

Abstract
Global ischemia, catecholamine surge, and rapid heart rhythm (RHR) due to ventricular tachycardia or ventricular fibrillation (VF) are the three major factors of sudden cardiac arrest (SCA). Loss of excitability culminating in global electrical failure (asystole) is the major adverse outcome of SCA with increasing prevalence worldwide. The roles of catecholamines and RHR in the electrical failure during SCA remain unclear. We hypothesized that both β-adrenergic stimulation (βAS) and RHR accelerate electrical failure in the globally ischemic heart. We performed optical mapping of the action potential (OAP) in the right ventricular (RV) and left (LV) ventricular epicardium of isolated rabbit hearts subjected to 30-min global ischemia. Hearts were paced at a cycle length of either 300 or 200 ms, and either in the presence or in the absence of β-agonist isoproterenol (30 nM). 2,3-Butanedione monoxime (20 mM) was used to reduce motion artifact. We found that RHR and βAS synergistically accelerated the decline of the OAP upstroke velocity and the progressive expansion of inexcitable regions. Under all conditions, inexcitability developed faster in the LV than in the RV. At the same time, both RHR and βAS shortened the time to VF (TVF) during ischemia. Moreover, the time at which 10% of the mapped LV area became inexcitable strongly correlated with TVF (R2 = 0 .72, P < 0.0001). We conclude that both βAS and RHR are major factors of electrical depression and failure in the globally ischemic heart and may contribute to adverse outcomes of SCA such as asystole and recurrent/persistent VF.
Keywords: β-adrenergic stimulation, inexcitability, myocardial ischemia, optical mapping, ventricular fibrillation
sudden cardiac arrest (SCA) remains one of the leading causes of death in developed countries (4, 32). The most common sequence of events leading to SCA is the degeneration of ventricular tachycardia (VT) into ventricular fibrillation (VF), often followed by asystole or pulseless electric activity (PEA) (27). In the context of out-of-hospital SCA (OHSCA) cardiac arrest, asystole is a nonshockable rhythm associated with a very poor prognosis of survival to hospital discharge (21, 22, 37). In recent years the reported incidence of asystole as the first recorded rhythm during OHSCA has increased compared with the incidence of VF (14, 41), which at least in part reflects faster degeneration of VF into asystole (26). This adverse phenomenon is expected to have an increasing impact on survival in OHSCA (21). Another complication of VF-induced SCA is refibrillation, which occurs in the majority of cases during OHSCA and is also negatively associated with survival (55).
Basic mechanisms determining the speed at which VF degenerates into asystole remain poorly understood. Presumably, asystole reflects global loss of excitability. The three major components of VF-induced SCA are global ischemia, catecholamine surge (30), and rapid heart rhythm (RHR). It is well known that myocardial ischemia will eventually lead to loss of excitability. However, the effects of catecholamines and RHR on the development of inexcitability in the ischemic heart have not been systematically investigated. The effect of β-adrenergic stimulation (βAS) is particularly relevant because during ischemia it may support so-called slow response action potential driven predominantly or exclusively by L-type Ca2+ current when fast Na channels are fully inactivated due to ischemic depolarization (16). In addition, generally pro-arrhythmic and pro-fibrillatory effects of βAS are well established (53). Thus one could expect that βAS would sustain electrical activity (normal or abnormal) in the ischemic heart for a longer time. On the other hand, there is limited, but intriguing evidence that βAS depresses conduction in the ischemic myocardium (17). Whether βAS can also accelerate the loss of excitability in the ischemic myocardium remains unknown. There is also limited evidence that heart rate modulates conduction in the ischemic ventricles. Specifically, RHR accelerated (17, 24), whereas bradycardia delayed (36) conduction slowing in ischemic myocardium. Collectively, these previous findings have led us to the hypothesis that RHR and βAS may synergistically promote loss of excitability in the globally ischemic heart. We used an isolated heart model of global ischemia, in which the propagation of electrical waves can be scrutinized using high resolution optical mapping and the effects of RHR and βAS can be dissected with relative ease.
Our study provides strong evidence that βAS and RHR - individually and combined - accelerate heterogeneous electrical depression and conduction failure in the globally ischemic heart. Moreover, both βAS and RHR also accelerated VF initiation during global ischemia, which apparently correlated with emergence of locally inexcitable regions. We propose that the observed synergism between RHR and βAS with respect to electrical depression may contribute to adverse outcomes of VF-induced SCA, including asystole and recurrent VF, and may also be relevant to VF initiation in the context of acute myocardial infarction.
METHODS
This investigation conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th Edition, 2011) and was approved by the Institutional Animal Care and Use Committee of the University of Utah (Protocols No. 12-09005 and 13-0005).
Langendorff-perfused rabbit hearts.
Adult New Zealand white rabbits of either sex (15 male and 16 female, weight 1.5 to 3.0 kg) were euthanized by pentobarbital sodium (130 mg/kg iv), and heparin (1 ml, 10,000 USP) was given to prevent clotting. Hearts were rapidly excised, cannulated on a Langendorff apparatus, and perfused with Tyrode's solution containing 130 mM NaCl, 24 mM NaHCO3, 1.2 mM NaH2PO4, 1.0 mM MgCl2, 5.6 mM glucose, 4.0 mM KCl, 1.8 mM CaCl2, and 0.1 g/l albumin, gassed with an O2/CO2 mixture (adjusted to maintain pH at 7.4) at a fixed rate of 30 ml/min. The mitral valve was disrupted by inserting a drainage tube into the LV via a small cut in the left atrial appendage to prevent buildup of LV pressure due to venous efflux through Thebesian veins. Hearts were immersed in Tyrode's superfusion solution and temperature in the right ventricular (RV) cavity and the superfusion solution was maintained at 37.0 ± 0.5°C. The volume-conducted ECG was monitored continuously throughout the experiment.
After 15 min of stabilization, the electromechanical uncoupler 2,3-butanedione 2-monoxime (BDM; 20 mM; Sigma-Aldrich, St. Louis, MO) was administered and continued for the rest of the duration of the experiment. About 10 min before the ischemic episode, hearts were stained with a bolus of the voltage-sensitive dye di-4-ANEPPS (3 ml of 10.4 μM solution; Life Technologies, Carlsbad, CA). Global ischemia was initiated by cessation of aortic perfusion and maintained for 30 min or until the initiation of VF (whichever was longer), followed by reperfusion. During global ischemia the temperature of the superfusate was maintained at 37.0 ± 0.5°C and the oxygen pressure in the superfusate was maintained below 40 mmHg by gassing the solution with a N2/CO2 gas mixture (adjusted to maintain pH at 7.4).
Pacing at either normal [cycle length (CL) = 300 ms] or fast (CL = 200 ms) rates was performed using two pairs of Ag/AgCl plate electrodes (diameter, 3 mm) positioned at the endocardial and the epicardial surface of the lateral RV and left ventricular (LV) free wall. Simultaneous RV and LV pacing was started 1–3 min before ischemia. The excitation threshold was 0.25 - 1.1 mA in the LV and 0.5–2.5 mA in the RV. Large variation in the threshold current was most likely due to variable contact of the electrode with the tissue, leading to variable leak of current into the superfusion solution. The stimulus current was set at 3× pre-ischemic threshold and was maintained at this level throughout the ischemic episode. The relatively large size of the pacing electrodes, the application of the current across the ventricular wall, and the relatively large amplitude of the stimulus ensured efficient capture during ischemia, when excitability was progressively diminished. Only in a few cases 1:1 capture was lost at some point during ischemia before VF initiation; in those cases the current was immediately increased to 10× pre-ischemic threshold, which restored 1:1 capture. Responses other than 1:1 (2:1 or 3:1, etc.) were not observed; however, some regions became completely inexcitable either during paced rhythm or after spontaneous initiation of VF, as described in results.
Experimental groups.
A total of five experimental groups were used in this study. In the 300ms group (n = 5), which served as the control, the hearts were paced at a CL = 300 ms and no drugs (except BDM) were used. In the 200ms group (n = 5), the hearts were paced at a CL = 200 ms. In the 300ms_Iso and 200ms_Iso groups (n = 5 and 6, respectively), the hearts were paced at CLs of either 300 or 200 ms in the presence of the β-adrenergic agonist isoproterenol (30 nM; Sigma-Aldrich). Perfusion with isoproterenol started 10 min before the onset of ischemia. Because in the 300ms_Iso hearts application of isoproterenol would preclude pacing at 300 ms due to increased intrinsic rate, the sinoatrial node was ablated. This was achieved by cooling a hemostat with liquid nitrogen and simultaneously crushing and freezing the nodal region near the junction of the right atrial appendage and the superior vena cava. In the VF_initiation group (n = 4) the hearts were treated the same way as in the 200ms_Iso group, but the optical recordings taken were much longer than in the main 200ms_Iso group, as explained in detail below. The purpose of these experiments was to maximally increase the chance of capturing spontaneous VF initiation, which was accomplished in three out of these four experiments.
Optical mapping.
