

Gemcitabine offers modest benefit in treatment of metastatic pancreatic cancer. Rigosertib, a small-molecule inhibitor of multiple effector pathways important in tumorgenesis and metastasis, has shown synergistic effects in combination with gemcitabine in preclinical pancreatic cancer models. This study investigated the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone.
Keywords: pancreatic cancer, phase II/III, PI3K inhibitor, PLK1 inhibitor, Ras mimetic, rigosertib
Abstract
Background
Rigosertib (ON 01910.Na), a first-in-class Ras mimetic and small-molecule inhibitor of multiple signaling pathways including polo-like kinase 1 (PLK1) and phosphoinositide 3-kinase (PI3K), has shown efficacy in preclinical pancreatic cancer models. In this study, rigosertib was assessed in combination with gemcitabine in patients with treatment-naïve metastatic pancreatic adenocarcinoma.
Materials and methods
Patients with metastatic pancreatic adenocarcinoma were randomized in a 2:1 fashion to gemcitabine 1000 mg/m2 weekly for 3 weeks of a 4-week cycle plus rigosertib 1800 mg/m2 via 2-h continuous IV infusions given twice weekly for 3 weeks of a 4-week cycle (RIG + GEM) versus gemcitabine 1000 mg/m2 weekly for 3 weeks in a 4-week cycle (GEM).
Results
A total of 160 patients were enrolled globally and randomly assigned to RIG + GEM (106 patients) or GEM (54). The most common grade 3 or higher adverse events were neutropenia (8% in the RIG + GEM group versus 6% in the GEM group), hyponatremia (17% versus 4%), and anemia (8% versus 4%). The median overall survival was 6.1 months for RIG + GEM versus 6.4 months for GEM [hazard ratio (HR), 1.24; 95% confidence interval (CI) 0.85–1.81]. The median progression-free survival was 3.4 months for both groups (HR = 0.96; 95% CI 0.68–1.36). The partial response rate was 19% versus 13% for RIG + GEM versus GEM, respectively. Of 64 tumor samples sent for molecular analysis, 47 were adequate for multiplex genetic testing and 41 were positive for mutations. The majority of cases had KRAS gene mutations (40 cases). Other mutations detected included TP53 (13 cases) and PIK3CA (1 case). No correlation between mutational status and efficacy was detected.
Conclusions
The combination of RIG + GEM failed to demonstrate an improvement in survival or response compared with GEM in patients with metastatic pancreatic adenocarcinoma. Rigosertib showed a similar safety profile to that seen in previous trials using the IV formulation.
introduction
Pancreatic cancer is a devastating disease with limited treatment options that offer modest benefit in the metastatic setting [1]. In the United States, pancreatic cancer is the fourth leading cause of cancer-related death with an estimated 39 590 deaths and 46 420 new diagnoses in 2014 [1]. Approximately 80%–85% of these patients will have unresectable disease at diagnosis, and the prognosis is dismal as the 5-year survival rate in the United States is <6% [1, 2].
Few treatments for advanced pancreatic cancer have shown efficacy. In 1997, gemcitabine became the standard first-line therapy in advanced pancreatic cancer based on a phase III trial demonstrating improved survival with gemcitabine versus 5-fluoruracil (5-FU) (5.65 versus 4.41 months, P = 0.0025) [3]. A subsequent phase III study evaluating the addition of erlotinib to gemcitabine resulted in a small but significant improvement in overall survival (OS) versus single-agent gemcitabine (6.24 versus 5.91 months, P = 0.038) [4]. The phase III ACCORD/PRODIGE trial compared the combination of 5-FU, leucovorin, oxaliplatin, and irinotecan (FOLFIRINOX) with gemcitabine alone and showed a significant improvement in median OS (11.0 versus 6.8 months, respectively; P < 0.0001) [5]. Widespread use of FOLFIRINOX has certain limitations, including its toxicity profile (higher incidence of grade 3/4 neutropenia, febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy). More recently, the combination of nab-paclitaxel and gemcitabine has been approved for first-line metastatic pancreatic cancer based on a phase III trial showing improved survival and response rates compared with gemcitabine alone (median OS of 8.5 versus 6.7 months, respectively; HR = 0.72; 95% CI 0.62–0.83; P < 0.001) [6].
