

Abstract
The fact that over 30% of current pharmaceuticals target heptahelical G protein–coupled receptors (GPCRs) attests to their tractability as drug targets. Although GPCR drug development has traditionally focused on conventional agonists and antagonists, the growing appreciation that GPCRs mediate physiologically relevant effects via both G protein and non–G protein effectors has prompted the search for ligands that can "bias" downstream signaling in favor of one or the other process. Biased ligands are novel entities with distinct signaling profiles dictated by ligand structure, and the potential prospect of biased ligands as better drugs has been pleonastically proclaimed. Indeed, preclinical proof-of-concept studies have demonstrated that both G protein and arrestin pathway-selective ligands can promote beneficial effects in vivo while simultaneously antagonizing deleterious ones. But along with opportunity comes added complexity and new challenges for drug discovery. If ligands can be biased, then ligand classification becomes assay dependent, and more nuanced screening approaches are needed to capture ligand efficacy across several dimensions of signaling. Moreover, because the signaling repertoire of biased ligands differs from that of the native agonist, unpredicted responses may arise in vivo as these unbalanced signals propagate. For any given GPCR target, establishing a framework relating in vitro efficacy to in vivo biologic response is crucial to biased drug discovery. This review discusses approaches to describing ligand efficacy in vitro, translating ligand bias into biologic response, and developing a systems-level understanding of biased agonism in vivo, with the overall goal of overcoming current barriers to developing biased GPCR therapeutics.

Introduction
Early models of G protein–coupled receptor (GPCR) signaling envisioned the receptor as existing in equilibrium between discrete "off" and "on" states distinguished by their ability to trigger downstream responses. Ligands were thought to act by perturbing this equilibrium and were classified as agonists if they elicited a maximal response, partial agonists if they generated a submaximal response at saturating ligand concentration, antagonists if they lacked intrinsic efficacy but competitively inhibited agonist responses, and inverse agonists if they preferentially stabilized the "off" state, leading to a suppression of basal receptor activity (Black and Leff, 1983; Black et al., 1985; Samama et al., 1993; Weiss et al., 1996). This conceptualization began to evolve in the mid-1990s, driven by the recognition that many GPCRs couple to several different G protein families, enabling a single receptor to engage multiple signaling pathways simultaneously (Offermanns et al., 1994; Laugwitz et al., 1996) or to activate them differentially in different tissues (Jin et al., 2001; Mahon et al., 2002). As appreciation of the complexity of GPCR signaling grew, so did evidence that changes in ligand structure can affect the efficiency of receptor coupling to different downstream effectors (Sagan et al., 1996; Berg et al., 1998; Maudsley et al., 1998; Takasu et al., 1999; Holloway et al., 2002). The finding that structurally distinct ligands can activate the same GPCR in different ways indicated that most, if not all, GPCRs possess more than one "active" receptor state and that ligand structure can "bias" downstream signaling relative to that of the native agonist (Kenakin, 1995; Christopoulos and Kenakin, 2002).
Adding to the complexity was the recognition that GPCRs signal through both G protein and non–G protein effectors. The first non–G protein effectors to be described were the arrestins, a small family of cytosolic proteins originally characterized for their role in GPCR desensitization (Ferguson, 2001). Beginning in 1999 with the report that arrestin 2 bound the nonreceptor tyrosine kinase, c-Src, and recruited it to activated β2 adrenergic receptors (Luttrell et al., 1999), studies linking arrestins to GPCR activation of novel enzymatic effectors began appearing with regularity. Putative arrestin-regulated effectors include Src family kinases (Barlic et al., 2000; DeFea et al., 2000a), components of the extracellular signal-regulated kinase 1/2 and c-Jun N-terminal kinase 3 mitogen-activated protein kinase cascades (DeFea et al., 2000b; McDonald et al., 2000; Luttrell et al., 2001), the E3 ubiquitin ligase Mdm2 (Shenoy et al., 2001), the cAMP phosphodiesterases PDE4D3/5 (Perry et al., 2002), diacylglycerol kinase (Nelson et al., 2007), the inhibitor of nuclear factor-κB IκBα (Witherow et al., 2004), the Ral-GDP dissociation stimulator Ral-GDS (Bhattacharya et al., 2002), the actin filament-severing protein cofilin (Zoudilova et al., 2007), and the Ser/Thr protein phosphatase 2A (Beaulieu et al., 2004, 2005). With time, it became clear that GPCRs signal via a variety of non–G protein effectors besides arrestins, including PDZ domain- and non-PDZ domain–containing scaffolds (Walther and Ferguson, 2015), and that interaction with accessory proteins is a critical determinant of both the ligand binding and effector coupling specificity of many GPCRs (Kenakin and Miller, 2010). The current model of GPCR pharmacodynamics recognizes that efficacy arises from the bidirectional interplay of ligands that stabilize unique ensembles of receptor states, and the complement of intracellular effectors, accessory proteins, and environmental influences that translate those conformations into cellular responses while at the same time modulating the conformational ensemble through allosteric interaction (Kenakin and Miller, 2010; van der Westhuizen et al., 2015). With respect to the dichotomous G protein– and arrestin-mediated pathways, GPCR signaling may be thought of as the minute-to-minute balance between highly amplified G protein–mediated signals that are rapidly desensitized by arrestin binding, and more durable, but less amplified, arrestin-mediated signals that arise from stoichiometric GPCR-arrestin "signalsome" complexes (Ferguson, 2001; Luttrell and Lefkowitz, 2002). Whereas second messengers generated via G protein–dependent activation of enzymatic effectors account for most of the classic short-term consequences of GPCR activation, arrestin-mediated signals appear to perform numerous functions, among them enhancing second messenger degradation, regulating cytoskeletal dynamics controlling vesicle trafficking, exocytosis and cell migration, and promoting cell survival, growth, and hyperplasia (Luttrell and Gesty-Palmer, 2010; Luttrell, 2013).
Importantly for our story, G protein– and arrestin-coupled components of the receptor ensemble are pharmacologically dissociable, such that highly efficacious activators of G protein signaling may not promote engagement of arrestins (Walters et al., 2009; Zhou et al., 2013), rendering them effectively nondesensitizing, whereas ligands displaying no efficacy for G protein coupling may yet serve as agonists for arrestin-mediated signaling events (Wei et al., 2003; Azzi et al., 2003; Gesty-Palmer et al., 2006; Zimmerman et al., 2012;). This conjunction of "pluridimensional efficacy" (Galandrin and Bouvier, 2006) and ligand "bias" has set the stage for development of novel pharmaceutical agents that act in vivo as pathway-selective agonists or as mixed agonist-antagonists that promote the activation of some pathways while inhibiting the capacity of the endogenous ligand to stimulate others.