Hearts were fully immersed in Tyrode's solution inside the imaging chamber to maintain the temperature at 37°C both during normoxic perfusion and no-flow ischemia. Optical mapping of changes in transmembrane voltage (optical action potential, OAP) was performed using an EMCCD camera (iXon DU-860D; Andor Technology, Belfast, UK) with a 6 mm objective lens, at a resolution of 64 × 64 pixels and frame interval of 2.06 ms. The voltage sensitive dye di-4-ANEPPS was excited by a 532 nm green solid-state laser (Coherent, Santa Clara, CA). The imaging area was 3 × 3 cm encompassing the entire anterior view of the right and left ventricles (see Fig. 1). Emitted fluorescence was collected using either a 600 ± 40 nm or 605 ± 32 nm bandpass filter. In four experimental groups (300ms, 200ms, 300ms_Iso, 200ms_Iso) 6-s-long movies were acquired 2 to 3 min before ischemia, at each minute from 0 to 30 min of ischemia, and during 20 min of reperfusion (reperfusion movies were used only to check recovery of electrical activity). The zero-minute ischemic movie was acquired at 5–10 s after interruption of the aortic flow. In the VF_initiation group the duration of optical recordings during ischemia was increased to 14.4 s (7,000 frames) interposed by 5.6-s intervals during the phase of ischemia when VF initiation was expected based on the data obtained from 200ms_Iso group. In this mode 72% of the time was filmed, which increased the chance of capturing spontaneous initiation of VF close to the maximum technically achievable in our set-up. Data acquisition was performed using a custom software package (Alexey Zaitsev and Paul Venable) written in Java and using libraries from Micro-Manager open source microscopy software (Vale Lab, University of California in San Francisco, San Francisco, CA) and NetBeans open source program development framework (netbeans.org, Oracle, Redwood City, CA).
Fig. 1.
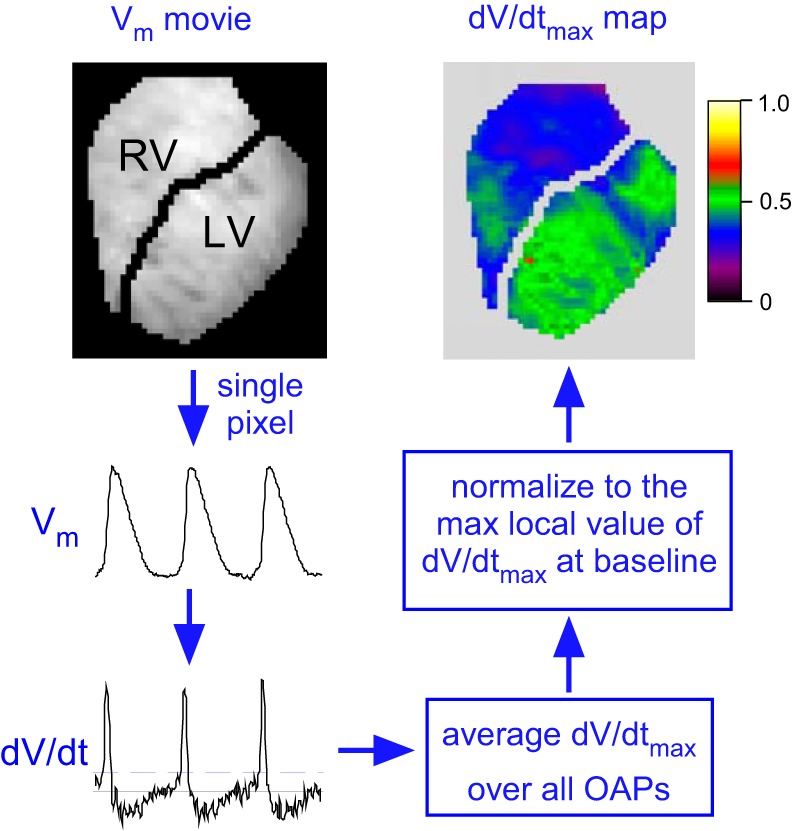
An overview of the dV/dtmax analysis. The voltage-sensitive fluorescence of di-4-ANEPPS was filtered and inverted yielding representation of the transmembrane potential (Vm). In each pixel, the maximum time derivative of Vm (dV/dtmax) was computed in each action potential. The average value of dV/dtmax over all action potentials was computed for each pixel in 6-s movies. The dV/dtmax values thus obtained were corrected for the background fluorescence (see methods), normalized to the maximum local dV/dtmax in the pre-ischemic movie, and superimposed on the images of the right ventricle (RV) and the left ventricle (LV). Pixels in which the average dV/dtmax values fell below the predefined threshold of detection (twice the level of dV/dt noise in the end-ischemic movies showing no activity) were assigned zero values and were counted as inexcitable. OAP, the optical action potential.
Data processing and analysis.
The software program Scroll (Sergey Mironov, Center for Arrhythmia Research, University of Michigan) was used for interactive analysis of movies and single pixel recordings, and also for manual selection of the mapped regions exclusive to RV and LV in each heart. All other analysis routines were written by Alexey Zaitsev using PV-WAVE software (Rogue Wave Software, Boulder, CO). We defined the area to the right of the LAD as RV and the area to the left of LAD as LV (see Fig. 1). The optical recordings were background-subtracted, inverted, and filtered in time and space as previously described (57), yielding a movie reflecting spatiotemporal distribution of the transmembrane potential (Vm). Specifically, a boxcar smoothing filter (box size, 5 points) was applied to each pixel followed by a two-dimensional convolution of frames with a cone-shaped 5-point kernel. The maximal time derivative of the OAP upstroke (dV/dtmax) was determined in each OAP of each single pixel recording in each movie (see Fig. 1). The average values of dV/dtmax in each pixel were corrected for the background fluorescence. Specifically, dV/dtmax in the brightest pixel (fluorescence level Fmax) remained unchanged, whereas in all other pixels the value of dV/dtmax was multiplied by the ratio of Fmax over F in the given pixel. This procedure compensated for the variation in dV/dtmax due to non-uniform illumination and staining as well as curvature of the heart. The corrected values of dV/dtmax in each pixel of each movie were normalized to the highest dV/dtmax value in the pre-ischemic movie. The dV/dtmax values that were corrected, normalized, and averaged over all action potentials for each pixel (ranging from 0 to 1) were then color-coded and projected on the images of the RV and the LV to generate dV/dtmax maps (see Fig. 1). Finally, the average values of dV/dtmax were computed for the RV and the LV in each movie and were plotted as a function of ischemia duration.
To detect the loss of excitability in individual locations in RV and LV, we applied a minimum dV/dtmax criterion. The criterion was selected to be just above twice the level of the dF/dt of noise in the movies in which the full loss of excitability was undoubted, as was the case in all movies recorded in the 200ms_Iso group at 30 min of ischemia. All movies analyzed for this study were carefully inspected to validate that the computed inexcitable areas matched well to the visually observable areas lacking propagating waves. The percent of pixels with dV/dtmax values above the threshold (percent of excitable areas) was computed in the RV and the LV and plotted as the function of ischemia duration. Action potential duration was computed in OAPs at the level of 80% of repolarization (APD80). Average values of APD80 were computed for RV and LV and plotted as a function of ischemia duration.
In all experiments VF spontaneously occurred at some point in ischemia. As expected, after the onset of VF the values of both APD and dV/dtmax immediately dropped consequent to a large increase in excitation rate. In addition, large regions of both ventricles eventually became inexcitable. A significant degree of inexcitability introduces ambiguity in the interpretation of the average values: neither inclusion (as 0 values) nor exclusion of the inexcitable regions from the calculation of the average dV/dtmax is fully appropriate. Because of the above, the comparison of the average RV and LV values of dV/dtmax between groups was performed only during the phase of ischemia when at least three hearts in each group remained in regular paced rhythm and maintained excitability in at least 80% of mapped regions in both the RV and the LV. This time point was 8 min of ischemia. In addition, in experiments using isoproterenol a residual motion artifact was present up to 3 min of ischemia despite the presence of BDM. This motion artifact had little effect on dV/dtmax but significantly distorted the measurement of APD80. Therefore, the comparison of the average RV and LV values of APD80 between groups was performed between 3 and 8 min of ischemia. For analysis of VF, we applied the Hilbert Transform to movies of voltage-sensitive fluorescence and identified singularity points and rotors as points where all OAP phases converged (20) (9). Time-space plots were created by stacking intensity profiles along a selected line of pixels in consecutive frames of the voltage-sensitive fluorescence (39).
It should be noted that in one heart from the 300ms group VF did not occur during the first 30 min of ischemia and therefore the ischemia duration was prolonged until VF was noted (at 33 min of ischemia). In all other experiments VF did occur within the 30-min ischemic episode. Thus, despite this deviation is a single 300ms experiment, we deemed it was fair to compare the time of VF initiation (TVF) between the groups including this experiment.
Statistical analysis.
