several atrophy models and when transgenically upregulated to be sufficient to activate proatrophy pathways (autophagy and proteolysis) and reduce protein synthesis (30). In contrast, ribosomal proteins are upregulated in the Sol but not in the Quad, suggesting increased protein synthesis in this muscle type. Although the Sol is not hypertrophied at the age when the transcriptome was studied (5 weeks), as mice get older the Sol does hypertrophy (Fig. 2) and the changes in transcription might lead that process. Interestingly, a population of hypertrophic fibers was also found in NEM patients, and in particular in ambulant patients (29). The hypertrophy that we found is likely to play a role in symptomatic stabilization in Neb cKO mice as it allows total force values to be similar to Ctrl in some Neb cKO muscles despite dramatic reductions in specific force levels (Fig. 6C and D, Sol). The ability of some muscle types to prevent atrophy or even hypertrophy is likely to be important for the survival of adult Neb cKO mice.
Structural basis of specific force reduction in Neb cKO mice
Specific maximal tetanic force was greatly reduced in the Neb cKO model (Fig. 6D). It is unlikely that this large force reduction is due to a depressed maximal myofilament activation level (e.g. less calcium released from the SR) or fiber-type switching, as large deficits in specific force were observed in skinned fiber bundles (Table 1) and when evaluating only type 1 fibers (Fig 8B). Ultrastructural studies showed that myofibrillar integrity of muscle was well-preserved overall (Fig. 5) and no gross disarray was found that could readily explain the large force deficit. However, nemaline rod bodies were present—either continuous with the Z-disk or as structures independent of the myofibril (Fig. 5G and H). Rod bodies are a hallmark of nemaline myopathies and may cause a reduction in force as they occupy CSA but are non-contractile. The total fiber area that is occupied by rod bodies is small in both EDL and Sol cKO muscle (∼3%, Fig. 5I, right) and it seems unlikely, therefore, that the presence of rod bodies results in a major force deficit. This is supported by several studies in NEM patients in which the prevalence of rod bodies also did not correlate well with the severity of disease (28,29,31,32).
A likely source for the force deficit is the reduction in thin filament length detected in both Sol and EDL muscles (Fig. 7B, Table 1). Shorter thin filaments are also present in the Neb KO and NebΔex55 models (15–17), but, surprisingly, thin filament length is normal in the KLHL40 KO mouse in which nebulin expression is also reduced (20). A possible explanation is that nebulin is reduced by only ∼50% in the KLHL40 model (20), and this might be adequate to maintain thin filament length. EM also showed that H-zones are absent in the Neb cKO and instead A-band protein density gradually decreases toward the M-band (Fig. 5F), indicating that thin filaments are variable in length. The non-uniform, thin filament length is likely to result in a force imbalance across the Z-disk, explaining the wavy Z-disks that were typically observed in Neb cKO muscle (Fig. 5). Wavy Z-disks are expected to result in a similarly wavy location of the pointed ends of the thin filaments, but this phenomenon was hard to visualize even in ultra-high magnification transmission electron micrographs. A likely reason for this is the non-uniform thin filament length of Neb cKO fibers. Future studies using, for example electron tomography (33) are needed to accurately visualize the ends of the thin filaments. Nebulin is thought to stabilize a large portion of the actin filament by binding to actin (1,8), and our findings support the notion that when this stabilizing effect is lost, thin filaments vary in length and are on average shorter than in Ctrl muscle.
The effect of shorter thin filaments on force development will depend on sarcomere length. On the descending limb of the force-sarcomere length relation, force will be reduced, but on the ascending limb force will be increased (34). A meta-analysis of the sarcomere length range during animal locomotion has shown that the physiological range typically includes part of the plateau and the descending limb (35). Assuming that the length range does not change in Neb cKO mice, it can be calculated that force will be reduced by ∼15% due to their shorter thin filaments. Although this reduction is functionally important, it is much less than the force deficit that was measured. Thus, another source must exist as well.
Altered cross-bridge cycling kinetics as a mechanism of specific force reduction
The finding that force and stiffness are equally reduced in Neb cKO mice (Fig. 8B) suggests that the force per cross-bridge is not affected by nebulin. However, ktr is decreased in Neb cKO fibers (Fig. 8C), a finding consistent with work on fibers from nebulin-based NEM patients (14,36). A reduced ktr indicates that when nebulin is absent either fapp or gapp (or both) is reduced. [In the two-state cross-bridge model, the rate of force redevelopment (ktr) is proportional to fapp + gapp, and tension cost is proportional to gapp (37).] Tension cost was found to be increased in cKO mice (Fig. 8D). One caveat is that unlike the ktr study, tension cost measurements were on fiber bundles (the single cKO fibers are too small for reliable ATPase measurements) and the fiber-type composition is mixed type I and IIA in Ctrl and nearly pure type I in cKO fiber bundles (Fig. 8A, right). How such fiber-type switch alone affects tension cost can be determined from previous tension cost measurements on single type I and IIA fibers from the rat, resulting in a tension cost reduction of 35% (for details, see Materials and Methods ‘ktr and tension cost analysis'). Assuming that fiber-type switching per se affects tension cost similarly in the mouse results in a tension cost of 0.92 pmol s−1 mN−1 mm−1 in cKO fiber bundles. Because the measured cKO tension cost is 2.2 pmol s−1mN−1mm−1, we conclude that nebulin deficiency increases tension cost (and thus gapp) 2.4-fold. The fraction of cross-bridges that generates force (α) corresponds to fapp/(fapp + gapp) (37). The decrease in ktr of Neb cKO muscle fibers together with their increased tension cost (gapp) indicates that α is reduced in Neb cKO fibers. From our measurements, we can estimate α in Ctrl fibers at 0.8 and in Neb cKO fibers at 0.13 (for details, see Materials and Methods ‘ktr and tension cost analysis’), a reduction of ∼80%. Thus, our work supports that nebulin plays an important role in regulating force development in skeletal muscle and that an important source for muscle weakness in nebulin-deficient muscle is the altered cross-bridge cycling kinetics that lowers the number of force-generating cross-bridges.