The combination of poor prognosis and limited treatment options with modest efficacy make pancreatic cancer a major focus in cancer research and treatment. Targeted therapy aimed at cell-cycle arrest offers a promising approach to improved treatment efficacy. A potential therapeutic target involved in pancreatic oncogenesis is the polo-like kinase 1 (PLK1) pathway [7]. Rigosertib, a first-in-class small-molecule inhibitor of multiple signaling pathways including phosphoinositide 3-kinase (PI3K) and PLK1 pathways, showed promising anti-tumor activity in solid tumor malignancies, including advanced pancreatic cancer in two phase I trials [8, 9]. Rigosertib is believed to have dual inhibitory activity in the PI3K and PLK1 signaling pathways, leading to cell-cycle arrest and apoptosis. Recent preclinical data have shown that rigosertib achieves these pleotropic effects through the inhibition of Ras activity. Specifically, rigosertib appears to bind to the Ras-binding domain (RBD) of downstream effector kinases such as Raf and PI3K, leading to their inactivation [10].
Preclinical studies have demonstrated that PLK1 is an important mitotic checkpoint molecule in G2 DNA damage-induced arrest, and is involved in multiple steps during mitosis [11]. In addition to PI3K, the PLK1 signaling pathway has been implicated in treatment resistance in a variety of tumor types [12, 13]. Jimeno et al. examined 50 different gene expression profiles in pancreatic adenocarcinoma post-gemcitabine exposure and showed that PLK1 signaling activity was the only gene that correlated with resistance to gemcitabine [12]. The addition of rigosertib to in vitro models showed reversal of gemcitabine resistance. Thus, the addition of rigosertib to gemcitabine may potentiate the effects of gemcitabine by synergistic inhibition mitosis promotion through PLK1.
A phase I trial studying rigosertib in combination with gemcitabine showed tolerability and efficacy in 40 patients with advanced solid malignancies [9]. Partial responses were seen in patients with pancreatic adenocarcinoma, thymic cancer, and Hodgkin lymphoma. The pharmacokinetic (PK) profile of rigosertib was not altered by gemcitabine. The toxicity profile was favorable for the combination treatment. The established dose was rigosertib 1800 mg/m2 as a 2-h continuous intravenous infusion (CIV) infusion on days 1, 4, 8, 11, 15, and 18, and gemcitabine 1000 mg/m2 as a 30-min CIV infusion on days 1, 8, and 15 of a 28-day cycle. Owing to these results, rigosertib plus gemcitabine treatment was further evaluated in patients with metastatic pancreatic cancer in a phase II/III trial.
materials and methods
study population
Eligible patients for this study were ≥18 years old with histologically or cytologically confirmed previously untreated metastatic adenocarcinoma of the pancreas and measurable disease, defined as a lesion ≥20 mm by conventional techniques in at least one dimension or ≥10 mm with spiral computed tomography (CT) scan. All patients must have had a serum creatinine ≤2.0 mg/dl and transaminase levels no higher than 3.0 times the institutions upper limit of normal (ULN) unless they had hepatic metastases, in which they were allowed to have transaminase levels of up to 5.0 times the ULN. Patients must also have had adequate bone marrow function, an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2, and a life expectancy of at least 12 weeks. Written informed consent was obtained from each patient after institutional review board approval of the clinical trial.
study design
This open-label, randomized, multicenter study involved 52 sites in multiple countries. Patients were randomized in a 2:1 fashion to receive either rigosertib + gemcitabine (RIG + GEM) or gemcitabine only (GEM). Using permuted block design, the patients were randomized and stratified according to ECOG performance status (0–1 versus 2). Dosing in the RIG + GEM group consisted of rigosertib 1800 mg/m2 via 2-h CIV infusions on days 1, 4, 8, 11, 15, and 18 of a 4-week cycle plus gemcitabine 1000 mg/m2 weekly for 3 weeks of a 4-week cycle. Dosing in the GEM group consisted of gemcitabine 1000 mg/m2 weekly for 3 weeks of a 4-week cycle. Patients in the GEM group were not permitted to cross over to the RIG + GEM group. Palliative radiotherapy was not allowed during the trial.
