

Abstract
A male infant at 36 weeks gestation was born by section. At 20 weeks of gestation, congenital diaphragmatic hernia and sacrococcygeal teratoma had been seen on ultrasound. At birth, the infant had features suggestive of Cornelia de Lange syndrome (CdLS). He remained hypoxic despite aggressive ventilatory manoeuvres and was palliated. At postmortem, the lungs were hypoplastic. In CdLS, mutations in NIPBL are found in around 50% of cases. Mutation analysis, including multiplex ligation dependent probe amplification of the NIPBL gene from the DNA extracted from peripheral blood lymphocytes was negative, but microarray comparative genomic hybridisation on DNA from skin fibroblast showed a 0.13Mb deletion on chromosome 5p13. The deleted region includes exons 42–47 of the NIPBL gene. It is important to perform NIBPL mutation analysis on DNA from more than one tissue when testing for CdLS.
Background
Cornelia de Lange syndrome (CdLS) (OMIM 122470) is a rare genetic condition characterised by a broad spectrum of clinical abnormalities, prenatal and postnatal growth deficiency, major malformations, developmental delay and behavioural disturbances. The prevalence is estimated to be 1 in 10 000.1 CdLS is genetically heterogeneous. In ‘classical’ CdLS, de novo mutations in NIPBL are found in around 50% of cases. More recently, mutations in other genes encoding other structural or regulatory components of the cohesion ring, SMC1A, SMC3, HDAC8 and RAD21, have been found in patients with ‘mild’ CdLS.2 The phenotypic and genotypic correlations and wide spectrum of clinical features are well described in the literature.3–6 To the best of our knowledge, sacrococcygeal teratoma has never been reported in CdLS.7
Case presentation
A male infant at 36+5 weeks gestation, birth weight 2.78 kg, was born by elective section after spontaneous onset of labour. Antenatally, at 12 weeks gestation, there were concerns due to increased nuchal thickness, which resolved. At 20 weeks, a left-sided congenital diaphragmatic hernia and a sacrococcygeal teratoma were seen on ultrasound and further delineated on MRI scan (figure 1). There was no history of exposure to teratogens. There were no other concerns during the pregnancy.
Figure 1.
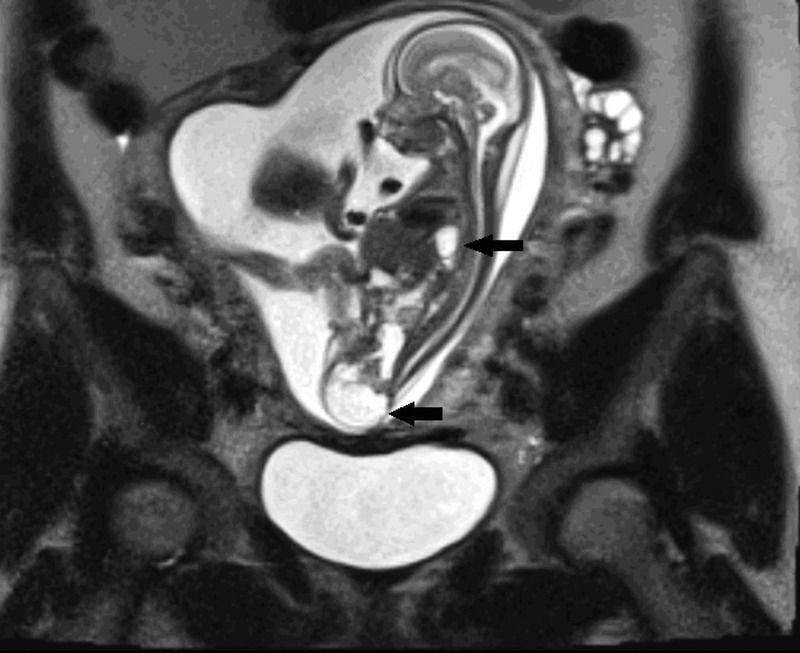
Fetal MRI sagittal T2 HASTE image showing sacrococcygeal teratoma and stomach bubble in the chest.
At birth, the infant was hypotonic and bradycardic, and remained cyanosed despite intubation and ventilation in 100% oxygen. Subsequent examination revealed microcephaly, broad forehead, hypertelorism, synophrys, long eyelashes, low set ears, thin upper lip, cleft palate, long philtrum, excess body hair and low anterior hairline. The upper limbs had mesomelic shortening of the forearms with bilateral ulnar hypoplasia and absence of third, fourth and fifth digits. The infant remained hypoxic despite aggressive ventilatory manoeuvres and, after developing a refractory bradycardia, was palliated at 4 h of age after discussion with his family. At postmortem, the lungs were hypoplastic (6 g). There was a left anterior diaphragmatic hernia; the spleen, stomach and distal small bowel had herniated into the right hemithorax through a defect behind the aorta. The sacrococcygeal teratoma was detected on antenatal scans. Postnatally, it was 90×85×55 mm in dimension and 510 g on postmortem examination. It had a variegated appearance with solid and cystic areas on cross section. Histology demonstrated an admixture of immature tissues of ectodermal, endodermal and mesodermal origin, consistent with an immature teratoma (type 2).
