

Abstract
Microcirculatory dysfunction may cause tissue malperfusion and progression to organ failure in the later stages of sepsis, but the role of smooth muscle contractile dysfunction is uncertain. Mice were given intraperitoneal LPS, and mesenteric arteries were harvested at 6-h intervals for analyses of gene expression and contractile function by wire myography. Contractile (myosin and actin) and regulatory [myosin light chain kinase and phosphatase subunits (Mypt1, CPI-17)] mRNAs and proteins were decreased in mesenteric arteries at 24 h concordant with reduced force generation to depolarization, Ca2+, and phenylephrine. Vasodilator sensitivity to DEA/nitric oxide (NO) and cGMP under Ca2+ clamp were increased at 24 h after LPS concordant with a switch to Mypt1 exon 24− splice variant coding for a leucine zipper (LZ) motif required for PKG-1α activation of myosin phosphatase. This was reproduced by smooth muscle-specific deletion of Mypt1 exon 24, causing a shift to the Mypt1 LZ+ isoform. These mice had significantly lower resting blood pressure than control mice but similar hypotensive responses to LPS. The vasodilator sensitivity of wild-type mice to DEA/NO, but not cGMP, was increased at 6 h after LPS. This was abrogated in mice with a redox dead version of PKG-1α (Cys42Ser). Enhanced vasorelaxation in early endotoxemia is mediated by redox signaling through PKG-1α but in later endotoxemia by myosin phosphatase isoform shifts enhancing sensitivity to NO/cGMP as well as smooth muscle atrophy. Muscle atrophy and modulation may be a novel target to suppress microcirculatory dysfunction; however, inactivation of inducible NO synthase, treatment with the IL-1 antagonist IL-1ra, or early activation of α-adrenergic signaling did not suppressed this response.
Keywords: myosin phosphatase, mesenteric artery, endotoxemia
sepsis is characterized by the induction of nitric oxide (NO) synthase [inducible NO synthase (iNOS)], oxidants, and cytokines that function to increase blood flow and recruitment of inflammatory cells to the site of infection. A vigorous endogenous response and early treatment are most critical to good patient outcomes (29, 41a). If unchecked, sepsis may progress to severe hypotension and shock with multiorgan failure. Despite intensive study in animal models and humans, the mortality for septic shock remains high, and therapy remains supportive with the administration of fluids and vasopressors such as norepinephrine and vasopressin (1, 23). Restoration of systemic blood pressure to “high” versus “low” levels (mean arterial pressures of 75–85 vs. 65–70 mmHg, respectively) did not affect patient outcomes (2), supporting the premise that microcirculatory dysfunction with tissue malperfusion is a major contributor to the poor outcomes in septic shock (12, 34, 47).
The etiology of the microcirculatory dysfunction in the later stages of sepsis is not well understood (9). A recent report (45) demonstrated a novel vasodilatory signaling pathway in the early stages of sepsis, oxidative activation of PKG-1α (45). Mice with a redox dead version of PKG-1α [mutation of cysteine 42 to serine (Cys42Ser)] had a suppressed early hypotensive response to endotoxin (LPS) or cecal ligation and puncture but exhibited a delayed response such that by 16–24 h they had hypotension similar to that of wild-type mice. We hypothesized that other changes in NO/PKG signaling or more generalized changes in arterial smooth muscle contractility may underlie the sustained vasodilation of the later stages of endotoxemia. One possibility is a general reduction in arterial smooth muscle contractility due to activation of atrophic pathways, as occurs in skeletal muscle in sepsis and other inflammatory conditions (4). Alternatively, the late hypotension of sepsis unaffected by PKG-1α Cys42Ser could reflect specific changes in NO/PKG or other vasodilator signaling pathways. Myosin phosphatase (MP) by dephosphorylation of myosin is the primary mediator of smooth muscle relaxation and a key target of NO and other signals that regulate vascular smooth muscle tone (11, 44). Expression of MP isoforms determines the sensitivity of smooth muscle to NO/PKG-mediated relaxation. Smaller resistance-type arteries predominately express the exon 24 (E24)-included isoform of the MP regulatory subunit of MP-targeting protein 1 (Mypt1), which codes for an isoform that lacks a COOH-terminal leucine zipper (LZ) motif (40, 55). This LZ motif is required for PKG-1α LZ-mediated heterodimerization and activation of MP (Refs. 21, 25, 43, and 51; for a review, see Ref. 17). The E24−/LZ+ isoform of Mypt1 predominates in the slow smooth muscle of the aorta and accounts for the ability of NO/PKG-1α to activate MP and relax aortic smooth muscle even under maximal Ca2+ concentrations, i.e., desensitization of force production to Ca2+. The expression of these isoforms is tissue specific and developmentally regulated (25, 39, 43) and modulates in disease (31, 40, 55) but has not been examined in models of endotoxemia.
In the present study, we examined MP subunits and other contractile proteins in relation to mesenteric arterial smooth muscle contractility in a mouse endotoxin model of endotoxemia. Endotoxin induced atrophy, as evidenced by reductions in contractile proteins and force generation, specific to small artery smooth muscle in the later stages of endotoxemia. NO/PKG-1 signaling was specifically modified by distinct mechanisms in early versus late endotoxemia. In the later stages (24 h) a switch to the Mypt1 E24−/LZ+ isoform increased sensitivity to NO- and cGMP-mediated Ca2+ desensitization of force production. Genetic deletion of E24 in smooth muscle phenocopied this effect, whereas PKG-1α mutation had no effect. At the early stage of endotoxemia (6 h), increased sensitivity to NO was abrogated in mice with the redox dead version of PKG-1α, whereas sensitivity to cGMP-mediated Ca2+ desensitization of force production was unaffected. This demonstrates different molecular mechanisms by which vasodilator reserve in resistance arteries is lost in early versus later stages of endotoxemia and suggests a target for abrogation of sustained vasodilation and malperfusion in later stages of endotoxemia. However, neither genetic inactivation of iNOS nor treatment of mice with the α1-adrenergic agonist phenylephrine (PE) or IL-1 receptor antagonist (IL-1ra) prevented the phenotypic and atrophic changes of resistance artery smooth muscle in the later stages of endotoxemia.
MATERIALS AND METHODS
Animal model.
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Maryland and adhered to National Institutes of Health guidelines. A total of 96 mice were used in this study. Adult male C57bl/6J mice (Jackson Laboratories) and iNOS knockout mice were purchased from Jackson Labs [Nos2tm1Lau/J, stock no. 002596 (27)]. SMMHCCreERT2//Mypt1 F/+ (heterozygotes) were generated as previously described (43). Briefly, LoxP sites were inserted to flank E24 of Mypt1 in the C57bl/6J background. Mice were bred into SMMHCCreERT2 mice (53). SMMHCCreERT2 mice without floxed alleles served as controls. SMMHCCreERT2//Mypt1 F/+ (heterozygotes; designated as Cre+//F/+) and SMMHCCreERT2//Mypt1 +/+ (controls; designated as Cre+//+/+) were injected with tamoxifen (50 mg/kg in sunflower oil, intraperitoneal) for 5 consecutive days at 3 wk of age for smooth muscle-specific deletion of Mypt1 E24 and studied at 8 wk of age. Adult PKG-1α male mice (4–7 mo of age) with the Cys42Ser mutation (redox dead) in PKG-1α (42) were generously provided by Philip Eaton (King's College).