Statistical analyses were performed in four groups (300ms, 200ms, 300ms_Iso, 200ms_Iso). The time course of dV/dtmax, percentage of excitable area, and APD80 were compared using a two-way ANOVA with Tukey post hoc test for multiple comparisons. The statement of significant difference in this case meant the significant separation between the entire curves. Analysis of differences between individual time points was not performed. The time of VF initiation (TVF) was compared between groups using a one-way ANOVA followed by Ryan, Einot, Gabriel, and Welsh Studentized Range Q (REGWQ) post hoc test for multiple comparisons. Data are given as means ± SE. Differences of P < 0.05 were considered statistically significant.
RESULTS
RHR and βAS mutually promote the progressive decline in dV/dtmax and the emergence of inexcitable areas.
Figure 2 shows dV/dtmax maps from representative individual experiments from 300ms, 200ms, 300ms_Iso, and 200ms_Iso groups. In Fig. 2, columns represent different experimental groups (labeled at the top), whereas rows represent different time points (labeled on the left-hand side). Colors in the dV/dtmax maps (from black to yellow-white) indicate local values of dV/dtmax normalized in each experiment to the largest value recorded at baseline. Black color indicates sites in which dV/dtmax fell below the threshold value, which was set just above the level of noise (see methods for more detail). All movies were carefully inspected to verify that the regions identified as inexcitable based on the dV/dtmax threshold criterion match reasonably well with those regions in which propagating waves could not be seen.
Fig. 2.

Representative examples of the progressive decline in dV/dtmax over time of ischemia in 4 tested groups: 300ms, 300ms_Iso, 200ms, and 200ms_Iso (see methods for definitions). Each column represents an experimental group; each row represents a time point in ischemia. The values of dV/dtmax in each pixel are corrected for the background fluorescence and normalized to the maximal value in the baseline map. The horizontal lines in each column indicate the 5-min interval in which ventricular fibrillation (VF) initiation took place. In the 300ms experiment (the leftmost column) VF started at 33 min of ischemia. Note that both the higher pacing frequency (the shorter pacing cycle) and β-adrenergic stimulation with isoproterenol tended to accelerate electrical depression and the onset of VF.
As can be seen in Fig. 2, at baseline dV/dtmax was higher in the LV than in the RV in all four groups. The left-to-right difference was the largest in the 200ms group. This is an interesting physiological observation but perhaps of no relevance to the subject of this study. The dV/dtmax progressively decreased with time of ischemia in all groups, but at different rates. The rate of decline progressively increased between the groups from 300ms to 300ms_Iso to 200ms to 200ms_Iso (from left to right in Fig. 2). In particular, note that at 10 min of ischemia there was only moderate decrease of dV/dtmax in both the 300ms and 300ms_Iso hearts by about 25%), whereas in the 200ms heart dV/dtmax decreased by about 50%, and in the 200ms_Iso heart the decrease was more than 50% in the RV and more than 80% in the most of the LV (exclusive of the apex).
By 15 min of ischemia (the 4th row in Fig. 2), there was a dramatic deterioration of dV/dtmax in the LV of the 300ms_Iso heart, with a sharp demarcation between the highly depressed LV (exclusive of the apical part adjacent to the RV) and the much better preserved (electrically) RV. Also, there was a large decrease of dV/dtmax in the 200ms heart. However, the most dramatic change had occurred by 15 min of ischemia in the 200ms_Iso heart, which completely lost excitability in most of the RV and LV, with only a few pockets of resistance in the apical LV and in the apical and basal RV. By 20 min of ischemia (the 5th row from the top in Fig. 2), the 300ms_Iso heart lost excitability in the entire mapped region of the LV. Finally, at 30 min of ischemia, the 300ms heart remained fully excitable (albeit electrically depressed), the 300ms_Iso heart had only a sliver of active tissue in the RV, and both 200ms and 200ms_Iso hearts were electrically silent.
The horizontal lines in Fig. 2 demarcate the dV/dtmax maps recorded before and after spontaneous initiation of VF. Note that here we show maps only at 5-min steps. More precisely, VF was first seen in the 19th-min movie in the 300ms_Iso heart and in the 11th-min movies in both 200ms and 200ms_Iso hearts. In the 300ms heart VF had not occurred within the 30-min ischemic period. One can see that in the three hearts in which VF did occur, at least in some regions of the heart a significant decrease in dV/dtmax (to below 25% of the baseline value, magenta) or a full loss of excitability (black) took place before VF initiation. It can also be seen that after the VF initiation, large regions became fully inexcitable in the 300ms_Iso and 200ms_Iso hearts. The same happened in the 200ms heart, but after a longer delay.
Figure 3 summarizes the dV/dtmax and inexcitability data in the four experimental groups: 300ms, 200ms, 300ms_Iso, and 200ms_Iso. All left-hand panels describe RV, all right-hand panels describe LV. Figure 3, A and B, shows the time course of dV/dtmax during ischemia in RV and LV, respectively. The values are normalized to the largest value of dV/dtmax measured in each heart before ischemia. Note that on average baseline values of dV/dtmax in the RV were lower (by 20–25%) than in the LV (recall that this could be seen in the baseline dV/dtmax maps shown in Fig. 2). There is an abrupt drop in the average dV/dtmax values in all groups and in both chambers at 0 min of ischemia (for practical purposes, this recording occurred within a few seconds of the interruption of aortic flow). The reason for this is not clear and might reflect changes in myocardial optical properties (e.g., absorbance or scattering of either exciting light or the fluorescence) associated with acute ischemia. Whatever the reason, this phenomenon had similar magnitude in all groups and is unlikely to affect the main findings of this study. Inspecting the time course of dV/dtmax from 0 min of ischemia onward, one can appreciate that the dV/dtmax decline becomes progressively faster between the groups in the order 300ms group < 300ms_Iso group < 200ms group < 200ms_Iso group. It also can be appreciated that the dV/dtmax decline occurs faster in the LV than in the RV (compare the slopes of the respective curves in Fig. 3, A and B). The vertical dashed lines indicate the upper limit of the time window in which the time course of dV/dtmax decline during ischemia was compared between different experimental groups. (As explained in methods, the upper limit of 8 min of ischemia excluded hearts, which experienced either a significant loss of excitable sites or developed VF. The lower limit is the pre-ischemic movie, taken 2 to 3 min before ischemia). In the RV, the difference was statistically significant in all pairwise comparisons excluding that between 200ms and 300ms_Iso groups and between 200ms and 200ms_Iso groups. In the LV, only the difference between 200ms and 300ms_Iso groups was not statistically significant. Of interest, the value of dV/dtmax transiently increased between 2 and 5 min of ischemia in the 300ms (control) group, most likely reflecting the phenomenon of supernormality whereby excitability is transiently increased due to a mild depolarization in early ischemia, bringing the resting potential closer to the excitation threshold (18). This phenomenon is much less visible or nonexistent in the three other groups.
Fig. 3.

The effects of increased excitation rate and β-adrenergic stimulation (βAS) on the progressive decline in dV/dtmax and the loss of excitability during ischemia. The data is from 4 groups: 300ms (black), 200ms (red), 300ms_Iso (blue), and 200ms_Iso (green). A and B: the time course of the average dV/dtmax in the RV and the LV, respectively. The data in each experiment are normalized to the maximum local value at the baseline (pre-ischemia). Only those data point are shown at which at least 3 hearts from each group remained in paced rhythm and maintained excitability in at least 80% of the mapped area (see methods). Note that the dV/dtmax decline during ischemia is synergistically promoted by shorter pacing interval and βAS with isoproterenol (30 nM). The vertical dashed line indicates the upper limit of the time window in which statistical comparisons between groups were performed. C and D: percentage of excitable area vs. the duration of ischemia in the RV and LV, respectively. The data from each individual experiments are shown. Solid lines with symbols indicate the time points at which the hearts were in paced rhythm. Dashed lines without symbols indicate the time points at which the hearts were in VF or irregular rhythm. Horizontal dashed line in D indicates 10% inexcitability level in the LV. Note that in the majority of experiments VF initiation coincided with the onset of rapid decline in the percent of the excitable LV area. See text for more detail. *P < 0.05.
Figure 3, C and D, shows the progressive decrease in the percent of excitable area in the RV and the LV, respectively. Curves represent individual experiments from each of the four groups. In each curve the solid lines with symbols represent time points before VF initiation, whereas dashed and symbol-less lines represent the time points after VF initiation. Note that although the first movie in VF could be reliably identified, at later stages of ischemia when the loss of excitability was almost complete in the mapped regions of most of the hearts, the notion of VF became somewhat ambiguous; however, once VF started, the ensuing rhythm never returned to a regular rhythm responding to stimulation in a one-to-one manner. The horizontal dashed line in Fig. 3D shows the level at which 90% of the LV mapped area remained excitable (or 10% of the area became inexcitable). The time points at which the LV percent excitability curves in individual experiments crossed that line (by linear interpolation) were used for the graphs shown in Fig. 7B below.
Fig. 7.

VF initiation during ischemia correlates with the emergence of inexcitable regions in the LV. Data are from 4 groups (300ms, 200ms, 300ms_Iso, and 200ms_Iso). A: average time at which 10% of the mapped region in the LV lost excitability (T10% inexcitability). B: average VF initiation time (TVF). C: TVF plotted vs. T10% inexcitability. (the same data as in A and B). *P < 0.05 vs. 300ms; #P < 0.05 vs. 200ms_Iso.
