

Phase I clinical trials are now complex trials that investigate toxicity, activity, biological endpoints on several dozens of patients. The 3 + 3 design—proposed to select a dose with less than two DLTs—lacks flexibility. Model-based methods have been tailored for these new challenges. Statisticians and investigators need to work together to carry out successful trials.
Keywords: continual reassessment method, dose finding, efficiency, targeted agents
Abstract
Background
More than 95% of published phase I trials have used the 3 + 3 design to identify the dose to be recommended for phase II trials. However, the statistical community agrees on the limitations of the 3 + 3 design compared with model-based approaches. Moreover, the mechanisms of action of targeted agents strongly challenge the hypothesis that the maximum tolerated dose constitutes the optimal dose, and more outcomes including clinical and biological activity increasingly need to be taken into account to identify the optimal dose.
Patients and methods
We review key elements from clinical publications and from the statistical literature to show that the 3 + 3 design lacks the necessary flexibility to address the challenges of targeted agents.
Results
The design issues raised by expansion cohorts, new definitions of dose-limiting toxicity and trials of combinations are not easily addressed by the 3 + 3 design or its extensions.
Conclusions
Alternative statistical proposals have been developed to make a better use of the complex data generated by phase I trials. Their applications require a close collaboration between all actors of early phase clinical trials.
introduction
Two main statistical approaches are used in phase I trials to identify the dose recommended for phase II trials, usually taken as the maximum tolerated dose (MTD) [1]. Algorithm-based methods, mainly represented by the 3 + 3 design (formerly called Fibonacci), the rolling six and the accelerated titration design, can be distinguished from model-based methods such as the continual reassessment method (CRM) and its variations (escalation with overdose control [2], time to event CRM [3] etc.).
Recent reviews of published phase I clinical trials have shown that the vast majority of trials (98.4%) use the former [4] and that the overall duration of the trials is not markedly reduced when using the CRM. It is also well known that the 3 + 3 design is much easier to implement as basically no computation is required.
Is there any reason to question the relative performance of these two competing approaches? Yes, if we consider that 25% of the oncology agents registered by the FDA are labeled at a dose different from that identified in phase I trials [5]. Not to mention the numerous drug developments that may have been prematurely interrupted due to the use of an inadequate dose level. Furthermore, six patients are clearly insufficient to provide an accurate estimate and the statistical community agrees on the limitations of the 3 + 3 method [6]. More conceptually, the mechanisms of action of targeted agents strongly challenge the hypothesis that the MTD systematically constitutes the optimal dose, which is the basis of the 3 + 3 design.
Using various statistical contributions and reviews, we show that the 3 + 3 design gives a lower chance of identifying the right dose compared with the CRM, and importantly it has major limitations to address the challenges of targeted agents for which the optimal dose may be lower than the MTD. Objectives of phase I trials are increasingly complex and the DLT after the first cycle may not be the only relevant endpoint. CRM and extensions cover the needs for dose finding and allows addressing more complex questions.
methods
In oncology, the dose at which >33% of patients experience dose-limiting toxicity (DLT) is deemed too toxic. With the 3 + 3 method, the MTD is then the highest dose at which at most one patient out of six patients (i.e. 17%) experience DLT. With the CRM, this is the dose associated with a prespecified rate of DLT, which is usually set between 20% and 30% by physicians to match the previous definition. Another target could be selected if we are willing to accept a higher or lower risk of toxicity.
3 + 3 design and extensions (‘algorithm-based methods’)
This design proceeds in cohorts of three patients treated at increasing dose levels [7]. Dose escalation stops as soon as at least two out of three or six patients experience DLT at that dose level. The following criticisms have been raised: too many patients tend to be treated at low and probably ineffective doses; dose escalation may be too slow because of an excessive number of escalation steps; too few patients tend to be treated close to the MTD, which may result in a substantial residual uncertainty about the MTD and the safety of the dose [7]. To address some of the limitations of the 3 + 3 design, Simon et al. proposed several accelerated titration designs based on the idea, introduced by Storer [8], of initially treating only one patient per dose level, and reverting to a 3 + 3 design as soon as a first DLT, or two grade 2 toxicities are observed [9].