Mice were given a single intraperitoneal injection of LPS (Esherichia coli serotype O111:B4 in 0.87% sterile saline) at varying concentrations (1, 10, and 20 mg/kg). Control animals were injected with vehicle (0.87% sterile saline). Animals were euthanized at 6-h intervals from 6 to 24 h after injection, and blood vessels were isolated for analyses of mRNA, protein, and vascular contractility. In separate experiments, wild-type mice were injected with LPS (20 mg/kg ip) followed by intraperitoneal injection of 1) PE (1 or 5 mg/kg) 3 h later (n = 8 total) or 2) IL-1ra (80 μg/kg, Sigma) 1 or 3 h later (n = 8 total). All mice of the different genotypes appeared severely ill at 24 h after LPS and did not survive beyond 36 h.
mRNA and protein assays.
mRNA and protein were assayed as previously described (18, 43) with minor modifications. In brief, the aorta, portal vein, femoral artery, and entire mesenteric arterial arcade (stripped from the superior mesenteric artery to third-order arteries) were isolated in RNALater, homogenized, and total RNA column purified (RNEasy, Qiagen, Valencia, CA). Total RNA (100 ng) was reverse transcribed with Superscript III enzyme (Invitrogen) followed by PCR. Mypt1 E24 splice variants were quantified in a single PCR using primers that flank the alternative exon. E24+ and E24− products were separated by gel electrophoresis, band intensities were directly quantified with LI-COR Odyssey, and data are reported as percentages of Mypt1 E24+ (E24/total). mRNAs were quantified by quantitative PCR using Taqman probes (Applied Biosystems) and normalized to cyclophilin A (Ppia), which was invariant between control and experimental groups. Fold changes of transcripts were calculated via the 2ΔΔCt method (where Ct is threshold cycle).
For protein assays, mesenteric arteries and thoracic aortas were homogenized using a Next Advance Bullet Blender with ∼10 times volume of lysis buffer containing 125 mM Tris·HCl (pH 6.8), 20% sucrose, 10% SDS, and 1% proteinase inhibitor cocktail (Sigma). Homogenization of mesenteric arteries yielded ∼100 μg protein and aortas yielded ∼500 μg protein. Protein lysates (10 μg) were loaded to Mini-PROTEAN TGX 4–15% Tris-glycine gels (Bio-Rad), separated at 80 V for 1.5 h, and then transferred to nitrocellulose membranes at 25 V for 2 h. Membranes were blocked and hybridized in LI-COR Odyssey blocking buffer (927-40000, LI-COR). The following primary antibodies were used: rabbit polyclonal total MYPT1 (Ab24670, Abcam, 1:3,000), rabbit polyclonal MYPT1 LZ+ (1:3,000) and LZ- (1:3,000) (39, 57), rabbit polyclonal C-kinase-activated protein phosphatase-1 inhibitor [CPI-17; 1:5,000, a gift from M. Eto (15)], mouse monoclonal myosin light chain kinase (MLCK; M7905, Sigma, 1:3,000), and rabbit monoclonal cyclophilin A (Ab131334, Abcam, 1:3,000). IRDye 800CW and 680LT (LI-COR) goat anti-rabbit or mouse IgG were used as secondary antibodies (1:10,000). Blots were scanned in an Odyssey digital scanner and quantified in Image Studio 3.0 (LI-COR).
Vascular function.
Vascular function was assayed as previously described (43, 55) with minor modifications. First-order mesenteric arteries (2-mm length, 0.15- to 0.25-mm diameter) were isolated, cleaned of all fat and debris, and mounted on a wire myograph (model 610M, Danish Myo Technology). Arteries were normalized and set to IC90 (36). HEPES-bicarbonate buffer solution contained the following (in mM): 112 NaCl, 25.7 NaHCO3, 4.9 KCl, 2.0 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 11.5 glucose, and 10.0 HEPES. The solution was equilibrated with a mixture of 95% O2-5% CO2 at pH 7.4 at 37°C. Vessels were primed with 10 μM PE as previously described. Force was measured in intact vessels in response to KCl depolarization (100 mM) or to PE (α-adrenergic agonist) and diethylamine (DEA)/NO (NO donor) at cumulative concentrations of 1 nM-100 μM. A subset of mesenteric arteries was permeabilized with α-toxin (1,000 U/ml, Sigma) as previously described (3, 43). Vessels were fully relaxed in high relaxing solution (pCa 9) composed of (in mM) 60 potassium methanesulfonate, 5 EGTA, 0.02 CaCl2, 9.26 MgCl2, 5.2 Na2ATP, 25 creatine phosphate, and 25 N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES) with pH 7.1 (intracellular pH) by 1 N KOH based on established protocols (13, 37). The dose response to Ca2+ was assessed in mesenteric arteries from pCa 9-pCa 4.5, and force was measured at steady state. Relaxation to cumulative concentrations of 8-bromo-cGMP (8-Br-cGMP; 1 nM-100 μM) was measured under Ca2+ clamp (pCa 6, 1 μM). PE-induced Ca2+ sensitization was performed under Ca2+ clamp (pCa 6, 1 μM), and intracellular Ca2+ stores were depleted by preincubation with 10 μM A-23187. Data are presented as force (in mN) and for experiments of relaxation as percentages of maximum force. EC50 was calculated for concentration curves using standard curve analysis. All reagents for experiments of vascular function were from Sigma.
Telemetry blood pressure measurements.
Blood pressure was measured by telemetry in conscious mice using PA-C10 transmitters (Data Sciences, St. Paul, MN) and detected using individual telemetry receiver platforms and Dataquest software. Transmitters were surgically implanted into the left carotid artery and were turned on 1 wk after surgery. Baseline blood pressures were averaged over the course of 3 days before LPS injection. After the LPS injection (20 mg/kg ip), blood pressures were measured continuously for an additional 24 h and are reported as the reduction in mean arterial pressure versus baseline.
Statistical analysis.
Data were analyzed with Sigma Plot software and are presented as means ± SE (SYSTAT, Chicago, IL). Isolated mesenteric artery responses were analyzed using one-way ANOVA with a post hoc Bonferroni test. Kruskal-Wallis ANOVA on ranks was used where applicable. EC50 values were calculated using standard curve analysis. One-way ANOVA and Student's t-test were used for Mypt1 splice variant PCR, quantitative PCR, protein analyses, and mean arterial pressures. Statistical significance was assumed with P < 0.05.
RESULTS
Shift to Mypt1 E24− mRNA variant and downregulation of contractile mRNAs.
The ratio of Mypt1 E24+ to E24− splice variants in the mature mouse mesenteric arterial arcade was ∼60:40 (Fig. 1A) (18). A shift to the E24− variant was first evident at 12 h after intraperitoneal injection of LPS (20 mg/kg) and was maximal by 18 h (Fig. 1A). This shift was not restricted to this regional circulation as it also occurred in the femoral artery (Fig. 1B) as well as in the portal vein (Fig. 1C), a prototypical fast smooth muscle, each with a slightly delayed time course. In contrast, the prototypical slow smooth muscle of the aorta was predominately Mypt1 E24− at baseline with no change after LPS (Fig. 1D).
Fig. 1.

Mouse vessels switch to the myosin phosphatase (MP)-targeting protein (Mypt1) exon 24 (E24)− mRNA splice variant after intraperitoneal injection of LPS. Vessels were harvested at the times indicated after intraperitoneal injection with LPS (20 mg/kg) or control (vehicle; 24 h postinjection). Total RNA was purified from the mesenteric artery (MA), femoral artery (FA), portal vein (PV), and aorta (Ao). PCR was performed with a single set of primers to amplify both Mypt1 E24+ and E24− variants. Products were separated by gel electrophoresis, directly quantified, and graphed as percent Mypt1 E24+ (Mypt1 E24/total), as described in materials and methods. A–D: representative gels of PCR products from mesenteric arteries (A), femoral arteries (B), portal veins (C), and aortas (D). All data are expressed as means ± SE; n = 4/group. *P < 0.05 vs. control.
mRNAs of the regulatory (Mypt1) and inhibitory (CPI-17) subunits of MP as measured by quantitative PCR were decreased in mesenteric arteries at 6 and 24 h after LPS (Fig. 2A). CPI-17 mRNA was also reduced in the femoral artery and portal vein at 6 and 24 h after LPS (Fig. 2A), whereas Mypt1 mRNA was reduced to a lesser magnitude in femoral arteries only at 12 and 18 h (0.3 ± 0.3 and 0.4 ± 0.3 vs. control, n = 4, P < 0.05 vs. control) and in portal veins at 12 and 18 h (Mypt1: 0.3 ± 0.1- and 0.3 ± 0.3-fold vs. control, n = 4, P < 0.05 vs. control) and 24 h (Fig. 2A) after LPS. Mypt1 and CPI-17 mRNAs were paradoxically increased approximately fourfold in the thoracic aorta at 6 h after LPS (Fig. 2A) and gradually declined thereafter (Fig. 2A and data not shown) such that at 24 h, Mypt1 had returned to control levels, whereas CPI-17 was significantly decreased.
