

Abstract
By exploring a new mode of Ni-catalyzed cross-coupling, we have developed a protocol to transform both aromatic and aliphatic aldehydes into either esters or amides directly. The success of this oxidative coupling depends on the appropriate choice of catalyst and organic oxidant, including the use of either α,α,α-trifluoroacetophenone or excess aldehyde. We present mechanistic data that supports a catalytic cycle involving oxidative addition into the aldehyde C–H bond.
Keywords: dehydrogenation, cross-coupling, nickel catalysis, amides, esters
Developing nickel catalysts for the selective oxidation of C–H bonds is an emerging strategy that overcomes the need for precious metals and enables new transformations.[1] Our laboratory has previously focused on the oxidation of aldehydes under Rh, Ru, and Co-catalysis.[2] Inspired by the promise of base metals, we considered that Ni-catalyzed cross-coupling of aldehydes could be classified into three general types: redox neutral, reductive, and oxidative (Figure 1). For example, Ogoshi's cross-coupling between two aldehydes is a redox neutral method that generates ester bonds.[3] In comparison, reductive coupling reactions generate carbon-carbon bonds in the presence of an external reductant (e.g., Et2Zn).[4] A complementary Ni-catalyzed cross-coupling in the presence of an external oxidant, however, represents a mode of reactivity that warrants study.[5] Herein, we showcase a unified approach for transforming aldehyde C–H bonds into both C–O and C–N bonds using Ni-catalyzed dehydrogenative cross-coupling.[6]
Figure 1.

Three general modes of Ni-catalyzed cross-coupling with aldehydes.
The transformation of aldehydes to esters and amides in onestep is an attractive goal that has been pursued using precious metal-catalysts,[7] base-metal catalysts with peroxide oxidants or elevated temperatures,[8] and NHC-catalysis.[9] While these methods are promising, a protocol that couples aromatic and aliphatic aldehydes with alcohols, anilines, and amines has yet to be achieved. Towards this challenge, we chose to examine carbonyl compounds as mild oxidants by hydrogen transfer.[10]
Our initial studies focused on cross-coupling benzaldehyde (1a) and isopropanol (2a) in the presence of various hydrogen acceptors. We discovered that NHC ligands in 1,4-dioxane produced the most promising results (Table 1).[11] In the absence of any Ni-salts, we observed no reactivity. However, in the presence of nickel with benzaldehyde as both the substrate and hydrogen acceptor we observed the desired ester 4a and Tishchenko homodimer 5a in a 5.9:1 ratio. To suppress the Tishchenko pathway, we sought an acceptor that undergoes reduction faster than benzaldehyde (1a). While acetone (3a) and cyclobutanone (3b) decreased the rate of the desired crosscoupling, both benzophenone (3c) and α,α,α- trifluoroacetophenone (3d) showed a remarkable enhancement in rate and selectivity for 4a. As a result, we were able to use equimolar quantities of the coupling partners and 1.1 equivalents of oxidant 3d, with no observation of dimer 5a. Related oxidations typically require excess alcohol,[12] or alcohol as the solvent.[13]
Table 1.
Examining organic hydrogen acceptors for oxidative esterification.[a]

| ||||
|---|---|---|---|---|
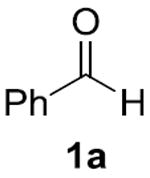
|
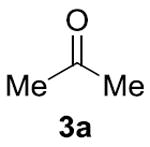
|

|

|
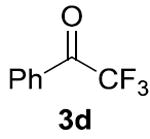
|
| 47% (5.9:1) | 35% (4.4:1)[c] | 12% (2.4:1) | 78% (8.7:1)[e] | 95% (48:1)[f] |
| 98% (5.5:1)[b] | 10% (>99:1)[c, d] | |||
Conditions: 0.1 mmol 1a, 0.1 mmol 2a, 0.2 M. Yields were determined by GC analysis using dodecane as an internal standard. IPr = 1,3-Bis(2,6- diisopropylphenyl)imidazole-2-ylidene. Yield of product 4a is shown, and the ratio of 4a:5a is shown in parentheses.