CRM and extensions (‘model-based methods’)
The CRM is a statistical approach based on the principle that each patient should be allocated to the dose most likely to be the MTD, in other words, the dose for which the estimated risk of DLT is closest to the target. Estimates of the risk of DLT are based on a model that relates the probability of DLT to the dose level [10]. Although numerous implementation of the CRM exist, the sketch is as follows: (a) the first patient or group of patients is treated at the lowest level; (b) the DLT outcome is measured; (c) the model of the dose–toxicity relationship is fitted to previous observation(s), which in turn provides estimates of the risk of DLT at each dose level; (d) the process is repeated from step (b) for the next patients and the knowledge of the dose–toxicity relation is continuously updated until a stopping rule is met [11]. A final estimate of the risk of DLT together with some measure of precision are computed. Starting dose may not be the first dose, as it is common practice to define levels −1 and −2 that are visited only if the starting dose is excessively toxic. Figure 1 illustrates the steps (b and c) of reassessment of the dose–toxicity relationship of a hypothetical trial in which the first five patients tolerated doses 1, 2, 3, 3 and 3, respectively, and patient 6 experienced DLT at dose 4. The proportion of DLT at each dose is 0 for doses 1–3 and 1 for dose 4; they are represented by dots. The solid line indicates the model fit; dose 3 associated with 14% risk of DLT, which is closest to the 20%-target—is the best current estimate of the MTD and is allocated to patient 7. In this example, we suppose that patient 7 tolerates dose 3; the model is fit again and the risk of DLT is updated leading to recommend dose 4 for the next patient. The dose–toxicity relationship is continuously reassessed.
Figure 1.

Estimated dose–DLT curves in a simulated trial with five dose levels (d1–d5) after patients 6 (black line in A and B) and 7 (gray line in B). Dark gray (red online) dots represent the proportion of DLT at each dose level (empirical frequencies), the black (blue online) line corresponds to the targeted percentile of the dose–toxicity relation (20%); circled doses are the doses closest to the target, corresponding to the best current estimate of the MTD; this dose is recommended for the next patient.
A complete description of implementation of this model can be found elsewhere [7, 12]. The main aspects that interest us here are that all data collected at a given timepoint are used to reassess the dose–toxicity relationship, and not only the last cohort, as with the 3 + 3 model; therefore, the more data are collected the more precise becomes the estimate of the MTD, which provides a powerful tool to reassess the risk of DLT in expansion cohorts. Furthermore, the model can be easily enriched to control the risk of overdosing, to account for late/delayed events [3, 13], to identify an MTD adjusted to patient characteristics as in phase I trials on different populations [14], or to incorporate intermediate grades in the MTD evaluation [15].
results
3 + 3 versus CRM
A recent review compared the efficiency of the 3 + 3 and CRM in first-in-man phase I trials of targeted agents administered as single agents [16]. The median number of dose levels escalated in trials using a standard ‘3 + 3’ design was 6 compared with the 10 levels explored in trials that used the CRM. This range corresponded to a median ratio between the highest dose and the starting dose of 9 and 30 for the 3 + 3 and the CRM, respectively. However, the mean number of patients enrolled in trials using the CRM was lower (44 versus 38). Analysis of the allocated doses showed that the mean number of patients exposed to doses exceeding the MTD was twice as high in trials using a standard ‘3 + 3’ design or an accelerated titration design compared with trials using a CRM (9 versus 4). The 3 + 3 was then associated with slower dose escalations, larger sample size and, more importantly, an increased risk of overdosing. A review of all trials using model-based methods (single agents or combinations) showed that implementation of the CRM and derivatives was safe for the patients [17].
These results should be interpreted cautiously, as the investigators may have chosen to use the CRM when they expected that a large range of dose levels would need to be escalated. Comparisons based on published data are instructive, but should be completed by comparisons of the same clinical setting. However, it is not possible to strictly apply a method that would require say 44 patients on retrospective data where only 38 patients had actually been treated. Simulations carried out on virtual patients are therefore preferred. Statistical simulations have consistently demonstrated that the 3 + 3 design is outperformed by new dose-escalation designs such as the CRM: the chance of correctly identifying the MTD was 20% lower with 3 + 3 method compared with CRM for various scenarios and comparable sample sizes except when the MTD was the first or second dose level [18]. The 3 + 3 method tended to be overly conservative and to identify a dose lower than the MTD [19]. The risk of overdosing was slightly higher with the CRM compared with the 3 + 3. Lastly, the CRM appears to be very sensitive to DLT occurring early in the trial, which may slow down dose escalation and lead to conservative operating characteristics [12]. The 3 + 3 design, are much more robust to this type of early events that are sometimes due to misclassified clinical symptoms related to disease progression.