Fig. 2.

Changes in MP subunits and other contractile mRNAs after LPS. RNA samples (A and B) after 20 mg/kg LPS, as described in Fig. 1, or after LPS dose response (1–20 mg/kg; C and D) were analyzed by quantitative PCR using Taqman probes and normalized to invariant cyclophilin A (CycloA). The 2−ΔΔCt (where Ct is threshold cycle) was used to calculate fold changes versus the saline (vehicle) control. A: fold changes in Mypt1 and C-kinase-activated protein phosphatase-1 inhibitor (CPI-17) mRNAs in the different vessels at 6 and 24 h after LPS (20 mg/kg). B: fold changes in contractile mRNAs in mesenteric arteries 24 h after intraperitoneal injection of LPS (20 mg/kg). C and D: fold changes in mRNAs from mesenteric arteries and aortas from mice injected intraperitoneally with LPS at doses of 1, 10, or 20 mg/kg versus the vehicle (saline) control at 6 and 24 h after LPS. All data are expressed as means ± SE; n = 4/group. *P < 0.05 vs. control. MLCK, myosin light chain kinase; smMHC, smooth muscle myosin heavy polypeptide 11; Ppp1cγ, catalytic subunit of protein phosphatase-1 (γ-isozyme); PHI-1, inhibitory subunit 14b of protein phosphatase-1.
Additional contractile gene mRNAs were measured for a broader assessment of the effect of LPS on the mesenteric arterial smooth muscle gene program. At 24 h after LPS (20 mg/kg), mRNAs for smooth muscle-specific myosin heavy chain, smooth muscle α-actin, smooth muscle γ-actin, and smooth muscle MLCK were significantly decreased (Fig. 2B). In contrast, there were no changes in mRNAs for the catalytic subunit of MP, Ppp1cγ, and a second member of the inhibitory subunit family of MP, phosphatase inhibitor-1, nor in cyclophilin A, the mRNA used for internal normalization.
As LPS injected intraperitoneally at 20 mg/kg is a sublethal dose, we examined the dose response to injection of LPS from 1–20 mg/kg ip. At the 6-h time point, there was a dose-response effect for the decrease of Mypt1 mRNA in mesenteric arteries (Fig. 2C) and for the increase in Mypt1 and CPI-17 mRNAs in the aorta (Fig. 2D). At the 24-h time point, CPI-17 and Mypt1 mRNAs were each similarly decreased in mesenteric arteries at all doses of LPS (Fig. 2C). In the aorta at 24 h after LPS, CPI-17 mRNA was increased twofold at the lowest dose of 1 mg/kg and decreased at doses of 10 and 20 mg/kg, whereas Mypt1 mRNA was unchanged at all doses (Fig. 2D).
Shifts in Mypt1 LZ+/− protein isoforms and downregulation of contractile proteins.
Mypt1, CPI-17, and MLCK proteins were decreased in mesenteric arteries at 24 h after intraperitoneal injection of LPS at 20 mg/kg (Fig. 3). Antibodies specific to the COOH-terminal LZ+ and LZ− isoforms were used to measure isoform-specific levels of Mypt1 protein. The Mypt1 LZ+ signal was increased by 1.5-fold and the LZ− signal was decreased to 0.4-fold of control at 24 h after LPS. Thus, the ratio of Mypt1 LZ+ to LZ− was increased by 3.6-fold at 24 h after LPS, corresponding well with the increase in the Mypt1 E24− splice variant encoding the LZ+ isoform. LPS did not alter the expression of Mypt1, CPI-17, or MLCK proteins in the thoracic aorta (Fig. 3).
Fig. 3.

Switch to the Mypt1 leucine zipper (LZ)+ isoform and decrease in MP subunits and other contractile proteins in mesenteric arteries 24 h after LPS (20 mg/kg). Protein lysates were prepared and processed for Western blot analysis as described in materials and methods. Membranes were probed with the indicated antibodies, and bands were imaged, quantified, and normalized to invariant cyclophilin A using LI-COR Odyssey. Data are expressed as fold changes of LPS-injected mice versus saline (vehicle)-injected control mice. Rabbit polyclonal antibody recognized the COOH-terminal LZ motif present in Mypt family members Mypt1 (130 kDa) and p85 (85 kDa). A second membrane was probed with rabbit polyclonal antibody specific for the Mypt1 COOH-terminal LZ− isoform, and the ratio of the Mypt1 LZ+ to LZ− signal was calculated. All data are expressed as means ± SE; n = 4–9/group. *P < 0.05 vs. control.
Tests of signaling pathways that may mediate vascular smooth muscle phenotypic changes.
We next examined inflammatory signals that might mediate atrophy and phenotypic modulation of mesenteric arterial smooth muscle. iNOS mRNA, as an indicator of the inflammatory response, was significantly increased at 24 h after intraperitoneal injection of LPS (20 mg/kg) in the mesenteric arteries (8.4 ± 0.2-fold, n = 4, P < 0.05) and portal vein (55.7 ± 0.6-fold, P < 0.05) but was unchanged in the femoral artery (1.0 ± 0.3-fold) and thoracic aorta (0.9 ± 0.5-fold). To determine if the induction of iNOS was required for the phenotypic modulation of mesenteric arterial smooth muscle, LPS was administered to mice in which iNOS was inactivated by deletion of the calmodulin-binding domain (27). Intraperitoneal injection of LPS (20 mg/kg) to homozygous iNOS knockout mice caused a switch to the Mypt1 E24− splice variant (Fig. 4A) and decreases in Mypt1 and CPI-17 mRNAs at 24 h (Fig. 4B), similar to mesenteric arteries of wild-type mice. We next tested two other signaling pathways that are activated in models of sepsis, adrenergic signaling, which we have shown controls the mesenteric artery gene program and MP subunit expression in developmental contexts (43), and the cytokine IL-1, a mediator of sepsis-induced changes in gut smooth muscle gene expression (38). Treatment with the α-agonist PE (1 or 5 mg/kg ip) 3 h after LPS administration had no effect on the LPS-induced changes in MP subunit mRNAs in mesenteric arteries at 24 h (Fig. 4, A and B). IL-1ra given at a dose of 80 μg/kg ip 1 or 3 h after endotoxin administration also had no effect on the LPS-induced changes in MP subunit mRNAs in mesenteric arteries at 24 h (Fig. 4, A and B).
Fig. 4.

Inducible nitric oxide (NO) synthase (iNOS) inactivation, IL-1 receptor antagonist (IL-1ra), or treatment with phenylephrine (PE) did not suppress LPS-induced mesenteric artery phenotypic modulation. Total RNA was isolated from mesenteric arteries from iNOS knockout (KO) or wild-type mice 24 h after injection with LPS (20 mg/kg) or saline (vehicle) control. A second set of wild-type mice was injected with LPS (20 mg/kg) followed by PE at 1 or 5 mg/kg 3 h later. There was no difference between these groups at 24 h, and they were combined for analysis (n = 8). A third set of mice was injected with LPS (20 mg/kg) followed by IL-1ra (80 μg/kg) 1 or 3 h later. There was no difference between these groups at 24 h, and they were combined for analysis (n = 8). A: Mypt1 E24+-to-E24− ratios as measured by PCR and described in Fig. 1. B: Mypt1 and CPI-17 mRNAs measured by quantitative PCR as described in Fig. 2. Data are reported as fold changes versus saline-injected control mice; n = 4–8/group. All data are expressed as means ± SE. *P < 0.05 vs. control.