Using 2.5 equivalents of 1a.
Reaction performed at 80 °C.
Using 100 equivalents of 3a.
Using 3 equivalents of 3c.
Reaction performed at 30 °C.
Under our optimized conditions, aromatic aldehydes couple with primary (4b, 97%), secondary (4c, 98%) and tertiary (4d, 79%) alcohols at 30 °C (Table 2). Our protocol is the first intermolecular oxidative esterification to achieve high yields using only one equivalent of tertiary alcohol nucleophiles.[14] Comparatively less nucleophilic partners such as benzyl alcohol also work well (4e, 96%). Both electron rich (4f, 96%) and electron deficient (4g, 72%) aromatic aldehydes can be transformed into esters. While many NHC-catalyzed esterification reactions are limited to aromatic aldehyde substrates,[15] we found that aliphatic aldehydes undergo oxidative functionalization with Ni-catalysis. Citronellal, a natural product, can be readily converted to hindered ester 4h (83%), or methyl ester 4i (97%). Hindered α-branched aldehydes are also well tolerated (4j, 91%).
Table 2.
Dehydrogenative cross-coupling of aldehydes and alcohols.[a]
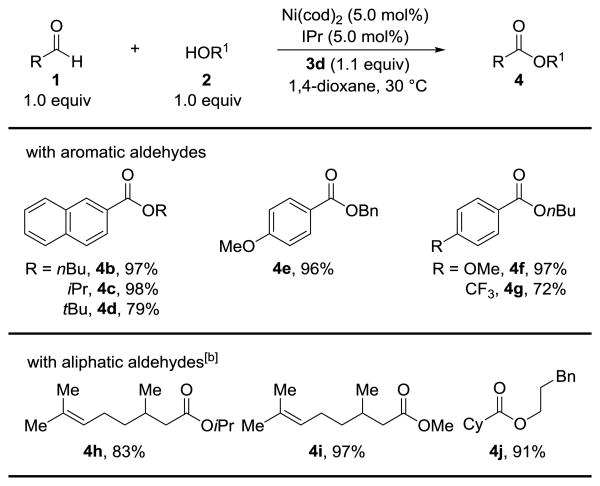
|
Conditions: 0.5 mmol 1, 0.5 mmol 2, 0.2 M, 8 h. Isolated yields are shown.
Reactions performed using 10 mol% of the catalyst.
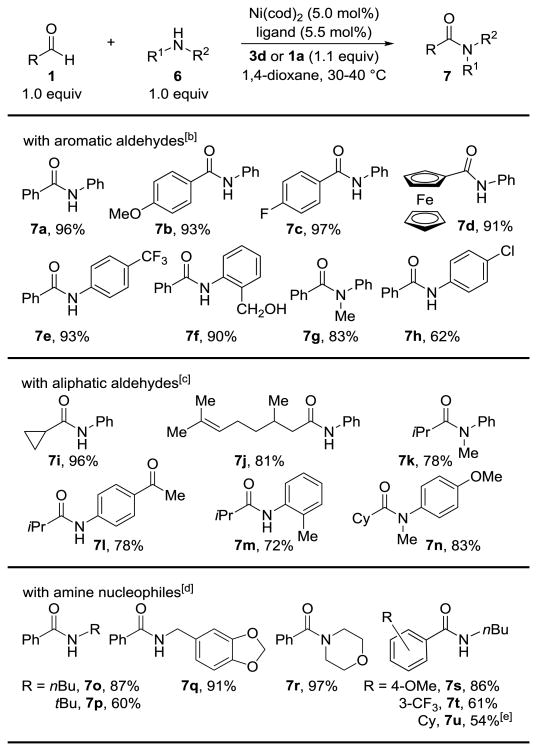
In principle, coupling amines should present a greater challenge due to the possibility of condensation or catalyst inhibition. However, we found that using the same protocol at slightly elevated temperatures (40 °C) amide bond formation was observed using aniline nucleophiles (Table 3). Thus, we can convert aldehydes to amides using base metal catalysis without relying on highly reactive reagents, such as peroxide[8] or arylazide[16] substrates. Under these conditions, aldehydes containing electron donating (7b, 93%) or withdrawing (7c, 97%) groups react efficiently, as well as aldehydes which would be sensitive to peroxide oxidants (7d, 91%).[17] Aniline nucleophiles with electron withdrawing (7e, 93%) and electron donating (7f, 90%) groups undergo coupling with similar efficiency. Hindered anilines are suitable partners, including those with orthosubstitution (7f, 90%) or N-substitution (7g, 83%). Few byproducts are observed in these transformations, even in the presence of an aryl chloride (7h, 62%).
