

Abstract
(E)-4-(3,5-dimethoxystyryl)phenyl acetate (Cmpd1) is a resveratrol analog that preferentially inhibits glioma, breast, and pancreatic cancer cell growth, with IC50 values of 6–19 μM. Notably, the human U251MG glioblastoma tumor line is the most sensitive, with an IC50 of 6.7 μM, compared with normal fibroblasts, which have an IC50 > 20 μM. Treatment of U251MG cells that harbor aberrantly active signal transducer and activator of transcription (Stat) 3 with Cmpd1 suppresses Stat3 tyrosine705 phosphorylation in a dose-dependent manner in parallel with the induction of pserine727 Stat3 and extracellular signal-regulated kinase/mitogen-activated protein kinase 1/2 (pErk1/2MAPK). Inhibition of pErk1/2MAPK induction by the mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] blocked both the pserine727 Stat3 induction and ptyrosine705 Stat3 suppression by Cmpd1, indicating dependency on the mitogen-activated protein/extracellular signal-regulated kinase kinase–Erk1/2MAPK pathway for Cmpd1-induced modulation of Stat3 signaling. Cmpd1 also blocked epidermal growth factor–stimulated pStat1 induction, whereas upregulating pSrc, pAkt, p-p38, pHeat shock protein 27, and pmammalian target of rapamycin levels. However, pJanus kinase 2 and pEpidermal growth factor receptor levels were not significantly altered. Treatment of U251MG cells with Cmpd1 reduced in vitro colony formation, induced cell cycle arrest in the G2/M phase and cleavage of caspases 3, 8, and 9 and poly(ADP ribose) polymerase, and suppressed survivin, myeloid cell leukemia 1, Bcl-xL, cyclin D1, and cyclin B1 expression. Taken together, these data identify a novel mechanism for the inhibition of Stat3 signaling by a resveratrol analog and suggest that the preferential growth inhibitory effects of Cmp1 occur in part by Erk1/2MAPK-dependent modulation of constitutively active Stat3.
Introduction
Resveratrol is a stilbenoid phytoalexin present in red grapes, red wine, peanuts, and other plant sources. Numerous studies in cardiovascular, neurologic, immune regulation, and cancer chemoprevention models and anticancer efficacy evaluations have all shown significant biologic activity in vitro and in vivo for resveratrol and have highlighted the potential benefits of this agent in diverse human diseases (Jang et al., 1997; Rimando et al., 2002; Baur and Sinclair, 2006; Pervaiz and Holme, 2009). In both chemo-preventative and anticancer efficacy studies using models of solid and hematologic malignancies, resveratrol inhibited tumor cell proliferation, survival, and invasion, as well as tumor angiogenesis and tumor formation (Jang et al., 1997; Baur and Sinclair, 2006; Shankar et al., 2007; Pervaiz and Holme, 2009). It has been well recognized that the multitude of effects and the notable health benefits of resveratrol are due to its activities on multiple targets and processes. Among others, resveratrol affects cyclooxygenase (COX)-1 and COX-2, NAD+-dependent histone deacetylase sirtuin 1 (SIRT1), quinone reductase (QR) 2, and ribonucleotide reductase enzymes and extracellular signal-regulated kinase/mitogen-activated protein kinase 1/2 (Erk1/2MAPK), Src, protein kinase C, nuclear factor B, and phosphoinositide 3-kinase signaling pathways and alters DNA synthesis (Yu et al., 2001; Jeong et al., 2004; Fröjdö et al., 2007; Pirola and Fröjdö, 2008; Pervaiz and Holme, 2009; Calamini et al., 2010). Despite the clinical and anticancer potential of resveratrol, its development for clinical application has been hampered by its low potency and limited bioavailability (Jang et al., 1997; Baur and Sinclair, 2006; Shankar et al., 2007). Efforts have therefore been placed on discovering analogs that have enhanced potency and appreciable bioavailability suitable for clinical application.
Previous reports also showed that resveratrol suppresses signal transducer and activator of transcription (Stat) 3 signaling via the inhibition of Src or Janus kinase (Jak) 2 induction, thereby inducing growth inhibitory and apoptotic effects against cultured human breast, prostate, and pancreatic cancer cells and the v-Src–transformed mouse fibroblasts and malignant natural killer cells (Kotha et al., 2006; Quoc Trung et al., 2013). The Stat family of cytoplasmic transcription factors is normally activated transiently via Tyr phosphorylation by Jaks, Src, and growth factor receptor Tyr kinases. Phosphorylation leads to Stat/Stat dimerization, nuclear translocation, and gene transcription that promote cell growth and proliferation, differentiation, inflammation, and other physiologic responses to growth factors and cytokines (Darnell, 1997; Bromberg, 2000). In contrast, constitutively active Stat3 is prevalent in many human cancers, including glioblastoma, breast, and pancreatic cancers and represents a critical mediator of malignant transformation and tumor progression (Bowman et al., 2000; Miklossy et al., 2013). Aberrantly active Stat3 promotes tumorigenesis in part via dysregulation of gene expression, leading to uncontrolled growth and survival of cells, enhanced tumor angiogenesis, and metastasis (Bowman et al., 2000; Bromberg and Darnell, 2000; Turkson and Jove, 2000; Turkson, 2004; Yu and Jove, 2004; Miklossy et al., 2013) and the repression of tumor immune surveillance (Wang et al., 2004; Yu and Jove, 2004).
Although evidence suggests the inhibition of Stat3 signaling represents one of the mechanisms for the antitumor cell response to resveratrol (Kotha et al., 2006; Quoc Trung et al., 2013), the mechanism of resveratrol-induced Stat3 inhibition is less understood. Initial reports suggested the inhibition of Stat3 activity might occur indirectly through inhibition of Src and Jaks induction (Kotha et al., 2006; Quoc Trung et al., 2013). Several resveratrol analogs were screened for improved activity against human tumor cells that harbor aberrantly active Stat3, leading to the identification of one potentially more active agent. The analog (E)-4-(3,5-dimethoxystyryl)phenyl acetate (Cmpd1) more potently inhibited constitutive Stat3 tyrosine705 phosphorylation while enhancing Stat3 serine727 phosphorylation in malignant cells. These events were associated with the induction of pErk1/2MAPK. Although the inhibition of mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK)–Erk1/2MAPK induction suppressed Cmpd1-induced pserine727 Stat3, it reversed the inhibitory effects of Cmpd1 on ptyrosine705 Stat3, suggesting that Erk1/2MAPK induction is required for Cmpd1-dependent modulation of both Stat3 serine727 and tyrosine705 phosphorylation events. Cmpd1 further induced pSrc, pmammalian target of rapamycin (mTOR), pAkt, pHsp27, and p-p38; suppressed pStat1; and had no significant effects on pJanus kinase (JAK) 2 and pepidermal growth factor receptor (EGFR). The activities of Cmpd1 resulted in the loss of cell viability, growth, and survival of human glioma, breast, or pancreatic cancer cells and induced cell cycle block at the G2/M phase and apoptosis with human glioma cells in culture.