Several notable features can be observed in Fig. 3, C and D. First, that despite a relatively large variability in the time course of excitability loss in individual experiments, there was a clear overall trend that both RHR and βAS (and the combination thereof) promoted earlier emergence and progression of inexcitability. Second, in many (but not all) cases VF initiation occurred when the inexcitable regions just started to emerge. Third, the loss of excitability was apparently accelerated after VF initiation, but it cannot be said that it was always the case. Also, it cannot be said that VF was necessary to achieve a significant degree of inexcitability, since in two out five 300ms hearts, three out of five 300ms_Iso hearts, and two out five 200ms_Iso hearts at least 10% of the mapped LV area was inexcitable before the onset of VF. Curiously, the 200ms group showed the least amount of inexcitability before VF onset; the largest percentage of inexcitable area before VF in this group was 9% (in the LV). Finally, it can be seen that the progression of inexcitability was overall faster in the LV than in the RV.
Because there was no clear indication in Fig. 3 that the percent of inexcitable area is strongly dependent on the presence of VF, we performed statistical analysis of the percent inexcitable area curves over the entire course of ischemia irrespective of VF presence. The results are shown in Fig. 4 (the data is exactly the same as shown in Fig. 3, C and D). Figure 4, A and B, shows the dynamics of excitability loss in the four experimental groups arranged by the chamber (RV and LV, respectively). One can readily appreciate that in both chambers RHR and βAS synergistically accelerated the emergence of inexcitable areas. In fact, all of the groups were significantly different between each other. Figure 4, C–F, shows the same data as in Fig. 4, A and B, but plots the RV versus LV curve for each experimental group to visualize the right- to left difference in each group. One can see that under all experimental conditions the inexcitable regions emerged sooner in the LV than in the RV. The inter-chamber difference did not reach statistical significance in the 300ms group because both chambers were fully excitable for the majority of the ischemic episode; analysis limited to the last 7 min of ischemia yielded statistical significance (not shown). In all hearts from all groups excitability was recovered in all mapped regions within 2 to 3 min of reperfusion (not shown).
Fig. 4.

The time course of excitability loss during ischemia. Averaged data from 300ms, 200ms, 300ms_Iso, and 200ms_Iso groups (the same as in Fig. 3, C and D) are shown disregarding the presence or absence of VF. A and B: percentage of the excitable area in the RV and the LV, respectively, is plotted for each group as a function of ischemia time. Note that rapid pacing and βAS - independently and in combination - promote electrical failure during ischemia. C–F: RV and the LV curves of excitability loss in individual experimental groups (as indicated) are superimposed to highlight the inter-chamber differences. (The curves are the same as shown in A and B, grouped differently). Note that under all experimental conditions the LV started losing excitability earlier than the RV. *P < 0.05.
The effects of RHR and βAS on APD shortening and heterogeneity during ischemia.
It can be seen from Figs. 2 and 3 above that RHR and βAS accelerated the onset of VF during ischemia. Both APD shortening and increased APD heterogeneity are generally thought to facilitate reentrant arrhythmias and VF. Hence, it was important to assess the effects of RHR and βAS on the changes in APD during ischemia.
For the reasons explained in methods, comparison of APD80 between different experimental groups was limited to the time window between 3 and 8 min of ischemia. Figure 5 shows representative examples of individual APD maps and single pixel recordings at the 3rd and 8th min of ischemia in the four experimental groups (300ms, 200ms, 300ms_Iso, 200ms_Iso). Figure 6 summarizes the statistical analysis of APD changes in these groups. Figure 6, A and B, shows the time course of APD80 during ischemia in the RV and LV, respectively. The comparisons of APD80 between different groups in each chamber were carried out for the time window between 3 and 8 min of ischemia (vertical dashed lines in Fig. 6, A and B). One can see that both RHR and βAS (independently and in combination) promoted APD shortening during ischemia. The rate of APD shortening between 3 and 8 min of ischemia was the fastest when RHR and βAS were combined (green curves in Fig. 6). In both chambers, the APD80 time course was significantly different in all pairwise comparisons between groups with the exception of the comparison between the 200ms and 300ms_Iso, which did not yield statistical significance.
Fig. 5.

Representative examples of action potential duration at the level of 80% of repolarization (APD80) maps and individual OAPs in 4 groups (300ms, 300ms_Iso, 200ms, and 200ms_Iso) at 3 and 8 min of ischemia. Left: APD80 maps. Right: individual OAPs recorded from the pixels indicated in the respective APD maps. Rows represent experimental groups; columns represent the RV and the LV, respectively; black and red color represent 3 and 8 min of ischemia, respectively. See article for more detail.
Fig. 6.

The time course of the progressive APD80 shortening during ischemia. The data are from 4 groups: 300ms (black), 200ms (red), 300ms_Iso (blue), and 200ms_Iso (green). In each experiment, the APD80 values for the RV and the LV were obtained by averaging over all pixels in the respective mapped regions. Note that APD80 measurements were not performed fort the time points before 3 min of ischemia, due to the presence of motion artifact in the optical signals in isoproterenol-treated groups, despite the use of the electromechanical uncoupler BDM. Starting from 3 min of ischemia, the motion artifact was minimal in all groups due to ischemic suppression of contractility. For all time points after 3 min of ischemia, the APD80 data are shown only if at least 3 hearts in a given experimental group remained in regular paced rhythm and maintained excitability in at least 80% of mapped regions in both the RV and the LV. A and B: time course of APD80 decline during ischemia in the RV and the LV, respectively. The comparison between groups in each chamber was performed for the time interval between 3 and 8 min of ischemia. C–F: RV and the LV curves of APD80 decline in individual experimental groups (as indicated) are superimposed to highlight the inter-chamber differences. (The curves are the same as shown in A and B, just grouped differently). In each group, the inter-chamber difference in APD80 was analyzed for all time points satisfying the criteria stated above. Note that a prominent right-to-left APD80 gradient developed in all groups except 200ms_Iso. *P < 0.05. See article for more detail.
Figure 6, C–F, shows the same APD80 curves as in Fig. 6, A and B, but rearranged to highlight the difference between the RV and LV in each group (300ms, 200ms, 300ms_Iso and 200ms_Iso, respectively). Note that the comparisons between the RV and LV in individual groups were carried out taking into account all ischemic time points, which met the criteria mentioned above. Thus, for example, more ischemic time points were used for comparison between the RV and LV in 300ms group than in the 200ms_Iso group. One can see that a large RV-to-LV APD80 difference develops in the 300ms group starting from 7 min of ischemia (Fig. 6C). In the 200ms group, a RV-to-LV APD80 difference of the same direction develops earlier, starting from 4 to 5 min of ischemia (Fig. 6D). In the 300ms_Iso group the RV-to-LV APD80 difference is blunted as compared with the 300ms group (compare Fig. 6, C and E). Finally, the interchamber APD80 difference was fully eliminated it in the 200ms_Iso group (see Fig. 6F). The right-to-left APD80 difference was statistically significant in all groups except for the 200ms_Iso group.
The relationship between VF initiation and electrical depression under different experimental conditions.
In our experimental model VF occurred during ischemia in 100% of experiments. Overall, the conditions that accelerated electrical depression also promoted earlier initiation of VF. We tested the relationship between the early signs of electrical failure in the LV (which occurred earlier than in the RV under all conditions) and VF initiation in four experimental groups (300ms, 200ms, 300ms_Iso, and 200ms_Iso). Figure 7A shows the time at which 10% of the mapped region of the LV became inexcitable (T10% inexcitability). Note that in some cases the time points of 10% inexcitability were obtained by linear interpolation (i.e., when the inexcitability curves crossed the dashed line in Fig. 3D). Figure 7B shows TVF. One can see that both RHR (200ms group) and βAS (300ms_Iso group) applied individually significantly shortened both T10% inexcitability and TVF as compared with the control (300ms group). The combination of RHR and βAS (200ms_Iso group) further shortened both T10% inexcitability and TVF. In Fig. 7C, we plotted TVF versus T10% inexcitability in individual experiments. One can see that overall the emergence of inexcitable regions in the LV is strongly correlated with the time of VF onset when all the groups are pooled together (R2 = 0.72; P < 0.0001). It is of interest that the 200ms group supplied the largest number of points deviating from the general trend. Removal of 200ms group from the correlation analysis increased R2 value to 0.94 (not shown).
The superfusion solution used during ischemia in this study was severely hypoxic, but it lacked other components of an ischemic environment (expected during cardiac arrest) such as hyperkalemia and acidosis. To exclude the possibility that these factors affected the outcomes, we performed five additional experiments in which hearts were subjected to pacing at 200 ms and isoproterenol as in the 200ms_Iso group, but the ischemic superfusion solution was acidic (pH = 6.8) and hyperkalemic ([K+]o = 8 mM) in addition to being hypoxic (pO2 = 25 mm). These modifications of the superfusion solution did not significantly affect the studied phenomena. In particular, the time at which 10% and 50% of excitability was lost in the RV and the LV, and TVF were not affected by the presence or the absence of the acidosis and hyperkalemia in the superfusate (see Table 1).