Table 3.
Dehydrogenative cross-coupling of aldehydes and amines.[a]
|
Conditions: 0.5 mmol 1, 0.5 mmol 6, 0.2 M, 8 h. Isolated yields are shown.
The reactions were performed at 40 °C using IPr as the ligand and 3d as the hydrogen acceptor.
The reactions were performed at 30 °C using IPr as a ligand and 3d as the hydrogen acceptor.
The reactions were performed at 40 °C using ItBu as the ligand and an extra equivalent of the aldehyde as the hydride acceptor.
Using 1a rather than cyclohexanecarboxaldehyde as the hydrogen acceptor.
Aliphatic aldehydes are excellent coupling partners for amide synthesis (Table 3). At 30 °C the reaction proceeds to full conversion with cyclopropyl carboxaldehyde without any ring opening (7i, 96%). β-Branching (7j, 81%) is well tolerated, albeit pivaldehyde only provided the N-phenyl amide in 36% yield (not shown). An aniline with a ketone in the para position gives the desired amide (7l) in 78% yield illustrating that ketone functional groups are tolerated despite the transfer hydrogenation nature of this reaction. Hindered amide 7m (72%) and tertiary amides 7k (78%) and 7n (83%) highlight the efficiency of this transformation.
In preliminary studies that focused on amine nucleophiles the reactions were plagued by condensation side products. To avoid condensation with ketone 3d, we used a second equivalent of the aldehyde as the oxidant. We were also able to suppress Ni(cod)2 catalyzed condensation between the amine and the aldehyde by using ItBu as the ligand and a Ni:ligand ratio of 1:1.1 (Table 3).[11] Under these conditions the dehydrogenative coupling proceeds with primary alkyl amines (7o, 87%), hindered amines (7p, 60%), benzylic amines (7q, 91%), or even cyclic amines (7r, 97%). The synthesis of 7r represents a facile way to convert aldehydes to morpholine amides that are versatile reagents for further functionalizations.[18] Amidation with amine nucleophiles is not limited to benzaldehyde substrates and can be extended to electron donating (7s, 86%) or electron withdrawing (7s, 61%) aldehydes by using two equivalents of the aldehyde. Aliphatic aldehydes can also be converted to N-alkyl amides by using benzaldehyde as an inexpensive oxidant (7u, 97%).
To understand the selectivity between nucleophiles we performed a competition experiment between aniline (6a) and butanol (2b) with benzaldehyde (1a) (scheme 1, equation 1). The N-aryl amide could be formed in preference to the ester in a 16:1 ratio and 86% overall yield. Performing a similar experiment between butanol (2b) and N-butyl amine (6o) (scheme 1, equation 2), revealed a preference for amides in a 24:1 ratio, and 72% yield with respect to nBuNH2.
Scheme 1.

Exploring selectivity for amidation over esterification.