Dose adjustments were made for all non-hematologic grade 3 adverse events (AEs) or more. Rigosertib dose was reduced to 75% of the starting dose (1350 mg/m2) after the first grade 3 AE or more and to 50% of the starting dose (900 mg/m2) after the second. Gemcitabine dose was reduced to 750 mg/m2 after the first grade 3 AE or more, and to 600 mg/m2 after the second. Rigosertib was discontinued after the third occurrence of a grade 3 AE or more, and patients were removed from the study after the third grade 3 AE or more due to gemcitabine.
The use of granulocyte growth factors (G-CSF or GM-CSF), erythropoietin, erythropoietin-like substances, or blood transfusions was permitted at the discretion of the treating investigator. Imaging and measurement of CA19-9 were done every 8 weeks after study enrollment. Patients were treated until disease progression, withdrawal of consent, unacceptable toxicities, or death. AE grading was done using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03. Safety was assessed using AE reports, laboratory tests, vital signs/physical examination and weight, and toxicity assessments.
tissue collection and biomarker analysis
Archival tissue from resection or biopsy obtained before enrollment in the study were collected and later analyzed to identify molecular characteristics and potential biomarkers of treatment susceptibility or resistance. Adequate tissue samples (minimum 2 mm × 2 mm) in paraffin block from resection or core biopsy of metastasis or primary tumor were sent to the Colorado Molecular Correlates Laboratory (CMOCO) for analysis. In cases where only slides were available, 15 unstained 5-µm sections mounted on slides were used. CMOCO carried out molecular testing of samples using a multiplex single-nucleotide base extension assay (SNaPshot®, Life Technologies), designed to detect >60 point mutations in 15 gene targets, including PIK3CA, EGFR1, BRAF, KRAS, NRAS, PTEN, TP53, NOTCH1, AKT, APC, KIT, JAK2, FLT3, CTNNB1, and MEK1.
statistical analysis
The primary end point of this study was OS, defined as the time from study enrollment until death. The secondary objectives included progression-free survival (PFS), overall response rate (ORR) using RECIST v.1.1, and safety. PFS was defined as the time from randomization until disease progression. Additional secondary objectives involved biomarker analysis (including PI3K/AKT and PLK1 pathways) carried out on archival tissue from all patients. The target accrual of 150 patients provided a 90% power to detect a one-sided P-value of <0.15 and treatment effect size of at least 10 weeks. Patients were followed until 125 deaths occurred, at which point the unadjusted, unstratified log-rank test was used to compare the Kaplan–Meier survival curves. If the one-sided long-rank P-value was >0.50, the study would be permanently discontinued for futility. This study was initially powered for continuation into a phase III trial. The last patient was randomized on 7 March 2013 and the data cut-off date was 9 December 2013.
results
baseline characteristics
Accrual was discontinued after 160 patients were enrolled and 132 deaths had occurred (Table 1 and Figure 1). The last patient was randomized in March 2013. There remains two patients on-treatment (one in each arm). The mean age was 62.7 years old (range 29–87), and 63% of patients were male. Seventy-one percent were self-reported as Caucasian, followed by 21% Asian, and 6% Black. ECOG performance status was 0, 1, and 2 in 26%, 69%, and 5%, respectively. There were no major differences in demographic or tumor characteristics between the two treatment groups. Sixty-four cases were submitted for DNA analysis using SNaPshot®, and 47 samples were adequate for SNaPshot testing. The variation between sample submission and adequate testing was due to insufficient tissue or suboptimal SNaPshot® analysis.
Table 1.