Table 1.
No effect of acidosis and hyperkalemia in the superfusate on the loss of excitability and the time of VF onset during global ischemia in 200ms_Iso group
| Superfusion Composition | Hypoxic | Hypoxic, Acidic, Hyperkalemic |
|---|---|---|
| Time of 10% excitability loss | ||
| RV | 10.5 ± 0.6 | 10.6 ± 2.4 |
| LV | 8.9 ± 0.6 | 7.1 ± 0.9 |
| Time of 50% excitability loss | ||
| RV | 13.5 ± 0.9 | 14.0 ± 3.0 |
| LV | 10.9 ± 0.9 | 10.9 ± 2.11 |
| Time to VF | 9.9 ± 0.9 | 10.2 ± 1.1 |
Time values are means ± SE and are in minutes; n = 6 for Hypoxic group, and n = 5 for Hypoxic, Acidic, Hyperkalemic group. The hearts were subjected to rapid pacing (cycle length = 200 ms), isoproterenol (30 nM), and global no-flow ischemia. In hypoxic superfusion, pO2 was below 40 mmHg, but otherwise the solution had normal composition. In hypoxic, acidic, hyperkalemic solution, pO2 was reduced to 25 mmHg, pH was reduced to 6.8, and [K+]o was elevated to 8 mM. VF, ventricular fibrillation; RV, right ventricular; LV, left ventricular. All differences, P > 0.1 by unpaired 2-tailed Student test.
Patterns of initiation and maintenance of ischemic VF.
In this study we had a total of six cases when a transition from paced rhythm (at 300 or 200 ms) to VF occurred during optical recording (see Supplemental Movies S1–S6). We saw a variety of patterns at the onset of VF. However, the typical case of VF initiation was a breakthrough pattern in the apical half of the anterior LV including the LV/RV junction, which eventually stabilized to a mother source pattern (Supplemental Movies S1–S4).
A representative example is shown in Fig. 8 (corresponds to Supplemental Movie S1). Figure 8A shows OAPs from a single pixel whose location is shown in Fig. 8D. Figure 8B shows activation maps in the last paced beat (P) and selected arrhythmic beats at the initiation of VF indicated in Fig. 8A. The red color denotes the sites of the earliest activation; magenta denotes the sites of the latest activation. All activation maps are limited to the first 50 ms from the earliest activation in the field of view. In the paced beat (P) the earliest activation is seen at the RV apex and adjacent RV free wall. The first premature excitation (A1) occurs ∼150 ms after the last paced excitation and features a large area of almost simultaneous activation in the anterio-apical LV, with a second focus appearing slightly later in mid-anterior LV (arrows in Fig. 8B, map A1). Beats A7 and A8 reveal shifting pattern of epicardial breakthroughs compared with beat A1 (see arrows in maps A1, A7, and A8). Note that apparently early activation in the basal LV in A8 shows in fact the sites of the latest activation from the previous excitation (A7). In beat A10 the site of the earliest activation remained the same as in A8 (in the apical RV), but a dramatic slowing of conduction occurred, and a line of a full conduction block appeared in the mid-anterior RV (white arrow in map A10). The apparently early activations in the basal RV and LV in map A10 reflect the propagation of the wavefront started during previous excitation (A9).
Fig. 8.

A typical case of VF initiation during ischemia with the first beat of VF appearing as an epicardial breakthrough (see Supplemental Movie S1). A: a recording from a single pixel whose position is marked with a red square in D. The last paced beat (P) and selected arrhythmic beats (A1, A7, A8, A10, A23, A40, A56) are labeled above the recording. B: activation maps for selected beats indicated in A. Red indicates the earliest activation; magenta, the latest. All activation maps are limited to the first 50 ms in the optically mapped region. C: phase maps obtained using Hilbert Transform for selected beats indicated in A. A stable spiral wave with the center of rotation (white circle) anchored on the RV/LV junction was first seen during beat A23 and maintained its position for at least next 23 cycles. D: image of the heart showing the mapped RV and LV regions and the position of the pixel used to generate optical signal shown in A. See article for more detail.
The pattern of activation remained similar between A10 and A22 (not shown). However, starting from beat A23 and at least for the next 25 cycles a stable rotor was present at the RV/LV junction, apparently driving VF. Figure 8C shows phase maps for beats A23, A40, and A56. Note the presence of a rotor anchored at the same location and having a counterclockwise direction of rotation.
Figure 9 shows the unique but interesting case from the VF_initiation group in which the first beat of VF was due to reentry (see Supplemental Movie S6). Figure 9A shows individual frames (snapshots) of the fluorescence movie recorded at instances of time marked under the snapshots. The contrast in the snapshots is greatly increased, so the sites with relatively large amplitude of OAP are saturated to highlight OAPs with low amplitude. Red dots with numbers in Fig. 9A indicate the locations of selected pixels from which the OAP recordings were collected and presented in Fig. 9B. The gray rectangle in Fig. 9B indicates the time window encompassing the snapshots shown in Fig. 9A. One can see that the last paced wavefront (Plast in Fig. 9B) propagated from site 1 towards RV base (sites 10 and 9) and LV apex (sites 2 and 3), but failed to activate site 8 in the LV base (see frames at 41, 52, and 62 ms). The impulse, however, was able to propagate, at a very low amplitude, from site 3 to site 7 around a large area in the mid-anterior LV which was inexcitable (see frames at 52, 62, 82, and 93 ms). Finally, a faint impulse could be recognized at site 8 (frame at 113 ms) from which the impulse re-entered site 9 (frame at 124 ms). From there the wavefront gained amplitude and propagated counterclockwise from RV base to RV apex in the direction opposite to that during the paced beats (P and Plast in Fig. 9B), forming the first beat of VF (A1 in Fig. 9B).
Fig. 9.

A case of VF initiation during ischemia by epicardial reentry involving inexcitable and poorly excitable regions (see Supplemental Movie S6). A: selected single frame snapshots taken during the formation of the first reentrant circuit. The contrast in the snapshots is greatly increased to highlight OAPs with low amplitude. Red dots with numbers indicate the locations of selected pixels. B: single pixel recordings from the numbered pixels shown in A. Gray rectangle indicates the time window encompassing the snapshots shown in A. P, a regular paced beat, represents the relatively constant activation pattern before VF initiation. Plast, the last paced wavefront giving rise to reentry; A1, the first nonpaced beat. Recordings from the sites critical for the formation of unidirectional block (pixels nos. 7–9) are shown in green. T-shaped red lines indicate the sites of conduction block. Red line with arrowhead indicates the full reentrant circuit initiating VF. C: a phase map during the stable phase of VF. D: time-space plot along the line y-y′ shows the characteristic “Christmas tree” pattern, indicating a stable spiral wave source in the basal RV. E: image of the heart demarcating the mapped regions in the RV and the LV. See article for more detail.
The original reentrant circuit was still visible during the next two cycles of VF, but the overall activation pattern quickly became more complex and disorganized, eventually losing any association with the activation pattern of the first arrhythmic beat (see Supplemental Movie S6). Figure 9C shows a phase map from the Hilbert-transformed phase movie, which corresponds to ∼10 s after VF initiation. One can see that several wavefronts (visible as transitions from magenta to red) are present simultaneously, typical of VF. The fine-grained pattern in the mid-anterior LV is due to the lack of activation in that region, leading to the random distribution of phase derived from background noise. Figure 9D shows the time-space plot generated from the pixel line y-y′ indicated in Fig. 9C and spanning ∼1 s. The time-space plot reveals a “Christmas three” pattern indicative of a stable spiral wave in the basal RV (at the level of outflow tract), which was apparently the dominant source of activation (mother rotor) for at least several seconds.
DISCUSSION
The most important findings of this study are: 1) βAS and RHR, individually and synergistically, promote heterogeneous electrical depression and loss of excitability during total (no-flow) ischemia; and 2) the time when inexcitable regions start to emerge correlates with the time of spontaneous VF initiation. Despite this correlation, however, a direct mechanistic link between heterogeneous electrical depression and the onset of VF could be demonstrated only in a single case of VF initiation (Fig. 9). In a typical case the first arrhythmic beat manifested in the form of a broad epicardial breakthrough, suggesting an intramural or endocardial source of activation. Because those locations were inaccessible using epicardial optical mapping, neither abnormal automaticity nor reentry could be excluded.
βAS and electrical depression during myocardial ischemia.
Myocardial ischemia is associated with large increases in the interstitial levels of epinephrine and norepinephrine. The excessively high local norepinephrine concentrations are combined with an enhanced responsiveness of the myocyte to catecholamines. According to Schömig et al. (45), myocardial ischemia of 15 min duration may result in a 100-fold increase in the interstitial catecholamine concentrations, a twofold increase in functionally coupled α-adrenoceptors, and a 30% increase in β-adrenoceptors. In a study involving a porcine model of VF-induced cardiac arrest, interstitial norepinephrine increased sixfold during 8 min of VF, and progressed to an over 100-fold increase after defibrillation and reperfusion (30), suggesting that during life-saving procedures the heart is exposed to high catecholamine concentrations even without exogenous administration as recommended by current resuscitation guidelines.