Towards elucidating the mechanism, we performed a stoichiometric experiment between NiIPr, 2-naphthaldehyde (1b), and ketone 3d. In this experiment, we observed reduction of the ketone and decarbonylation of the aldehyde at 30 °C (Scheme 2, eq 3). This result supports the intermediacy of an acyl Ni-hydride species which can undergo decarbonylation in the absence of a coupling partner. Moreover, a KIE measurement of 6.0 suggests that C–H bond activation is rate determining (Scheme 2, eq 4).[19]
Scheme 2.

Mechanism experiments.
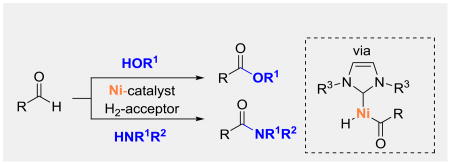
On the basis of initial studies and literature reports, we propose the mechanism shown in Scheme 3.[20] Ketone 3d binds to nickel to form complex 8a, which can coordinate to an aldehyde (1) to give intermediate 8b. Oxidative addition to the aldehyde C–H bond generates 8c which reduces the hydrogen acceptor 3d to yield acyl-nickel alkoxide 8d.[21] Ligand exchange with the nucleophile affords 8e, and reductive elimination provides the final product. Coordination of the ketone 3d may occur prior to,[22] or immediately after reductive elimination.[23]
Scheme 3.

Proposed mechanism for Ni-catalyzed cross dehydrogenative coupling via C–H bond activation.
Computational studies by Fu support the concept of electron deficient π-ligands on nickel promoting oxidative addition into aldehyde C–H bonds.[24] As empirical support for this claim we found that there was a significant increase in the reaction rate when using ketone 3d when compared to aldehyde 1a as the hydrogen acceptor.[11] We propose that strong binding of the ketone accelerates oxidative addition.[25] This coordination also inhibits the Tishchenko byproduct by disrupting the formation of a cycloisomerization intermediate between two aldehydes.[26]
We have communicated a novel nickel-catalyzed dehydrogenative cross-coupling of aldehydes. Our method generates hindered esters and provides a needed complement to NHC catalyzed oxidative esterification. Secondary or tertiary amides can be accessed using both aniline and amine nucleophiles in a unified approach. The discovery of 3d as an oxidant allows cross-couplings to proceed at 30–40 °C in nearly equimolar amounts of aldehyde, nucleophile, and acceptor. Future studies will include the use of Ni(II) precatalysts[11] and nucleophiles that can result in C-C bond formation. We are also currently investigating the reaction mechanism in greater detail. Finally, the insights from this study will contribute to the expanding field of Ni-catalyzed transfer hydrogenation.[27]
Supplementary Material
Communication.
Nickel for a change
By exploring a new mode of Ni-catalyzed cross-coupling, we have developed a protocol to transform both aromatic and aliphatic aldehydes into either esters or amides directly. The success of this oxidative coupling depends on the appropriate choice of catalyst and organic oxidant, including the use of either α,α,α-trifluoroacetophenone or benzaldehyde. We present mechanistic data that supports a catalytic cycle involving oxidative addition into the aldehyde C–H bond.
Acknowledgments
Funding was provided by UC Irvine and the National Institutes of Health (GM105938). We acknowledge David George for preliminary screens with aliphatic aldehydes and Sarah Kurtoic for purification of products 7h, 7m, and 7n.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Select examples: Liu D, Liu C, Li H, Lei A. Angew Chem. 2013;125:4549.; Angew Chem Int Ed. 2013;52:4453.; Shiota H, Ano Y, Aihara Y, Fukumoto Y, Chatani N. J Am Chem Soc. 2011;133:14952. doi: 10.1021/ja206850s.; Ogata K, Atsuumi Y, Shimada D, Fukuzawa SI. Angew Chem. 2011;123:6018. doi: 10.1002/anie.201101468.; Angew Chem Int Ed. 2011;50:5896.; Nakao Y, Idei H, Kanyiva KS, Hiyama T. J Am Chem Soc. 2009;131:5070. doi: 10.1021/ja901153s.; Donets PA, Cramer N. J Am Chem Soc. 2013;135:11772. doi: 10.1021/ja406730t.; Aihara Y, Chatani N. J Am Chem Soc. 2014;136:898. doi: 10.1021/ja411715v.; Bair JS, Schramm Y, Sergeev AG, Clot E, Eisenstein O, Hartwig JF. J Am Chem Soc. 2014;136:13098. doi: 10.1021/ja505579f..