Demographics
| RIG + GEM (n = 106) | GEM (n = 54) | Total (n = 160) | |
|---|---|---|---|
| Demographic and baseline characteristics ITT population (n = 160) | |||
| Age (years) | |||
| Mean (range) | 63.2 (29–83) | 61.8 (30–87) | 62.7 (29–87) |
| Male, n (%) | 69/106 (65) | 31/54 (57) | 100/160 (63) |
| Hispanic or Latino, n (%) | 7/106 (7) | 2/54 (4) | 9/160 (6) |
| Race, n (%) | |||
| White | 75/106 (71) | 39/54 (72) | 114/160 (71) |
| Black or African American | 8 (8) | 2 (4) | 10 (6) |
| Asian | 20 (19) | 13 (24) | 33 (21) |
| Other | 3 (3) | 0 | 3 (2) |
| ECOG performance status, n (%) | |||
| 0 | 28/106 (26) | 13/54 (24) | 41/160 (26) |
| 1 | 71 (67) | 40 (74) | 111 (69) |
| 2 | 7 (7) | 1 (2) | 8 (5) |
| Metastasis, n (%) | 103/106 (97) | 53/54 (98) | 156/160 (98) |
| Liver | 88 (83) | 48 (89) | 136 (85) |
| Lung | 28 (26) | 11 (20) | 39 (24) |
| Peritoneum | 11 (10) | 5 (9) | 16 (10) |
| Retroperitoneal | 11 (10) | 2 (4) | 13 (8) |
| Weight (kg) | |||
| Mean (range) | 70.1 (33–110) | 68.6 (37–112) | 69.6 (33–112) |
Figure 1.

CONSORT diagram.
treatment exposure
The mean duration of treatment was 3.6 cycles (range, 1–14) for RIG + GEM and 4.0 cycles (range, 1–12) for GEM. In the RIG + GEM group, 58% of patients had adjustments of the rigosertib dose and 62% required dose adjustments of gemcitabine. In the GEM group, 47% of patients had dose adjustments. The mean cumulative dose of gemcitabine was 16.2 g in the RIG + GEM group and 18.7 g in the GEM group.
Sixty-one patients went on to receive subsequent therapy (38 in the RIG + GEM group versus 23 in the GEM group). Specific chemotherapy regimens were not captured.
safety
In the RIG + GEM group, the most frequently reported treatment-related non-hematologic AEs were fatigue (27%), nausea (25%), and hyponatremia (25%) (Table 2). Grade 3 or higher treatment-related AEs observed more frequently in the RIG + GEM group were hyponatremia, fatigue, diarrhea, nausea, vomiting, hyopabluminemia, thrombocytopenia, and neutrophil count decreased. Anemia was the most common hematologic AE reported and was similar in both groups, followed by neutropenia and thrombocytopenia. Only hyponatremia (RIG + GEM = 25%, GEM = 6%) and AST increased (RIG + GEM = 15%, GEM = 9%) were notably higher in the RIG + GEM group. Serious AEs were reported in 49 patients (46%) in the RIG + GEM group versus 15 (32%) in the GEM group. AEs led to the study regimen discontinuation in 35 patients (33%) in the RIG + GEM group versus 9 (19%) in the GEM group. Between the first dose and up until 30 days after the last dose of drug given, there were 17 deaths in the RIG + GEM group versus 14 in the GEM group.
Table 2.
Toxicity
| MedDRA system organ class/preferred term | RIG + GEM (N = 106) |
GEM (N = 47) |
||
|---|---|---|---|---|
| Any grade [n (%)] | Grade 3–4 [n (%)] | Any grade [n (%)] | Grade 3–4 [n (%)] | |
| AEs deemed related to study regimen by NCI CTCAE grade (>10% of patient population) Safety population (N = 153) | ||||
| GI | ||||
| Nausea | 26 (25) | 2 (2) | 13 (28) | 0 |
| Diarrhea | 17 (16) | 4 (4) | 7 (15) | 1 (2) |
| Vomiting | 14 (13) | 1 (1) | 10 (21) | 0 |
| Metabolism | ||||
| Hyponatremia | 26 (25) | 18 (17) | 3 (6) | 2 (4) |
| Decreased appetite | 13 (12) | 0 | 5 (11) | 0 |
| Hypoalbuminemia | 11 (10) | 1 (1) | 4 (9) | 0 |
| Blood | ||||
| Anemia | 30 (28) | 8 (8) | 13 (28) | 2 (4) |
| Neutropenia | 11 (10) | 8 (8) | 7 (15) | 3 (6) |
| Thrombocytopenia | 12 (11) | 3 (3) | 6 (13) | 1 (2) |
| General | ||||
| Fatigue | 29 (27) | 5 (5) | 12 (26) | 1 (2) |
| Investigations | ||||
| ALT increased | 15 (14) | 1 (1) | 5 (11) | 1 (2) |
| increased | 16 (15) | 1 (1) | 4 (9) | 1 (2) |
| Neutrophil count decreased | 13 (12) | 10 (9) | 5 (11) | 2 (4) |
Column header counts and denominators are the number of patients randomized who have received at least one dose of study drug. A patient is counted at most once in each cell of the table.