Our study revealed a depressant effect of βAS on conduction and excitability in ischemic myocardium. Of interest, the depressant effect of βAS was additive to that of the increased pacing rate (i.e., RHR). The pattern of electrical depression was highly nonuniform but with a similar pattern in all groups, such that LV experienced earlier depression than RV. This is reminiscent of highly heterogeneous loss of excitability in globally ischemic, fibrillating canine hearts (57). In the present study we analyzed the dV/dtmax as an index of local excitability. dV/dtmax reflects primarily the availability of Na+ channels (11), but in the syncytium depends also on the source-sink relationship, so that a decreased sink (e.g., due to partial cellular uncoupling) may cause an increase in dV/dtmax (49). The canonical mechanism for reduction in Na+ channel availability and dV/dtmax, as well as the complete loss of excitability during ischemia, is the increase in the extracellular [K+] leading to membrane depolarization (47, 48). However, in our previous study we showed that the level of [K+]o actually attained in the globally ischemic fibrillating canine heart is too low to explain the observed degree of electrical depression and inexcitability (52). In fact, the inadequacy of [K+]o elevation to explain conduction slowing during global ischemia has been known for a very long time [e.g., (23)]. Also, the depressant effect of isoproterenol observed in this study is difficult to explain in terms of accelerated [K+]o elevation, since in previous studies catecholamines either did not change or even reduced [K+]o elevation during global ischemia (59, 61). Regarding the possible effect of heart rate on ischemic [K+]o elevation, it was shown to be virtually absent at heart rates above 100 beats/min (23, 60). Thus it is highly unlikely the change in pacing rate from 180 to 300 beats/min in our study would cause an additional increase in the ischemic level of [K+]o. As a summary of what is mentioned above, the accelerated onset of electrical depression and emergence of inexcitable regions due to βAS and/or RHR cannot be explained in terms of increased extracellular potassium accumulation.
βAS increases peak ICa,L and slows inactivation, both effects leading to an increased Ca++ loading of cardiomyocytes. Importantly, RHR also leads to increased Ca++ loading; thus the observed synergism between βAS and RHR suggests that the mechanism of electrical depression might be mediated via increased [Ca]i. In a series of studies, Clusin and his colleagues investigated the effects of heart rate, elevated extracellular Ca++, and β-adrenergic stimulation on ischemic cellular depolarization, conduction velocity, and ventricular arrhythmias (5, 6, 13, 17). Overall, their results suggest that various conditions which increase cellular Ca++ loading promote cellular depolarization, conduction slowing, and arrhythmias during ischemia. Of particular interest is the observation that Ca++-loading conditions enhanced the component of cellular depolarization independent of [K+]o (estimated as the difference between the estimated resting potential and the K+ equilibrium potential) (5, 6). The mechanisms mediating Ca++-dependent ischemic depolarization not related to hyperkalemia remain unknown to this day and warrant further studies.
How βAS and RHR promote ischemic VF?
It is well established that βAS increases incidence of VF in the context of myocardial ischemia and infarction and that β-adrenergic receptor antagonists reduce the incidence of ischemic arrhythmias and sudden cardiac death (2, 11, 34, 45, 50). The pro-arrhythmic effects of βAS are usually explained in terms of nonreentrant mechanisms (EADs and DADs) downstream of increased ICa,L and/or prolonged APD, or increased SR Ca++ load and/or spontaneous SR Ca++ release (for the sake of space, we will not discuss these diverse mechanisms in more detail here) (8, 19, 42, 43, 44, 50, 51). It should be noted, however, that when it comes to the conditions of acute ischemia, the evidence that EADs or DADs are responsible for arrhythmogenesis mediated by βAS is limited and controversial. Essentially, the promotion of EADs and DADs by β-AS was observed only under conditions of mild hypoxia (19, 43), which poorly represents real ischemia since at least two critical factors (KATP activation and hyperkalemia) are absent. More severe simulated ischemic conditions, including one or more of the following: severe hypoxia, hyperkalemia, and a relatively complete metabolic inhibition (causing KATP activation), abolish EADs and DADs (15, 43) (35). Using dual optical mapping of transmembrane potential and intracellular [Ca++], Lakkireddy et al. (31) demonstrated epicardial OAP sequences resembling EADs during reperfusion. However, with our best effort we did not find in the literature any direct evidence of EADs or DADs under conditions of acute no-flow ischemia, comparable with experimental conditions used in our study. We did not observe apparent EADs (or DADs) as standalone phenomena during paced beats either before or during ischemia.
Our study shows that βAS and RHR synergistically accelerated heterogeneous electrical depression and the initiation of VF during ischemia. Although not quantified in this study, the conduction velocity is in general proportional to dV/dtmax and it was visibly reduced in regions subsequently losing excitability. In addition, βAS and RHR accelerated the ischemic decline in APD (see Fig. 6). Conduction slowing and APD shortening facilitate formation of reentrant circuits. However, only in one out of six cases of documented VF initiation was the first beat of VF due to identifiable reentry involving inexcitable and highly depressed regions in the LV epicardium (Fig. 9). In other cases the direct cause-effect relationship between the heterogeneous electrical depression and VF initiation could not be established. However, the heterogeneous electrical depression culminating in formation of mosaic nonconducting regions may create favorable substrate for initiation of reentry in other myocardial layers not accessible with epicardial optical mapping. It is possible that the substrate for unidirectional block is formed in the subendocardium, perhaps involving the Purkinje network. With the assumption that the inexcitable or poorly excitable regions spread across the ventricular wall, the Purkinje-muscle interface may be the site of the initial reentrant circuit. On the other hand, we cannot exclude that βAS and RHR promote EADs or DADs in Purkinje fibers, especially given the fact that at least some of them may experience milder ischemic conditions due to better exchange of oxygen and waste products with the solution in the ventricular cavities. Indeed, the propensity of isolated Purkinje fibers for DADs is the largest at a moderate degree of hypoxia (35). Multifocal epicardial activation observed in some cases of VF initiation (see Fig. 8) strongly suggests that the origin of the first non-paced beat was in the Purkinje network. It is possible that electrophysiological changes caused by βAS and/or RHR in the working myocardium created favorable conditions for EADs in Purkinje fibers to occur. In fact, depolarization and partial uncoupling of working myocardium can play permissive roles for development of EADs or DADs in Purkinje fibers, reducing the repolarizing junctional current, which otherwise suppresses afterdepolarizations (25, 58). Thus the VF-promoting effect of βAS and/or RHR could at least in part be due to creating necessary source/sink relationship at the Purkinje-myocardium interface, permitting abnormal impulses formed in Purkinje fibers to invade working myocardium and interact with a highly heterogeneous conductive substrate.
A few words have to be said regarding the possible role of slow response type of propagation in VF initiation. This term refers to the propagation of electrical impulse in the heart driven exclusively by (slow) Ca current, as opposed to the fast Na current. Experimentally, the slow response was observed under conditions of elevated extracellular [K+] (>15 mM) and high catecholamine concentrations (>1 μM) (16). The presence of this phenomenon under realistic conditions of ischemia has never been demonstrated. However, it is not impossible that in our experimental model βAS, while suppressing the Na+ current-dependent fast response conduction, at some point promoted the slow response conduction. This could explain cases when conduction in a highly depressed region suddenly improved despite ongoing ischemia (such as in locations 7 and 8 in Fig. 9B). However, such events were extremely rare; thus, even if the slow response-mediated conduction was present, it was a low probability phenomenon in our model. Alternatively, the transient improvements of conduction in highly depressed regions could be explained by fluctuations in metabolism, which could translate into fluctuations of excitability in ischemic myocytes (1, 38).
It is of interest that the stable phase of VF was usually driven by a mother source (28) [corresponding to Type II VF (12)], which always had a different location than the first beat initiating VF. Thus the mechanism of VF trigger was different from the mechanism of VF maintenance. Whereas we cannot ascertain the predominant mechanism of the ischemic VF trigger in this experimental model, it is likely that at least in part βAS and RHR accelerated the onset of VF due to promotion of spatially heterogeneous electrical depression during ischemia.
Relevance to the outcomes of VF-induced cardiac arrest.
The combination of global ischemia, RHR and βAS reproduce major pathophysiological factors of VF-induced cardiac arrest. We speculate that the dependence of electrical depression and VF initiation on RHR and βAS observed in this study is relevant to the three major adverse outcomes of VF-induced cardiac arrest: asystole, pulseless electrical activity (PEA), and recurrent VF. Even though we did not determine true global asystole from the volume conductor ECG (due to severe distortion of the ECG by stimulus artifacts generated by large pacing electrodes), the full lack of activation in the optical maps most likely indicated global inexcitability. Also, the lack of activation in large parts of LV as observed in this study (see Fig. 2) could create conditions for PEA after a successful defibrillation shock. Finally, the combination of RHR and βAS promoted VF, meaning that a longer time spent in VF and larger catecholamine concentrations would increase the chance of VF recurrence after defibrillation shock. These considerations emphasize that catecholamine administration during cardiac arrest can be counterproductive and may worsen the outcomes, which is in line with growing concerns regarding the appropriateness of epinephrine use during life-saving interventions. It is of interest that in observational studies, epinephrine worsened long-term outcomes mostly in patients found in VF (10).