What Bias Is—And What It Isn’t
Translating orthosteric ligand bias into novel therapeutics requires both a formal understanding of what functional selectivity is and a means to quantify it so that the efficacy profiles of different ligands can be meaningfully compared. The current evolution of the Black-Leff operational model as applied to receptor allostery envisions GPCRs not as binary switches but as "ensembles" of tertiary conformations (Black and Leff, 1983; Christopoulos and Kenakin, 2002; Kenakin, 2007; Griffin et al., 2007; Kenakin, 2009; Ehlert, 2008; Kenakin and Miller, 2010). Because there is no a priori reason that a receptor conformation that links it to one downstream effector would necessarily couple it with equal efficiency to all possible effectors, one can posit that a finite number of discrete active and inactive states exist within the sterically permissible conformational ensemble. Thus, the biologic activity of the system at any instant is a reflection of the distribution of the receptor population among the component microstates that make up the ensemble. Within this framework, there is likewise no a priori reason that the distribution of receptor conformations stabilized by one ligand should be identical to that generated by a structurally distinct ligand. In this case "functional selectivity" may arise from differences in the efficiency with which ligands stabilize different active states.
Evidence generated using intramolecular fluorescence probes to monitor the effect of ligand binding on receptor conformation supports the concept that ligands with different structure stabilize unique receptor ensembles (Yao et al., 2006, 2009). One-dimensional 19F NMR spectroscopic analysis of conventional and biased ligand binding to the β2 adrenergic receptor indicates that G protein activation correlates with movement of transmembrane helix VI, whereas arrestin-biased ligands predominantly affect the conformation of helix VII (Liu et al., 2012). In this study, helix VI and helix VII were found to move independently, providing a physical basis for biased agonism and a conformational "signature" predictive of arrestin selectivity. Although to date there is little direct X-ray crystallographic structure of GPCRs bound to conventional and biased agonist ligands, computational modeling of agonist docking within the ligand binding pocket of family A GPCRs suggests that efficacy correlates with engagement of certain residues and exclusion of interaction with others (Katritch et al., 2012; Jacobson and Costanzi, 2012; Costanzi, 2014).
Historically, the experimental hallmark of orthosteric ligand bias has been "reversal of potency," where two ligands exhibit opposite rank order of potency for two downstream responses measured in the same system (Sagan et al., 1996; Berg et al., 1998; Takasu et al., 1999), or "reversal of efficacy," where a single ligand exhibits opposing efficacy toward two different downstream responses (Gray and Roth, 2001; Holloway et al., 2002; Sneddon et al., 2004). Such phenomena can only be explained by the existence of more than one active receptor conformation and thus offered proof that different ligands can activate the same receptor in different ways. Importantly for drug discovery, the existence of bias dictates that ligand classification is both assay and context dependent; a biased ligand may be classified as an inverse agonist, antagonist, or agonist depending on the assay or cell/tissue system used to define it. Biased ligands are novel pharmacological entities, and they cannot be adequately described using low-dimensionality screening techniques.
Central to this definition is that bias must be demonstrable when two measures of receptor activity are examined in a common cellular background. Because GPCRs that couple to multiple downstream effectors may do so with different efficiencies, increasing ligand occupancy will cause the most efficiently coupled response(s) to be activated first, followed by less efficiently activated processes. In this case, variations in the level of receptor or effector expression between systems can cause the appearance of "new" signaling processes (Zhu et al., 1994; Cordeaux et al., 2000; Nasman et al., 2001) and even create the illusion of unique functional states (Kenakin, 1995). In the case of partial agonists, responses that are tightly coupled or highly amplified will persist because fractional receptor activation is sufficient to generate a maximal response, whereas responses that are weakly coupled or unamplified, where full receptor activation is necessary for a maximal response, will be very sensitive to changes in receptor or effector expression and will appear or disappear depending on cellular background. This form of variability, arising from differences in "signal strength," does not require the existence of multiple active states, and the rank order of potency of a series of conventional full and partial agonists will not vary when compared in different systems (Kenakin, 1995, 2007).
The efficiency with which any given orthosteric agonist will promote receptor coupling to downstream signaling pathways may vary between effectors, e.g., heterotrimeric G protein species or arrestins. Although the observed signaling output may vary in different cell backgrounds, intrinsic efficacy is an innate property of the ligand, i.e., a ligand can only be biased in relation to the intrinsic efficacy of some other ligand acting on the same receptor. For classification purposes, this reference ligand is usually the native hormone or neurotransmitter. Yet ligand bias is not merely a product of synthetic pharmacology. Nature has been exploiting bias far longer than pharmaceutical scientists have been working to discover clinically useful biased drugs. This is apparent in the case of chemokine signaling where there is more than twice the number of endogenous chemokines than chemokine receptors. For example, two endogenous ligands for the CC chemokine receptor 7, CCL19 and CCL21, while retaining similar G protein coupling efficacy, differ in their capacity to promote receptor desensitization, arrestin recruitment, and ERK1/2 activation (Kohout et al., 2004; Zidar et al., 2009). Differential activation of serotonin receptors by metabolic derivatives of serotonin and "trace amines" is another physiologically relevant example of endogenous bias. In response to serotonin, the serotonin 5HT2A receptor (5HT2AR) activates Akt in mouse cortex and cortical neurons via an arrestin3-Src–dependent mechanism. However, this signaling complex does not form when the 5HT2AR is activated by N-methyltryptamines (Schmid and Bohn, 2010). These differences in neuronal signaling translate into agonist-induced behaviors, where the 5HT2AR-mediated activation of the head twitch response in mice requires the production of N-methyltryptamine metabolites to activate the nonarrestin-dependent pathway used by serotonin (Schmid et al., 2008; Schmid and Bohn, 2010). Thus, it appears that ligand bias a common solution to the need for fine regulation in complex signaling systems and that GPCR systems may already be poised to respond selectively to variations in ligand structure.
Quantifying Bias
When considering a single dimension of efficacy, the actions of a ligand can be described by two terms: the equilibrium dissociation constant of the ligand-receptor complex (Kd) and the maximal observed change in receptor activity (Vmax), which together specifies the relationship between receptor occupancy and system response. Any single parameter used to describe ligand efficacy in a given pathway needs to incorporate both of these factors. One approach, based on the Black-Leff operational model (Black and Leff, 1983), is to determine a "transduction coefficient," log(τ/KA), by fitting the dose-response curve to the operational model below, where τA is the efficacy of the ligand (A) for the pathway, KA is the equilibrium dissociation constant for the agonist-receptor complex, and EM is the maximum response capability of the system:
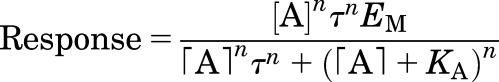 |
The term τ encompasses both the intrinsic efficacy of the ligand and system-dependent factors such as receptor density and coupling efficiency. Because the latter factors are constant for any dose-response curve determined in the same cell for any given signaling pathway, the ratio of τ values for any two agonists in the same system will yield an intrinsic efficacy ratio for activation of the pathway that is independent of receptor number or coupling efficiency. Once determined for each ligand of interest, the efficiency of ligands producing activation of a pathway relative to a reference agonist, e.g., the endogenous ligand, can be quantified by the normalized transduction coefficient Δlog(τ/KA) (Kenakin 2009, 2014; Kenakin et al., 2012; Stahl et al., 2015).