efficacy
Median OS, the primary end point of the study, was 6.1 months for RIG + GEM versus 6.4 months for GEM (HR = 1.24, 95% CI 0.85–1.81) (Figure 2a). Median PFS was 3.4 months for both groups (HR = 0.96, 95% CI 0.68–1.36) (Figure 2b). The overall best response per RECIST criteria was partial response in 19% versus 13% of patients for RIG + GEM and GEM, respectively. Stable disease was found in 50% of patients in the RIG + GEM group versus 56% in the GEM group. There were no complete responses in either group. Mutational analysis showed no correlation with efficacy between treatment arms (supplementary Table S1, available at Annals of Oncology online).
Figure 2.

(a) OS Kaplan–Meier estimates of OS ITT population (n = 160). (b) PFS Kaplan–Meier estimates of PFS ITT population (n = 160).
SNaPshot mutational analysis
Of the 64 total cases submitted, 49 samples were prepared for DNA extraction, 47 of which had adequate tissue to proceed with testing. Forty-one samples showed detectable mutations (Figure 3). The majority of cases had KRAS gene mutations (40 of the 41 samples with mutations). The KRAS mutations detected included c.35G>A, p.G12D (21 cases), c.35G>T, p.G12V (12 cases), c.34G>C, p.G12R (4 cases), c.183C>A, p.Q61H (2 cases), and c.34G>T, p.G12C (1 case). Other mutations detected included TP53 (13 cases) and PIK3CA (1 case). Twelve of the 13 cases with TP53 gene mutation and the 1 sample with PIK3CA mutation also harbored a KRAS mutation.
Figure 3.
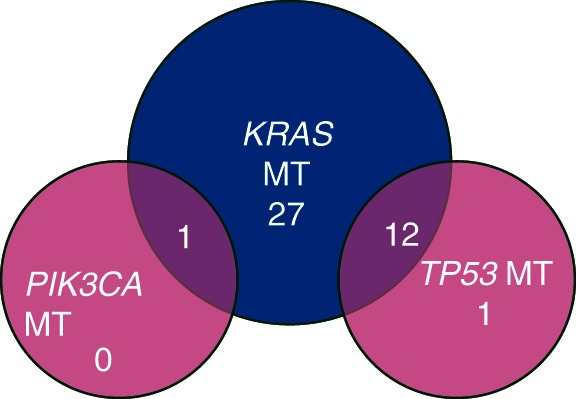
Mutation analysis—41 samples positive for a mutation.
discussion
This was a randomized, open-label, multicenter study of rigosertib, a small-molecule Ras mimetic and inhibitor of the PI3K and PLK1 pathways, administered in combination with gemcitabine versus gemcitabine alone in patients with chemo-naïve metastatic pancreatic adenocarcinoma.
The toxicity profile of the combination arm was similar to that of the previous phase I study aside from hyponatremia, which occurred in ∼25% of patients compared with an incidence of ∼5% in phase 1 trials [9]. The etiology of this increased incidence in hyponatremia remains unclear. Based on previous phase 1 data, the most common non-hematologic AEs of rigosertib were fatigue, nausea, vomiting, anorexia, and diarrhea. The most common AEs in the RIG + GEM group included fatigue, nausea, diarrhea, hyponatremia, and anemia. The hematologic toxicity profile was similar between the two treatment groups. There were more serious AEs and treatment discontinuations due to AE in the RIG + GEM group.