Notably, the probability of correct selection of the MTD was usually <60% for all methods explored in simulations [18]. In other words, we expect that, in 40% of trials, the identified dose is higher than or less than the MTD. Performance may be much better in special scenarios with very few dose levels for instance. On average, it is unreasonable to expect a miracle solution, as shown by the performance of a theoretical ‘optimal’ method that assumes that all patients could be treated at all dose levels [20]. These statistical works show that even if the 3 + 3 is inferior to the CRM, the main limit of dose finding is not the design or the method but the sample size and the endpoint. As strikingly illustrated on the database of 54 trials collected by the European Organization for Research and Treatment of Cancer (EORTC) [21], 25 000 adverse events possibility related to treatment were reported, but only the 161 DLTs were used to define the MTD, resulting in a massive loss of information.
safety evaluation in the expansion cohort
To address the concern of insufficient experience after six patients regarding toxicity and activity, cohorts of additional patients are commonly treated at the MTD. The main objective of these ‘expansion cohorts’ is to investigate the toxicity of the agent(s) with sample sizes ranging from four to several hundred patients [22]. However, no statistical methods are used to monitor toxicity, reassess the risk of DLT and possibly refine the MTD if more toxicity is reported. The 3 + 3 that uses only the three last observations is not adapted to address these objectives. For instance, if two grade 3–4 toxicities are observed after eight additional patients, the 3 + 3 is helpless to determine whether or not the risk of severe toxicity is too high.
A clear distinction between the dose escalation and the expansion cohort is not optimal and a seamless transition with continuous monitoring of the risk of DLT is more natural and more effective. The CRM and derivatives can be applied during the expansion cohort. They afford a simple way to continuously reassess the risk of toxicity without suspending accrual and hence without extending the trial duration; all data collected at given timepoints (for instance every five patients) are analyzed even when some evaluations are pending. If the dose tested in the expansion cohort appears to be excessively toxic, the dose level can be refined. This approach would improve patient safety and optimize the accuracy of the final recommendation. The 3 + 3 is not sufficiently flexible to conduct early phase trials in which several dozens of patients may be treated.
MTD and optimal dose
To document the optimal dose and not only the MTD, clinical and biological activity endpoints are increasingly investigated in addition to DLT. Recent reviews have even reported that ∼25% of the published single-agent phase I trials do not identify an MTD due to the lack of dose-limiting toxicities [21]. Algorithm-based approaches such as the ‘5/6 design’ proposed to target a biological endpoint do not integrate both toxicity and biological endpoints [23]. Model-based methods have therefore been developed to identify a dose associated with a certain activity rate and an acceptable toxicity risk [24–26]. Relationships between the dose and both toxicity and activity are investigated, which allows evaluating the trade-off between toxicity and activity. Integration of more than one endpoint in the analysis is better achieved using model-based methods. Currently efficacy is mainly evaluated during the expansion cohort treated at the MTD but the optimal dose is usually not reassessed.
chemoradiation trials and cumulative risk of toxicity
Due to the specificities of radiotherapy, delayed severe toxicity may commonly occur. In chemoradiation trials, the recommended DLT assessment period is 4–6 months after treatment initiation [27]. When the 3 + 3 method is used, accrual has to be suspended until the three patients have completed the DLT assessment period. This considerably increases the trial duration. Consequently, most phase I chemoradiation trials use a relatively short DLT assessment method (<3 months) in order to apply the 3 + 3, which may lead to underestimation of the overall risk of severe toxicity [27].