An alternative approach that is valid for dose-response data described by curves with Hill coefficients near unity is to determine "intrinsic relative activity" (RAi) from EC50 and Emax data according to the method of Ehlert (2005), where A and B are the reference and test ligands, respectively:
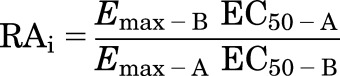 |
Because EC50 and Kd are equal when the Hill slope of the dose response curve is 1.0, the two approaches yield similar results as long as this condition is fulfilled. Similar to the operational model, when applied correctly Δlog(RAi) provides a measure of ligand efficiency relative to a reference agonist.
To quantify ligand bias, it is then necessary to compare the Δlog(RAi) value obtained for each agonist/pathway of interest. For any two pathways j1 and j2, a "bias factor" can be derived as the ΔΔlog(τ/KA) or ΔΔlog(RAi) (Kenakin et al., 2012):
where
Because GPCR efficacy can be profiled in as many "dimensions" as there are assays, a convenient way to visualize efficacy is by preparing multiaxial graphic displays of bias factors determined using a series of ligands in a panel of assays. As shown in Fig. 1, such plots provide a depiction of ligand bias that is easily comprehended and permits identification of ligands with similar efficacy profiles.
Fig. 1.

Example of ligand bias described using the operational model. Transduction efficiencies [Δlog(τ/KA)] for five different assays of κ opioid receptor signaling were used to calculate bias factors (ΔΔlog(τ/KA)j1–j2) that are presented in a multiaxial graphic format. The κ opioid receptor-selective agonist, U69,593 [(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide], was used as the reference ligand, hence its bias factor conforms to unity in all assays. The pathways represented are: membrane [35S]GTPγS binding (mG protein); βarr2 enzyme fragment complementation (βarr2 EFC), βarr2 fluorescence imaging (βarr2 imaging), cellular impedance (impedance), and whole cell [35S]GTPγS binding (wcG protein). As depicted, the two test compounds, 2-(4-(furan-2-ylmethyl)-5-((4-methyl-3-(trifluoromethyl)benzyl)thio)-4H-1,2,4-triazol-3-yl)pyridine (1.1) and 2-(2-fluorobenzyl)-N-(4-methyl-3-(trifluoromethyl)phenyl)-1-oxo-octahydroisoquinoline-8-carboxamide (2.1), display bias for G protein signaling over arrestin recruitment. This research was originally published in Zhou et al. (2013).
An important advantage of the operational model is its capability to quantify the full range of agonism from submaximal effects to amplified effects in very sensitive systems with effective receptor reserve (Black and Leff, 1983; Black et al., 1985). Furthermore, log(τ/KA) ratios determined in one system are applicable to all systems without the need to independently quantify functional selectivity in each (Kenakin, 2009; Kenakin et al., 2012). Variation in receptor density and coupling efficiency between systems might change the ability of all agonists targeting a given receptor to activate a particular pathway, but it will not change the pathway selective bias of different ligands relative to one another or to a reference agonist run in parallel in all assays. On the other hand, using the operational model to describe bias requires generating extensive dose response data and KA measurements for the test agonists and reference agonist in multiple assays. Although clearly useful, it is not readily adaptable to high-throughput screening programs, which historically depend on "single shot" screens of vast numbers of compounds in single assays of cellular response. Moreover, quantifying bias by assessing cell-based responses alone provides no information about the degree of bias needed to produce a physiologically relevant change in signaling activity nor does it help in defining the type of bias needed to dissociate therapeutic from deleterious ligand effects in vivo.
Translating Bias
The therapeutic promise of biased agonism resides in its capacity to elicit biologic responses in vivo that cannot be obtained via conventional agonist/antagonist ligands. In effect, a GPCR bound to a biased ligand is a different functional entity, with different signaling characteristics, than the same receptor bound to a conventional agonist or partial agonist. This presents both an opportunity and a major challenge to developing biased therapeutics. If the appropriate in vitro efficacy profile is known, then judicious multiplex screening should identify compounds that produce the desired physiologic effects. On the other hand, because biased ligands activate GPCRs in "unnatural" ways compared with their conventional agonist counterparts, the relationship between the in vitro efficacy profile and the desired in vivo biologic response is not necessarily obvious and cannot simply be inferred based on prior knowledge of the native ligand. In most cases, the effect of ligand bias in vivo has to be determined empirically (Appleton and Luttrell, 2013; Luttrell, 2013; Schmid et al., 2013).
Several examples illustrate both the promise and pitfalls encountered in translating ligand bias to complex in vivo systems. One very promising area for biased drug development involves G protein pathway–selective opioid receptor agonists (Dewire et al., 2013; Schmid et al., 2013; Zhou et al., 2013). From the initial discovery that the impaired μ opioid receptor (MOR) desensitization observed in arrestin3 null mice led to enhanced morphine analgesia with less tolerance (Bohn et al., 1999, 2000), it was apparent that nondesensitizing opioid analogs might be superior analgesics. The additional benefit became evident when it was realized that the side effects of constipation, respiratory suppression, and physical dependence were lessened in the absence of arrestin3, suggesting that MOR-arrestin3 signaling may underlie some of these physiologic responses (Raehal et al., 2005, 2011; Bohn and Raehal, 2006; Raehal and Bohn, 2011). Although efforts are still ongoing to try to capture the endogenous signalsomes using arrestins downstream of MOR, the generation of G protein–biased ligands has allowed for testing the role of G protein– versus arrestin-mediated effects in vivo.
The first generation of G protein–biased MOR agonists has shown promise in mouse models. This compound, TRV-130 [[(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine], is weakly biased (bias factor of 3-fold for G protein over arrestin signaling), yet the authors reported a favorable separation between analgesic efficacy and gastrointestinal and respiratory side effects when tested in rodent models (Dewire et al., 2013). Currently in clinical trials, TRV-130 has proven to be a very potent analgesic in humans (Soergel et al., 2014). However, the results of the phase II clinical trial have not revealed a significant reduction in side effects, raising the question of whether biased agonists at MOR can dissociate analgesia from respiratory depression/constipation or if a greater degree of bias is necessary to translate into human therapeutic responses. To fully test this hypothesis, compounds of different chemical structures and that display more bias for G protein signaling over arrestin signaling should be developed and tested.
Selective κ opioid receptor (KOR) agonists also produce antinociceptive effects in animal models through activation of Gi/o family heterotrimeric G proteins (Gullapalli and Ramarao, 2002), but unlike MOR agonists do not cause physical dependence or respiratory depression (Charbogne et al., 2014). However, their clinical utility is hampered by an array of undesirable side effects, including dysphoria, sedation, diuresis, hallucination, and depression (Pfeiffer et al., 1986; Togashi et al., 2002; Land et al., 2008; Van’t Veer and Carlezon, 2013). Interestingly, it has been proposed that the dysphoric component of KOR agonism is mediated via a process involving arrestins (Bruchas et al., 2006; Redila and Chavkin, 2008), suggesting that G protein biased KOR agonists may deliver the desired analgesic effect while circumventing deleterious mood disturbances (Schmid et al., 2013; Zhou et al., 2013; White et al., 2014).