Materials and Methods
Cell Lines and Reagents.
The human glioma lines U251MG and SF-295 were obtained from the Division of Cancer Treatment and Diagnosis, Tumor Repository of the National Cancer Institute (Frederick, MD) and cultured in Roswell Park Memorial Institute medium containing 10% fetal bovine serum (FBS) or Dulbecco’s modified Eagle’s medium containing 10% FBS and supplemented with 1% nonessential amino acids (Corning Inc., Corning, NY). The human breast (MDA-MB-231 and MCF-7) and pancreatic (Panc-1) cancer cells and the normal mouse fibroblasts (NIH3T3) and their v-Src–transformed (NIH3T3/v-Src) or v-Ras–transformed (NIH3T3/v-Ras) counterparts have all been previously reported (Kotha et al., 2006; Zhao et al., 2010; Zhang et al., 2012). These cells were grown in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated FBS. Resveratrol was purchased from Sigma-Aldrich (St. Louis, MO). Pterostilbene and PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] were purchased from Cayman Chemical (Ann Arbor, MI). Cmpd1, Cmpd2, and Cmpd3 have all been previously described (Sun et al., 2010; Kondratyuk et al., 2011). Except for anti–β-actin, glyceraldehyde 3-phosphate dehydrogenase (Santa Cruz Biotechnology, Inc., Dallas, TX), and cyclin D1 (SPM587) (Novus Biologicals, Littleton, CO), antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Recombinant human epidermal growth factor (EGF) was purchased from Invitrogen/Life Technologies (Carlsbad, CA). Where appropriate, cells were stimulated with recombinant human EGF (10 ng/ml) for 12 minutes. PD98059, SB202190 [4-[4-(4-fluorophenyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]phenol], and LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride] (Calbiochem, EMD Millipore, Billerica, MA), wortmannin and staurosporine (Sigma-Aldrich), and dasatinib (ChemieTEK, Indianapolis, IN) were obtained from the designated sources.
CyQuant Cell Proliferation Assay.
The CyQuant cell proliferation assay (Invitrogen/Life Technologies) was used following the manufacturer’s instructions to evaluate the biologic activities of compounds as previously reported (Zhang et al., 2012). Relative cell viability of the treated cells was normalized to the dimethyl sulfoxide–treated control cells.
Trypan Blue Cell Counting.
Cells in culture were treated once with compounds at increasing concentrations for 0–72 hours. Treated and untreated cells were harvested every 24 hours, and viable cells were stained with Trypan blue dye and counted under a microscope with a hemocytometer.
Colony Survival Assay.
Five hundred single cells were seeded on a 6-cm dish, cultured for 24 hours, and treated once with Cmpd1. Cells were allowed to grow for 14–21 days until colonies were visible, which were stained with crystal violet for 1–3 hours, imaged, and counted.
Electrospray Ionization Tandem Mass Spectrometry Analysis of Cmpd1 and Pterostilbene.
Studies were performed by the Analytical Biochemistry Shared Resource at the University of Hawaii Cancer Center. The pterostilbene standard [10 μM in methanol (MeOH)] was serially diluted in MeOH/H2O (1:1 v/v) to calibrate the system. Cell lysates were extracted with ethyl acetate. The organic phase was dried under nitrogen and reconstituted in MeOH/H2O for liquid chromatography–mass spectrometry analysis. Liquid chromatography–mass spectrometry analysis was carried out on a Q-Exactive mass spectrometer coupled to an HTC autosampler and Accela pump (all from Thermo Fisher Scientific, Waltham, MA). Twenty-five microliters of the standard or extracts were injected onto an Ascentis Express C18 column (15 × 3 mm; 2.7 µm; Supelco, Sigma-Aldrich). Chromatographic separation was achieved with a linear gradient consisting of (A) 0.1% formic acid in water, (B) 0.1% formic acid in MeOH, and (C) 0.1% formic acid in acetonitrile at 0.3 ml/min; A/B/C= 80:10:10 to 20:40:40 in 10 minutes and keeping at 20:40:40 for another 10 minutes. Mass spectrometric measurements were performed in full-scan electrospray ionization (ESI) mode with positive and negative switching. Quantitation of the pterostilbene ([M+H]+ 257.11722; [M−H]− 255.10212) and acetyl-pterostilbene ([M+H]+ 299.12779; [M−H]− 297.11268) was performed with Xcalibur software (Thermo Fisher Scientific) using a 5-ppm window from the exact masses.
Western Blot Analysis.
Immunoblotting analysis was performed as previously reported (Kotha et al., 2006; Zhang et al., 2010, 2012). Briefly, whole-cell lysates were prepared using ice-cold radioimmunoprecipitation assay buffer containing phenylmethylsulfonyl fluoride, dithiothreitol, sodium orthovanadate (Na3VO4), and aprotinin, leupeptin, and antipain. Lysates were spun down at 13,300 rpm for 20 minutes at 4°C using a microcentrifuge, and the supernatants were collected. Protein concentrations were determined by the Bradford assay (Pierce Biotechnology, Rockford, IL). Equivalent amounts of total protein were separated by SDS-PAGE and transferred to a nitrocellulose or polyvinylidene fluoride membrane (BioRad, Hercules, CA). The membranes were blocked for 1 hour with 5% milk in Tris-buffered saline with Tween 20, incubated with primary antibody overnight, washed three times, and then incubated with secondary antibody at room temperature for 1 hour. Membranes were then treated with an enhanced chemiluminescence reagent (BioRad) for imaging analysis.
Immunoprecipitation Assay.
Immunoprecipitation/SDS-PAGE analyses were performed as previously reported (Zhang et al., 2012). Briefly, following treatment, cells were washed with 1× phosphate-buffered saline (PBS) followed by 1× 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.05% Nonidet P-40 and 1× low-salt HEPES buffer [10 mM HEPES (pH 7.8), 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol]. Cells were lysed with 1× low-salt buffer containing 10% Nonidet P-40, and cell lysates were spun down at 13,300 rpm for 20 minutes at 4°C to collect the supernatants. Lysates were incubated with protein A/G beads (Santa Cruz Biotechnology, Inc.) in the cold room on an orbital rocker for 30 minutes and subsequently centrifuged at 3000 rpm at 4°C for 3 minutes. The supernatants were collected and incubated with the primary antibody for 12 hours overnight in a cold room and then with protein A/G beads for 1 hour. Subsequently, samples were spun down at 3000 rpm at 4°C for 3 minutes to collect the bead pellets, which were washed four times with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.05% Nonidet P-40 added to 30 µl of 1× SDS loading buffer and boiled for 5 minutes. Samples were subjected to SDS-PAGE and immunoblotting analysis.