Reduction in mesenteric arterial smooth muscle contractile function after LPS.
We next tested how the changes in contractile protein expression induced by LPS affect the contractile performance of mesenteric arterial smooth muscle. First-order mesenteric arteries were studied ex vivo under isometric conditions from mice 6 or 24 h after injection with LPS or vehicle (saline). Maximum force induced by depolarization with 100 mM KCl was significantly reduced in mesenteric arteries at 24 h but not at 6 h after LPS injection (Fig. 5A). Maximum force and sensitivity to the α-adrenergic agonist PE were reduced in mesenteric arteries at 24 h but not at 6 h after LPS injection (EC50: control, 0.3 ± 0.1 μM; 6-h LPS, 0.5 ± 0.2 μM, P > 0.05; 24-h LPS, 3.5 ± 1.4 μM, P < 0.05; Fig. 5B). Preincubation of mesenteric arteries with N-nitro-l-arginine methyl ester (l-NAME; 0.1 mM) to block NOS production did not affect PE-induced force generation in mesenteric arteries from control or 6-h LPS-treated mice (Fig. 6, A and B) and significantly increased but did not completely normalize force production in mesenteric arteries from 24-h LPS-treated mice (Fig. 6, C compared with A).
Fig. 5.

Reduced contractile function of mesenteric arteries 24 h after LPS. First-order mesenteric arteries were harvested from mice 6 and 24 h after injection of LPS (20 mg/kg) or saline (vehicle). Force (in mN) was measured in intact (A and B) or α-toxin-permeabilized (C and D) mesenteric arteries under isometric conditions on a wire myograph as described in materials and methods. A: maximal force generation to 100 mM KCl depolarization. B: dose response to PE. C: mesenteric arteries were α-toxin permeabilized and subjected to a dose response to Ca2+ (pCa). D: mesenteric arteries were α-toxin permeabilized and force activated at submaximal Ca2+ (pCa 6, 1μM) followed by the α-agonist PE (10 μM) with and without N-nitro-l-arginine methyl ester (l-NAME; 0.1 mM) preincubation. All data are expressed as means ± SE; n = 4–6/group. *P < 0.05 vs. control; †P < 0.05 vs. 6-h LPS.
Fig. 6.
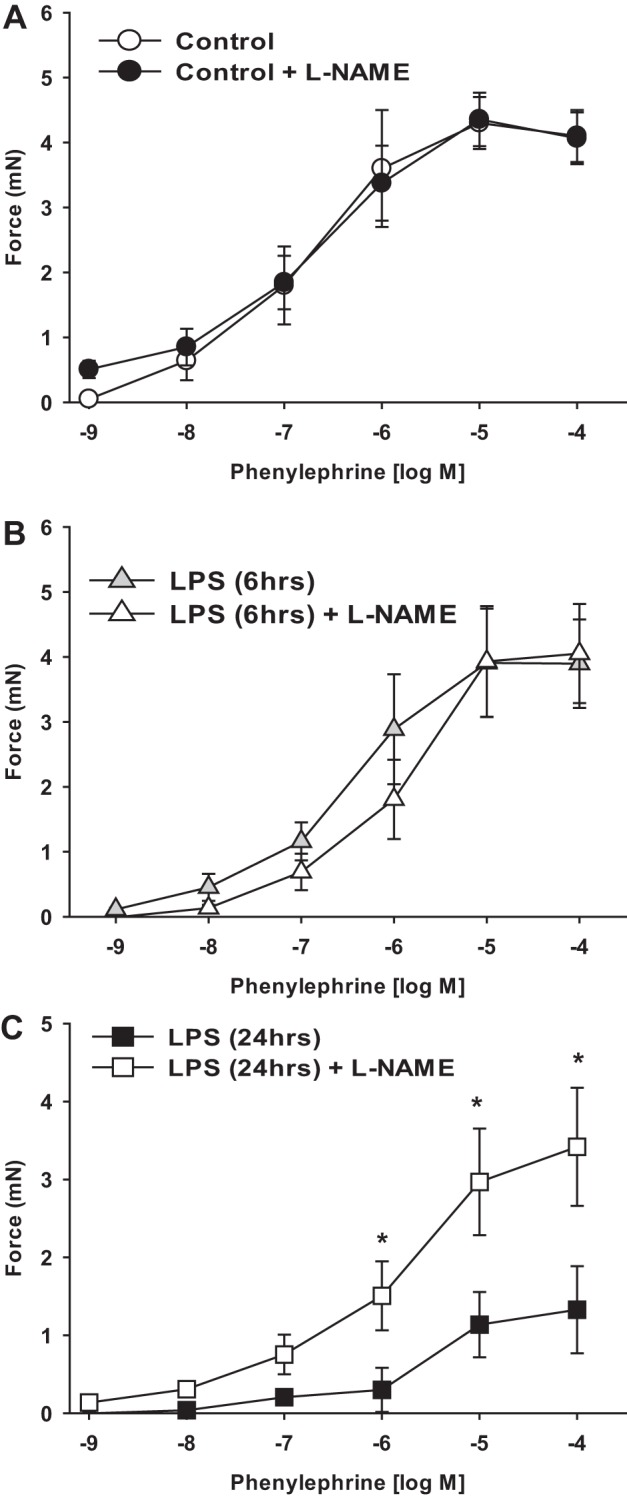
Effect of NO synthase inhibition with l-NAME on mesenteric arterial contractile function. First-order mesenteric arteries were harvested from mice 6 and 24 h after injection of LPS (20 mg/kg) or saline (vehicle). Force (in mN) was measured in intact mesenteric arteries under isometric conditions on a wire myograph as described in materials and methods. A–C: dose responses to PE with and without preincubation with l-NAME (0.1 mM) in control (A), 6-h LPS-injected (B), and 24-h LPS-injected (C) mice. All data are expressed as means ± SE; n = 5–6/group.
To more directly test the function of myofilaments in this LPS model of endotoxemia, mesenteric arteries were α-toxin permeabilized and studied under Ca2+ clamp. Maximum force to Ca2+ (pCa 4.5) was significantly reduced in mesenteric arteries at 24 h after LPS injection (Fig. 5C), whereas the sensitivity to Ca2+ (EC50) was unchanged (pCa: control, 5.6 ± 0.2; 6-h LPS, 6.2 ± 0.4; and 24-h LPS, 5.5 ± 0.2, P = 0.2 by one-way ANOVA). Next, permeabilized vessels were activated with submaximal Ca2+ (pCa 6, 1 μM) followed by 10 μM PE, a maneuver that sensitizes the filaments to Ca2+ by inhibiting MP activity. In this context, mesenteric arteries from LPS-injected mice generated <50% of the force of control mesenteric arteries (Fig. 5D); the PE-induced increment in force was similarly reduced by 50% (control: 0.6 mN vs. 24-h LPS: 0.3 mN, P < 0.05). In these permeabilized preparations, preincubation with l-NAME to block NO signaling had no effect on force production in either group of mesenteric arteries (Fig. 5D). There were no differences in responses to Ca2+ or Ca2+ plus PE in mesenteric arteries from mice 6 h after LPS (Fig. 5, C and D).
Increase in mesenteric arterial vasorelaxation after LPS.