In animal models, certain properties, such as antinociception, are retained with G protein signaling–biased KOR agonists; however the first reports from in vivo testing show that biased agonists derived from Salvinorin A analogs still promote aversive behaviors in mouse conditioned place preference tests (White et al., 2015). Because aversion can be produced by any substance that an animal associates with an unpleasant sensation, whether due to direct neurologic stimulation or to visceral discomfort, it is difficult to determine whether such findings disprove the hypothesis that arrestin-dependent signaling mediates the KOR aversive response. Although other behavioral models, such as altering reward thresholds using intracranial self-stimulation, offer better models of dysphoria, it is clear that the development of additional ligands across diverse chemical scaffolds with conserved bias properties will be necessary to assign causality. Expanding the spectrum of bias to include ligands with different degrees of bias among downstream effectors, such as ERK activation, will provide valuable tools for navigating the complex effects of signalsome engagement. Recent examples of this approach have been described for KOR agonists, where a triazole series was modified to maintain G protein over arrestin bias while altering selectivity for ERK1/2 activation (Lovell et al., 2015).
The in vivo effects of arrestin pathway-selective biased agonists are even less predictable. Because loss of function in arrestin null animals could arise either as a consequence of enhanced G protein pathway activation or due to loss of arrestin-mediated signaling, determining the role of arrestin signaling in vivo is best accomplished by empirically examining the effects arrestin-biased ligands in a wild-type background. Arrestin-dependent signaling by the type 1 parathyroid hormone receptor (PTH1R) is a case in point. The native hormone, PTH(1-84), and its conventional agonist N-terminal fragment human PTH(1-34), promote intestinal calcium absorption, renal calcium retention, and osteoblastic bone formation. Parathyroid hormone (PTH)–dependent osteoblast activation produces coupled activation of bone-resorbing osteoclasts through G protein–dependent elaboration of osteoclast activating cytokines, like RANKL, the receptor activator of nuclear factor κB ligand. Although hPTH(1-34) is Food and Drug Administration approved as an anabolic treatment to build new bone in patients with severe osteoporosis, its beneficial effects depend on the kinetics of administration, because continuous exposure to PTH leads to hypercalcemia and net bone resorption, whereas intermittent exposure stimulates bone formation in excess of resorption (Qin et al., 2004). Thus, wild-type C57BL/6 mice given daily injections of PTH(1-34) experience a net increase in bone mass, characterized by increases in trabecular bone volume and cortical thickness, osteoblast number, bone matrix deposition, and mineral apposition rate. Reflective of osteoblast-osteoclast coupling, osteoclast numbers also increase, along with markers of bone resorption, and treated mice develop hypercalciuria from PTH1R regulated Gs signaling in the proximal renal tubule (Mohan et al., 2000; Sebastian et al., 2008).
Surprisingly, wild-type mice treated with (d-Trp12,Tyr34)-bPTH(7-34), an arrestin biased PTH1R agonist (Gesty-Palmer et al., 2006), also exhibit increased bone formation with greater trabecular bone volume, increased osteoblast number, matrix deposition, and mineral apposition (Gesty-Palmer et al., 2009, 2013). Unlike the conventional agonist, (d-Trp12,Tyr34)-bPTH(7-34) fails to activate the G protein–mediated processes regulating osteoclast number, bone turnover, and calciuresis, thereby accomplishing the potentially beneficial feat of "uncoupling" PTH1R-mediated bone formation from bone resorption. All responses to (d-Trp12,Tyr34)-bPTH(7-34) are either absent or reversed in arrestin3 null mice, suggesting its actions in vivo result from arrestin biased signaling. Given that prior work had attributed the bone-forming capacity of PTH solely to its ability to activate Gs-cAMP-protein kinase A signaling (Mohan et al., 2000; Lian et al., 2006; Sebastian et al., 2008; Deng et al., 2008), the finding that arrestin-dependent signaling is sufficient to promote osteoblastic bone formation is both paradoxical and not predicted from the study of conventional PTH1R agonism.
Understanding Bias at a Systems Level
Orthosteric GPCR agonists, whether conventional or biased, mediate their cellular effects by interacting with the ligand-binding pocket and changing the distribution of conformations within the receptor ensemble. Everything that follows, whether it is short-term modulation of intermediary metabolism, cellular contractility, or membrane potential, or long-term changes in cell proliferation, differentiation, and survival, is entrained and functionally encrypted at that point in time. The concept of conformational selection, upon which allosteric models of GPCR function are based, posits that biased ligands may stabilize active receptor conformations in different proportions than the native ligand but do not force receptors into sterically unfavorable conformations or couple them to novel effectors. If true, then the actions of a biased ligand, at least in the short term, must comprise a subset of those produced by a conventional pluripotent agonist. In fact, experimental data examining the actions of biased agonists in vitro using a wide range of readouts tend to bear this out (Swaminath et al., 2005; Rajagopal et al., 2006; Aplin et al., 2007; Kendall et al., 2011; Liu et al., 2012; Sauliere et al., 2012; Gesty-Palmer et al., 2013). Even "unbiased" comparisons of conventional and arrestin biased GPCR signaling in vitro using quantitative global phosphoproteomics suggest that biased agonist responses are subsumed within the larger whole of conventional agonism. For example, a phosphoproteomic comparison of the conventional angiotensin AT1A receptor agonist, angiotensin II (AngII), and the arrestin biased agonist, [Sar1,Ile4,Ile8]AngII, which identified 1183 regulated protein phosphorylation sites after 5 minutes of ligand exposure, found that 756 (64%) were unique to AngII, 369 (34%) were regulated by both AngII and [Sar1,Ile4,Ile8]AngII, and only 58 (5%) were unique to [Sar1,Ile4,Ile8]AngII (Christensen et al., 2010).