The time-to-event CRM addresses these issues, as patients with partial follow-up can still be analyzed to reassess the risk of DLT [3]. It allows continual enrollment of patients, assigning fractional weights proportional to follow-up to patients who have not experienced toxicity. At the cost of a slightly increased risk of overdosing, the overall duration of the trial is drastically reduced. For instance, inclusion of 25 patients at four levels, with the MTD being the third one that would require 36 months with the 3 + 3, can be achieved in 13 months with the tite-CRM [28]. The risk of overdosing can be high if the accrual rate is very rapid compared with the timing of DLTs.
incorporating multiple cycles in the evaluation of the RP2D
The EORTC set up the DLT and toxicity assessment recommendation group for early trials of targeted therapies (DLT-TARGETT) [21, 29]. A key element was that toxicity at all cycles should be used to recommend a dose for the phase II trials. The recommended dose for ceritinib could have been different if such a comprehensive approach had been used, as 62% of the patients treated at the MTD required a dose reduction after cycle 3 [30]. Once again, the 3 + 3 is not really suitable to achieve this goal, as any data beyond cycle 1 are discarded. As illustrated by Doussau et al., the CRM can be extended to evaluate the risk of DLT at each cycle using models for longitudinal data [31]. In a reanalysis of three trials, the authors obtained a more accurate estimate of the MTD and in one trial they detected an increased risk of DLT after repeated treatment cycles, suggesting late or cumulative toxicity. However, the challenge of incorporating dose modification and dose delay in such an assessment requires further statistical developments in close collaboration with pharmacologist and investigators.
combination trials
The 3 + 3 is the method most commonly used to find the optimal dosage of two agents [32]. However, in order to apply this method, only a limited preselected set of increasing dose levels can be studied. For two agents, the median ratio of the number of planned combinations to the number of possible combinations was 0.67 [32], with no guarantee that the optimal combination is included in the preselected set (see Figure 2 that describes two possible sets of ordered doses). Much current statistical research aims at either determining the complex interrelationship between the two agents [33] or at taking advantage of the ordering of dose levels [34]. As several combinations may match the MTD definition, an activity endpoint is then interesting to select the better of the two combinations. Model-based methods have been extended to relate the risk of DLT to the dose of each agent and to incorporate an activity endpoint [26]. Although few trials using these approaches have been published to date, they appear to be very promising.
Figure 2.
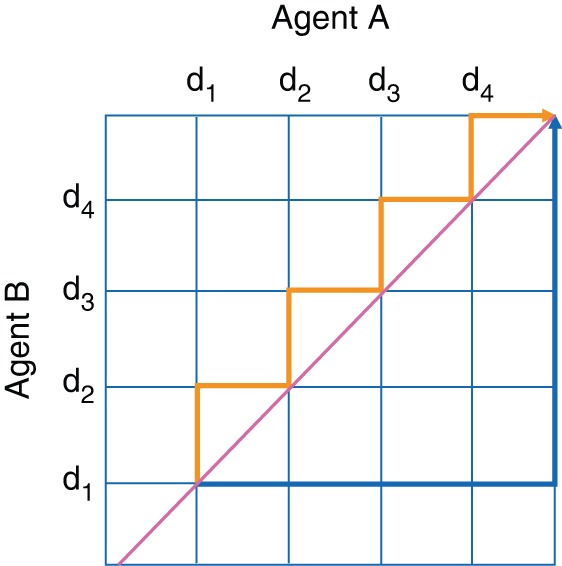
Two possible sets of predefined combinations of agents A and B to be explored in a dose-escalation trial using the 3 + 3; combinations outside the dark gray (orange online) and black (blue online) lines are not tested.
discussion
The fundamental assumption ‘the more the better’ is strongly debated for several classes of agents such as small molecules or antibodies [35, 36]. In this case, we cannot generally identify the optimal dose with the 3 + 3 method, which is a simple algorithm to identify a dose with two DLTs. While phase I trials were formerly limited experiments designed to describe the toxicity profile, they are now expected to provide a more comprehensive evaluation of the benefit–risk ratio. Compared with previous decades, phase I trials now address more objectives and more complex questions. In this respect, they more closely resemble dose-ranging trials carried out in other diseases. As shown in clinical examples and statistical simulations, model-based methods outperform the 3 + 3 design in terms of their ability to identify the correct MTD, the percentage of patients treated at a dose close to the MTD and the range of dose levels explored. More importantly, model-based methods can be considerably enriched in order to be adapted to new objectives. The DLT as the primary endpoint of phase I trials is clearly a major limitation to the statistical efficiency of any dose-finding method. Development of strong translational research with repeat biopsies, as well as functional imaging or immune monitoring in early phase trials, should rapidly provide much more sensitive indicators of the treatment effect. Data integration is not possible with the 3 + 3.