We next tested how the changes in contractile protein expression induced by LPS affect the relaxation properties of mesenteric arterial smooth muscle. Mesenteric arteries preconstricted with PE (10 μM) were significantly more sensitive to relaxation to the NO donor DEA/NO at 6 and 24 h after LPS injection (EC50: control, 16.0 ± 1.6 μM; 6-h LPS, 0.03 ± 0.02 μM; and 24-h LPS, 0.02 ± 0.01 μM, n = 4–5, P < 0.05; Fig. 7A). All arteries completely relaxed at the highest concentrations of DEA/NO. The dose responses to the NO donor were not altered by preincubation of arteries with 0.1 mM l-NAME to block synthesis of endogenous NO (data not shown). To test the extent to which increased sensitivity to DEA/NO may reflect increased activation of MP, arteries were permeabilized and force activated with submaximal Ca2+ (pCa 6, 1 μM) followed by dose response to 8-Br-cGMP, the classic assay for MP activity in situ (25, 28). Relaxation to 8-Br-cGMP was increased in mesenteric arteries at 24 h but not at 6 h after LPS injection (EC50: control, 37.5 ± 7.1 nM; 6-h LPS: 39.3 ± 11.6 nM; and 24-h LPS, 4.7 ± 1.6 nM, P < 0.05; Fig. 7B). The increased sensitivity of 24-h LPS mesenteric arteries to DEA/NO and cGMP under Ca2+ clamp is consistent with the switch to the Mypt1 E24−/LZ+ isoform at 24 h, as the COOH-terminal LZ motif is thought to be required for cGMP/PKG-1α activation of MP. To more directly test this, we examined mice in which E24 was specifically deleted in smooth muscle using a tamoxifen-inducible Cre-lox strategy, as described above in materials and methods. This induced a switch in E24 flox heterozygous mice mesenteric arteries to the E24− variant of Mypt1 (Fig. 8A) of a magnitude similar to that induced by LPS treatment of wild-type mice (Fig. 1A) with a corresponding increase in the Mypt1 LZ+ isoform (Fig. 8B). The genetic switch to the Mypt1 E24−/LZ+ variant increased the sensitivity of mesenteric arteries preconstricted with PE (10 μM) to the NO donor DEA/NO (EC50: 0.01 ± 0.001 μM, P < 0.05 vs. control; Fig. 7A) and to 8-Br-cGMP under Ca2+ clamp (EC50: 5.3 ± 3.0 nM, P < 0.05; Fig. 7B) to the same magnitude as occurred 24 h after LPS treatment of wild-type mice. Treatment of control mice (Cre+//+/+) with tamoxifen at the age of 3 wk had no effect on contraction and relaxation properties of their mesenteric arteries studied at the age of 8 wk (data not shown).
Fig. 7.

Increased relaxation of mesenteric arteries to NO and cGMP after LPS and genetic models. First-order mesenteric arteries were harvested from wild-type and PKG-1α knockin (KI) mice 6 and 24 h after injection of LPS (20 mg/kg) or saline (vehicle). First-order mesenteric arteries were harvested from adult mice 5 wk after tamoxifen-induced smooth muscle-specific heterozygous deletion of E24 from Mypt1 (Cre+//f+). Force (in mN) was measured in intact (A and C) or α-toxin-permeabilized (B and D) mesenteric arteries. A and C: intact mesenteric arteries activated with 10 μM PE followed by dose response to the NO donor diethylamine (DEA)/NO. B and D: mesenteric arteries were α-toxin permeabilized and Ca2+ clamped (pCa 6, 1μM) followed by dose response to 8-bromo-cGMP (8-Br-cGMP). Data are plotted as percentages of maximum force generated. All data are expressed as means ± SE; n = 4–6/group, *P < 0.05, control vs. 24-h LPS; †P < 0.05, control vs. 6-h LPS; ‡P < 0.05, control vs. Cre+//F/+; §P < 0.05, 6-h LPS vs. 24-h LPS.
Fig. 8.

Cre-Lox-mediated deletion of E24 shifts Mypt1 to the E24−/LZ+ isoform and lowers blood pressure. Mypt1 E24 Cre+//+/+ (control) and Cre+//F/+ (E24 heterozygous) mice were treated with tamoxifen at 3 wk of age, and mesenteric arteries were harvested at 8 wk of age. Mypt1 E24 splice variants (A) and LZ+ protein (B) were assayed as described in Figs. 1 and 3, respectively. Rabbit polyclonal antibody recognized the COOH-terminal LZ motif present in Mypt family members Mypt1 (130 kDa) and p85 (85 kDa) on the same blot. Data are expressed as the ratio of Mypt1 LZ+ to p85. Blood pressure was continuously monitored by telemetry in conscious mice from 3 days before to 24 h after the injection of LPS. Data are reported as mean arterial pressures at baseline (C), 6 h (D), and 24 h (E) after injection of LPS in control (Cre+//+/+) versus Cre+//F/+ mice. All data are expressed as means ± SE; n = 3–6/group. *P < 0.05 vs. control.
At 6 h after LPS, the mesenteric arterial sensitivity to DEA/NO was increased but the sensitivity to 8-Br-cGMP under Ca2+ clamp was unchanged, consistent with the absence of a Mypt1 isoform switch at this early stage and suggesting a different mechanism. A previous study (45) suggested that redox activation of PKG-1α mediates the early hypotension of sepsis, as demonstrated by the suppression of this response by a single amino acid substitution in PKG-1α Cys42 to serine (Cys42Ser, redox dead mice). To test the role of redox signaling in the time-dependent changes in arterial function, these mice were injected with LPS (20 mg/kg), and mesenteric arterial function was studied at 6 and 24 h. The loss of reactive Cys42 in PKG-1α abolished the increased sensitivity of mesenteric arteries to DEA/NO at 6 h after LPS but had no effect on the increased sensitivity to DEA/NO at 24 h after LPS (EC50: PKG knockin control, 1.2 ± 0.4 μM; PKG knockin 6-h LPS, 2.3 ± 0.4 μM; and PKG knockin 24-h LPS, 0.003 ± 0.001 μM, n = 4, P < 0.05; Fig. 7, C compared with A). The increased sensitivity of permeabilized mesenteric arteries to 8-Br-cGMP under Ca2+ clamp 24 h after LPS was similarly unaffected by the PKG-1α mutation (EC50: PKG knockin control, 2.8 ± 0.4 μM; and PKG knockin 24-h LPS, 0.3 ± 0.1 μM, n = 4, P < 0.05; Fig. 7, D compared with B), while, as in wild-type mice, there was no difference in response to 8-Br-cGMP at 6 h after LPS. PKG-1α knockin mice did not differ from wild-type mice in their contractile responses to PE (10 μM) under any of the conditions tested (PKG-1α knockin: control, 5.2 ± 1.2 mN; 6-h LPS, 5.5 ± 1.0 mN; and 24-h LPS, 2.0 ± 0.5 mN, P < 0.05 vs. control).
Conditional deletion of Mypt1 E24 lowers blood pressure.
Mean arterial pressures measured via telemetry in conscious mice were significantly lower at baseline in E24 conditional knockout mice (Cre+//F/+) compared with control mice (Cre+//+/+; 90.1 ± 2.4 vs. 106.2 ± 3.8 mmHg, P < 0.05; Fig. 8C). Control and Cre+//F/+ mice showed similar reductions in mean arterial pressure at 6 h (Fig. 8D) and 24 h (Fig. 8E) after LPS injection.
DISCUSSION
In the present study, we demonstrated time-dependent changes in mouse mesenteric arterial smooth muscle contractile gene expression and function in an LPS model of peritoneal endotoxemia. The reduction in mRNAs for MP subunits Mypt1 and CPI-17 at 6 h suggests accelerated degradation of the mRNAs. This effect is specific as mRNAs for cyclophilin A, Ppp1cγ, and phosphatase inhibitor-1 were not affected. Downregulation of CPI-17 and Mypt1 has also been shown to occur in colonic smooth muscle in models of sepsis or in response to the inflammatory mediator IL-1β and has been proposed to mediate the ileus of sepsis (20, 38). The mechanism for this rapid and specific reduction in these mRNAs is not known but could involve LPS induction of micro-RNAs destabilizing transcripts, as occurs in immune cells in inflammation (for a review, see Ref. 30). A switch from the E24+ to E24− splice variant of Mypt1 occurs with a time course that is delayed relative to the reduction in Mypt1 mRNA, suggesting that the two processes are independent of one another. A switch to the Mypt1 E24− variant occurs in a number of animal models of vascular disease, including portal hypertension (40), mesenteric artery ligation (high-flow/low-flow) (55), and hypertension of pregnancy (31). While some cis-elements and trans-acting factors that regulate splicing of Mypt1 E24 have been defined (10, 18, 48), the control mechanisms for splicing of this or other alternative exons in these models has not been determined. Alternatively, the shift to the Mypt1 E24− variant could, in part, reflect selective degradation of the Mypt1 E24+ transcript as this contains a premature termination codon that could trigger nonsense-mediated decay (for reviews, see Refs. 24 and 56).