In vivo, however, where drugs must exert their therapeutic effects, the limited systems level data available suggest that conventional and biased agonists may be dramatically different (Appleton and Luttrell, 2013). Figure 2A illustrates this point using the three-dimensional data visualization application Omnimorph (http://www.ott.nih.gov/technology/e-143-2010) to depict the transcriptomic fingerprint of the conventional PTH1R agonist hPTH(1-34) and the arrestin biased agonist (d-Trp12,Tyr34)-bPTH(7-34) in the kidneys of mice exposed to vehicle or ligand for 4 weeks (Maudsley et al., 2015a). As shown, at a gestalt level the global transcriptomic effects of hPTH(1-34) and (d-Trp12,Tyr34)-bPTH(7-34) appear to be markedly different in wild-type mice, and although the effects of the G protein–competent conventional ligand are largely conserved between wild-type and arrestin3 null backgrounds, the loss of arrestin3 profoundly disrupts the effects of the arrestin-biased ligand. Similar divergence is evident when the transcriptomic effects of the two ligands are compared at the pathway level by geneset enrichment analysis (Gesty-Palmer et al., 2013; Maudsley et al., 2015a). Figure 2B illustrates the substantial degree of nonoverlap observed in calvarial bone after long-term hPTH(1-34) or (d-Trp12,Tyr34)-bPTH(7-34) treatment. Unlike data from short-term phosphoproteomic surveys, transcriptomic datasets from six different tissues show only 1.4 to 38.9% commonality at the pathway level between conventional and arrestin biased PTH1R agonists (Maudsley et al., 2015a). Whereas in bone hPTH(1-34) primarily affects pathways classically associated with enhanced bone turnover, including collagen synthesis, matrix mineralization, and osteoclast activation, (d-Trp12,Tyr34)-bPTH(7-34) primarily affects pathways regulating cell cycle progression, cell survival, and migration (Gesty-Palmer et al., 2013).
Fig. 2.

Systems level characterization of conventional and arrestin biased PTH1R agonism in vivo. (A) Omnimorph structures depicting a high-dimensionality z score representation of the transcriptional responses to hPTH(1-34) (panels 1 and 2, WT; panels 3 and 4, Arrb2 KO) or (d-Trp12,Tyr34)-bPTH(7-34) (panels 5 and 6, WT; panels 7 and 8, Arrb2 KO) in wild-type (WT) and arrestin3 null (Arrb2 KO) murine kidney. (B) Significantly-populated canonical signaling pathways induced by (D-Trp12,Tyr34)-bPTH(7-34) (red bars) or by hPTH(1-34) (black bars) in murine calvarial bone identified using Ingenuity Systems Pathways Analysis. Each histogram bar represents the signaling pathway score. Signaling pathways within the yellow block represent coherently regulated common pathways between the two ligands. The associated Venn diagram indicates the functional signaling separation between hPTH(1-34) and (d-Trp12,Tyr34)-bPTH(7-34). (C) Word cloud interpretation of the individual Textrous! output (dismantled noun-phrases) performed using the most cross-tissue conserved (d-Trp12,Tyr34)-bPTH(7-34) regulated transcripts. (D) Word cloud interpretation of the collective Textrous! output performed using the most cross-tissue conserved (d-Trp12,Tyr34)-bPTH(7-34) regulated transcripts. Textrous! is a latent semantic indexing–based analytical tool that correlates the strength of association between specific genes in a dataset with user-defined interrogation terms, in this case biomedical words and noun-word phrases extracted from PubMed, Online Mendelian Inheritance in Man, and the Mammalian Phenotypes Database at the Jackson Laboratories Mouse Genomic Informatics portal (Chen et al., 2013a). Research originally published in Maudsley et al. (2015a).
This divergence may not be surprising. In vitro, biased agonists activate a subset of the signaling pathways regulated by a conventional agonist, whereas in vivo they might be expected to exert mixed agonist-antagonist effects, generating some signals while antagonizing activation of others by the endogenous ligand. The individual components that comprise downstream signaling may not be qualitatively different, but they will be quantitatively and stoichiometrically "unbalanced" compared with the native ligand. As these unbalanced signals propagate, differences in the tissue response arising from even small differences in signal strength or the kinetics of pathway activation/inhibition have the potential to introduce unpredicted, and perhaps unpredictable, biologic outcomes.
Given the myriad signaling events attributed to arrestins (Luttrell and Gesty-Palmer, 2010; Whalen et al., 2011) and the relative paucity of information about which might be physiologically relevant, developing a systems level appreciation of arrestin biased agonism in vivo might facilitate the rational development of biased GPCR ligands. One attempt to develop a comprehensive description of the core activities of (d-Trp12,Tyr34)-bPTH(7-34) through bioinformatic analysis of in vivo transcriptomic data are illustrated in Fig. 2, C and D (Maudsley et al., 2015b). For this analysis, the most conserved transcripts populating the most conserved signaling pathways from a comparison of the actions of (d-Trp12,Tyr34)-bPTH(7-34) in six different murine tissues were analyzed using the reverse latent semantic indexing application Textrous! (Chen et al., 2013a). Latent semantic indexing–based analysis correlates the strength of association between specific genes in a dataset with user-defined interrogation terms. Individual processing investigates the links between scientifically relevant words and individual transcripts, while collective processing generates a hierarchical word cloud indicating the word groups most strongly associated with the entire input dataset (Chen et al., 2013b). As illustrated, at the individual processing level, arrestin biased signaling correlates most prominently with regulation of protein kinase activity and phosphorylation (Fig. 2C), whereas at the collective level a bias toward cellular growth, apoptosis/survival, remodeling, histone regulation, and cell cycle control is evident (Fig. 2D). Adding credence to the analysis, the major functions identified are consistent with the in vitro characterization of (d-Trp12,Tyr34)-bPTH(7-34) actions in primary calvarial osteoblasts (Gesty-Palmer et al., 2013) and data emerging from the cancer field suggesting that arrestins function as regulators of tumor cell proliferation, survival, and metastasis (Buchanan et al., 2006; Dasgupta et al., 2006; Moussa et al., 2008; Chun et al., 2009; Lakshmikanthan et al., 2009; Li et al., 2009; Rosanò et al., 2009; Liu et al., 2011; Bonnans et al., 2012; Fereshteh et al., 2012).
Systems level transcriptomic studies of PTH1R agonism in vivo suggest that an arrestin biased ligand, with its limited signaling repertoire, has narrower and more cross-tissue conserved effects than its pluripotent conventional agonist counterpart. Thus, arrestin biased ligands may possess greater stability and predictability of activity across multiple cell types and perhaps across diverse pathologic contexts (Maudsley et al., 2012). It remains to be determined, however, if the transcriptomic "signature" of arrestin biased PTH1R agonism is transferrable across different GPCRs or whether each GPCR uses arrestins to its own purpose. Nonetheless, current evidence suggests that systems level studies may contribute to a conceptual framework within which the in vivo outcomes of arrestin-biased signaling may be generalized.
Conclusions
Table 1 summarizes some of the opportunities and challenges currently facing efforts to translate biased GPCR agonism into the clinic. Clearly, the field possesses a sound theoretical basis for coping with ligand bias, numerous assays capable of detecting both G protein and arrestin signaling that are amenable to high-throughput screening, and mathematical tools to quantitatively compare ligands across multiple dimensions of efficacy. The major challenge from the screener’s perspective is that we do not have a good idea what to screen for, i.e., the link between in vitro efficacy and biologic activity is at best tenuous for many potential biased agonist targets. Even in settings where a sound rationale exists for favoring G protein or arrestin signaling, the extent to which a biased ligand must favor one pathway over another to produce a therapeutic benefit is unknown. True selective agonism, i.e., opposing efficacy toward different effectors, is rare. Most ligand bias is more subtle, i.e., quantitative variance from the native ligand in the efficiency of coupling to different effectors. In most cases, neither the direction of bias, e.g., G protein versus arrestin selectivity, nor the magnitude of bias needed for optimal clinical outcomes, is necessarily obvious. In such a near vacuum, the cost and complexity of screening every possible dimension of GPCR efficacy is prohibitive.