Considerable experience has been acquired with the 3 + 3 design, and it has been shown to work reasonably well empirically for chemotherapies with narrow therapeutic indexes. However, the lack of flexibility of the method makes the 3 + 3 poorly adapted to conduct contemporary complex early phase trials of targeted agents. Several statistical methods have been tailored for these new challenges that include methods for combination therapy or immunotherapy. Statisticians and investigators need to work together to carry out successful trials.
funding
This work was partially funded by the French National Cancer Institute (INCa) via the SIRIC program (grant no. INCa-DGOS-4654).
disclosure
All authors declare they have no conflict of interest for this presented work.
references
- 1.Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 2009; 101: 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tighiouart M, Rogatko A, Babb JS. Flexible Bayesian methods for cancer phase I clinical trials. Dose escalation with overdose control. Stat Med 2005; 24: 2183–2196. [DOI] [PubMed] [Google Scholar]
- 3.Cheung YK, Chappell R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics 2000; 56: 1177–1182. [DOI] [PubMed] [Google Scholar]
- 4.Rogatko A, Schoeneck D, Jonas W et al. . Translation of innovative designs into phase I trials. J Clin Oncol 2007; 25: 4982–4986. [DOI] [PubMed] [Google Scholar]
- 5.Le Tourneau C, Stathis A, Vidal L et al. . Choice of starting dose for molecularly targeted agents evaluated in first-in-human phase I cancer clinical trials. J Clin Oncol 2010; 28: 1401–1407. [DOI] [PubMed] [Google Scholar]
- 6.O'Quigley J, Zohar S. Experimental designs for phase I and phase I/II dose-finding studies. Br J Cancer 2006; 94: 609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paoletti X, Buyse M. Statistical designs for first-in-man phase I cancer trials. In Eisenhauer EA, Twelves C, Buyse M (eds), Phase I Cancer Clinical Trials: A Practical Guide, 2nd edition Oxford University Press, 2015. [Google Scholar]
- 8.Storer BE. Design and analysis of phase I clinical trials. Biometrics 1989; 45: 925–937. [PubMed] [Google Scholar]
- 9.Simon R, Freidlin B, Rubinstein L et al. . Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst 1997; 89: 1138–1147. [DOI] [PubMed] [Google Scholar]
- 10.O'Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics 1990; 46: 33–48. [PubMed] [Google Scholar]
- 11.O'Quigley J, Reiner E. A Stopting rule for the continual reassesssment method. Biometrika 1998; 85: 7. [Google Scholar]
- 12.Paoletti X, Baron B, Schoffski P et al. . Using the continual reassessment method: lessons learned from an EORTC phase I dose finding study. Eur J Cancer 2006; 42: 1362–1368. [DOI] [PubMed] [Google Scholar]
- 13.Doussau A, Thiebaut R, Paoletti X. Dose-finding design using mixed-effect proportional odds model for longitudinal graded toxicity data in phase I oncology clinical trials. Stat Med 2013; 32: 5430–5447. [DOI] [PubMed] [Google Scholar]
- 14.O'Quigley J, Shen LZ, Gamst A. Two-sample continual reassessment method. J Biopharm Stat 1999; 9: 17–44. [DOI] [PubMed] [Google Scholar]
- 15.Van Meter EM, Garrett-Mayer E, Bandyopadhyay D. Proportional odds model for dose-finding clinical trial designs with ordinal toxicity grading. Stat Med 2011; 30: 2070–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Tourneau C, Gan HK, Razak AR et al. . Efficiency of new dose escalation designs in dose-finding phase I trials of molecularly targeted agents. PLoS One 2012; 7: e51039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iasonos A, O'Quigley J. Adaptive dose-finding studies: a review of model-guided phase I clinical trials. J Clin Oncol 2014; 32: 2505–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iasonos A, Wilton AS, Riedel ER et al. . A comprehensive comparison of the continual reassessment method to the standard 3 + 3 dose escalation scheme in Phase I dose-finding studies. Clin Trials 2008; 5: 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiner E, Paoletti X, O'Quigley J. Operating characteristics of the standard phase I clinical trial design. Comput Stat Data Anal 1999; 30: 12. [Google Scholar]