Conclusions
Neb cKO mice survive to adulthood while expressing low levels of nebulin. Nebulin deficiency causes sarcomere defects that lower specific force, including a reduction in force-generating cross-bridges and shortening of thin filaments. Neb cKO mice display nemaline rod formation, glycolytic fiber atrophy, fiber-type switching to oxidative fiber types and in older mice hypertrophy of oxidative muscle. It is unknown what drives certain muscles toward hypertrophy and others in the opposite direction. We propose that the activity level is a factor. Type IIB fibers are recruited infrequently and have a low base activity level (18), which combined with the dramatically reduced specific force when nebulin is absent might be detected by biomechanical sensing/signaling pathways as a disuse state, triggering atrophy. Type I and IIA rich muscles also suffer from reduced specific force, but they might escape an atrophic fate as a result of their higher baseline activity level (e.g. the Sol is an anti-gravity muscle and the Diaph is cyclically activated at the respiration rate). Additionally, hypertrophy might be explained by the increased load due to hypotrophy of synergist muscles [e.g. the hypotrophy of the gastrocnemius is expected to increase the load on the Sol, analogous to what occurs following synergist ablation surgery (38)]. Hypertrophy of oxidative muscles is expected to play a critical role in symptomatic stabilization in Neb cKO mice, as it allows normalization of total force despite dramatic reductions in specific force. The novel Neb cKO mouse provides long-awaited opportunities for developing therapeutic strategies in nebulin-based NEM. Our work suggests a focus on ameliorating the force deficit of the sarcomere and increasing the trophic state of muscle (e.g. through exercise).
Materials and Methods
Animals and genotyping
To create Neb cKO mice, a targeting vector was made with loxP sites inserted downstream of exon 3 [which contains the start codon for Nebulin and corresponds to exon 1 of Bang et al. (15 )] and in the 5′ untranslated region of exon 2 upstream of the ATG. Floxed mice were bred to a MCK-Cre strain (#6475 Jackson Laboratory) that expresses Cre recombinase under control of the Muscle Creatine Kinase (MCK) promotor that is expressed in striated muscle (39). Genotyping was used to determine the presence of the MCK-Cre transgene (following the protocol provided by the Jackson Laboratory) and the floxed nebulin allele (for details, see Supplementary Material, Fig. S1). Mice homozygous for the floxed nebulin allele were bred to mice that were hemizygous for MCK-Cre and heterozygous for the floxed allele (MCKCre+, Neb+/flox). Offspring that was hemizygous for MCK-Cre and homozygous for the floxed allele (MCKCre+, Neb flox/flox) was deficient in nebulin and is referred to as Neb cKO. We used as control offspring that had at least one nebulin WT allele and that were either MCK-Cre positive or negative. Since no differences were found among the control genotypes [as expected because mice that have one WT nebulin allele contain a normal level of nebulin (16 )], they were grouped as control (Ctrl) mice. We also studied mice that were hemizygous for MCK-Cre and that contained one floxed allele and the other was a conventional KO allele [exons 2 and 3 were permanently deleted (16)], and their results were indistinguishable from the MCK-Cre+, Neb flox/flox mice. All animal experiments were approved by the University of Arizona Institutional Animal Care and Use Committee and followed the US National Institutes of Health ‘Using Animals in Intramural Research’ guidelines for animal use.
Tissue collection
Mice were weighed, anesthetized with isoflurane and sacrificed by cervical dislocation. The following muscle was dissected and rapidly weighed and flash-frozen in liquid nitrogen. Hind limb: ttibialis cranialis (TC, soleus (Sol, extensor digitorum longus (EDL, Diaphragm (Diaph, gastrocnemius (Gast), quadriceps (Quad; forelimb: Biceps (B), Triceps medialis (Tm), Triceps lateralis (Tla) and Triceps longus (Tlo). Tibias were removed, and tibia length was measured using a caliper. Muscles were stored at −80°C for later use.
Exercise testing
We used 6-month-old mice that were individually housed in a 13.9″L × 9.25″W × 7.9″H pan outfitted with a freely rotating 12.7 cm diameter wheel with a 5.7 cm wide running surface (Lafayette Instruments, Lafayette, IN, USA). Running wheels were connected to a computer that recorded activity in 5s intervals, for a total of 21 days. Mean running time, speed and duration per 24 h period were determined for the last 8 days of the testing period (when running performance was stable), and results were plotted in Figure 6A. The mice were provided food and water ad libitum under a 12:12 h light/dark cycle.