Cell Cycle Analysis by Flow Cytometry.
Following treatment with Cmpd1 for 24 or 72 hours, cells in culture in 6-cm plates were trypsinized, centrifuged at 1000 rpm at 4°C for 10 minutes, and washed with 1× PBS. Cells were fixed with fixation buffer (70% ethanol and 3% FBS in PBS) overnight at −20°C. Subsequently, cells were washed two times with 1× PBS and once with propidium iodide/RNase staining buffer (BD Biosciences, San Jose, CA) and stained by incubating with propidium iodide/RNase buffer at room temperature for 15 minutes. Samples were placed on ice and analyzed using a FACS Calibur (BD Biosciences). Data were collected with CellQuest Pro (BD Biosciences) and FlowJo (Ashland, OR).
Statistical Analysis.
Statistical analysis was performed on the mean values using Prism (GraphPad Software, Inc., La Jolla, CA). The significance of differences between groups was determined by the paired t test at *P < 0.01.
Results
Resveratrol Analogs Cmpd1, Cmpd2, and Cmpd3 More Potently Inhibit Human Tumor Cell Growth.
Initial screening of 98 resveratrol analogs (unpublished data) identified three analogs, Cmpd1, Cmpd2, and Cmpd3 (Fig. 1A), which showed stronger activity against the viability of the human glioma U251MG and breast MDA-MB-231 cancer cells than resveratrol. Of these, Cmpd1 inhibited tumor cell viability the greatest (Fig. 1B).
Fig. 1.

Resveratrol, analogs Cmpd1, Cmpd2, and Cmpd3, and pterostilbene differentially suppress tumor cell growth and survival. (A) Structures of Cmpd1, Cmpd2, Cmpd3, resveratrol (Res), and pterostilbene (PTE). CyQuant cell proliferation assay for the effects of 72-hour treatment with (B) 0–20 µM (i) Cmpd1, (ii) Cmpd2, and (iii) Cmpd3 or (C) (i) 0–80 µM Res or (ii) 0–50 µM PTE on human glioma (U251MG and SF295), breast (MDA-MB-231), or pancreatic (Panc-1) cancer cells harboring activated Stat3 or breast cancer (MCF7) cells and normal mouse fibroblasts (NIH3T3) that do not. Viable cell numbers as a percentage of control are plotted. U251MG cells (D) or normal NIH3T3 fibroblasts (E) growing in culture were treated once with 0–20 µM Cmpd1 for 0–72 hours, and viable cells were counted at the indicated times by Trypan blue exclusion/phase-contrast microscopy. Viable cell numbers are plotted. (F) Cultured human glioma (U251MG and SF295), breast (MDA-MB-231 and MCF7), and pancreatic (Panc-1) cancer cells and normal NIH3T3 mouse fibroblasts were seeded as single cells, treated once with 0–20 μM Cmpd1, and allowed to culture until large colonies were visible, which were stained with crystal violet, counted, and plotted. Values are mean ± S.D.; n = 3–4. *P < 0.01. DMSO, dimethyl sulfoxide.
Human glioma (U251MG and SF295), breast (MDA-MB-231 and MCF7), and pancreatic (Panc-1) cancer cells and normal mouse fibroblasts (NIH3T3) in culture were treated once with 0–20 µM of Cmpd1, Cmpd2, or Cmpd3 for 72 hours and processed for the CyQuant cell proliferation assay to determine viable cell numbers. Cmpd1 showed a dose-dependent inhibition of cell viability that is strongest against U251MG, moderate against MCF7, Panc-1, and MDA-MB-231, and weak against SF295 cells, with IC50 values of 6.7, 10.8, 18.1, 18.5, and >20 µM, respectively (Fig. 1Bi; Table 1). By contrast, the inhibitory effect of Cmpd1 against normal NIH3T3 was insignificant, except at 20 µM, with an IC50 > 20 µM (Fig. 1Bi; Table 1). Cmpd2 showed moderate activity against MDA-MB-231 and no effect on U251MG up to 20 µM (Fig. 1Bii), whereas Cmpd3 was weak against both cell lines at concentrations up to 20 µM (Fig. 1Biii). Similar Cyquant cell proliferation assay results obtained with U251MG cells showed only a moderate effect with resveratrol (Fig. 1Ci), with an IC50 > 50 µM, whereas its structural analog, pterostilbene (Rimando et al., 2002; Pan et al., 2008) induced a strong and dose-dependent response (Fig. 1Cii), with an IC50 of 6.3 µM. From literature reports and our studies, the activities observed for Cmpd1 are significantly superior to that of resveratrol against the same cell lines (Fig. 1, Bi and Ci; Table 1), whereas Cmpd1 and pterostilbene are equally active against U251MG cell growth.
TABLE 1.
Inhibitory constants on the effects of Cmpd1 or resveratrol on the growth of human tumor and normal cells
Trypan blue exclusion/phase-contrast microscopy for viable cells treated once with Cmpd1 (0–20 µM) for 0–72 hours confirmed the dose-dependent suppression of human glioma U251MG cell growth (Fig. 1D) compared with a marginal decline in cell growth induced against normal NIH3T3 (Fig. 1E). In the clonogenic assay, one time treatment with Cmpd1 (0–20 µM) of single-cell cultures for 14–21 days significantly suppressed colony numbers for U251MG, SF-295, and MCF7 while only moderately affecting that of Panc-1 cells, except at a concentration of 20 µM (Fig. 1F). Unlike the limited effects on cell proliferation and cell growth (Fig. 1, B and E), treatment of single-cell cultures of NIH3T3 with Cmpd1 in the clonogenic assay reduced the colony numbers (Fig. 1F). However, we note the larger relative effects against U251MG, SF295, and MCF7 colonies at 15 and 20 µM compared with that against NIH3T3, which suggest higher tumor-cell sensitivity. We infer that the reduced colony numbers of normal NIH3T3 cells could be due to small colony sizes that were the result of apparent slowness in the growth rate in response to Cmpd1 (Fig. 1E). Small colonies may be excluded from staining and quantification (NIH3T3) (Supplemental Fig. 1).
Cmpd1 Inhibits Stat3 Tyrosine705 Phosphorylation While Upregulating Stat3 Serine727 Phosphorylation.