The corresponding contractile proteins were also specifically and significantly reduced 24 h after intraperitoneal injection of endotoxin. This also suggests activation of degradation of the myofibrillar proteins in the microvasculature, as has been observed in animal models of mesenteric artery ligation (55) and myocardial infarction (19). In contrast to their coupled synthesis, degradation of mRNAs and proteins are independent of one another and may explain the variance between changes in mRNA and corresponding protein in this study. The reduced protein levels coupled with reduced force generation at 24 h support that these changes are a form of muscle atrophy, similar to the more studied skeletal muscle wasting that occurs in sepsis and other inflammatory disorders due to activation of the ubiquitin proteasome and other proteolytic mechanisms (for reviews, see Refs. 49 and 52). The extent to which these proteolytic pathways may be activated in mesenteric arteries and therapeutically targeted in sepsis requires further study.
In contrast to peripheral vessels, Mypt1 and CPI-17 mRNAs were increased severalfold in the aorta early after intraperitoneal injection of LPS. The aorta contains prototypical slow (tonic) smooth muscle, whereas peripheral arteries contains phenotypically mixed (slow-fast) smooth muscle, so this result is similar to skeletal muscle, in which the fast twitch muscle is more susceptible than the slow-twitch muscle to atrophy induced by sepsis and other inflammatory conditions (for a review, see Ref. 52). Interestingly, the induction of CPI-17 was also reported in response to inflammatory mediators such as IL-1β and TNF-α in cultured aortic smooth muscle cells (26), though this and other in vitro studies were limited by basal downregulation of smooth muscle contractile gene expression. This may suggest an intrinsic difference in the signaling response dependent on the smooth muscle phenotype or, alternatively, their embryological origin, as has been demonstrated for responses to transforming growth factor-β signaling (7, 32). Alternatively, the differences between the aorta and mesenteric arteries could also reflect exposure to differing signals in this model. We speculate that the early increase in Mypt1 and CPI-17 in the aorta is a stress response, whereas their subsequent decline is due to progression of the inflammatory response. Given the differing basal and time- and dose-dependent responses of gene expression in the aorta in this model, as well as its minimal contribution to vascular resistance, studies of the aorta and other conduit vessels are of limited utility for understanding vascular dysfunction in sepsis and other inflammatory conditions.
Whether sepsis induces an intrinsic contractile defect in resistance-type arterial smooth muscle is controversial and may depend on the model, timing, vascular bed, and measures of contractility (8, 14, 16, 33, 41). Certainly, specific molecular mechanisms have not been identified. The present study shows that mesenteric artery smooth muscle intrinsic contractile function is unchanged at 6 h after intraperitoneal injection of LPS but significantly changed at 24 h. Reduced expression of the contractile (myosin and actin) and regulatory (MLCK and myosin light chain phosphatase) mRNA and proteins is consistent with the reduced maximum force to KCl depolarization and Ca2+ without a change in sensitivity to the latter, suggesting no change in Ca2+ activation of MLCK. Sensitivity and maximum force to the α-agonist PE were decreased and partially normalized by inhibition of NO synthesis with l-NAME, consistent with increased NO signaling as well as an intrinsic defect in the contractile response to the α-adrenergic agonist. The specific nature of this defect was not defined in the present study, as we focused on the vasodilator pathway, but could involve reduced expression of CPI-17 or Mypt1 subunits, targets of PKC and Rho kinase-dependent inhibition of MP activity, respectively [Ca2+ sensitization (for a review, see Ref. 11)]. A previous study (8) of a similar model in the rat showed reduced phosphorylation of Mypt1 as a marker of MP inhibition in mesenteric arteries at 6 and 24 h after LPS and increased vasorelaxant efficacy of the Rho kinase inhibitor Y-27632, which was normalized by the guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one but not by inhibition of NO synthesis (at 24 h). The authors concluded that changes in mechanisms of Ca2+ sensitization play a role in the sustained vasodilation of septic shock. However, this study was limited by an absence of primary measures of MP activity, i.e., permeabilized preparations under Ca2+ clamp, and a reliance on chemical inhibitors that may not be specific for the presumed targets in this or other models. In the prior study, Mypt1 and Rho kinase proteins were increased, whereas CPI-17 was not measured, whereas in the present study, Mypt1 and CPI-17 mRNA and proteins were reduced at 24 h. Other than the use of different species, the reasons for these discrepant findings are not clear. More definitive evidence for the role of reduced CPI-17 (or Mypt1) expression in sepsis-induced hypocontractility will require rescue experiments via transgenic forced expression. Such an approach has demonstrated the role of CPI-17 in determining sensitivity to α-agonist-mediated vasoconstriction and blood pressure (50), while the vascular function of these mice in sepsis or other disease models has not been reported.
Mesenteric arterial smooth muscle dilator function was increased at both 6 and 24 h after LPS with nearly 100-fold increased sensitivity of relaxation to the NO donor DEA/NO. Only at 24 h, was there also a 10-fold increased sensitivity to cGMP-mediated relaxation under Ca2+ clamp, the classic assay indicating activation of MP in situ (25, 28). Differences in relaxation were largest at concentrations of 10 nM cGMP (50% vs. 80%), approximating the Km of cGMP for its target, PKG-1α. This increased sensitivity to NO and cGMP is concordant with the shift to the E24−/LZ+ isoform of Mypt1. The genetic Cre-lox-induced shift of Mypt1 to the E24−/LZ+ isoform specifically in smooth muscle caused a similar increase in sensitivity to DEA/NO and cGMP under Ca2+ clamp, supporting biochemical and physiological studies showing that the Mypt1 LZ is required for PKG-1α binding and activation of MP (21, 25, 51). The 100-fold increase in sensitivity to NO in mesenteric arteries indicates the “NO reserve” and is consistent with the concept of receptor reserve as demonstrated in a study (35) of guanylate cyclase-deficient mice. Precisely how much of the NO reserve in this and other disease models is contained within the toggling of Mypt1 from E24+(LZ−) to E24−(LZ+) will require a model with forced expression of the E24+/LZ− isoform.
The nearly 100-fold increase in mesenteric arterial smooth muscle relaxation to the NO donor DEA/NO at 6 h after LPS was completely abrogated by a redox dead version of PKG-1α, demonstrating that reactive Cys42 of PKG-1α is required for this response. In contrast, Cys42 mutation had no effect on the increased vasorelaxation to DEA/NO or cGMP at 24 h after LPS, consistent with a previous study (45) in which the early fall in blood pressure was significantly suppressed by PKG-1α Cys42Ser with similar levels of hypotension at later stages (12–24 h). This suggests different mechanisms underlying the early versus late hypotension of sepsis and septic shock. As discussed above, the late changes are, at least in part, due to a change in the expression of a critical target of MP, Mypt1 splice variants, increasing its affinity for PKG-1α. In contrast, the PKG-1α redox dead version had no effect on activation of MP in control vessels at 6 or 24 h after LPS, raising the possibility of differing targets dependent on how PKG-1α is activated or the redox milieu, with previous studies (42, 54) suggesting K+ channels mediating hyperpolarization as a target of oxidation/oxidized PKG-1α. One limitation of the present study is that assays of MP activity were performed in permeabilized arteries; the resulting large volume of distribution or some other effect may cause loss of oxidation of 1) PKG-1α or 2) targets that mediate vasorelaxation. Future studies could examine H2O2 and cGMP analogs in combination in activation of MP under Ca2+ clamp to test for synergistic activation of PKG-1α and its targets, as suggested by the dependence on PKG-1α Cys42Ser for increased sensitivity to DEA/NO at 6 h after LPS as well as the severalfold lower sensitivity of PKG-1α redox dead mouse arteries to nitroglycerin-mediated relaxation, as demonstrated in a previous study (46).