Targeted mutational analysis demonstrated that the majority of tumor samples adequate for analysis harbored a KRAS mutation (40 out of 47 samples). There was a single PIK3CA mutation and 13 TP53 mutations. These findings are consistent with known genetic mutation profiles in pancreatic cancer, as historically PIK3CA mutations are rare in pancreatic cancer [14]. Of note, the testing methodology implemented only tested for known hotspot mutations, thus other mutations may have been present in PIK3CA or TP53 which would not have been detected.
Pancreatic cancer is believed to be a KRAS-driven malignancy [15]. Recent data have shown that rigosertib binds to the RBD of downstream Ras effector proteins, thus preventing their activation. In addition, preclinical data in cell lines and patient-derived xenografts have shown that rigosertib may act synergistically with gemcitabine in resistant pancreatic cancer cell lines [11, 13, 16]. Due to this, we hypothesized that rigosertib plus gemcitabine may in fact lead to clinical benefit in an unselected pancreatic cancer population. However, in our trial, the combination of rigosertib and gemcitabine did not improve the treatment outcome (median OS of 6.1 versus 6.4 months; HR = 1.24, 95% CI 0.85–1.81).
While the specific mechanisms for the lack of improved efficacy with the combination of rigosertib and gemcitabine in this study remain unclear, there are likely multiple contributing factors. For example, independent gain-in-function mutations of Ras effector proteins would be downstream of rigosertib intervention. Furthermore, while mutations such as KRAS occur in up to 90% of pancreatic adenocarcinomas, multiple studies have identified human pancreatic cancer cells with varying levels of sensitivity to KRAS inactivation [17, 18]. Strong evidence for this concept comes from a recent study in which investigators classified tumors from pancreatic cancer patients into three distinct subtypes based on gene expression profiles: ‘classical’ (41.2%), ‘quasi-mesenchymal’ (36.5%), and ‘exocrine-like’ (22.3%) [18]. The relationship between KRAS dependence and subtype was explored using KRAS RNAi. Importantly, ‘classical’ tumors proved to be more dependent upon KRAS than the other subtypes, suggesting that inhibition of this highly mutated oncogene may only be effective in a subset of pancreatic cancer patients. This and other studies highlight the inherent heterogeneity of pancreatic cancer and may help to explain the lack of clinical activity for rigosertib in KRAS mutant pancreatic cancer patients. Finally, while no detailed PK data were collected in this study, the dosages and administration of gemcitabine and rigosertib were based on phase I PK data and we believe that PK s are unlikely to explain the results from this study [8].
Multiple trials investigating rigosertib as a single agent and in combination for hematologic malignancies including myelodysplastic syndromes (MDSs) have shown promising results [19–21]. The reasons for the difference in clinical efficacy between pancreatic cancer and MDS remain unclear. In a recent study by Hyoda et al., rigosertib was shown to induce apoptosis in MDS cancer cells lines through the inhibition of the PI3K/Akt pathway and promoted the phosphorylation of histone H2AX leading to DNA damage-induced G2/M arrest [22]. This argues that at least a portion of MDS cases may rely on the PI3K/Akt pathway for oncogenesis, and this inhibition may lead to induction of apoptosis and improved clinical outcomes. In contrast, pancreatic cancer may achieve resistance through aberrant protein upregulation in alternative pathways, such as c-MET and HER-2, that in turn activate the RAS pathway downstream of KRAS inhibition by rigosertib.
Clinical trials using rigosertib in MDSs and other hematologic neoplasms are ongoing. Based on the results of our trial, there are no current plans to perform further trials with the use of rigosertib in pancreatic cancer. Rigosertib was well tolerated and exhibited a similar safety profile to that observed in other trials using the IV formulation.
funding
The trial was supported by Onconova Therapeutics, Inc., and conducted by the Academic Gastrointestinal Cancer Consortium (AGICC; www.agicc.org). This trial was also supported in part by the Histology Service Center of the University of Colorado Cancer Center Support Grant (P30 CA046934) from the National Cancer Institute.
disclosure
RM, FW, and MM work for or have worked for Onconova Therapeutics Inc. The other co-authors' institutions received research funding from Onconova Therapeutics Inc. to support clinical trial research costs.