A previous report showed that resveratrol treatment suppresses Stat3 tyrosine705 phosphorylation in v-Src–transformed mouse fibroblasts (NIH3T3/v-Src) (Kotha et al., 2006). The most sensitive U251MG cell line and NIH3T3/v-Src fibroblasts, both of which harbor constitutively activated Stat3, were used to investigate the effects of Cmpd1 on Stat3 signaling. Cells (U251MG and NIH3T3/v-Src) in culture were treated with 0–20 µM Cmpd1 for 3 hours or 15 µM for 0–24 hours, and whole-cell lysates were prepared for immunoblotting analysis. Cmpd1 inhibited constitutive Stat3 tyrosine705 phosphorylation at 5 µM and higher (Fig. 2A; Supplemental Fig. 2A) and as early as 1 hour (Fig. 2B) in U251MG cells or at a later time in 10–24 hours in NIH3T3/v-Src fibroblasts (Supplemental Fig. 2A). Notable differences in the effectiveness and kinetics of inhibition of ptyrosine705 Stat3 were observed between the two cell lines. For U251MG cells, ptyrosine705 Stat3 inhibition by Cmpd1 was complete at 1–3 hours and was followed by a gradual recovery at 10–24 hours (Fig. 2B). By contrast, ptyrosine705 Stat3 inhibition by Cmpd1 in NIH3T3/v-Src fibroblasts was only moderate and occurred at a later time of 10–24 hours (Supplemental Fig. 2A).
Fig. 2.

Effects of Cmpd1, resveratrol, and pterostilbene on Stat3 tyrosine and serine phosphorylation and Stat3 acetylation in human glioma cells. Immunoblots of pYStat3, pSer727Stat3, Stat3, acetylated Stat3 (aStat3), and β-actin from whole-cell lysate preparation from the human glioma U251MG cells harboring aberrantly active Stat3 untreated or treated for (A) 3 hours with 0–20 µM Cmpd1, (B) 0–24 hours with 15 µM Cmpd1, (C) 0–24 hours with 20 µM resveratrol (Res), (D) 0–24 hours with 20 µM pterostilbene (PTE), or (E and F) 0–24 hours with 15 µM Cmpd1. The positions of the proteins in the gel are labeled. Bands corresponding to the phospho-Stat3 or acetylated Stat3 protein levels in the gel were quantified by ImageQuant and calculated as a percentage of control (dimethyl sulfoxide) relative to the total proteins and β-actin levels. Control lane (0) represents whole-cell lysates prepared from 0.025% dimethyl sulfoxide–treated cells. Data are representative of three independent determinations.
For comparison, 20 µM resveratrol mediated a time-dependent, progressive suppression of ptyrosine705 Stat3 that occurred at 1–10 hours, followed by a recovery at 24 hours in both U251MG and NIH3T3/v-Src lines (Fig. 2C; Supplemental Fig. 2B). These data indicate that unlike Cmpd1, resveratrol inhibits Stat3 tyrosine705 phosphorylation, with nearly similar kinetics and potencies in both U251MG and NIH3T3/v-Src lines. However, Cmpd1 shows improved inhibitory activity against ptyrosine705 Stat3, which reflects its structural modifications. Surprisingly, pterostilbene (20 µM) only moderately inhibited Stat3 tyrosine705 phosphorylation in U251MG cells, albeit with similar kinetics as Cmpd1 (Fig. 2D). Therefore, the subtle, acetoxy group change at the 4′ position between Cmpd1 and pterostilbene (Fig. 1A) is sufficient to alter the inhibitory activity against Stat3 tyrosine705 phosphorylation, despite the fact that both agents are equally active against U251MG cell growth (Fig. 1, Bi and Cii). These results suggest that ptyrosine705 Stat3 inhibition may only moderately contribute to the effects of pterostilbene against U251MG cell growth. On the other hand, ESI tandem mass spectrometry employed to qualitatively analyze the levels of Cmpd1 (acetyl pterostilbene) in triplicate cellular samples of Cmpd1 (15 µM, 1 hour) treated cells, relative to dimethyl sulfoxide–treated (control) cells, showed detectable levels of pterostilbene (retention time of 12.5 minutes) and minimal detection of acetyl pterostilbene (unpublished data), suggesting that by 1 hour, Cmpd1 has converted to and exists predominantly as pterostilbene in tumor cells. We hypothesize that the early, more potent effect of Cmpd1 on pYStat3 is likely due to the combined activities of both the acetylated and deacetylated forms.
Stat3 is phosphorylated on serine727, and this modification is associated with an increased transcriptional function (Wen et al., 1995). Unexpectedly, Stat3 serine727 phosphorylation was induced in U251MG cells in response to treatment with Cmpd1 as early as 1–3 hours and returned to near baseline levels by 10–24 hours (Fig. 2E). We note that the kinetics of the pserine727 Stat3 induction are parallel to the inhibition of ptyrosine705 Stat3 (Fig. 2B versus Fig. 2E). Studies have also shown that resveratrol directly or indirectly activates the cellular metabolism master regulator, 5′-adenosine monophosphate-activated protein kinase, and the protein deacetylase SIRT1 (Pervaiz and Holme, 2009; Calamini et al., 2010; Fullerton and Steinberg, 2010) and that the activities of SIRT1 leads to the suppression of Stat3 lysine685 acetylation (Jain et al., 1998). Immunoblotting analysis, however, showed no significant change in acetylated Stat3 in U251MG cells at 3 and 10 hours post-treatment with Cmpd1 (Fig. 2F) and no significant alteration in the expression of SIRT1 levels (unpublished data). To determine whether Cmpd1 could inhibit other Stat family members, mouse fibroblasts overexpressing the human EGFR (NIH3T3/hEGFR) were treated for 1–24 hours and stimulated with EGF and whole-cell lysate prepared and subjected to immunoblotting analysis. Pretreatment with Cmpd1 diminished both EGF-induced pY705Stat3 and pY701Stat1 (Supplemental Fig. 1C) with similar kinetics. Despite the inhibition of pY701Stat1, Cmpd1 did not show evidence of general cytotoxicity against NIH3T3 cells at 15–20 µM (Fig. 1, Bi and E).
Cmpd1 Induces Src, Erk1/2MAPK, Akt, Hsp27, p38, and mTOR Phosphorylation and Has No Effects on pJak2 or pEGFR Induction.
Resveratrol was previously reported to inhibit Stat3 tyrosine705 phosphorylation via inhibition of upstream kinases, including Src and Jak2 (Kotha et al., 2006; Quoc Trung et al., 2013). We asked whether Cmpd1 could similarly inhibit tyrosine kinases, such as pSrc, pJak2, or pEGFR. Surprisingly, pY416Src was strongly induced in U251MG cells in response to Cmpd1 (15 µM) at 30 minutes to 10 hours and downregulated by 24 hours, with no significant change in total protein levels (Fig. 3A). The enhanced pY416Src, which is a reflection of its autophosphorylation, suggests Src could not be the responsible target site for the inhibition of pY705Stat3 that is observed at 1–10 hours in Cmpd1-treated U251MG cells (Fig. 2B). However, the pattern of effect of Cmpd1 on Src was different in v-Src–transformed NIH3T3/v-Src fibroblasts, where pSrc levels were marginally induced at 30 minutes to 3 hours post-treatment with the compound and then suppressed or returned to baseline level at 10–24 hours (Supplemental Fig. 3A). The decrease in pSrc levels in NIH3T3/v-Src at 10–24 hours occurred with similar kinetics as the suppression of Stat3 tyrosine705 phosphorylation in the same cell line (Supplemental Fig. 2A), suggesting the two events may be causally related in the Cmpd1-treated NIH3T3/v-Src, as was similarly reported for the effects of resveratrol in the same cell line (Kotha et al., 2006). By comparison, treatment with 20 µM resveratrol induced an early (30 minutes) and sustained inhibition of pSrc in both the U251MG (Fig. 3B) and NIH3T3/v-Src cell lines (Supplemental Fig. 3B). The early and sustained decline in pSrc levels correlates with the inhibition of ptyrosine705 Stat3 at 1 hour and later in both cell lines (Fig. 2C) as was previously reported (Kotha et al., 2006). Data herein therefore reveal differences between Cmpd1 and resveratrol, with regards to their respective effects on pSrc induction in U251MG and NIH3T3/v-Src lines and of the possible mechanisms for their inhibitory effects on Stat3 tyrosine705 phosphorylation.