- 20.Paoletti X, Kramar A. A comparison of model choices for the continual reassessment method in phase I cancer trials. Stat Med 2009; 28: 3012–3028. [DOI] [PubMed] [Google Scholar]
- 21.Postel-Vinay S, Collette L, Paoletti X et al. . Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents. DLT-TARGETT, an EORTC-led study. Eur J Cancer 2014; 5012: 2040–2049. [DOI] [PubMed] [Google Scholar]
- 22.Manji A, Brana I, Amir E et al. . Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single-agent phase I cancer trials. J Clin Oncol 2013; 31: 4260–4267. [DOI] [PubMed] [Google Scholar]
- 23.Hunsberger S, Rubinstein LV, Dancey J et al. . Dose escalation trial designs based on a molecularly targeted endpoint. Stat Med 2005; 24: 2171–2181. [DOI] [PubMed] [Google Scholar]
- 24.Houede N, Thall PF, Nguyen H et al. . Utility-based optimization of combination therapy using ordinal toxicity and efficacy in phase I/II trials. Biometrics 2009; 66(2): 532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thall PF, Cook JD. Dose-finding based on efficacy-toxicity trade-offs. Biometrics 2004; 60: 684–693. [DOI] [PubMed] [Google Scholar]
- 26.Mandrekar SJ, Cui Y, Sargent DJ. An adaptive phase I design for identifying a biologically optimal dose for dual agent drug combinations. Stat Med 2007; 26: 2317–2330. [DOI] [PubMed] [Google Scholar]
- 27.Pijls-Johannesma M, van Mastrigt G, Hahn SM et al. . A systematic methodology review of phase I radiation dose escalation trials. Radiother Oncol 2010; 95: 135–141. [DOI] [PubMed] [Google Scholar]
- 28.Polley MY. Practical modifications to the time-to-event continual reassessment method for phase I cancer trials with fast patient accrual and late-onset toxicities. Stat Med 2011; 30: 2130–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paoletti X, Le Tourneau C, Verweij J et al. . Defining dose-limiting toxicity for phase I trials of molecularly targeted agents: results of a DLT-TARGETT international survey. Eur J Cancer 2014; 5012: 2050–2056. [DOI] [PubMed] [Google Scholar]
- 30.Shaw AT, Kim DW, Mehra R et al. . Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med 2014; 370: 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doussau A, Thiebaut R, Geoerger B et al. . A new approach to integrate toxicity grade and repeated treatment cycles in the analysis and reporting of phase I dose-finding trials. Ann Oncol 2015; 26(2): 422–428. [DOI] [PubMed] [Google Scholar]
- 32.Riviere MK, Le Tourneau C, Paoletti X et al. . Designs of drug-combination phase I trials in oncology: a systematic review of the literature. Ann Oncol 2015; 26: 669–674. [DOI] [PubMed] [Google Scholar]
- 33.Riviere MK, Dubois F, Zohar S. Competing designs for drug combination in phase I dose-finding clinical trials. Stat Med 2015; 34(1): 23–26. [DOI] [PubMed] [Google Scholar]
- 34.Wages NA, Conaway MR, Slingluff CL Jr et al. . Recent developments in the implementation of novel designs for early-phase combination studies. Ann Oncol 2015; 26(5): 1036–1037. [DOI] [PubMed] [Google Scholar]
- 35.Robert C, Ribas A, Wolchok JD et al. . Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384(9948): 1109–1117. [DOI] [PubMed] [Google Scholar]
- 36.Gupta S, Hunsberger S, Boerner SA et al. . Meta-analysis of the relationship between dose and benefit in phase I targeted agent trials. J Natl Cancer Inst 2012; 104(24): 1860–1866. [DOI] [PubMed] [Google Scholar]