In the present study, we used two different lines of genetically modified mice to model and study the effects of endotoxemia on mesenteric arterial function. The Mypt1 E24 Cre-Lox line enabled us to specifically and conditionally delete E24 in smooth muscle with tamoxifen treatment. LoxP sites were inserted into the introns flanking E24 outside of the previously defined splicing regulatory sequences (10, 48) and thus, by themselves, had no effect on the splicing of E24 or expression of Mypt1. When crossed into the highly smooth muscle-specific and inducible SMMHCCreER line(53), treatment with tamoxifen caused highly efficient deletion of E24, thereby converting Mypt1 from the LZ− to LZ+ isoform (Fig. 9). There were no observed nonspecific effects of Cre or tamoxifen as the arteries from Cre+ but E24 wild-type mice (Cre+//+/+) treated with tamoxifen had contractile properties that were not different from untreated wild-type mice. The lower blood pressure and increased sensitivity of E24 Cre+//F/+ mice to NO- and cGMP-mediated relaxation reported here is consistent with previous physiological and biochemical studies (21, 25, 43) indicating that the LZ motif is required for NO/cGMP/PKG-1α activation of MP. Further characterizations of Mypt1 E24 heterozygous (reported here) and homozygous mice are in progress. The PKG-1α redox dead mice developed by Eaton and co-workers have a substitution of Cys42 for serine within the NH2-terminal LZ motif that is unique to the α-isoform (5, 6, 42, 45). They demonstrated that this mutation blocks ROS-triggered homodimerization, which has been biochemically shown to be required for ROS-mediated activation of PKG-1α. As this is a germline substitution, it is possible that the change in arterial contractility observed in these mutant mice is not autonomous to arterial smooth muscle. Further studies are required to determine if MP or some other target of PKG-1α mediates the ROS-dependent activation of arterial relaxation in endotoxemia and the specific molecular mechanism by which Cys42 is required for increased sensitivity to NO in endotoxemia.
Fig. 9.
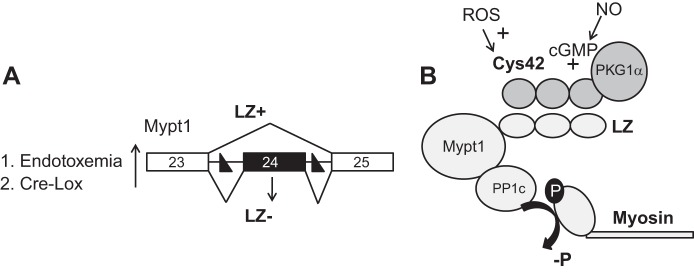
Model of MP isoforms and vasorelaxant signaling. A: skipping of E24 of the Mypt1 codes for a COOH-terminal LZ motif (LZ+). Inclusion of this 31-nt exon alters the reading frame and codes for the Mypt1 LZ− isoform. In mice treated with endotoxin, mesenteric and other blood vessels switch to Mypt1 E24-skipped isoform mRNA coding for the Mypt1 LZ+ isoform. In the present study, we modeled this by inserting LoxP sites (designated as solid triangles) in the introns flanking E24. Tamoxifen activation of smooth muscle-specific Cre causes deletion of Mypt1 E24, thereby converting the muscle to the Mypt1 E24−/LZ+ isoform. B: the working model derived from this and previous studies is that LZ-mediated heterodimerization between PKG-1α and Mypt1 is required for NO/cGMP-mediated activation of MP and dephosphorylation of myosin. This results in arterial smooth muscle relaxation even in the presence of activating Ca2+, i.e., Ca2+ desensitization of force production. Using various mutant mice, data from the present study support a model in which the vasodilation of early endotoxemia (6 h) is mediated by ROS activation of PKG-1α signaling through Cys42, whereas the vasodilation of later sepsis (24 h) is mediated by the switch to the Mypt1 E24−/LZ+ isoform mediating increased activation of MP by NO/cGMP/PKG-1α.
A brisk early response with increased local blood flow and recruitment of inflammatory cells is the best predictor of a good outcome in sepsis (29, 41a). If unresolved, the more prolonged inflammatory state leads to high mortality. Despite intensive study, there remains no specific therapies to improve the outcome of septic shock. The present study demonstrated intrinsic changes in mesenteric arterial function in the later stages of endotoxemia, including a switch in MP isoforms that increases vasodilator sensitivity to NO by 100-fold and diminution of vasoconstrictor function as part of generalized arterial muscle atrophy. Efforts to suppress these later changes of endotoxemia by targeting single mediators such as iNOS, IL-1β, or early treatment with the α-adrenergic agonist PE were unable to suppress the modulation of arterial smooth muscle in this model of septic shock. Further studies are indicated to determine if blockade of inflammatory mediators singly or in combination can suppress resistance-type arterial smooth muscle contractile modulation and thereby prevent microcirculatory dysfunction, tissue malperfusion, organ failure, and other late sequelae of sepsis.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-066171, 7R37-HL-023081, T32-HL-072751, and T32-AR-007592.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.J.R. and S.A.F. conception and design of research; J.J.R., X.Z., and L.D.A. performed experiments; J.J.R. and X.Z. analyzed data; J.J.R., X.Z., and S.A.F. interpreted results of experiments; J.J.R. and X.Z. prepared figures; J.J.R. and S.A.F. drafted manuscript; J.J.R., X.Z., and S.A.F. edited and revised manuscript; J.J.R., X.Z., L.D.A., and S.A.F. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Alex Lloyd and Shivani Kapoor for technical expertise as well as Dr. Philip Eaton and Dr. David Kass for providing the PKG-1α mutant mice.