Fig. 3.

Cmpd1 promotes phospho-Src, -Erk1/2MAPK, -Akt, -mTOR, -p38, and -Hsp27 induction and has no significant effects on Jak2 and EGFR in glioma U251MG cells. Immunoblotting analysis of whole-cell lysate preparation from the human glioma U251MG cells untreated or treated with (A and D–J) 15 µM Cmpd1, (B) 20 µM resveratrol (Res), or (C) 100 nM dasatinib (Das) for 0–24 hours and probing for pY416Src, Src, pYStat3, Stat3, pJak2, Jak2, pY1173EGFR, EGFR, pErk1/2MAPK, Erk1/2MAPK, p-p38, p38, pmTOR, mTOR, pAkt, Akt, pHsp27, Hsp27, β-actin, or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The positions of proteins in the gel are labeled. Bands corresponding to the phospho-protein levels in the gel were quantified by ImageQuant and calculated as a percentage of control (dimethyl sulfoxide) relative to the total proteins and β-actin or GAPDH levels. Control lane (0) represents whole-cell lysates prepared from 0.025% dimethyl sulfoxide–treated cells. Data are representative of three independent determinations.
In light of the observation that Src may not be responsible for Cmpd1-mediated inhibition of ptyrosine705 Stat3, we sought to investigate if Src activity could contribute to promoting Stat3 tyrosine705 phosphorylation in U251MG cells by treating cells with the small-molecule Src inhibitor dasatinib (100 nM). Immunoblotting analysis showed complete suppression of both pSrc and ptyrosine705 Stat3 levels (Fig. 3C), suggesting that Src activity promotes pYStat3 induction in the glioma U251MG cell line. However, ptyrosine705 Stat3 levels rebounded at 18–24 hours, despite the fact that pSrc levels remained attenuated, suggesting that other tyrosine kinases contribute to Stat3 tyrosine705 phosphorylation and act as compensatory mechanisms. Tyrosine kinases responsible for Stat3 tyrosine705 phosphorylation include Jaks and EGFR. Immunoblotting analysis showed no change in pJak2 and an apparent moderate induction of pEGFR at 30 minutes in U251MG cells treated with Cmpd1 (15 µM) (Fig. 3, D and E). These results together suggest that Jaks, EGFR, and Src are not involved in the mechanisms leading to the reduction of ptyrosine705 Stat3 levels in Cmpd1-treated U251MG cells. By contrast, resveratrol inhibits Stat3 tyrosine705 phosphorylation, in part, through its suppressive effects on Src activity (Kotha et al., 2006). Treatment with Cmpd1 (15 µM) also promoted early (30 minutes to 3 hours) induction of pErk1/2MAPK, p-p38, pmTOR, and pAkt, whereas pHsp27 was moderately induced at a later (24 hours) time (Fig. 3, F–J), indicating that Cmpd1 also triggers the induction of multiple cellular signaling pathways.
Erk1/2MAPK Activation by Cmpd1 Leads to Pserine727 Stat3 Induction and the Inhibition of Stat3 Tyrosine705 Phosphorylation.
It was previously reported that Erk1/2MAPK induction led to the suppression of Stat3 tyrosine705 phosphorylation (Jain et al., 1998). Of significance, Cmpd1-mediated induction of pErk1/2MAPK at 30 minutes (Fig. 3F) occurred prior to Stat3 serine727 phosphorylation at 1 hour (Fig. 2E), raising the possibility that pErk1/2MAPK could be responsible for inducing Stat3 serine727 phosphorylation in response to Cmpd1. To assess whether pErk1/2MAPK induction has a causal role in the parallel ptyrosine705 Stat3 inhibition and pserine727 Stat3 induction, U251MG cells were treated with the MEK inhibitor PD98059 for 30 minutes prior to treating the cells with Cmpd1 for 15 or 30 minutes and preparing whole-cell lysates for immunoblotting analysis. The results showed parallel suppression of ptyrosine705 Stat3 by Cmpd1 (Fig. 4A, lane 2, pY705Stat3), together with the induction of both pErk1/2MAPK and pserine727 Stat3 in the absence of PD98059 (Fig. 4A, lane 2; and Fig. 4B, lanes 2 and 3; pErk1/2MAPK and pS727Stat3), which were all prevented when cells were pretreated with the MEK inhibitor PD98059 (Fig. 4A, lane 4; and Fig. 4B, lanes 6 and 7; pY705Stat3, pS727Stat3, and pErk1/2MAPK). The MEK inhibitor alone also strongly suppressed the background pErk1/2MAPK levels and induced ptyrosine705 Stat3 (Fig. 4A, lane 3; and Fig. 4B, lanes 4 and 5; pErk1/2MAPK and pY705Stat3). Taken together, these results indicate that Cmpd1 promotes pErk1/2MAPK induction that is sensitive to MEK inhibitor, which in turn phosphorylates serine727 Stat3, and these events are associated with the suppression of ptyrosine705 Stat3.
Fig. 4.

Cmpd1-mediated inhibition of pYStat3 is associated with Erk1/2MAPK induction, which promotes pS727Stat3 and is reversed by the MEK inhibitor PD98059. Immunoblotting analysis of whole-cell lysate preparation from U251MG cells untreated (–) or treated with 15 µM Cmpd1 for 15 or 30 minutes following pretreatment with or without 50 µM PD98059 for the indicated times and probing for pYStat3, pS727Stat3, Stat3, pErk1/2MAPK, Erk1/2MAPK, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The positions of the proteins in the gel are labeled. Bands corresponding to the phospho-protein levels in the gel were quantified by ImageQuant and calculated as a percentage of control (dimethyl sulfoxide) relative to the total proteins and GAPDH levels. Control lane (–) represents whole-cell lysates prepared from 0.025% dimethyl sulfoxide–treated cells. Data are representative of three independent determinations.