- 2.a) Kou KGM, Le DN, Dong VM. J Am Chem Soc. 2014;136:9471. doi: 10.1021/ja504296x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oldenhuis NJ, Guan Z, Dong VM. Tetrahedron. 2014;70:4213. doi: 10.1016/j.tet.2014.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Murphy SK, Dong VM. J Am Chem Soc. 2013;135:5553. doi: 10.1021/ja4021974. [DOI] [PubMed] [Google Scholar]; d) Chen QA, Kim DK, Dong VM. J Am Chem Soc. 2014;136:3772. doi: 10.1021/ja500268w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoshimoto Y, Ohashi M, Ogoshi S. J Am Chem Soc. 2011;133:4668. doi: 10.1021/ja109908x.; Related reactions: Hoshimoto Y, Hayashi Y, Suzuki H, Ohashi M, Ogoshi S. Angew Chem. 2012;124:10970. doi: 10.1002/anie.201206186.; Angew Chem Int Ed. 2012;51:10812.; Taniguchi H, Ohmura T, Suginome M. J Am Chem Soc. 2009;131:11298. doi: 10.1021/ja9046894.; Tekavec TN, Louie J. J Org Chem. 2008;73:2641. doi: 10.1021/jo702508w.; Tsuda T, Kiyoi T, Saegusa T. J Org Chem. 1990;55:2554.; Ogoshi S, Hoshimoto Y, Ohashi M. Chem Commun. 2010;46:3354. doi: 10.1039/b926866a..
- 4.Recent reviews: Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274.; Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun. 2007:4441. doi: 10.1039/b707737h.; Kimura M, Tamaru Y. Top Curr Chem. 2007;279:173.; Iida H, Krische MJ. Top Curr Chem. 2007;279:77.; Montgomery J. Top Curr Chem. 2007;279:1.; Montgomery J. Angew Chem. 2004;116:3980.; Angew Chem Int Ed. 2004;43:3890..
- 5.Montgomery reported a redox neutral Ni-catalyzed transformation of α,β-unsaturated aldehydes to methyl esters. Herath A, Li W, Montgomery J. J Am Chem Soc. 2008;130:469. doi: 10.1021/ja0781846.; Buchwald has noted Ni-catalyzed amide formation as a side reaction (<26% yield) in the cross-coupling of 4-chlorobenzaldehyde and pyrrolidine: Wolfe JP, Buchwald SL. J Am Chem Soc. 1997;119:6054..
- 6.Two-step, one-pot procedures: Ueda T, Konishi H, Manabe K. Org Lett. 2013;15:5370. doi: 10.1021/ol4026815.; Iwahana S, Iida H, Yashima E. Chem Eur J. 2011;17:8009. doi: 10.1002/chem.201100737.; De Sarkar S, Studer A. Org Lett. 2010;12:1992. doi: 10.1021/ol1004643..