Gel electrophoresis and western blotting
Flash-frozen muscle tissues were prepared as previously described (40–42). Briefly, the tissues were flash-frozen in liquid nitrogen and solubilized between glass pestles cooled in liquid nitrogen. Tissues were primed at −20°C for a minimum of 20 min, then suspended in 50% urea buffer [(in mol/L) 8 urea, 2 thiourea, 0.05 Tris–HCl, 0.075 dithiothreitol with 3% SDS and 0.03% bromophenol blue pH 6.8] and 50% glycerol with protease inhibitors [(in mmol/L) 0.04 E64, 0.16 leupeptin and 0.2 PMSF] at 60°C for 10 min. Then the samples were centrifuged at 13 000 revolutions per minute (rpm) for 5 min, aliquoted and flash-frozen in liquid nitrogen and stored at −80°C. Nebulin was visualized on 1.0% agarose gels stained with coomassie blue, as described previously (40,43). Myosin heavy chain isoform composition was visualized using 8% acrylamide gels stained with coomassie blue (44) and Atrogin-1 and MuRF-1 with 12% SDS–PAGE gel and run for 2 h at 100 V. For western blot experiments, gels ran for 3 h at 15 mA/gel and proteins were transferred to PVDF membrane using a semi-dry transfer unit (Bio-Rad, Hercules, CA, USA) for 2.5 h at 150 mA. All transferred blots were stained with Ponceau S to visualize total transferred protein. The blots were then probed with primary antibodies at 4° overnight [anti-Nebulin N-term (1:750 rabbit 1357L, kindly provided by Dr Carol Gregorio]; anti-MuRF-1(1:1000 chicken IgY, kindly provided by Dr Siegfried Labeit); anti-Atrogin-1 (1:1000 rabbit AP2041, ECM Biosciences); GAPDH Loading Control Antibody (1:5000 mouse #GA1R, ThermoScientific). To normalize for loading differences, MHC from the Ponceau S-stained membrane was used for nebulin western blots; GAPDH expression was used to normalize other western blots. Secondary antibodies conjugated with fluorescent dyes with infrared excitation spectra were used for detection. IR western blots were analyzed using Odyssey Infrared Imaging System (Li-Cor Biosciences, NE, USA). Ponceau S images were analyzed with One-D scan EX (Scanalytics Inc., Rockville, MD, USA).
Intact muscle mechanics
Intact muscle mechanics was performed using the Aurora 1200A ex vivo test system that has been described previously (45,46). Briefly, muscles were attached between a combination servomotor-force transducer and fixed hook via silk suture in a bath containing oxygenated Ringer solution (145 mm NaCl, 2.5 mm KCl, 1.0 mm MgSO4, 1.0 mm CaCl2, 10.0 mm HEPES, 10 mm glucose, pH 7.4, 30°C). The optimal length (L0) was determined by adjusting muscle length until optimal maximal twitch force was produced (pulse duration of 200 μs with biphasic polarity). Active force was determined from a force–frequency protocol. The Sol muscle was stimulated at incremental stimulation frequencies 1, 10, 20, 30, 50, 70, 100 and 150 Hz waiting 30, 30, 60, 90, 120, 120, 120 and 120 s, respectively, in between each stimulation. Measured force in mN was normalized to CSA [muscle mass (mg)/[(L0 (mm) × 1.056)]] to obtain specific force (mN/mm2). The EDL protocol matched that of the Sol, except for an additional force measurement at a frequency of 200 Hz.
Fiber typing and CSA analysis
Detailed immunofluorescence methods have been described previously (47). Briefly, Sol and EDL muscles were dissected from control or NEB cKO mice, quick frozen and sectioned in the mid-belly of the muscle. Sections were stained with anti-laminin (L9393 Sigma Aldrich) to mark the cell boundary and with monoclonal anti-MHC I (MHC-I) (M8421 Sigma Aldrich), MHC-IIA (SC-71 DSHB) or MHC-IIB (BF-F3 DSHB) antibody. Images were collected on a Zeiss Axio Imager M.1 microscope utilizing a Zeiss Axio Cam MRC. CSA and MinFeret diameter (smallest diameter of the myofiber) were measured using the Image-J program. We also performed a study on Quad muscle that was sectioned and stained with antidystrophin and anti-MHC-IIB; analysis was via an automated fiber-type frequency and fiber-type-specific CSA assessment algorithm [for details, see Ref. (48)].
Myofiber size in the quadriceps muscle was quantified by automated measurement of MinFeret diameter, as previously described (49). Briefly, eight micron frozen sections of quadriceps muscle were double-immunostained with rabbit antidystrophin antibodies (ab15277; Abcam, Cambridge, MA, USA) and mouse monoclonal antibodies against MHC type 2b (clone BF-F3; Developmental Studies Hybridoma Bank). Immunofluorescence whole-slide scanning was performed in the Children's Hospital of Wisconsin Research Institute's Imaging Core facility using a Olympus VS120 fluorescent whole-slide scanner, and images were sent to the laboratory of Dr Lin Yang. Minferet diameter for type 2b positive (glycolytic) or negative (oxidative) fibers was calculated on regions of interest from these scanned slides using previously described algorithms developed in the Yang laboratory (49).