Although treatment with the MEK inhibitor eliminated the background pErk1/2 levels (Fig. 4A, lane 3; Fig. 4B, lanes 4 and 5), it failed to completely abrogate pErk1/2 induction in the presence of Cmpd1 (Fig. 4A, lane 4; Fig. 4B, lanes 6 and 7), leaving a residual pErk1/2 activation that may be MEK independent. To determine whether other pathways are involved in Cmpd1-mediated pErk1/2 induction, we pretreated U251MG cells with the inhibitors of p38 (SB202190), Src (dasatinib), mTOR (rapamycin), PI3-kinase/Akt (LY294002), or the general protein kinase inhibitor staurosporine prior to treatment with Cmpd1, and whole-cell lysates were prepared for immunoblotting analysis. As noted, pretreatment with PD98059 substantially, albeit not completely, suppressed Cmpd1-induced pErk1/2 levels (Supplemental Fig. 4A). Pretreatment with the other inhibitors, on the other hand, had no suppressive effects on Cmpd1-induced pErk1/2 levels (Supplemental Fig. 4A). Pterostilbene similarly promoted pErk1/2 induction, which was sensitive to the MEK inhibitor PD98059 but not to the inhibitors of p38, PI3-kinase, Src, mTOR, or general protein kinase inhibitors (Supplemental Fig. 4B). We sought to further investigate the induction of pErk1/2 by Cmpd1 in the context of v-Ras transformation (NIH3T3/v-Ras), in which Erk1/2 signaling is under the regulation of oncogenic Ras, compared with normal cells (NIH3T3). Cells were pretreated for 1 hour with PD98059 (20 µM) prior to treatment with Cmpd1 (15 µM, 30 minutes). Immunoblotting analysis of whole-cell lysates showed minimal to moderate modulation by Cmpd1 of pY705Stat3, pS727Stat3, or pErk1/2 in both cell lines (Supplemental Fig. 5). The results further showed the suppression by PD98059 of the resting levels of pErk1/2 in both lines and the background levels of pS727Stat3 in the transformed NIH3T3/v-Ras line (Supplemental Fig. 5). These results together indicate that Cmpd1 is unable to substantially promote pErk1/2 and pS727Stat3 induction in normal fibroblasts and in transformed cells where pErk1/2 is constitutively induced by the upstream oncogenic Ras.
Cmpd1 Treatment Suppresses Cell Cycle Regulatory and Apoptotic Genes, Arrests the Cell Cycle, and Induces Apoptosis of Glioma Cells.
With respect to the importance of constitutively active Stat3 in tumor cell growth and survival (Miklossy et al., 2013), inhibition of Stat3 activity in tumor cells leads to induction of cell cycle arrest and apoptosis by downregulating Stat3-regulated genes (Zhang et al., 2010, 2012). Resveratrol was previously shown to induce cell cycle arrest at the G0/G1 or S phase (Kotha et al., 2006). Western blotting analysis shows 24-hour treatment with Cmpd1 leads to suppression of survivin, myeloid cell leukemia 1, Bcl-xL, cyclin D1, and cyclin B1 expression in U251MG cells (Fig. 5A). Analysis by flow cytometry revealed that similar treatment with Cmpd1 induced cell cycle arrest at the G2/M phase as early as 24 hours and at 72 hours post-treatment, which is associated with decreased populations at the G0/G1 and/or S phases (Fig. 5B, upper panel). By contrast, treatment with Cmpd1 only moderately induced a temporary G2/M block in normal NIH3T3 fibroblasts at 24 hours, which was reversed by 72 hours (Fig. 5B, lower panel). Moreover, Cmpd1 treatment induced the cleavage of caspases 3, 8, and 9 and poly(ADP ribose) polymerase at 48 hours post-treatment (Fig. 5C).
Fig. 5.

Cmpd1 suppresses the expression of cell cycle and apoptotic regulatory genes, inhibits the cell cycle, and induces caspases and poly(ADP ribose) polymerase (PARP) cleavage. Immunoblotting analysis of whole-cell lysate preparation from U251MG cells untreated (0) or treated with 15 µM Cmpd1 for 24 or 48 hours and probing for (A) survivin, myeloid cell leukemia 1, Bcl-xL, cyclin D1, cyclin B1, and β-actin or (C) caspase 8, 9, or 3, PARP, and β-actin. (B) Cell cycle distribution analysis of U251MG or NIH3T3 cells treated or untreated (dimethyl sulfoxide) with 15 µM Cmpd1 for 24 or 72 hours, processed by propidium iodide staining, and analyzed by flow cytometry for DNA content, which is plotted. The positions of the proteins in the gel are labeled. Bands corresponding to the phospho-protein levels in the gel were quantified by ImageQuant and calculated as a percentage of control (dimethyl sulfoxide) relative to the total proteins and β-actin or glyceraldehyde 3-phosphate dehydrogenase levels. Control lane (0) represents whole-cell lysates prepared from 0.025% dimethyl sulfoxide–treated cells. Data are representative of three independent determinations. Values are mean ± S.D.; n = 3–4. *P < 0.01.
Discussion
First reported as compound 55 (Sun et al., 2010), Cmpd1 was subsequently evaluated as compound 86 and found to exhibit no significant activity in QR1 activation, QR2 inhibition, nitric oxide production, aromatase, nuclear factor κB, COX-1/2, antiproliferative activity, and other in vitro assays (Kondratyuk et al., 2011). In the present study, however, it is notable that this analog is more potent than resveratrol in suppressing the viability and growth of the human glioma, breast, and pancreatic cancer cells, with a preferential effect against U251MG glioma cells, while only weakly affecting normal NIH3T3 at the highest dose tested. The cell growth inhibition, cell cycle block at the G2/M phase, and apoptosis induced by Cmpd1 in U251MG cells all parallel the reported inhibitory effects of resveratrol in human breast, pancreatic, prostate, rhabdomyosarcoma, and other tumor cells (Hadi et al., 2000; Chow et al., 2005; Kotha et al., 2006; Meeran and Katiyar, 2008; Quoc Trung et al., 2013). Cmpd1-induced biologic responses are preceded by decreases in the expression of cell cycle regulatory and apoptotic genes. Interestingly, Cmpd1 and pterostilbene showed a similar inhibitory potency against glioma cell growth, suggesting the acetoxy group change at the 4′ position has a minimal impact on in vitro activity. Given the susceptibility of ester groups to hydrolysis by intracellular esterases (Fukami and Yokoi, 2012), the 4′ acetoxy group on Cmpd1 appears to be hydrolyzed in tumor cells, thereby generating pterostilbene from Cmpd1 as ESI–tandem mass spectrometry studies show. It is proposed that the rapid modulation of Stat3 in response to Cmpd1 is likely the combined activities of the acetylated and deacetylated forms.