Relevance to arrhythmias in the context of acute regional ischemia.
Although only global ischemia was tested in our study, previous studies in models of regional ischemia showed qualitatively similar phenomena, namely, the dependence of the time to VT/VF initiation on the heart rate (3, 24, 54) and sympathetic stimulation (46). Moreover, in a canine model of regional ischemia VF initiation was quantitatively related to conduction delay in the center and border of the ischemic zone (40). Thus it is possible that the mutual dependence of electrical depression and VF initiation on RHR and βAS as demonstrated in our study is also relevant to mechanisms of arrhythmogenesis associated with regionally ischemic myocardium.
Conclusion
This may be the first study to show that rapid rhythm and β-adrenergic stimulation synergistically promote heterogeneous electrical failure in a globally ischemic heart. Moreover, there is a strong correlation between emergence of inexcitable regions and the initiation of VF, although the mechanistic relationship between the two phenomena cannot be fully ascertained without detailed endocardial and/or intramural mapping of activation. The synergistic effect of RHR and βAS with respect to ischemic electrical depression may be relevant to the adverse outcomes of VF-induced cardiac arrest, such as asystole, PEA, and recurrent VF.
Limitations.
An obvious limitation is the limited fraction of the ventricular myocardium accessible by optical mapping. Panoramic optical mapping (7, 33) could improve our ability to determine the mechanism of VF initiation and maintenance. It is possible that it would ascertain that all of the mother sources are due to stable rotors. However, it would not help to resolve the sources of broad LV epicardial breakthroughs predominantly initiating VF, most likely located in the mid- or endocardium. Endocardial mapping using an endoscope camera (29) in the LV could help to identify possible endocardial triggers of VF and would be a reasonable follow-up of this study. Use of BDM to immobilize the heart could distort the progressive electrophysiological changes during ischemia, but in two experiments using pacing at 200 ms and isoproterenol (30 nM) in the absence of uncouplers, the overall dynamics of electrical depression, emergence of inexcitable regions, and initiation of VF was similar to those observed in our 200ms_Iso group (not shown). In contrast, another uncoupler blebbistatin in three experiments postponed the adverse electrophysiological outcomes of ischemia (not shown) consistent with results from our recent study (56). Lack of mechanical load in the Langendorff-perfused hearts could also distort the ischemic changes (62); thus the effect of mechanical load should be investigated in future studies. Finally, even though an increase in [Ca++]i is highly expected under conditions of RHR and βAS, a quantitative imaging of [Ca++]i in this model could provide additional insights by revealing, for example, regional differences in cellular Ca load. However, any outcomes of [Ca++]i imaging would not change the main conclusion of this study that RHR and βAS mutually promote electrical failure and VF in the globally ischemic heart.
GRANTS
This work was supported by the United States National Institutes of Health (5R01HL088444 and 1RO1HL103877) and by a Research Grant from the Nora Eccles Treadwell Foundation to A. V. Zaitsev.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.G., T.G.T., and A.V.Z. conception and design of research; V.G., T.G.T., M.W., P.W.V., K.J.S., and J.S. performed experiments; V.G., T.G.T., M.W., P.W.V., and A.V.Z. analyzed data; V.G., T.G.T., and A.V.Z. interpreted results of experiments; V.G., T.G.T., and A.V.Z. prepared figures; V.G., T.G.T., and A.V.Z. drafted manuscript; V.G., T.G.T., and A.V.Z. edited and revised manuscript; V.G., T.G.T., M.W., P.W.V., K.J.S., J.S., and A.V.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Alicja Booth, Jayne H. Davis, and Nancy C. Allen for excellent technical support and Bruce Steadman, Dennis King, Wilson Lobania, and Phil Ershler for help in developing hardware and software necessary for this project.
Present address of V. Garg: Dept. of Physiology, Univ. of California - San Francisco, Genentech Hall, 600 16th St., San Francisco, CA 94158.
Present address of T. Taylor: Physio-Control, 11811 Willows Road NE, Redmond, WA 98073.
Present address of P. Venable: Blackrock Microsystems, 630 Komas Dr. Suite 200, Salt Lake City, UT 84108.
REFERENCES
- 1.Akar FG, Aon MA, Tomaselli GF, O′Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest 115: 3527–3535, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aupetit JF, Frassati D, Bui-Xuan B, Freysz M, Faucon G, Timour Q. Efficacy of a β-adrenergic receptor antagonist, propranolol, in preventing ischaemic ventricular fibrillation: dependence on heart rate and ischaemia duration. Cardiovasc Res 37: 646–655, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Aupetit JF, Timour Q, Freysz M, Loufoua-Moundanga J, Omar S, Chevrel G, Faucon G. Vulnerability to ventricular fibrillation related to ischaemia: comparison of the acute effects of beta-blockers and calcium antagonists. Arch Int Pharmacodyn Ther 327: 25–39, 1994. [PubMed] [Google Scholar]
- 4.Bayes de Luna A, Elosua R. Sudden death. Rev Esp Cardiol 65: 1039–1052, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Blake K, Clusin WT, Franz MR, Smith NA. Mechanism of depolarization in the ischaemic dog heart: discrepancy between T-Q potentials and potassium accumulation. J Physiol 397: 307–330, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blake K, Smith NA, Clusin WT. Rate dependence of ischaemic myocardial depolarisation: evidence for a novel membrane current. Cardiovasc Res 20: 557–562, 1986. [DOI] [PubMed] [Google Scholar]
- 7.Bourgeois EB, Reeves HD, Walcott GP, Rogers JM. Panoramic optical mapping shows wavebreak at a consistent anatomical site at the onset of ventricular fibrillation. Cardiovasc Res 93: 272–279, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 590: 3291–3304, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bray MA, Wikswo JP. Considerations in phase plane analysis for nonstationary reentrant cardiac behavior. Phys Rev E 65: 051902, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Callaway CW. Epinephrine for cardiac arrest. Curr Opin Cardiol 28: 36–42, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 79: 917–1017, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Chen PS, Wu TJ, Ting CT, Karagueuzian HS, Garfinkel A, Lin SF, Weiss JN. A tale of two fibrillations. Circulation 108: 2298–2303, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Clusin WT, Buchbinder M, Ellis AK, Kernoff RS, Giacomini JC, Harrison DC. Reduction of ischemic depolarization by the calcium channel blocker diltiazem. Correlation with improvement of ventricular conduction and early arrhythmias in the dog. Circ Res 54: 10–20, 1984. [DOI] [PubMed] [Google Scholar]
- 14.Cobb LA, Fahrenbruch CE, Olsufka M, Copass MK. Changing incidence of out-of-hospital ventricular fibrillation, 1980–2000. JAMA 288: 3008–3013, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Coetzee WA, Opie LH. Effects of components of ischemia and metabolic inhibition on delayed afterdepolarizations in guinea pig papillary muscle. Circ Res 61: 157–165, 1987. [DOI] [PubMed] [Google Scholar]
- 16.Cranefield PF, Wit AL, Hoffman BF. Conduction of the cardiac impulse: III. Characteristics of very slow conduction. J Gen Physiol 59: 227–246, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis SC, Coetzee WA, de Jong JW, Clusin W, Opie LH. Effects of coronary flow, pacing rate, isoprenaline and diltiazem on ischemic ventricular arrhythmias in guinea pig hearts. J Pharmacol Exp Ther 248: 372–377, 1989. [PubMed] [Google Scholar]
- 18.Dominguez G, Fozzard HA. Influence of extracellular K+ concentration on cable properties and excitability of sheep cardiac Purkinje fibers. Circ Res 26: 565–574, 1970. [DOI] [PubMed] [Google Scholar]
- 19.Gaur N, Rudy Y, Hool L. Contributions of ion channel currents to ventricular action potential changes and induction of early afterdepolarizations during acute hypoxia. Circ Res 105: 1196–1203, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray RA, Pertsov AM, Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature 392: 75–78, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Hallstrom A, Herlitz J, Kajino K, Olasveengen TM. Treatment of asystole and PEA. Resuscitation 80: 975–976, 2009. [DOI] [PubMed] [Google Scholar]
- 22.Herlitz J, Svensson L, Engdahl J, Silfverstolpe J. Characteristics and outcome in out-of-hospital cardiac arrest when patients are found in a non-shockable rhythm. Resuscitation 76: 31–36, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation 61: 768–778, 1980. [DOI] [PubMed] [Google Scholar]