Thin filament length measurements
Muscles were rapidly excised and placed in relaxing solution (in mm: 20 BES, 10 EGTA, 6.56 MgCl2, 5.88 NaATP, 1 DTT, 46.35 K-propionate, 15 creatine phosphate, pH 7.0 at 20°C) with 1% (w/v) Tritron X-100 and protease inhibitors for overnight on a 2D rocker at 4°C. The solution was then replaced with fresh relaxing solution (without Triton) followed by 5 h in 50% glycerol/relaxing solution before storing at −20°C. Skinned muscles were placed in a sylgard dish containing 50% glycerol solution and dissected into fiber bundles. The ends of the bundles were attached to aluminum T-clips and the solution replaced with fresh relaxing solution. Bundles were stretched ∼50% of their base length. Relaxing solution was then replaced with 4% formaldehyde solution and muscles were fixed for overnight. After fixation, muscles were washed with phosphate buffer saline (PBS) and embedded in Tissue-Tek O.C.T.compound (Ted Pella Inc) and stored at −80 °C. The O.C.T. embedded specimen was sectioned into 5 µm thick (Microm HM 550; Thermo Scientific) and placed on gelatin-coated cover glass. Fixed tissues were permeabilized again with 0.2% Triton X-100 in PBS for 20 min at room temperature on a light box to bleach out the background fluorescence. Then tissues were blocked with 2% bovine calf serum plus 1% normal donkey serum in PBS and incubated overnight with primary antibodies diluted in PBS. The primary antibodies were anti-α-actinin (for Z-disk localization, 1:5000 mouse monoclonal A7811, Sigma Aldrich), anti-Tmod (1:600 rabbit polyclonal, courtesy of Dr Carol Gregorio) and Alexa Fluor 488-conjugated Phalloidin (for actin staining 1:2000, A12379, Life Technologies). The tissues were washed with PBS for 30 min and incubated with secondary antibodies at room temperature in a dark box on 2D rocker for 2–4 h. The secondary antibodies were polyclonal Alexa Fluor 350-conjugated goat anti-mouse IgG(H + L) (1:200, A21049, Invitrogen) and Texas Red-conjugated goat anti-rabbit IgG (1:600, T2767, Life Technologies). Coverslips were mounted onto slides with Aqua Poly/Mount (Polysciences Inc.). Images were captured using a Deltavision RT system (Applied Precision) with an inverted microscope (IX70; Olympus), a ×100 objective, and a charge-coupled device camera (CoolSNAP HQ; Photometrics) using SoftWoRx 3.5.1 software (Applied Precision). The images were then deconvolved using SoftWoRx. An average of 10 areas was observed for each tissue section. Thin filament lengths and sarcomere lengths were obtained from deconvolved images of Sol and EDL muscles stained with fluorescently conjugated phalloidin and anti-Tmod antibodies. Deconvolved images were reopened in ImageJ (http://rsb.info.nih.gov/ij), then the 1D plot profile was calculated. The plot profile was analyzed using Fityk0.9.8 (http://fityk.nieto.pl). Well-defined peaks were fitted with Gaussian curves for T-mod and α-actinin. Sarcomere length was calculated from the distance between two adjacent α-actinin peak centers. A custom ‘rectangle + 2 half Gaussian’ function was used for analyzing phalloidin-stained images that consisted of a rectangle that was flanked by two half Gaussian curves. To account for actin overlapping in the Z-disk which creates a small bump in the center of the rectangle, we developed a special script designed for Fityk that de-activates the center points within the rectangle fit. This improved the subsequent fit for the ‘rectangle + 2 half Gaussian’ function. Thin filament length was calculated as half the width of the rectangle plus half the width of the Gaussian fit at half maximum height. SL was calculated from the distance between the centers of two adjacent Gaussian fits. We analyzed a large number of images and determined thin filament length within the SL range of 2.6–3.1 µm (Table 1).
Electron microscopy
For electron microscopy, skinned fiber bundles were used that were stretched in relaxing solution by ∼20% of their slack length. The fiber bundles were fixed in 3% paraformaldehyde (PF; Sigma P-6148) and 2% glutaraldehyde and 0.03% tannic acid in 1× PBS (0.01 m, pH 7.2) at 4°C for 45 min and postfixed in 1% OsO4 in PBS for 45 min at 4°C. The samples were then dehydrated and embedded in araldite/embed812. Ultrathin sections (60 nm) were cut with the diamond knife edge parallel to the muscle fiber direction; sections were contrasted with potassium permanganate and lead citrate followed by transmission electron microscopy (FEI Philips CM12). Digital images were processed by ImageJ software and density profiles along the myofibril direction were processed by Fityk software.
Skinned muscle mechanics
Force measurement
The procedures for skinned muscle contractility were as described previously (50). Soleus and EDL muscles from control and NEB cKO mice were skinned overnight at ∼4°C in relaxing solution (in mm: 20 BES, 10 EGTA, 6.56 MgCl2, 5.88 NaATP, 1 DTT, 46.35 K-propionate, 15 creatine phosphate, pH 7.0 at 20°C) containing 1% (w/v) Triton X-100 and protease inhibitors. Muscles were then washed thoroughly with relaxing solution and stored in 50% glycerol/relaxing solution at −20°C. Muscles were used for experiments within 1 week. On the day of an experiment, single fibers were dissected for experiments in Figure 8A–C and fiber bundles (CSA ∼0.05 mm2) for experiments in Figure 8D. After completion of the mechanical studies (below), fibers/bundles were fiber-typed (Fig. 8A shows examples). The fibers/fiber bundles were mounted using aluminum T clips between a length motor (ASI 403A, Aurora Scientific Inc., Ontario, Canada), and a force transducer element (ASI 315C-I, Aurora Scientific Inc.) in a skinned fiber apparatus (ASI 802D, Aurora Scientific Inc.) that was mounted on an inverted microscope (Zeiss Axio Observer A1). Sarcomere length was set using a high-speed VSL camera and ASI 900B software (Aurora Scientific Inc.). Muscle bundles were stretched and activated in pCa4.5 activating solution (in mm: 40 BES, 10 CaCO3 EGTA, 6.29 MgCl2, 6.12 Na-ATP, 1 DTT, 45.3 potassium-proprionate, 15 creatine phosphate) and protease inhibitors (in mm: 0.01 E64, 0.04 leupeptin and 0.5 PMSF) at sarcomere length 2.4 µm to record maximal active tension. After one cycle of full activation (pCa 4.5) and relaxation (pCa 8.5), the resting SL was readjusted to 2.4 μm. Fiber width and diameter were measured at three points along the fiber, and the CSA was determined assuming an elliptical cross-section. Specific force (or tension) was expressed as force per CSA.