- 7.Select examples of oxidative esterification: Liu C, Tang S, Lei A. Chem Commun. 2013;49:1324. doi: 10.1039/c2cc38086b.; Liu C, Tang S, Zheng L, Liu D, Zhang H, Lei A. Angew Chem. 2012;124:5760. doi: 10.1002/anie.201201960.; Angew Chem Int Ed. 2012;51:5662.; Liu C, Wang J, Meng L, Deng Y, Li Y, Lei A. Angew Chem. 2011;123:5250. doi: 10.1002/anie.201008073.; Angew Chem Int Ed. 2011;50:5144.; Gowrisankar S, Neumann H, Beller M. Angew Chem. 2011;123:5245. doi: 10.1002/anie.201008035.; Angew Chem Int Ed. 2011;50:5139.; Select examples of oxidative amidation: Zhou B, Yang Y, Shi J, Feng H, Li Y. Chem Eur J. 2013;19:10511. doi: 10.1002/chem.201301168.; Gunanathan C, Ben-David Y, Milstein D. Science. 2007;790:317. doi: 10.1126/science.1145295.; Ekoue-Kovi K, Wolf C. Org Lett. 2007;9:3429. doi: 10.1021/ol7014626.; Reviews: Tang S, Yuan J, Liua C, Lei A. Dalton Trans. 2014;43:13460. doi: 10.1039/c4dt01133c.; Miyamura H, Kobayashi S. Acc Chem Res. 2014;47:1054. doi: 10.1021/ar400224f.; Milstein D. Top Catal. 2010;53:915.; Ekoue-Kovi K, Wolf C. Chem Eur J. 2008;14:6302. doi: 10.1002/chem.200800353..
- 8.Select examples: Wang L, Priebbenow DL, Zou LH, Bolm C. Adv Synth Catal. 2013;355:1490.; Wu XF, Sharif M, Pews- Davtyan A, Langer P, Ayub K, Beller M. Eur J Org Chem. 2013:2783.; Zhu M, Fujita KI, Yamaguchi R. J Org Chem. 2012;77:9102. doi: 10.1021/jo301553v.; Yoo WJ, Li CJ. J Am Chem Soc. 2006;128:13064. doi: 10.1021/ja064315b.; For related reactions that use strong oxidants but are metal free see: Liu Z, Zhang J, Chen S, Shi E, Xu Y, Wan X. Angew Chem. 2012;124:3285. doi: 10.1002/anie.201108763.; Angew Chem Int Ed. 2012;51:3231.; Goh KS, Tan CH. RSC Adv. 2012;2:5536..
- 9.Select examples: Bera S, Samanta RC, Daniliuc CG, Studer A. Angew Chem. 2014;126:9776. doi: 10.1002/anie.201405200.; Angew Chem Int Ed. 2014;53:9622.; Finney EE, Ogawa KA, Boydston AJ. J Am Chem Soc. 2012;134:12374. doi: 10.1021/ja304716r.; Lee K, Kim H, Hong J. Angew Chem. 2012;124:5833. doi: 10.1002/anie.201201653.; Angew Chem Int Ed. 2012;51:5735.; De Sarkar S, Grimme S, Studer A. J Am Chem Soc. 2010;132:1190. doi: 10.1021/ja910540j.; Guin J, De Sarkar S, Grimme S, Studer A. Angew Chem. 2008;120:8855. doi: 10.1002/anie.200802735.; Angew Chem Int Ed. 2008;47:8727.; Reddy RS, Rosa JN, Veiros LF, Caddick S, Gois PMP. Org Biomol Chem. 2011;9:3126. doi: 10.1039/c1ob05151b.; For oxidative amidation with NHC catalysis, see ref 6 and an umpolung method employing nitroso compounds: Wong FT, Patra PK, Seayad J, Zhang Y, Ying JY. Org Lett. 2008;10:2333. doi: 10.1021/ol8004276.; Reviews: Mahatthananchai J, Bode JW. Acc Chem Res. 2014;47:696. doi: 10.1021/ar400239v.; De Sarkar S, Biswas A, Samanta RC, Studer A. Chem-Eur J. 2013;19:4664. doi: 10.1002/chem.201203707..