TABLE 1.
Opportunities and Challenges for Biased Drug Development
| Identification of Biased Agonists |
| Opportunities |
| • Sound conceptual understanding of ligand bias |
| • Available high-throughput screening platforms to detect G protein and arrestin efficacy |
| • Methods for quantifying bias allowing comparison of ligand efficacy profiles |
| Challenges |
| • Cost/complexity of employing multiplex ligand screening in early compound development |
| • Unclear relationship between magnitude of bias needed to produce a relevant change in physiologic response |
| • Limited understanding of the relationship between in vitro efficacy profile and in vivo drug effect for most targets |
| Linking In Vitro Efficacy to Therapeutic Effect |
| Opportunities |
| • Cellular and in vivo evidence that G protein and arrestin signaling pathways mediate different responses |
| • Proof-of-principle that biased agonists can elicit responses not attainable via conventional agonists/antagonists |
| Challenges |
| • No broad conceptual framework for arrestin-dependent efficacy in vivo |
| • Unknown phenotypic consequences of G protein– and arrestin-selective efficacy at most potential targets |
| • Potential for unexpected on-target effects due to "unbalanced" receptor activation |
Yet the potential of biased therapeutics seems clear. Solid evidence indicates that G proteins and arrestins mediate distinct physiologic processes and that the two GPCR signaling modes are pharmacologically dissociable, permitting the development of biased ligands with unique efficacy profiles in vivo. Moreover, in a handful of cases there is clear proof-of-principle that biased agonists can produce biologic effects that cannot be attained using conventional agonists or antagonists, at least in preclinical animal models. For most potential targets, the missing link is an understanding of how G protein– and arrestin-mediated signals both contribute to the biologic actions of specific GPCRs. Biased agonists are unique pharmacological entities, and the phenotypic consequences of treatment cannot necessarily be predicted from existing knowledge of receptor function. Early forays into probing arrestin biology at a systems level suggest there may be a conserved core of transposable arrestin-mediated functions, but it is still early days. Finally there is the specter of unanticipated "on-target" effects resulting from activating GPCRs in nonphysiologic ways.
Future progress will depend on knowing, for each potential GPCR target, what efficacy profile will deliver the most desirable physiologic response, whether that of a conventional agonist or antagonist or a G protein– or arrestin-selective biased agonist. Assuming that the number of proximal effectors, e.g., G protein and arrestin isoforms, engaged by any given GPCR is finite and that agonists simply select from this preset menu, then the number of "flavors" of biased agonism should likewise be finite (Maudsley et al., 2005). Initially, it may be helpful to generate multidimensional efficacy profiles encompassing receptor coupling to all G protein families, arrestin isoforms, second messengers, small G proteins, and functions like cell proliferation, survival, permeability, and migration (Sauliere et al., 2012; Schann et al., 2013) to cluster ligands into a smaller number of functional classes.
Even so, translating in vitro efficacy to in vivo biologic function is an endeavor that must be very carefully approached. Just as cellular context can determine whether a ligand appears as an agonist or antagonist and which intracellular signaling partners are engaged (Kenakin, 2007), the body as a whole presents multiple variables that can interfere with the valid comparison of a proposed “biased” agonist with the endogenous hormone or clinically validated agonist used for reference. Pharmacokinetic variables that affect the compound’s ability to reach its target, e.g., absorption, bioavailability, and metabolism, must be overcome to assure that the ligand has every opportunity to occupy the receptor before its biologic activity can be attributed to differential signaling arising from pharmacodynamic bias. Ultimately, primary cell cultures and tissue-based assays will provide the intermediary platforms for reducing the impact of such variables. Thus, for any given target, a thoughtful combination of high throughput screening to classify chemically diverse sets of compounds, followed by characterization in primary cells to determine the impact of different forms of bias in a native context, and finally testing in preclinical animal models to understand the physiologic impact of ligand bias, will be needed to empirically determine the most effective form of bias and the degree of bias factor necessary to translate in vitro efficacy to the in vivo state.
Acknowledgments
The authors thank E. L. Stahl and R. C. Tanner for critical reading of the manuscript.
Abbreviations
- AngII
angiotensin II
- GPCR
G protein–coupled receptor
- KOR
κ opioid receptor
- MOR
μ opioid receptor
- PTH
parathyroid hormone
- TRV-130
[(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bohn, Luttrell, Maudsley.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM095497], the National Institute of Diabetes, Digestive, and Kidney Diseases [Grant R01-DK055524], the Intramural Research Program of the National Institute on Aging, the National Institute on Drug Abuse [Grants R01-DA031927, R01-DA033073, P01-DA009158, and R01-DA038964], and the Research Service of the Charleston, SC Veterans Affairs Medical Center. The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjølbye AL, Sheikh SP, Hansen JL. (2007) Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin Pharmacol Toxicol 100:296–301. [DOI] [PubMed] [Google Scholar]
- Appleton KM, Luttrell LM. (2013) Emergent biological properties of arrestin pathway-selective biased agonism. J Recept Signal Transduct Res 33:153–161. [DOI] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Piñeyro G. (2003) Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA 100:11406–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, Kelvin DJ. (2000) Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat Immunol 1:227–233. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. (2005) An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122:261–273. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. (2004) Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA 101:5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. (1998) Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol 54:94–104. [PubMed] [Google Scholar]
- Bhattacharya M, Anborgh PH, Babwah AV, Dale LB, Dobransky T, Benovic JL, Feldman RD, Verdi JM, Rylett RJ, Ferguson SS. (2002) Beta-arrestins regulate a Ral-GDS Ral effector pathway that mediates cytoskeletal reorganization. Nat Cell Biol 4:547–555. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. (1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220:141–162 Biol. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P, Shankley NP, Wood J. (1985) An operational model of pharmacological agonism: the effect of E/[A] curve shape on agonist dissociation constant estimation. Br J Pharmacol 84:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. (2000) Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408:720–723. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Raehal KM. (2006) Opioid receptor signaling: relevance for gastrointestinal therapy. Curr Opin Pharmacol 6:559–563. [DOI] [PubMed] [Google Scholar]
- Bonnans C, Flacelière M, Grillet F, Dantec C, Desvignes JP, Pannequin J, Severac D, Dubois E, Bibeau F, Escriou V, et al. (2012) Essential requirement for β-arrestin2 in mouse intestinal tumors with elevated Wnt signaling. Proc Natl Acad Sci USA 109:3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem 281:18081–18089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. (2006) Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA 103:1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbogne P, Kieffer BL, Befort K. (2014) 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology 76 (Pt B):204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Martin B, Daimon CM, Maudsley S. (2013b) Effective use of latent semantic indexing and computational linguistics in biological and biomedical applications. Front Physiol 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Martin B, Daimon CM, Siddiqui S, Luttrell LM, Maudsley S. (2013a) Textrous!: extracting semantic textual meaning from gene sets. PLoS One 8:e62665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen GL, Kelstrup CD, Lyngsø C, Sarwar U, Bøgebo R, Sheikh SP, Gammeltoft S, Olsen JV, Hansen JL. (2010) Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics 9:1540–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol Rev 54:323–374. [DOI] [PubMed] [Google Scholar]