Muscle fiber stiffness
Fiber bundles were maximally activated (pCa 4.3). When the muscle fiber reached a steady-state isometric force (Fss), the muscle length was changed sinusoidially (amplitude 0.1%). Stiffness associated with the number of strongly bound cross-bridges was estimated from the relationship between length change (ΔL) and the peak force response, Fp.
Ktr measurements
To measure the rate of tension redevelopment (ktr), we used the large slack/release approach, originally described by Brenner (51,52), to disengage force-generating cross-bridges from the thin filaments, which were isometrically activated. Fast activation of the fiber was achieved by transferring the skinned muscle fibers from the pre-activation solution containing a low concentration of EGTA (0.5 mm) to a pCa 4.3 activating solution. Once the steady state was reached, a slack equivalent to 10% of the muscle length was rapidly induced using the motor resulting in unloaded shortening that was allowed to last for 15 ms. The remaining bound cross-bridges were mechanically detached by rapidly (∼0.5 ms) restretching the muscle fiber to its original length, after which tension redevelops. The rate constant of monoexponential tension redevelopment (ktr) was determined by fitting the rise of tension to the following equation: , where F is force at time t and ktr the rate constant of tension redevelopment. Figure 8C shows examples of Ctrl and Neb cKO fibers superimposed.
Force-ATPase measurement
Small strips (CSA ∼0.1 mm2) were used in a system similar to the one described by de Tombe and Stienen (53). To measure the ATPase activity, a near UV light was projected through the quartz window of the bath (30 μl volume and temp controlled at 20°C) and detected at 340 NEM. Maximum activation buffer (pCa 4.3) contained (mm): potassium propionate 31, Na2ATP 5.95, MgCl2 6.61, EGTA 10, CaCl2 10.11, BES 50 (pH 7.0), NaN3 5, NADH 0.9 and phosphoenol pyruvate 10, with 4 mg ml–1 pyruvate kinase (500 U mg–1), 0.24 mg ml–1 lactate dehydrogenase (870 U mg–1), 0.2 mm diadenosine-5′ pentaphosphate (A2P5) and 1.0 mm oligomycin as well as a cocktail of protease inhibitors. For efficient mixing, the solution in the bath was continuously stirred by means of motor-driven vibration of a membrane positioned at the base of the bath. ATPase activity of the skinned fiber bundles was measured as follows: ATP regeneration from ADP is coupled to the breakdown of phosphoenol pyruvate to pyruvate and ATP catalyzed by pyruvate kinase, which is linked to the synthesis of lactate catalyzed by lactate dehydrogenase. The breakdown of NADH, which is proportional to the amount of ATP consumed, is measured on-line by UV absorbance at 340 NEM. The ratio of light intensity at 340 NEM (sensitive to NADH concentration), and the light intensity at 410 NEM (reference signal), is obtained by means of an analog divider. After each recording, the UV absorbance signal of NADH was calibrated by multiple rapid injections of 0.5 NEMol of ADP into the bathing solution, with a motor-controlled calibration pipette. The slope of the [ATP] versus time trace during steady-state tension development of a calcium-induced contraction (see Fig. 8D, left for an example) was determined from a linear fit and the value divided by the fiber volume (in mm3) to determine the fiber's ATPase rate. ATPase rates were corrected for the basal ATPase in relaxing solution. The ATPase rate and tension at different pCa values were plotted against each other (see Supplementary Material, Fig. S8D, right), and the tension cost was determined from the slope of a linear fit to the data. All skinned fiber experiments were performed at 20°C.
Ktr and tension cost analysis
We used the two-state cross-bridge model with apparent rate constant fapp representing the transition from the non-force-generating state to the force-generating state, and gapp the transition from the force-generating state back to the non-force-generating state (37). In this model, the rate of force redevelopment (ktr) is proportional to fapp + gapp, and tension cost is proportional to gapp (37). The fraction of cross-bridges that generates force (α) corresponds to fapp/(fapp + gapp). Once gapp of type I fibers has been estimated and ktr has been measured (See Results), fapp can be determined as ktr − gapp, and then α can be calculated. In our work (see Results), tension cost could only be measured in fiber bundles (due to the small size and low force of cKO single fibers). Ctrl and cKO bundles used for experiments were found to not have the same fiber-type composition (Ctrl: 53% type I and 47% type IIA; cKO: 97% type I and 3% Type IIA). We estimated the tension cost differences due to the fiber-type switch per se, using previous measurements in the rat (their CSA is much larger than in the mouse and these experiments can be performed at the single fiber level in this species) where tension cost of type I and type IIA fibers is 0.66 and 1.51 pmol s−1 mN−1 mm−1, respectively (24). We can calculate from these values the tension cost in the rat in a bundle with a fiber-type composition 53% type I and 47% type IIA (as in the our Ctrl bundles) at 1.05 pmol s−1 mN−1 mm−1 and in bundles 97% type I and 3% Type IIA (as in the cKO) at 0.69 pmol s−1 mN−1 mm−1, that is a 35% reduction. We used this 35% value to correct the measured tension cost of our Ctrl fiber bundles. This results in a tension cost of 0.92 pmol s−1 mN−1 mm−1, that is the tension cost of cKO fibers if nebulin were not to affect tension. Because the measured cKO tension cost is 2.2 pmol s−1mN−1mm−1 we conclude that nebulin deficiency increases tension cost (and thus gapp) 2.2/0.92 or 2.4-fold. We estimated the absolute gapp of type I Ctrl fibers from the 2 s−1 gapp of type IIB fibers (37) and using the fiber-type dependence of tension cost (24). This results in 0.4 s−1 as gapp for type I Ctrl fibers. From the measured ktr, we can calculate fapp and from this α at 0.8. For Neb cKO type I fibers, gapp is 2.4 times that in Ctrl fibers (see above), or 0.96 s−1. This results in a Neb cKO α of 0.13. Thus, this analysis shows a reduction in the fraction of force-generating cross-bridges from 0.8 (Ctrl) to 0.13 (cKO), or a reduction of ∼80%. Although this is a best estimate only (direct measurements are needed), the analysis does reveal that a major source of the force deficit in the Neb cKO mouse is likely to be altered cross-bridge cycling kinetics. Finally, in the two-state cross-bridge model, the fraction of cross-bridges that generates force (α) corresponds to fapp/(fapp + gapp). Once gapp of type I fibers has been estimated and ktr has been measured (See Results), fapp can be determined as ktr − gapp and then α can be calculated. See Results and Discussion.