REFERENCES
- 1.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 369: 840–851, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Asfar P, Meziani F, Hamel JF, Grelon F, Megarbane B, Anguel N, Mira JP, Dequin PF, Gergaud S, Weiss N, Legay F, Le Tulzo Y, Conrad M, Robert R, Gonzalez F, Guitton C, Tamion F, Tonnelier JM, Guezennec P, Linden TVD, Vieillard-Baron A, Mariotte E, Pradel G, Lesieur O, Ricard JD, Hervé F, Cheyron DD, Guerin C, Mercat A, Teboul JL, Radermacher P. High versus low blood-pressure target in patients with septic shock. N Eng J Med 370, 1583–1593, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Basu S, Srinivasan DK, Yang K, Raina H, Banerjee S, Zhang R, Fisher SA, Proweller A. Notch transcriptional control of vascular smooth muscle regulatory gene expression and function. J Biol Chem 288: 11191–11202, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6: 25–39, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD, Eaton P. Cysteine redox Ssensor in PKGIa enables oxidant-induced activation. Science 317: 1393–1397, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne JR, Oka S, Ale-Agha N, Eaton P. Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system. Antioxid Redox Signal 18: 1042–1052, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheung C, Bernardo AS, Trotter MWB, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotech 30: 165–173, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.da Silva-Santos JE, Chiao CW, Leite R, Webb RC. The Rho-A/Rho-kinase pathway is up-regulated but remains inhibited by cyclic guanosine monophosphate-dependent mechanisms during endotoxemia in small mesenteric arteries. Crit Care Med 37: 1716–1723, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Backer D, Orbegozo Cortes D, Donadello K, Vincent JL. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence 5: 73–79, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dippold RP, Fisher SA. A bioinformatic and computational study of myosin phosphatase subunit diversity. Am J Physiol Regul Integr Comp Physiol 307: R256–R270, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dippold RP, Fisher SA. Myosin phosphatase isoforms as determinants of smooth muscle contractile function and calcium sensitivity of force production. Microcirculation 21: 239–248, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donati A, Domizi R, Damiani E, Adrario E, Pelaia P, Ince C. From macrohemodynamic to the microcirculation. Crit Care Res Pract 892710: 27, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dougherty PJ, Davis MJ, Zawieja DC, Muthuchamy M. Calcium sensitivity and cooperativity of permeabilized rat mesenteric lymphatics. Am J Physiol Regul Integr Comp Physiol 294: R1524–R1532, 2008. [DOI] [PubMed] [Google Scholar]
- 14.El-Awady MS. Voltage-independent calcium channels mediate lipopolysaccharide-induced hyporeactivity to endothelin-1 in the rat aorta. Am J Physiol Heart Circ Physiol 296: H1408–H1415, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Eto M, Senba S, Morita F, Yazawa M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: its specific localization in smooth muscle. FEBS Lett 410: 356–360, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Farmer MR, Roberts RE, Gardiner SM, Ralevic V. Effects of in vivo lipopolysaccharide infusion on vasoconstrictor function of rat isolated mesentery, kidney, and aorta. J Pharmacol Exp Ther 306: 538–545, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Fisher SA. Vascular smooth muscle phenotypic diversity and function. Physiol Genomics 42A: 169–187, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu K, Mende Y, Bhetwal BP, Baker S, Perrino BA, Wirth B, Fisher SA. Tra2β protein is required for tissue-specific splicing of a smooth muscle myosin phosphatase targeting subunit alternative exon. J Biol Chem 287: 16575–16585, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han YS, Brozovich FV. Altered reactivity of tertiary mesenteric arteries following acute myocardial ischemia. J Vasc Res 50: 100–108, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu W, Mahavadi S, Li F, Murthy KS. Upregulation of RGS4 and downregulation of CPI-17 mediate inhibition of colonic muscle contraction by interleukin-1β. Am J Physiol Cell Physiol 293: C1991–C2000, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang QQ, Fisher SA, Brozovich FV. Unzipping the role of myosin light chain phosphatase in smooth muscle cell relaxation. J Biol Chem 279: 597–603, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, Remick DG. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev 93: 1247–1288, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13: 700–712, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khatri JJ, Joyce KM, Brozovich FV, Fisher SA. Role of myosin phosphatase isoforms in cGMP-mediated smooth muscle relaxation. J Biol Chem 276: 37250–37257, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Kim JI, Urban M, Young GD, Eto M. Reciprocal regulation controlling the expression of CPI-17, a specific inhibitor protein for the myosin light chain phosphatase in vascular smooth muscle cells. Am J Physiol Cell Physiol 303: C58–C68, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA 92: 10688–10692, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MR, Li L, Kitazawa T. Cyclic GMP causes Ca2+ desnsitization in vascular smooth muscle by activating the myosin light chain phosphatase. J Biol Chem 272: 5063–5068, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Lilly CM. The ProCESS Trial–a new era of sepsis management. N Engl J Med 370: 1750–1751, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Liu G, Abraham E. MicroRNAs in immune response and macrophage polarization. Arterioscler Thromb Vasc Biol 33: 170–177, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y, Zhang H, Gokina N, Mandala M, Sato O, Ikebe M, Osol G, Fisher SA. Uterine artery myosin phosphatase isoform switching and increased sensitivity to SNP in a rat l-NAME model of hypertension of pregnancy. Am J Physiol Cell Physiol 294: C564–C571, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27: 1248–1258, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Martinez MC, Muller B, Stoclet JC, Andriantsitohaina R. Alteration by lipopolysaccharide of the relationship between intracellular calcium levels and contraction in rat mesenteric artery. Br J Pharmacol 118: 1218–1222, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuda N, Hattori Y. Vascular biology in sepsis: pathophysiological and therapeutic significance of vascular dysfunction. J Smooth Muscle Res 43: 117–137, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Mergia E, Friebe A, Dangel O, Russwurm M, Koesling D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest 116: 1731–1737, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 41: 19–26, 1977. [DOI] [PubMed] [Google Scholar]
- 37.Nishimura J, Moreland S, Ahn HY, Kawase T, Moreland RS, van Breemen C. Endothelin increases myofilament Ca2+ sensitivity in alpha-toxin- permeabilized rabbit mesenteric artery. Circ Res 71: 951–959, 1992. [DOI] [PubMed] [Google Scholar]
- 38.Ohama T, Hori M, Sato K, Ozaki H, Karaki H. Chronic treatment with interleukin-1β attenuates contractions by decreasing the activities of CPI-17 and MYPT-1 in intestinal smooth muscle. J Biol Chem 278: 48794–48804, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Payne MC, Zhang HY, Prosdocimo T, Joyce KM, Koga Y, Ikebe M, Fisher SA. Myosin phosphatase isoform switching in vascular smooth muscle development. J Mol Cell Cardiol 40: 274–282, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Payne MC, Zhang HY, Shirasawa Y, Koga Y, Ikebe M, Benoit JN, Fisher SA. Dynamic changes in expression of myosin phosphatase in a model of portal hypertension. Am J Physiol Heart Circ Physiol 286: H1801–H1810, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Price SA, Spain DA, Wilson MA, Harris PD, Garrison RN. Subacute sepsis impairs vascular smooth muscle contractile machinery and alters vasoconstrictor and dilator mechanisms. J Surg Res 83: 75–80, 1999. [DOI] [PubMed] [Google Scholar]
- 41a.ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, Barnato AE, Weissfeld LA, Pike F, Terndrup T, Wang HE, Hou PC, LoVecchio F, Filbin MR, Shapiro NI, Angus DC. A randomized trial of protocol-based care for early septic shock. N Engl J Med 370: 1683–1693, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 18: 286–290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reho JJ, Zheng X, Benjamin JE, Fisher SA. Neural programming of mesenteric and renal arteries. Am J Physiol Heart Circ Physiol 307: H563–H573, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reho JJ, Zheng X, Fisher SA. Smooth muscle contractile diversity in the control of regional circulations. Am J Physiol Heart Circ Physiol 306: H163–H172, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudyk O, Phinikaridou A, Prysyazhna O, Burgoyne JR, Botnar RM, Eaton P. Protein kinase G oxidation is a major cause of injury during sepsis. Proc Natl Acad Sci USA 110: 9909–9913, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rudyk O, Prysyazhna O, Burgoyne JR, Eaton P. Nitroglycerin fails to lower blood pressure in redox-dead Cys42Ser PKG1 knock-in mouse. Circulation 126: 287–295, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russell JA. Is there a good MAP for septic shock? N Engl J Med 370: 1649–1651, 2014. [DOI] [PubMed] [Google Scholar]
- 48.Shukla S, Fisher SA. Tra2β as a novel mediator of vascular smooth muscle diversification. Circ Res 103: 485–492, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith IJ, Lecker SH, Hasselgren PO. Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab 295: E762–E771, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su W, Xie Z, Liu S, Calderon LE, Guo Z, Gong MC. Smooth muscle-selective CPI-17 expression increases vascular smooth muscle contraction and blood pressure. Am J Physiol Heart Circ Physiol 305: H104–H113, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM, Mendelsohn ME. Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Iα. Science 286: 1583–1587, 1999. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Pessin JE. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care 16: 243–250, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind S, Offermanns S. G12–G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008. [DOI] [PubMed] [Google Scholar]
- 54.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res 110: 471–480, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Fisher SA. Conditioning effect of blood flow on resistance artery smooth muscle myosin phosphatase. Circ Res 100: 730–737, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z, Hu L, Kong X. MicroRNA or NMD: why have two RNA silencing systems? J Genet Genomics 40: 497–513, 2013. [DOI] [PubMed] [Google Scholar]
- 57.Zheng X, Heaps CL, Fisher SA. Myosin phosphatase isoforms and related transcripts in the pig coronary circulation and effects of exercise and chronic occlusion. Microvasc Res; doi: 10.1016/j.mvr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]