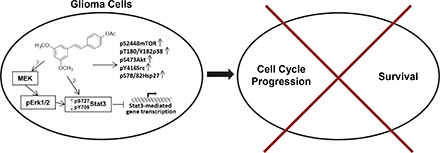
The resveratrol analog Cmpd1 promotes early inhibition of aberrantly active Stat3 in tumor cells, which would be expected to contribute to the growth inhibitory effects against U251MG, MDA-MB-231, and Panc-1 cells (Kotha et al., 2006; Miklossy et al., 2013). However, the present studies reveal differences between Cmpd1 and resveratrol in the mechanisms leading to the inhibition of Stat3 signaling in tumor cells. In contrast to inhibition of pSrc or Jak2 induction by resveratrol (Kotha et al., 2006; Quoc Trung et al., 2013), Cmpd1 promotes pSrc induction and has no substantial effects on Jaks or EGFR Tyr kinases. Worthy of note is the induction of pErk1/2MAPK, which has been previously reported to suppress Stat3 activity (Jain et al., 1998). Data provided herein show that Cmpd1 mediated pErk1/2MAPK induction, which in turn promotes Stat3 serine727 phosphorylation, and the two events have a causal relationship with the inhibition of Stat3 tyrosine705 phosphorylation (Fig. 6). Accordingly, the inhibition of MEK by PD98059 blocked Cmpd1-mediated pErk1/2MAPK and pserine727 Stat3 induction and prevented ptyrosine705 Stat3 suppression. The induction of pErk1/2MAPK may promote the formation of an Erk1/2MAPK/Stat3 complex, which would interrupt Stat3 recruitment to activating kinases, thereby suppressing its tyrosine705 phosphorylation (Fig. 6) as was previously reported (Tian and An, 2004).
Fig. 6.

Model of Cmpd1-mediated modulation of Stat3 signaling and the role of the MEK-Erk1/2 pathway and Cmpd1-dependent induction of mTOR, p38, Akt, Hsp27, and Src signaling in human glioma cells. Cmpd1 mediates inhibition of Stat3 tyrosine phosphorylation (pY705Stat3) and Stat3-dependent gene transcription in parallel with the upregulation of pS727Stat3, which is dependent on MEK-Erk1/2 induction, and Cmpd1 further upregulates mTOR(pS2448), p38 (pT180/Y182), Akt (pS473), Hsp27 (pS78/82), and Src (pY416) induction in glioma U251MG cells. ?, not definitively determined; ↑, induction; ↓, suppression.
Despite the fact that tumor cells harbor constitutively active Stat3, they exhibit differential responses that could not all be explained by the inhibition of aberrant Stat3 signaling. Furthermore, human breast cancer MCF7 cells that are also responsive to Cmpd1 do not harbor constitutive Stat3 activity, suggesting other factors contribute to the responsiveness of tumor cells to Cmpd1. Notably, in addition to Erk1/2MAPK activation, Akt, mTOR, p38, and Hsp27 are all induced in response to Cmpd1 (Fig. 6), which is indicative of pleiotropic effects, as has been observed for resveratrol and its metabolites (Yu et al., 2001; Fröjdö et al., 2007; Pirola and Fröjdö, 2008; Pervaiz and Holme, 2009; Calamini et al., 2010). To what extent the combination of these changes facilitates the growth inhibitory effects of Cmpd1 is presently unclear.
The increased potency of Cmpd1 as compared with resveratrol is likely due to the 3,5-dimethoxy groups (Fig. 1). This is supported by the similar antitumor potency of pterostilbene, which shares the 3,5-dimethoxy groups, and the published reports that methoxylated resveratrol analogs have increased activity against cell viability compared with the parent compound (Mazué et al., 2010). However, pterostilbene had a moderate effect on Stat3 tyrosine705 phosphorylation compared with Cmpd1, underscoring the differences in the underlying molecular mechanisms between the two compounds. It is notable that Cmpd1 lacks the side chain hydroxyl groups that are known to drive the antioxidant properties and the oxidative stress/redox mechanisms of resveratrol and pterostilbene, mechanisms known to promote biologic responses (Bhat et al., 2001; Rimando et al., 2002; Pan et al., 2008; Farghali et al., 2013; McCormack and McFadden, 2013), although we show that the 4′ acetoxy group becomes hydrolyzed inside tumor cells. Although Stat3 signaling has been reported to be influenced by redox changes (Li et al., 2010; Liu et al., 2012), whether Cmpd1 induces oxidative stress events that contribute to the inhibition of Stat3 activation and the induction of antitumor responses in vitro is unknown. The inhibitory activity of Cmpd1 against the Stat3 pathway suggests the potential that this agent could be useful as part of treatment strategies tailored to tumors harboring aberrantly active Stat3 following in vivo validation of antitumor efficacy in Stat3 relevant tumor models. The present study suggests that gliomas and, to a lesser extent, breast cancer harboring constitutive Stat3 activity may be responsive to Cmpd1.
Supplementary Material
Acknowledgments
The authors thank the members of our laboratory for stimulating discussions. Flow cytometry services were provided by the Molecular and Cellular Immunology Core of the John A. Burns School of Medicine, University of Hawaii. ESI–tandem mass spectrometry analysis studies to qualitatively determine the levels of Cmpd1 and pterostilbene were performed by the Analytical Biochemistry Shared Resource at the University of Hawaii Cancer Center.
Abbreviations
- Cmpd1
(E)-4-(3,5-dimethoxystyryl)phenyl acetate
- COX
cyclooxygenase
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ESI
electrospray ionization
- FBS
fetal bovine serum
- Jak
Janus kinase
- LY294002
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride
- MEK
mitogen-activated protein/extracellular signal-regulated kinase kinase
- mTOR
mammalian target of rapamycin
- PBS
phosphate buffered saline
- PD98059
2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one
- QR
quinone reductase
- SB202190
4-[4-(4-fluorophenyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]phenol
- Stat
signal transducer and activator of transcription
Authorship Contributions
Participated in research design: Chelsky, Yue, Turkson.
Conducted experiments: Chelsky, Yue.
Contributed new reagents or analytic tools: Kondratyuk, Pezzuto, Cushman, Turkson.
Performed data analysis: Chelsky, Yue, Paladino, Turkson.
Wrote or contributed to the writing of the manuscript: Chelsky, Paladino, Cushman, Pezzuto, Turkson.