Microarray
GeneChip: Quad and Sol tissues were dissected from six male mice (6 weeks old) of each Neb cKO and Ctrl mice and stored in RNAlater (Invitrogen). Total RNA was isolated using the RNeasy Fibrous Tissue Mini Kit (Qiagen). RNA quality was assessed by NanoDrop 1000 Spectrophotometer and 2100 Bioanalyzer (Agilent); all samples had RIN ≥ 9.1. Samples were hybridized with the Mouse Gene 1.0ST Array (Affymetrix); processing (labeling through scanning) was performed by the Genomics Core, University of Arizona, using Affymetrix protocols, supplies and equipment; Affymetrix software (Expression Console and Transcriptome Analysis Console) was used, incorporating RMA processing and Analysis of Variance with Benjamini–Hochberg False Discovery Rate (FDR) correction for multiple testing. A conservative cutoff of adjusted P-value <0.001 was used to identify differentially regulated genes. Current annotation for the ProbeSetIDs was retrieved from the Mouse Genome Informatics server (http://www.informatics.jax.org/). Proportional Venn diagrams were made using the BioVenn website (http://www.cmbi.ru.nl/cdd/biovenn/) (54) (54). Over-representation analysis used KEGGpathways at GeneCodis3 (http://genecodis.cnb.csic.es/) (55). The data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE70213 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70213).
Statistics
All data are represented as average ± SEM (standard error of the mean). For data with numbers of mice (n) greater than or equal to eight, an unpaired t-test with a P-value <0.05 was considered significant. For experiments performed with data <8, a Mann–Whitney test that does not assume normal distributions was performed (P < 0.05 significant). For experiments with multiple groups, we used ANOVA with Bonferroni post hoc tests corrected for multiple comparisons (P < 0.05 significant). The number of mice that were used are as follows. Figure 1 A: we used 53 mice per group; B: 10 mice per data point; C: 6 mice per data point; ANOVA with multiple testing correction (Bonferroni) was used to compare results versus the Quad muscle. Figure 2. At 3 weeks 6 mice per group; 5 weeks 30 mice for Ctrl and 27 for cKO; 6 months 32 mice per group. Figure 3A–H. 11 muscles from 11 mice per group (each genotype and each muscle type). Figure 4. Data from six muscles from six mice for each muscle type and genotypes. Figure 5. A: 8 mice per group; B–D: 8 mice per group (each genotype and each muscle type). Figure 6. A: 8 muscles from 8 mice for Sol and 9 EDL Ctrl and 6 EDL cKO; C: data from 8 muscles from 6 mice per group. Figure 7. 8 Muscles from 8 mice per group. Figure 8. A–C: 10 fibers from 6 muscles from 6 mice per group. D: 10 fibers from 6 muscles from 6 mice per group. Symbols: *, **, *** and **** P < 0.05, P < 0.01, P < 0.001 and P < 0.00001, respectively, in statistical significance test versus age-matched control (unless indicated otherwise).
Supplementary material
Funding
This work was supported by National Institutes of Health (5R01AR053897 and 5R01HL062881 to H.G, T32GM084905 to R.S., T32HL07249 to C.B., T32GM008659 to D.B., T34 GM008718 to Y.E., and K08 AR058750 to M.L.); Muscular Dystrophy Association (295195 to H.G. and M.L.); a Foundation Building Strength (to H.G.).
Supplementary Material
Acknowledgements
We are grateful to our lab members (Maya Adler, Matt Bull, Dr Carlos Hidalgo, Xiangdang Liu, Chandra Saripalli, Luann Wyly and Xiaoqun Zhou) and the GEMM core (Drs Tom Doetschman and Teodora Georgiev) for gene targeting mouse services.
Conflict of Interest statement. None declared.