- Chun KS, Lao HC, Trempus CS, Okada M, Langenbach R. (2009) The prostaglandin receptor EP2 activates multiple signaling pathways and beta-arrestin1 complex formation during mouse skin papilloma development. Carcinogenesis 30:1620–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Megson AE, McDonnell J, Dickenson JM, Hill SJ. (2000) Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A(1) receptor: evidence for signaling pathway-dependent changes in agonist potency and relative intrinsic activity. Mol Pharmacol 58:1075–1084. [DOI] [PubMed] [Google Scholar]
- Costanzi S. (2014) Modeling G protein-coupled receptors in complex with biased agonists. Trends Pharmacol Sci 35:277–283. [DOI] [PubMed] [Google Scholar]
- Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, Chellappan S. (2006) Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest 116:2208–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Vaughn ZD, O’Bryan EM, Nishijima D, Déry O, Bunnett NW. (2000a) The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc Natl Acad Sci USA 97:11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. (2000b) beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 148:1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng ZL, Sharff KA, Tang N, Song WX, Luo J, Luo X, Chen J, Bennett E, Reid R, Manning D, et al. (2008) Regulation of osteogenic differentiation during skeletal development. Front Biosci 13:2001–2021. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, et al. (2013) A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344:708–717. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. (2005) Analysis of allosterism in functional assays. J Pharmacol Exp Ther 315:740–754. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. (2008) On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn Schmiedebergs Arch Pharmacol 377:549–577. [DOI] [PubMed] [Google Scholar]
- Fereshteh M, Ito T, Kovacs JJ, Zhao C, Kwon HY, Tornini V, Konuma T, Chen M, Lefkowitz RJ, Reya T. (2012) β-Arrestin2 mediates the initiation and progression of myeloid leukemia. Proc Natl Acad Sci USA 109:12532–12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS. (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev 53:1–24. [PubMed] [Google Scholar]
- Galandrin S, Bouvier M. (2006) Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol 70:1575–1584. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, et al. (2006) Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem 281:10856–10864. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, Luttrell LM. (2009) A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med 1:1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesty-Palmer D, Yuan L, Martin B, Wood WH, 3rd, Lee MH, Janech MG, Tsoi LC, Zheng WJ, Luttrell LM, Maudsley S. (2013) β-arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol Endocrinol 27:296–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Roth BL. (2001) Paradoxical trafficking and regulation of 5-HT(2A) receptors by agonists and antagonists. Brain Res Bull 56:441–451. [DOI] [PubMed] [Google Scholar]
- Griffin MT, Figueroa KW, Liller S, Ehlert FJ. (2007) Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J Pharmacol Exp Ther 321:1193–1207. [DOI] [PubMed] [Google Scholar]
- Gullapalli S, Ramarao P. (2002) Role of L-type Ca(2+) channels in pertussis toxin induced antagonism of U50,488H analgesia and hypothermia. Brain Res 946:191–197. [DOI] [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ, Thomas WG. (2002) Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol 61:768–777. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Costanzi S. (2012) New insights for drug design from the X-ray crystallographic structures of G-protein-coupled receptors. Mol Pharmacol 82:361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LQ, Wang HY, Friedman E. (2001) Stimulated D(1) dopamine receptors couple to multiple Galpha proteins in different brain regions. J Neurochem 78:981–990. [DOI] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. (1995) Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci 16:232–238. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2007) Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol 72:1393–1401. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. (2009) '7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci 30:460–469. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2014) Quantifying biased β-arrestin signaling. Handbook Exp Pharmacol 219:57–83. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 3:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall RT, Strungs EG, Rachidi SM, Lee M-H, El-Shewy HM, Janech MG, Luttrell DK, Luttrell LM. (2011 The beta-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. J Biol Chem 286:19880–19891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. (2004) Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem 279:23214–23222. [DOI] [PubMed] [Google Scholar]
- Lakshmikanthan V, Zou L, Kim JI, Michal A, Nie Z, Messias NC, Benovic JL, Daaka Y. (2009) Identification of betaArrestin2 as a corepressor of androgen receptor signaling in prostate cancer. Proc Natl Acad Sci USA 106:9379–9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. (2008) The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci 28:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE, Schultz G. (1996) The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc Natl Acad Sci USA 93:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TT, Alemayehu M, Aziziyeh AI, Pape C, Pampillo M, Postovit L-M, Mills GB, Babwah AV, Bhattacharya M. (2009) Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol Cancer Res 7:1064–1077. [DOI] [PubMed] [Google Scholar]
- Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW. (2006) Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord 7:1–16. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335:1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Long J, Zhang PH, Li K, Tan JJ, Sun B, Yu J, Tu ZG, Zou L. (2011) Elevated β-arrestin1 expression correlated with risk stratification in acute lymphoblastic leukemia. Int J Hematol 93:494–501. [DOI] [PubMed] [Google Scholar]
- Lovell KM, Frankowski KJ, Stahl EL, Slauson SR, Yoo E, Prisinzano TE, Aubé J, Bohn LM. (2015) Structure activity relationship studies of functionally selective kappa opioid receptor (KOR) agonists that modulate ERK 1/2 phosphorylation while preserving G protein over βArrestin2 signaling bias. ACS Chem Neurosci, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM. (2013) Arrestin pathways as drug targets. Prog Mol Biol Transl Sci 118:469–497. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, et al. (1999) Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283:655–661. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Gesty-Palmer D. (2010) Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev 62:305–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. (2002) The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115:455–465. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. (2001) Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA 98:2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon MJ, Donowitz M, Yun CC, Segre GV. (2002) Na(+)/H(+ ) exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signalling. Nature 417:858–861. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Gent JP, Findlay JB, Donnelly D. (1998) The relationship between the agonist-induced activation and desensitization of the human tachykinin NK2 receptor expressed in Xenopus oocytes. Br J Pharmacol 124:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley S, Martin B, Gesty-Palmer D, Cheung H, Johnson C, Patel S, Becker KG, Wood WH, 3rd, Zhang Y, Lehrmann E, et al. (2015a) Delineation of a conserved arrestin-biased signaling repertoire in vivo. Mol Pharmacol 87:706–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley S, Martin B, Janssens J, Etienne H, Jushaj A, van Gastel J, Willemsen A, Chen H, Gesty-Palmer D, Luttrell LM. (2015b) Informatic deconvolution of biased GPCR signaling mechanisms from in vivo pharmacological experimentation. Methods, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley S, Martin B, Luttrell LM. (2005) The origins of diversity and specificity in g protein-coupled receptor signaling. J Pharmacol Exp Ther 314:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley S, Patel SA, Park SS, Luttrell LM, Martin B. (2012) Functional signaling biases in G protein-coupled receptors: Game Theory and receptor dynamics. Mini Rev Med Chem 12:831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin F-T, Davis RJ, Lefkowitz RJ. (2000) Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290:1574–1577. [DOI] [PubMed] [Google Scholar]