Supplementary Material
acknowledgements
The authors thank the patients and families who volunteered to participate in this clinical trial.
references
- 1.Society AC. Cancer Facts & Figures 2014. Atlanta, GA: American Cancer Society; 2014. [Google Scholar]
- 2.Li D, Xie K, Wolff R et al. Pancreatic cancer. Lancet 2004; 363(9414): 1049–1057. [DOI] [PubMed] [Google Scholar]
- 3.Burris HA, Moore MJ, Andersen J et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15(6): 2403–2413. [DOI] [PubMed] [Google Scholar]
- 4.Moore MJ, Goldstein D, Hamm J et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25(15): 1960–1966. [DOI] [PubMed] [Google Scholar]
- 5.Conroy T, Desseigne F, Ychou M et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364(19): 1817–1825. [DOI] [PubMed] [Google Scholar]
- 6.Al-Hajeili M, Azmi AS, Choi M. Nab-paclitaxel: potential for the treatment of advanced pancreatic cancer. OncoTargets Ther 2014; 7: 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray PJ Jr, Bearss DJ, Han H et al. Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther 2004; 3(5): 641–646. [PubMed] [Google Scholar]
- 8.Jimeno A, Li J, Messersmith WA et al. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J Clin Oncol 2008; 26(34): 5504–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma WW, Messersmith WA, Dy GK et al. Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin Cancer Res 2012; 18(7): 2048–2055. [DOI] [PubMed] [Google Scholar]
- 10.Divakar S, Vasqez-Del Caprio R, Baker SJ et al. Targeting the Ras-Binding domain of Ras effector proteins by a small molecule inhibitor, Rigosertib. Cancer Res 2014; 74(Suppl 19): abstr nr LB-108. [Google Scholar]
- 11.Van Vugt MA, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005; 24(17): 2844–2859. [DOI] [PubMed] [Google Scholar]
- 12.Jimeno A, Rubio-Viqueira B, Rajeshkumar NV et al. A fine-needle aspirate-based vulnerability assay identifies polo-like kinase 1 as a mediator of gemcitabine resistance in pancreatic cancer. Mol Cancer Ther 2010; 9(2): 311–318. [DOI] [PubMed] [Google Scholar]
- 13.Bowles DW, Jimeno A. New phosphatidylinositol 3-kinase inhibitors for cancer. Expert Opin Investig Drugs 2011; 20(4): 507–518. [DOI] [PubMed] [Google Scholar]
- 14.Jones S, Zhang X, Parsons DW et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (New York, N.Y.) 2008; 321(5897): 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eser S, Schnieke A, Schneider G et al. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014; 111(5): 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jimeno A, Chan A, Cusatis G et al. Evaluation of the novel mitotic modulator ON 01910.Na in pancreatic cancer and preclinical development of an ex vivo predictive assay. Oncogene 2009; 28(4): 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh A, Greninger P, Rhodes D et al. A gene expression signature associated with ‘K-Ras addiction’ reveals regulators of EMT and tumor cell survival. Cancer cell 2009; 15(6): 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collisson EA, Sadanandam A, Olson P et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011; 17(4): 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komrokji RS, Raza A, Lancet JE et al. Phase I clinical trial of oral rigosertib in patients with myelodysplastic syndromes. Br J Haematol 2013; 162(4): 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silverman LR, Greenberg P, Raza A et al. Clinical activity and safety of the dual pathway inhibitor rigosertib for higher risk myelodysplastic syndromes following DNA methyltransferase inhibitor therapy. Hematol Oncol 2014; 33(2): 57–66. [DOI] [PubMed] [Google Scholar]
- 21.Raza A, Mukherjee S, Eisenberger AJ et al. A phase II study of the orally administered rigosertib (ON 01910.Na) in transfusion-dependent lower-risk myelodysplastic syndrome (MDS) patients. J Clin Oncol 31 2013; 7031. [Google Scholar]
- 22.Hyoda T, Tsujioka T, Nakahara T et al. Rigosertib induces cell death of a myelodysplastic syndrome-derived cell line by DNA damage-induced G2/M arrest. Cancer Sci 2015; 106(3): 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.