- 10.a) Tschaen BA, Schmink JR, Molander GA. Org Lett. 2013;15:500. doi: 10.1021/ol303298g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dong ZR, Li YY, Yu SL, Sun GS, Gao JX. Chin Chem Lett. 2012;23:533. [Google Scholar]; c) Tang G, Cheng CH. Adv Synth Catal. 2011;353:1918. [Google Scholar]
- 11.See SI for details.
- 12.Zhang J, Leitus G, Ben-David Y, Milstein D. J Am Chem Soc. 2005;127:10840. doi: 10.1021/ja052862b. Ref 7–9. [DOI] [PubMed] [Google Scholar]
- 13.Jagadeesh RV, Junge H, Pohl MM, Radnik J, Brückner A, Beller M. J Am Chem Soc. 2013;135:10776. doi: 10.1021/ja403615c. Ref 7–9. [DOI] [PubMed] [Google Scholar]
- 14.For an NHC catalyzed method with an example of an isopropyl ester in good yield, see: Conan C, Baragwanath L, Connon SJ. Tetrahedron Lett. 2008;49:4003..
- 15.For a discussion of the aliphatic aldehyde challenges, see: Kuwano S, Harada S, Oriez R, Yamada K. Chem Commun. 2012;48:145. doi: 10.1039/c1cc15539c..
- 16.Jin LM, Lu H, Cui Y, Lizardi CL, Arzua TN, Wojtas L, Cui X, Zhang XP. Chem Sci. 2014;5:2422. doi: 10.1039/C4SC00697F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baciocchi E, Bietti M, Di Fusco M, Lanzalunga O. J Org Chem. 2007;72:8748. doi: 10.1021/jo071211q. [DOI] [PubMed] [Google Scholar]
- 18.Martin R, Romea P, Tey C, Urpi F, Vilarrasa J. Synlett. 1997:1414. [Google Scholar]
- 19.These values are beyond the typical Claisen-Tischenko range of 2.0- 3.0: Sharma M, Andrea T, Brookes NJ, Yates BF, Eisen MS. J Am Chem Soc. 2011;133:1341. doi: 10.1021/ja105696p.; Gallego MG, Sierra MA. Chem Rev. 2011;111:4857. doi: 10.1021/cr100436k..
- 20.In the absence of a nucleophile, we observe no ketone hydroacylation, and the transformation can be performed in EtOAc as a solvent with no crossover ruling out a Tishchenko-transesterification mechanism. See: Curran SP, Connon SJ. Org Lett. 2012;14:1074. doi: 10.1021/ol203439g..
- 21.In the case of amine nucleophiles 3d may be replaced with a second equivalent of 1a. This is possible according to reference 24a and consistent with the higher temperatures and longer reaction times.
- 22.Giovannini R, Knochel P. J Am Chem Soc. 1998;120:11186. [Google Scholar]
- 23.Johnson JB, Bercot EA, Rowley JM, Coates GW, Rovis T. J Am Chem Soc. 2007;129:2718. doi: 10.1021/ja067845g. [DOI] [PubMed] [Google Scholar]
- 24.Yu H, Fu Y. Chem Eur J. 2012;18:16765. doi: 10.1002/chem.201202623.; For a related computational paper, see: Kozuch S, Lee SE, Shaik S. Organometallics. 2009;28:1303..
- 25.We did not observe decarbonylation without the ketone present. See SI.
- 26.Ogoshi S, Arai T, Ohashi M, Kurosawa H. Chem Commun. 2008:1347. doi: 10.1039/b717261c. [DOI] [PubMed] [Google Scholar]
- 27.a) Nakai K, Yoshida Y, Kurahashi T, Matsubara S. J Am Chem Soc. 2014;136:7797. doi: 10.1021/ja500666h. [DOI] [PubMed] [Google Scholar]; b) Yang P, Xu H, Zhou J. Angew Chem. 2014;126:12406. [Google Scholar]; Angew Chem Int Ed. 2014;53:12210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.