- Mohan S, Kutilek S, Zhang C, Shen HG, Kodama Y, Srivastava AK, Wergedal JE, Beamer WG, Baylink DJ. (2000) Comparison of bone formation responses to parathyroid hormone(1-34), (1-31), and (2-34) in mice. Bone 27:471–478. [DOI] [PubMed] [Google Scholar]
- Moussa O, Ashton AW, Fraig M, Garrett-Mayer E, Ghoneim MA, Halushka PV, Watson DK. (2008) Novel role of thromboxane receptors beta isoform in bladder cancer pathogenesis. Cancer Res 68:4097–4104. [DOI] [PubMed] [Google Scholar]
- Nasman J, Kukkonen JP, Ammoun S, Akerman KE. (2001) Role of G-protein availability in differential signaling by α 2-adrenoceptors. Biochem Pharmacol 62:913–922. [DOI] [PubMed] [Google Scholar]
- Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ. (2007) Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science 315:663–666. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Wieland T, Homann D, Sandmann J, Bombien E, Spicher K, Schultz G, Jakobs KH. (1994) Transfected muscarinic acetylcholine receptors selectively couple to Gi-type G proteins and Gq/11. Mol Pharmacol 45:890–898. [PubMed] [Google Scholar]
- Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, et al. (2002) Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298:834–836. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233:774–776. [DOI] [PubMed] [Google Scholar]
- Qin L, Raggatt LJ, Partridge NC. (2004) Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol Metab 15:60–65. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. (2011) The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM. (2011) Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev 63:1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. (2005) Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther 314:1195–1201. [DOI] [PubMed] [Google Scholar]
- Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA, Lefkowitz RJ. (2006) Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA 103:16284–16289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redila VA, Chavkin C. (2008) Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 200:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosanò L, Cianfrocca R, Masi S, Spinella F, Di Castro V, Biroccio A, Salvati E, Nicotra MR, Natali PG, Bagnato A. (2009) Beta-arrestin links endothelin A receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc Natl Acad Sci USA 106:2806–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagan S, Chassaing G, Pradier L, Lavielle S. (1996) Tachykinin peptides affect differently the second messenger pathways after binding to CHO-expressed human NK-1 receptors. J Pharmacol Exp Ther 276:1039–1048. [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. (1993) A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem 268:4625–4636. [PubMed] [Google Scholar]
- Saulière A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altié MF, Seguelas MH, Pathak A, Hansen JL, et al. (2012) Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol 8:622–630. [DOI] [PubMed] [Google Scholar]
- Schann S, Bouvier M, Neuville P. (2013) Technology combination to address GPCR allosteric modulator drug-discovery pitfalls. Drug Discov Today Technol 10:e261–e267. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Bohn LM. (2010) Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ß-arrestin2/Src/Akt signaling complex in vivo. J Neurosci 30:13513–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Raehal KM, Bohn LM. (2008) Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proc Natl Acad Sci USA 105:1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. (2013) Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at κ-opioid receptors in striatal neurons. J Biol Chem 288:22387–22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian EM, Suva LJ, Friedman PA. (2008) Differential effects of intermittent PTH(1-34) and PTH(7-34) on bone microarchitecture and aortic calcification in experimental renal failure. Bone 43:1022–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. (2001) Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 294:1307–1313. [DOI] [PubMed] [Google Scholar]
- Sneddon WB, Magyar CE, Willick GE, Syme CA, Galbiati F, Bisello A, Friedman PA. (2004) Ligand-selective dissociation of activation and internalization of the parathyroid hormone (PTH) receptor: conditional efficacy of PTH peptide fragments. Endocrinology 145:2815–2823. [DOI] [PubMed] [Google Scholar]
- Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. (2014) Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155:1829–1835. [DOI] [PubMed] [Google Scholar]
- Stahl EL, Zhou L, Ehlert FJ, Bohn LM. (2015) A novel method for analyzing extremely biased agonism at G protein-coupled receptors. Mol Pharmacol 87:866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminath G, Deupi X, Lee TW, Zhu W, Thian FS, Kobilka TS, Kobilka B. (2005) Probing the β2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem 280:22165–22171. [DOI] [PubMed] [Google Scholar]
- Takasu H, Gardella TJ, Luck MD, Potts JT, Jr, Bringhurst FR. (1999) Amino-terminal modifications of human parathyroid hormone (PTH) selectively alter phospholipase C signaling via the type 1 PTH receptor: implications for design of signal-specific PTH ligands. Biochemistry 38:13453–13460. [DOI] [PubMed] [Google Scholar]
- Togashi Y, Umeuchi H, Okano K, Ando N, Yoshizawa Y, Honda T, Kawamura K, Endoh T, Utsumi J, Kamei J, et al. (2002) Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol 435:259–264. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. (2015) Endogenous allosteric modulators of G protein-coupled receptors. J Pharmacol Exp Ther 353:246–260. [DOI] [PubMed] [Google Scholar]
- Van’t Veer A, Carlezon WA., Jr (2013) Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 229:435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, Chen JR, Muehlbauer MJ, Whalen EJ, Lefkowitz RJ. (2009) beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest 119:1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik S, Hunyady L, Luttrell LM, Lefkowitz RJ. (2003) Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100:10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. (1996) The cubic ternary complex receptor-occupancy model. III. resurrecting efficacy. J Theor Biol 181:381–397. [DOI] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 17:126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther C, Ferguson SS. (2015) Minireview: role of intracellular scaffolding proteins in the regulation of endocrine g protein-coupled receptor signaling. Mol Endocrinol 29:814–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. (2015) The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352:98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Scopton AP, Rives ML, Bikbulatov RV, Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, Roth BL. (2014) Identification of novel functionally selective κ-opioid receptor scaffolds. Mol Pharmacol 85:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. (2004) beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci USA 101:8603–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, Farrens D, Kobilka B. (2006) Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol 2:417–422. [DOI] [PubMed] [Google Scholar]
- Yao XJ, Vélez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, Kobilka B. (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci USA 106:9501–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, et al. (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem 288:36703–36716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Gilbert S, Birnbaumer M, Birnbaumer L. (1994) Dual signaling potential is common among Gs-coupled receptors and dependent on receptor density. Mol Pharmacol 46:460–469. [PubMed] [Google Scholar]
- Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci USA 106:9649–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, Bouvier M, Laporte SA. (2012) Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci Signal 5:ra33. [DOI] [PubMed] [Google Scholar]
- Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. (2007) Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem 282:20634–20646. [DOI] [PubMed] [Google Scholar]