Footnotes
This work was supported by the National Institutes of Health/National Cancer Institute [Grant CA161931] and the University of Hawaii start-up funds. The Molecular and Cellular Immunology Core is supported in part by the National Institute of General Medical Sciences Centers of Biomedical Research Excellence [Grant P20-GM103516]. The Analytical Biochemistry Shared Resource is supported by the National Institutes of Health/National Cancer Institute P30 Cancer Center Support [Grant P30-CA71789].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Baur JA, Sinclair DA. (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506. [DOI] [PubMed] [Google Scholar]
- Bhat KPL, Kosmeder JW, 2nd, Pezzuto JM. (2001) Biological effects of resveratrol. Antioxid Redox Signal 3:1041–1064. [DOI] [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. (2000) STATs in oncogenesis. Oncogene 19:2474–2488. [DOI] [PubMed] [Google Scholar]
- Bromberg J. (2000) Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res 2:86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg J, Darnell JE., Jr (2000) The role of STATs in transcriptional control and their impact on cellular function. Oncogene 19:2468–2473. [DOI] [PubMed] [Google Scholar]
- Burns J, Yokota T, Ashihara H, Lean ME, Crozier A. (2002) Plant foods and herbal sources of resveratrol. J Agric Food Chem 50:3337–3340. [DOI] [PubMed] [Google Scholar]
- Calamini B, Ratia K, Malkowski MG, Cuendet M, Pezzuto JM, Santarsiero BD, Mesecar AD. (2010) Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Biochem J 429:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow AW, Murillo G, Yu C, van Breemen RB, Boddie AW, Pezzuto JM, Das Gupta TK, Mehta RG. (2005) Resveratrol inhibits rhabdomyosarcoma cell proliferation. Eur J Cancer Prev 14:351–356. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr (1997) STATs and gene regulation. Science 277:1630–1635. [DOI] [PubMed] [Google Scholar]
- Farghali H, Kutinová Canová N, Lekić N. (2013) Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol Res 62:1–13. [DOI] [PubMed] [Google Scholar]
- Fröjdö S, Cozzone D, Vidal H, Pirola L. (2007) Resveratrol is a class IA phosphoinositide 3-kinase inhibitor. Biochem J 406:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami T, Yokoi T. (2012) The emerging role of human esterases. Drug Metab Pharmacokinet 27:466–477. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Steinberg GR. (2010) SIRT1 takes a backseat to AMPK in the regulation of insulin sensitivity by resveratrol. Diabetes 59:551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadi SM, Asad SF, Singh S, Ahmad A. (2000) Putative mechanism for anticancer and apoptosis-inducing properties of plant-derived polyphenolic compounds. IUBMB Life 50:167–171. [DOI] [PubMed] [Google Scholar]
- Jain N, Zhang T, Fong SL, Lim CP, Cao X. (1998) Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene 17:3157–3167. [DOI] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, et al. (1997) Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275:218–220. [DOI] [PubMed] [Google Scholar]
- Jeong WS, Kim IW, Hu R, Kong AN. (2004) Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res 21:661–670. [DOI] [PubMed] [Google Scholar]
- Kondratyuk TP, Park EJ, Marler LE, Ahn S, Yuan Y, Choi Y, Yu R, van Breemen RB, Sun B, Hoshino J, et al. (2011) Resveratrol derivatives as promising chemopreventive agents with improved potency and selectivity. Mol Nutr Food Res 55:1249–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R. (2006) Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther 5:621–629. [DOI] [PubMed] [Google Scholar]
- Li L, Cheung SH, Evans EL, Shaw PE. (2010) Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res 70:8222–8232. [DOI] [PubMed] [Google Scholar]
- Liu X, Guo W, Wu S, Wang L, Wang J, Dai B, Kim ES, Heymach JV, Wang M, Girard L, et al. (2012) Antitumor activity of a novel STAT3 inhibitor and redox modulator in non-small cell lung cancer cells. Biochem Pharmacol 83:1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazué F, Colin D, Gobbo J, Wegner M, Rescifina A, Spatafora C, Fasseur D, Delmas D, Meunier P, Tringali C, et al. (2010) Structural determinants of resveratrol for cell proliferation inhibition potency: experimental and docking studies of new analogs. Eur J Med Chem 45:2972–2980. [DOI] [PubMed] [Google Scholar]
- McCormack D, McFadden D. (2013) A review of pterostilbene antioxidant activity and disease modification. Oxid Med Cell Longev 2013:575482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeran SM, Katiyar SK. (2008) Cell cycle control as a basis for cancer chemoprevention through dietary agents. Front Biosci 13:2191–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy G, Hilliard TS, Turkson J. (2013) Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov 12:611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Agarwal AK, Xu T, Feng Q, Baerson SR, Duke SO, Rimando AM. (2008) Identification of molecular pathways affected by pterostilbene, a natural dimethylether analog of resveratrol. BMC Med Genomics 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervaiz S, Holme AL. (2009) Resveratrol: its biologic targets and functional activity. Antioxid Redox Signal 11:2851–2897. [DOI] [PubMed] [Google Scholar]
- Pirola L, Fröjdö S. (2008) Resveratrol: one molecule, many targets. IUBMB Life 60:323–332. [DOI] [PubMed] [Google Scholar]
- Quoc Trung L, Espinoza JL, Takami A, Nakao S. (2013) Resveratrol induces cell cycle arrest and apoptosis in malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS One 8:e55183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimando AM, Cuendet M, Desmarchelier C, Mehta RG, Pezzuto JM, Duke SO. (2002) Cancer chemopreventive and antioxidant activities of pterostilbene, a naturally occurring analogue of resveratrol. J Agric Food Chem 50:3453–3457. [DOI] [PubMed] [Google Scholar]
- Shankar S, Singh G, Srivastava RK. (2007) Chemoprevention by resveratrol: molecular mechanisms and therapeutic potential. Front Biosci 12:4839–4854. [DOI] [PubMed] [Google Scholar]
- Sun B, Hoshino J, Jermihov K, Marler L, Pezzuto JM, Mesecar AD, Cushman M. (2010) Design, synthesis, and biological evaluation of resveratrol analogues as aromatase and quinone reductase 2 inhibitors for chemoprevention of cancer. Bioorg Med Chem 18:5352–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian ZJ, An W. (2004) ERK1/2 contributes negative regulation to STAT3 activity in HSS-transfected HepG2 cells. Cell Res 14:141–147. [DOI] [PubMed] [Google Scholar]
- Turkson J. (2004) STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets 8:409–422. [DOI] [PubMed] [Google Scholar]
- Turkson J, Jove R. (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19:6613–6626. [DOI] [PubMed] [Google Scholar]
- Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10:48–54. [DOI] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250. [DOI] [PubMed] [Google Scholar]
- Yu H, Jove R. (2004) The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 4:97–105. [DOI] [PubMed] [Google Scholar]
- Yu R, Hebbar V, Kim DW, Mandlekar S, Pezzuto JM, Kong AN. (2001) Resveratrol inhibits phorbol ester and UV-induced activator protein 1 activation by interfering with mitogen-activated protein kinase pathways. Mol Pharmacol 60:217–224. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. (2010) A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol 79:1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yue P, Page BDG, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT, et al. (2012) Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci USA 109:9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Jaganathan S, Turkson J. (2010) A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro. J Biol Chem 285:35855–35865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.