References
- 1.Gokhin D.S., Fowler V.M. (2013) A two-segment model for thin filament architecture in skeletal muscle. Nat. Rev. Mol. Cell. Biol., 14, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kruger M., Wright J., Wang K. (1991) Nebulin as a length regulator of thin filaments of vertebrate skeletal muscles: correlation of thin filament length, nebulin size, and epitope profile. J. Cell. Biol., 115, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pappas C.T., Bliss K.T., Zieseniss A., Gregorio C.C. (2011) The Nebulin family: an actin support group. Trends Cell. Biol., 21, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labeit S., Gibson T., Lakey A., Leonard K., Zeviani M., Knight P., Wardale J., Trinick J. (1991) Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Lett., 282, 313–316. [DOI] [PubMed] [Google Scholar]
- 5.Donner K., Sandbacka M., Lehtokari V.L., Wallgren-Pettersson C., Pelin K. (2004) Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur. J. Hum. Genet., 12, 744–751. [DOI] [PubMed] [Google Scholar]
- 6.Jin J.P., Wang K. (1991) Nebulin as a giant actin-binding template protein in skeletal muscle sarcomere. Interaction of actin and cloned human nebulin fragments. FEBS Lett., 281, 93–96. [DOI] [PubMed] [Google Scholar]
- 7.Marttila M., Hanif M., Lemola E., Nowak K.J., Laitila J., Gronholm M., Wallgren-Pettersson C., Pelin K. (2014) Nebulin interactions with actin and tropomyosin are altered by disease-causing mutations. Skelet. Muscle, 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pappas C.T., Krieg P.A., Gregorio C.C. (2010) Nebulin regulates actin filament lengths by a stabilization mechanism. J. Cell. Biol., 189, 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallgren-Pettersson C., Sewry C.A., Nowak K.J., Laing N.G. (2011) Nemaline myopathies. Semin. Pediatr. Neurol., 18, 230–238. [DOI] [PubMed] [Google Scholar]
- 10.Lehtokari V.L., Kiiski K., Sandaradura S.A., Laporte J., Repo P., Frey J.A., Donner K., Marttila M., Saunders C., Barth P.G. et al. (2014) Mutation update: the spectra of nebulin variants and associated myopathies. Hum. Mutat., 35, 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.North K.N., Laing N.G., Wallgren-Pettersson C. (1997) Nemaline myopathy: current concepts. The ENMC International Consortium and Nemaline Myopathy. J. Med. Genet., 34, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ottenheijm C.A., Witt C.C., Stienen G.J., Labeit S., Beggs A.H., Granzier H. (2009) Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum. Mol. Genet., 18, 2359–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawlor M.W., Ottenheijm C.A., Lehtokari V.L., Cho K., Pelin K., Wallgren-Pettersson C., Granzier H., Beggs A.H. (2011) Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet. Muscle, 1, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ochala J., Lehtokari V.L., Iwamoto H., Li M., Feng H.Z., Jin J.P., Yagi N., Wallgren-Pettersson C., Penisson-Besnier I., Larsson L. (2011) Disrupted myosin cross-bridge cycling kinetics triggers muscle weakness in nebulin-related myopathy. FASEB J., 25, 1903–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bang M.L., Li X., Littlefield R., Bremner S., Thor A., Knowlton K.U., Lieber R.L., Chen J. (2006) Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell. Biol., 173, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witt C.C., Burkart C., Labeit D., McNabb M., Wu Y., Granzier H., Labeit S. (2006) Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. Embo J., 25, 3843–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ottenheijm C.A., Buck D., de Winter J.M., Ferrara C., Piroddi N., Tesi C., Jasper J.R., Malik F.I., Meng H., Stienen G.J. et al. (2013) Deleting exon 55 from the nebulin gene induces severe muscle weakness in a mouse model for nemaline myopathy. Brain, 136, 1718–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiaffino S., Reggiani C. (2011) Fiber types in mammalian skeletal muscles. Physiol. Rev., 91, 1447–1531. [DOI] [PubMed] [Google Scholar]
- 19.Bloemberg D., Quadrilatero J. (2012) Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One, 7, e35273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg A., O'Rourke J., Long C., Doering J., Ravenscroft G., Bezprozvannaya S., Nelson B.R., Beetz N., Li L., Chen S. et al. (2014) KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Invest., 124, 3529–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sartori R., Schirwis E., Blaauw B., Bortolanza S., Zhao J., Enzo E., Stantzou A., Mouisel E., Toniolo L., Ferry A. et al. (2013) BMP signaling controls muscle mass. Nat. Genet., 45, 1309–1318. [DOI] [PubMed] [Google Scholar]
- 22.Bodine S.C., Baehr L.M. (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab., 307, E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandra M., Mamidi R., Ford S., Hidalgo C., Witt C., Ottenheijm C., Labeit S., Granzier H. (2009) Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: a novel mechanism for increasing tension and reducing tension cost. J. Biol. Chem., 284, 30889–30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bottinelli R., Canepari M., Reggiani C., Stienen G.J. (1994) Myofibrillar ATPase activity during isometric contraction and isomyosin composition in rat single skinned muscle fibres. J. Physiol., 481(Pt 3), 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tai P.W., Fisher-Aylor K.I., Himeda C.L., Smith C.L., Mackenzie A.P., Helterline D.L., Angello J.C., Welikson R.E., Wold B.J., Hauschka S.D. (2011) Differentiation and fiber type-specific activity of a muscle creatine kinase intronic enhancer. Skelet. Muscle, 1, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen M.A., Joya J.E., Kee A.J., Domazetovska A., Yang N., Hook J.W., Lemckert F.A., Kettle E., Valova V.A., Robinson P.J. et al. (2011) Hypertrophy and dietary tyrosine ameliorate the phenotypes of a mouse model of severe nemaline myopathy. Brain, 134, 3516–3529. [DOI] [PubMed] [Google Scholar]
- 27.Corbett M.A., Robinson C.S., Dunglison G.F., Yang N., Joya J.E., Stewart A.W., Schnell C., Gunning P.W., North K.N., Hardeman E.C. (2001) A mutation in alpha-tropomyosin(slow) affects muscle strength, maturation and hypertrophy in a mouse model for nemaline myopathy. Hum. Mol. Genet., 10, 317–328. [DOI] [PubMed] [Google Scholar]