- 24.Hope RR, Williams DO, El-Sherif N, Lazarra R, Scherlag BJ. The efficacy of antiarrhythmic agents during acute myocardial ischemia and the role of heart rate. Circulation 50: 507–514, 1974. [DOI] [PubMed] [Google Scholar]
- 25.Huelsing DJ, Spitzer KW, Pollard AE. Electrotonic suppression of early afterdepolarizations in isolated rabbit Purkinje myocytes. Am J Physiol Heart Circ Physiol 279: H250–H259, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Hulleman M, Berdowski J, de Groot JR, van Dessel PF, Borleffs CJ, Blom MT, Bardai A, de Cock CC, Tan HL, Tijssen JG, Koster RW. Implantable cardioverter-defibrillators have reduced the incidence of resuscitation for out-of-hospital cardiac arrest caused by lethal arrhythmias. Circulation 126: 815–821, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Ilkhanoff L, Goldberger JJ. Out-of-hospital cardiac arrest: getting beyond the tip of the iceberg. Circulation 126: 793–796, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Jalife J. Ventricular fibrillation: mechanisms of initiation and maintenance. Annu Rev Physiol 62: 25–50, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Kalifa J, Klos M, Zlochiver S, Mironov S, Tanaka K, Ulahannan N, Yamazaki M, Jalife J, Berenfeld O. Endoscopic fluorescence mapping of the left atrium: a novel experimental approach for high resolution endocardial mapping in the intact heart. Heart Rhythm 4: 916–924, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Killingsworth CR, Wei CC, Dell′Italia LJ, Ardell JL, Kingsley MA, Smith WM, Ideker RE, Walcott GP. Short-acting beta-adrenergic antagonist esmolol given at reperfusion improves survival after prolonged ventricular fibrillation. Circulation 109: 2469–2474, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Lakkireddy V, Bub G, Baweja P, Syed A, Boutjdir M, El-Sherif N. The kinetics of spontaneous calcium oscillations and arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm 3: 58–66, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121: e46–e215, 2010. [DOI] [PubMed] [Google Scholar]
- 33.Lou Q, Li W, Efimov IR. The role of dynamic instability and wavelength in arrhythmia maintenance as revealed by panoramic imaging with blebbistatin vs. 2,3-butanedione monoxime. Am J Physiol Heart Circ Physiol 302: H262–H269, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lubbe WF, Podzuweit T, Opie LH. Potential arrhythmogenic role of cyclic adenosine monophosphate (AMP) and cytosolic calcium overload: implications for prophylactic effects of beta-blockers in myocardial infarction and proarrhythmic effects of phosphodiesterase inhibitors. J Am Col Cardiol 19: 1622–1633, 1992. [DOI] [PubMed] [Google Scholar]
- 35.Molina-Viamonte V, Anyukhovsky EP, Rosen MR. An alpha-1-adrenergic receptor subtype is responsible for delayed afterdepolarizations and triggered activity during simulated ischemia and reperfusion of isolated canine Purkinje fibers. Circulation 84: 1732–1740, 1991. [DOI] [PubMed] [Google Scholar]
- 36.Ng FS, Shadi IT, Peters NS, Lyon AR. Selective heart rate reduction with ivabradine slows ischaemia-induced electrophysiological changes and reduces ischaemia-reperfusion-induced ventricular arrhythmias. J Mol Cell Cardiol 59: 67–75, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olasveengen TM, Samdal M, Steen PA, Wik L, Sunde K. Progressing from initial non-shockable rhythms to a shockable rhythm is associated with improved outcome after out-of-hospital cardiac arrest. Resuscitation 80: 24–29, 2009. [DOI] [PubMed] [Google Scholar]
- 38.O′Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science 265: 962–966, 1994. [DOI] [PubMed] [Google Scholar]
- 39.Pertsov AM, Davidenko JM, Salomonzs R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res 72: 631–650, 1993. [DOI] [PubMed] [Google Scholar]
- 40.Peter T, Fujimoto T, Hamamoto H, Mandel WJ. Comparative study of the effect of slow channel inhibiting agents on ischemia-induced conduction delay as relevant to the genesis of ventricular fibrillation. Am Heart J 106: 1023–1028, 1983. [DOI] [PubMed] [Google Scholar]
- 41.Polentini MS, Pirrallo RG, McGill W. The changing incidence of ventricular fibrillation in Milwaukee, Wisconsin (1992–2002). Prehosp Emerg Care 10: 52–60, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol Heart Circ Physiol 258: H1796–H1805, 1990. [DOI] [PubMed] [Google Scholar]
- 43.Priori SG, Yamada KA, Corr PB. Influence of hypoxia on adrenergic modulation of triggered activity in isolated adult canine myocytes. Circulation 83: 248–259, 1991. [DOI] [PubMed] [Google Scholar]
- 44.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 115: 2305–2315, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schömig A, Haass M, Richardt G. Catecholamine release and arrhythmias in acute myocardial ischaemia. Eur Heart J 12: 38–47, 1991. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz PJ, Priori SG, Vanoli E, Zaza A, Zuanetti G. Efficacy of diltiazem in two experimental feline models of sudden cardiac death. J Am Coll Cardiol 8: 661–668, 1986. [DOI] [PubMed] [Google Scholar]
- 47.Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia: a theoretical study of altered cell excitability and action potential duration. Cardiovasc Res 35: 256–272, 1997. [DOI] [PubMed] [Google Scholar]
- 48.Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia. A mechanistic investigation of action potential conduction and conduction failure. Circ Res 80: 124–138, 1997. [DOI] [PubMed] [Google Scholar]
- 49.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res 81: 727–741, 1997. [DOI] [PubMed] [Google Scholar]
- 50.Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res 114: 1004–1021, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Sirenko S, Maltsev VA, Maltseva LA, Yang D, Lukyanenko Y, Vinogradova TM, Jones LR, Lakatta EG. Sarcoplasmic reticulum Ca2+ cycling protein phosphorylation in a physiologic Ca2+ milieu unleashes a high-power, rhythmic Ca2+ clock in ventricular myocytes: relevance to arrhythmias and bio-pacemaker design. J Mol Cell Cardiol 66: 106–115, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor TG, Venable PW, Booth A, Garg V, Shibayama J, Zaitsev AV. Does the combination of hyperkalemia and KATP activation determine excitation rate gradient and electrical failure in the globally ischemic fibrillating heart? Am J Physiol Heart Circ Physiol 305: H903–H912, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tovar OH, Bransford PP, Jones JL. Probability of induction and stabilization of ventricular fibrillation with epinephrine. J Mol Cell Cardiol 30: 373–382, 1998. [DOI] [PubMed] [Google Scholar]
- 54.Vaillant F, Dehina L, Dizerens N, Bui-Xuan B, Tabib A, Lauzier B, Chevalier P, Descotes J, Timour Q. Ivabradine but not propranolol delays the time to onset of ischaemia-induced ventricular fibrillation by preserving myocardial metabolic energy status. Resuscitation 84: 384–390, 2013. [DOI] [PubMed] [Google Scholar]
- 55.van Alem AP, Post J, Koster RW. VF recurrence: characteristics and patient outcome in out-of-hospital cardiac arrest. Resuscitation 59: 181–188, 2003. [DOI] [PubMed] [Google Scholar]
- 56.Venable PW, Sciuto KJ, Warren M, Taylor TG, Garg V, Shibayama J, Zaitsev AV. Mitochondrial depolarization and asystole in the globally ischemic rabbit heart: coordinated response to interventions affecting energy balance. Am J Physiol Heart Circ Physiol 308: H485–H499, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venable PW, Taylor TG, Shibayama J, Warren M, Zaitsev AV. Complex structure of electrophysiological gradients emerging during long-duration ventricular fibrillation in the canine heart. Am J Physiol Heart Circ Physiol 299: H1405–H1418, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verkerk AO, Veldkamp MW, Coronel R, Wilders R, van Ginneken AC. Effects of cell-to-cell uncoupling and catecholamines on Purkinje and ventricular action potentials: implications for phase-1b arrhythmias. Cardiovasc Res 51: 30–40, 2001. [DOI] [PubMed] [Google Scholar]
- 59.Warner MR, Kroeker TS, Zipes DP. Sympathetic stimulation and norepinephrine infusion modulate extracellular potassium concentration during acute myocardial ischemia. Circ Res 71: 1078–1087, 1992. [DOI] [PubMed] [Google Scholar]
- 60.Weiss J, Shine KI. Effects of heart rate on extracellular [K+] accumulation during myocardial ischemia. Am J Physiol Heart Circ Physiol 250: H982–H991, 1986. [DOI] [PubMed] [Google Scholar]
- 61.Weiss J, Shine KI. Extracellular K+ accumulation during myocardial ischemia in isolated rabbit heart. Am J Physiol Heart Circ Physiol 242: H619–H628, 1982. [DOI] [PubMed] [Google Scholar]
- 62.Wengrowski AM, Kuzmiak-Glancy S, Jaimes R 3rd, Kay MW. NADH changes during hypoxia, ischemia, and increased work differ between isolated heart preparations. Am J Physiol Heart Circ Physiol 306: H529–H537, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]