Investigations
Mutation analysis of the NIPBL gene was performed on DNA extracted from peripheral blood lymphocytes. No pathogenic mutations were detected within exons 2–47 of the NIPBL gene either on direct sequencing or by multiplex ligation dependent probe amplification (MLPA). Subsequent microarray comparative genomic hybridisation on DNA from skin fibroblast culture was performed using an Agilent whole genome 8×60 k oligo array (ISCA V.2.0), Genome Build GRCh37/UCSC assembly hg19. This showed a 0.13Mb deletion on chromosome 5p13. (arr5p13.2 (37 054 991–37 187 879)×1). The deleted region includes exons 42–47 of the NIPBL gene and a second gene, C5orf42. The presence of the NIPBL deletion in skin fibroblasts was confirmed by MLPA (MLPA kits P141-A2 and P142-A2 from MRC-Holland) (figure 2). The discrepant results between the blood and skin samples suggested somatic mosaicism for the NIPBL mutation.
Figure 2.

(A) Multiplex ligation dependent probe amplification (MLPA) analysis of NIPBL exons 1–47 in DNA from blood; (B) from skin fibroblasts, showing deletion of exons 42–47.
Discussion
Cornelia de Lange syndrome is a rare but well characterised condition. The important diagnostic features are distinctive facial features, limb abnormalities and growth restriction. The clinical features vary widely in individual cases. Presence of upper limb abnormalities has been correlated with the severity of CdLS.8 The phenotype of our patient was concordant with the classic type of CdLS. Diaphragmatic hernia (although rare as a feature of CdLS) has been documented previously. The defect in our case was unusual, as it was left anterior, and, in addition, there was herniation of abdominal contents into the right hemithorax through the defect posterior to the aorta, which severely compromised lung development. Sacrococcygeal teratomas have an incidence of 1 in 35 000–40 000 live-births and have a female predominance.9 Their prognosis is usually poor when diagnosis is made less than 30 weeks gestation and they are predominantly solid with a high degree of vascularisation (the latter resulting in high output cardiac failure). This in turn results in development of hydrops fetalis, preterm delivery or intrauterine demise.10 Although the occurrence of sacrococcygeal teratoma could be coincidental, we believe that it is possibly a rare extension of the wide clinical spectrum of CdLS. The discrepant results between the blood and skin sample in this case suggested somatic mosaicism for the NIBPL mutation. Biallelic mutations in C5orf42 are known to cause Joubert syndrome. Parents of children with Joubert syndrome are phenotypically normal so it is highly unlikely that loss of one copy of this gene contributed to the phenotype in this case. Recently, Huisman et al11 also suggested that somatic mosaicism for NIBPL gene is relatively frequent in clinically diagnosed cases of CdLS. This case highlights the importance of performing NIPBL mutation analysis on DNA from more than one tissue when testing for CdLS.
Learning points.
Cornelia de Lange syndrome (CdLS) is a rare genetic syndrome characterised by a broad spectrum of clinical abnormalities.
CdLS can present with sacrococcygeal teratoma in addition to multiple congenital abnormalities.
It is important to perform NIBPL mutation analysis on DNA from more than one tissue when testing for CdLS.
NIPBL mutation, even in a mosaic condition, can underlay a severe clinical CdLS presentation.
Footnotes
Contributors: NB: Writing manuscript, case design, literature review. AF: case design, writing manuscript. MS: case design, literature review, writing manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Krantz ID, McCallum J, DeScipio C. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004;36:631–5. 10.1038/ng1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramos FJ, Puisac B, Baquero-Montoya C et al. . Clinical utility gene card for: Cornelia de Lange syndrome. Eur J Hum Genet 2014. e1–e4; 10.1038/ejhg.2014.270 10.1038/ejhg.2014.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson L, Kline AD, Barr MA et al. . Cornelia de Lange syndrome: a case review of 310 individuals. Am J Med Genet 1993;47:940–6. 10.1002/ajmg.1320470703 [DOI] [PubMed] [Google Scholar]
- 4.de León FC, Gordillo-Domínguez LF, González-Carranza V et al. . Brachmann-Cornelia de Lange syndrome with a papilloma of the choroid plexus: analyses of molecular genetic characteristics of the patient and the tumor. A single-case study. Childs Nerv Syst 2015;31:141–6. 10.1007/s00381-014-2504-6 [DOI] [PubMed] [Google Scholar]
- 5.Oliver C, Bedeschi MF, Blagowidow N et al. . Cornelia de Lange syndrome: extending the physical and psychological phenotype. Am J Med Genet A 2010;152A:1127–35. 10.1002/ajmg.a.33363 [DOI] [PubMed] [Google Scholar]
- 6.Schrier SA, Sherer I, Deardorff MA et al. . Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am J Med Genet A 2011;155A:3007–24. 10.1002/ajmg.a.34329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dundar M, Uzak A, Erdogan M et al. . Partial trisomy 3q in a child with sacrococcygeal teratoma and Cornelia de Lange syndrome phenotype. Genet Couns 2011;22:199–205. [PubMed] [Google Scholar]
- 8.Allanson JE, Hennekam RC, Ireland M. De Lange syndrome: subjective and objective comparison of the classical and mild phenotypes. J Med Genet 1997;34:645–50. 10.1136/jmg.34.8.645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabra HO, Jesudason EC, McDowell HP et al. . Sacrococcygeal teratoma—a 25- year experience in a UK regional centre. J Pediatr Surg 2006;41:1513–16. 10.1016/j.jpedsurg.2006.05.019 [DOI] [PubMed] [Google Scholar]
- 10.Hedrick HL, Flake AW, Crombleholme TM et al. . Sacrococcygeal teratoma: prenatal assessment, fetal intervention, and outcome. J Pediatr Surg 2004;39:430–8. 10.1016/j.jpedsurg.2003.11.005 [DOI] [PubMed] [Google Scholar]
- 11.Huisman SA, Redeker EJ, Maas SM et al. . High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet 2013;50:339–44. 10.1136/jmedgenet-2012-101477 [DOI] [PubMed] [Google Scholar]