- 28.Malfatti E., Lehtokari V.L., Bohm J., De Winter J.M., Schaffer U., Estournet B., Quijano-Roy S., Monges S., Lubieniecki F., Bellance R. et al. (2014) Muscle histopathology in nebulin-related nemaline myopathy: ultrastrastructural findings correlated to disease severity and genotype. Acta. Neuropathol. Commun., 2, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallgren-Pettersson C., Rapola J., Donner M. (1988) Pathology of congenital nemaline myopathy. A follow-up study. J. Neurol. Sci., 83, 243–257. [DOI] [PubMed] [Google Scholar]
- 30.Ebert S.M., Dyle M.C., Kunkel S.D., Bullard S.A., Bongers K.S., Fox D.K., Dierdorff J.M., Foster E.D., Adams C.M. (2012) Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem., 287, 27290–27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dahl D.S., Klutzow F.W. (1974) Congenital rod disease. Further evidence of innervational abnormalities as the basis for the clinicopathologic features. J. Neurol. Sci., 23, 371–385. [DOI] [PubMed] [Google Scholar]
- 32.Arts W.F., Bethlem J., Dingemans K.P., Eriksson A.W. (1978) Investigations on the inheritance of nemaline myopathy. Arch. Neurol., 35, 72–77. [DOI] [PubMed] [Google Scholar]
- 33.Burgoyne T., Muhamad F., Luther P.K. (2008) Visualization of cardiac muscle thin filaments and measurement of their lengths by electron tomography. Cardiovasc. Res., 77, 707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Granzier H.L., Akster H.A., Ter Keurs H.E. (1991) Effect of thin filament length on the force-sarcomere length relation of skeletal muscle. Am. J. Physiol., 260, C1060–C1070. [DOI] [PubMed] [Google Scholar]
- 35.Burkholder T.J., Lieber R.L. (2001) Sarcomere length operating range of vertebrate muscles during movement. J. Exp. Biol., 204, 1529–1536. [DOI] [PubMed] [Google Scholar]
- 36.Ottenheijm C.A., Hooijman P., Dechene E.T., Stienen G.J., Beggs A.H., Granzier H. (2009) Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J. Struct. Biol., 170, 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenner B. (1988) Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. USA, 85, 3265–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyazaki M., Esser K.A. (2009) Cellular mechanisms regulating protein synthesis and skeletal muscle hypertrophy in animals. J. Appl. Physiol., 106, 1367–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyons G.E., Muhlebach S., Moser A., Masood R., Paterson B.M., Buckingham M.E., Perriard J.C. (1991) Developmental regulation of creatine kinase gene expression by myogenic factors in embryonic mouse and chick skeletal muscle. Development, 113, 1017–1029. [DOI] [PubMed] [Google Scholar]
- 40.Lahmers S., Wu Y., Call D.R., Labeit S., Granzier H. (2004) Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ. Res., 94, 505–513. [DOI] [PubMed] [Google Scholar]
- 41.Hidalgo C., Hudson B., Bogomolovas J., Zhu Y., Anderson B., Greaser M., Labeit S., Granzier H. (2009) PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ. Res., 105, 631–638, 617 p following 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warren C.M., Jordan M.C., Roos K.P., Krzesinski P.R., Greaser M.L. (2003) Titin isoform expression in normal and hypertensive myocardium. Cardiovasc. Res., 59, 86–94. [DOI] [PubMed] [Google Scholar]
- 43.Warren C.M., Krzesinski P.R., Greaser M.L. (2003) Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis, 24, 1695–1702. [DOI] [PubMed] [Google Scholar]
- 44.Agbulut O., Noirez P., Beaumont F., Butler-Browne G. (2003) Myosin heavy chain isoforms in postnatal muscle development of mice. Biol. Cell., 95, 399–406. [DOI] [PubMed] [Google Scholar]
- 45.Labeit S., Kohl C.H., Witt C.C., Labeit D., Jung J., Granzier H. (2010) Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J. Biomed. Biotechnol., 2010, 693741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ottenheijm C.A., Hidalgo C., Rost K., Gotthardt M., Granzier H. (2009) Altered contractility of skeletal muscle in mice deficient in titin's M-band region. J. Mol. Biol., 393, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buck D., Smith J.E. 3rd, Chung C.S., Ono Y., Sorimachi H., Labeit S., Granzier H.L. (2014) Removal of immunoglobulin-like domains from titin's spring segment alters titin splicing in mouse skeletal muscle and causes myopathy. J. Gen. Physiol., 143, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu F., Fry C.S., Mula J., Jackson J.R., Lee J.D., Peterson C.A., Yang L. (2013) Automated fiber-type-specific cross-sectional area assessment and myonuclei counting in skeletal muscle. J. Appl. Physiol. (1985), 115, 1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawlor M.W., Viola M.G., Meng H., Edelstein R.V., Liu F., Yan K., Luna E.J., Lerch-Gaggl A., Hoffmann R.G., Pierson C.R. et al. (2014) Differential muscle hypertrophy is associated with satellite cell numbers and Akt pathway activation following activin type IIB receptor inhibition in Mtm1 p.R69C mice. Am. J. Pathol., 184, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuda N., Wu Y., Farman G., Irving T.C., Granzier H. (2005) Titin-based modulation of active tension and interfilament lattice spacing in skinned rat cardiac muscle. Pflugers Arch., 449, 449–457. [DOI] [PubMed] [Google Scholar]
- 51.Brenner B. (1986) The cross-bridge cycle in muscle. Mechanical, biochemical, and structural studies on single skinned rabbit psoas fibers to characterize cross-bridge kinetics in muscle for correlation with the actomyosin-ATPase in solution. Basic. Res. Cardiol., 81 (Suppl. 1), 1–15. [DOI] [PubMed] [Google Scholar]
- 52.Brenner B., Eisenberg E. (1986) Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. USA, 83, 3542–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Tombe P.P., Stienen G.J. (1995) Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ. Res., 76, 734–741. [DOI] [PubMed] [Google Scholar]
- 54.Hulsen T., de Vlieg J., Alkema W. (2008) BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics, 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tabas-Madrid D., Nogales-Cadenas R., Pascual-Montano A. (2012) GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res., 40, W478–W483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.