

Abstract
The depolymerization of the recalcitrant polysaccharides found in lignocellulose has become an area of intense interest due to the role of this process in global carbon cycling, human gut microbiome nutritional contributions, and bioenergy production. However, underdeveloped genetic tools have hampered study of bacterial lignocellulose degradation, especially outside model organisms. In this report, we describe an in-frame deletion strategy for the Gram-negative lignocellulose-degrading bacterium Cellvibrio japonicus. This method leverages optimized growth conditions for conjugation and sacB counterselection for the generation of markerless in-frame deletions. This method produces mutants in as few as 8 days and allows for the ability to make multiple gene deletions per strain. It is also possible to remove large sections of the genome, as shown in this report with the deletion of the nine-gene (9.4-kb) gsp operon in C. japonicus. We applied this system to study the complex phenotypes of cellulose degradation in C. japonicus. Our data indicated that a Δcel5B Δcel6A double mutant is crippled for cellulose utilization, more so than by either single mutation alone. Additionally, we deleted individual genes in the two-gene cbp2ED operon and showed that both genes contribute to cellulose degradation in C. japonicus. Overall, these described techniques substantially enhance the utility of C. japonicus as a model system to study lignocellulose degradation.
INTRODUCTION
Cellulose is the most abundant terrestrial source of fixed carbon on the planet, and the synthesis and depolymerization of this polysaccharide contribute significantly in the carbon cycle (1). As the main drivers of cellulose deconstruction, microorganisms use cellulases to break down the cellulose into soluble oligosaccharides that can be used as a carbon source (2). Industry efforts have long sought to harness these enzymes to degrade cellulose in order to utilize it for biotechnological purposes, such as biofuel production (3). As a result, biochemical research has been performed for decades on a multitude of bacterial and fungal enzymes (4, 5). However, recent studies have suggested that purely biochemical analysis might not be enough to further understanding of the complexity of cellulose degradation. As pointed out by Zhang et al., in vivo studies are needed because in vitro studies have been shown to be poor predictors of how the degradative enzymes work on physiologically relevant substrates (6). This sentiment has been echoed in studies of biochemically redundant enzymes, where there are questions as to whether these enzymes are also physiologically redundant or whether novel enzyme activities that are difficult to characterize by standard enzyme assay are important to lignocellulose degradation (7, 8). In addition, in vitro work cannot determine the regulation of protein production or assess the essentiality of the enzyme in a biological context. Absence of these data severely hinders biotechnological efforts as they are needed to elucidate performance on physiologically or industrially relevant substrates.
In order to perform physiological in vivo studies, researchers have attempted to use model organisms, with various degrees of success. A major hurdle for in vivo studies is developing a genetic system to manipulate genes and gene expression. An emerging model organism to study lignocellulose degradation is Cellvibrio japonicus, which possesses the ability to degrade not only cellulose but all of the insoluble polysaccharides that comprise plant cell walls (9). Previous work examining C. japonicus physiology used random and targeted gene inactivation systems that were based on vector integration (10). Using this technique, it was determined that a type II secretion system was the mechanism by which cellulases are exported out of C. japonicus (11). A more comprehensive study of the complement of cellulases possessed by C. japonicus showed that despite the presence of eight cellulases of the glycoside hydrolase class 5 (GH5) class in the chromosome, only one is critical for cellulose degradation. Additionally, the study found novel genes that did not encode known enzymes were essential for efficient cellulose degradation (12). While the vector integration method used in these studies is effective, it is limited because the 500-bp region of homology required for recombination results in an insertion that is not precisely placed. This technique also causes polar effects when used within operons, limiting the use to inactivating genes not within an operon or inactivating the entire operon. Additionally, while several cellulase mutants were found individually to have growth defects when grown on cellulose, it was not possible to test combinations of mutants as the vector integration system cannot be used to construct multiple mutations in a strain. Nevertheless, prior studies with C. japonicus have shown that it has the potential to be a powerful model system and that it warranted additional effort to establish improved genetic tools to study lignocellulose degradation.
In this report we describe a method for constructing markerless in-frame deletions in C. japonicus. This method can be completed very rapidly and with a successful recombinant frequency equal to that of the vector integration system. The new method takes advantage of the counterselectable marker sacB, which allows for scarless deletions (13). We benchmark this system by creating an in-frame deletion of the entire 9.4-kb gsp operon, which encodes the type II secretion system, and demonstrate that it is phenotypically identical to the published gsp vector integration strain (11). Furthermore, we show the utility of this system by using it to further dissect cellulose degradation in C. japonicus. A previous study indicated that the glycoside hydrolase class 5 (GH5) endoglucanase Cel5B and glycoside hydrolase class 6 (GH6) cellobiohydrolase Cel6A are critical for efficient cellulose degradation. These enzymes were shown biochemically to work synergistically to release soluble oligosaccharides from crystalline cellulose. In addition, the Cbp2E and Cbp2D proteins were found to be required for cellulose degradation. These proteins are not cellulases but are thought to play a role in electron transfer for the cellulose-specific lytic polysaccharide monooxygenase (LPMO) enzyme C. japonicus LPMO10B (CjLPMO10B) (12). Bioinformatic analysis suggests that the cbp2ED genes are part of an operon while the cel5B and cel6A genes are transcribed individually. Here, we delete individual members of the cbp2ED operon and determine that both genes are needed for efficient cellulose degradation. Moreover, we make a cel5B cel6A double mutant, the first multiple-mutant strain constructed in C. japonicus, and show that this mutant is severely crippled in cellulose degradation, more so than by either of the single mutants. Application of the described techniques to other aspects of recalcitrant polysaccharide degradation will accelerate in vivo analysis of C. japonicus, which we discuss specifically in regard to addressing the perceived functional redundancy of many carbohydrate-active enzymes found in lignocellulose-degrading bacteria.
MATERIALS AND METHODS
Growth conditions.
Cellvibrio japonicus strains were grown in MOPS [3-(N-morpholino)propanesulfonic acid] defined medium (14) containing glucose (0.25%, wt/vol), carboxymethylcellulose (CMC; 0.5%, wt/vol), or filter paper (0.5%, wt/vol) as the sole source of carbon and energy. Escherichia coli strains were grown in lysogenic broth (LB) (15, 16). Plate medium was solidified with 1.5% agar. Cultures were grown at 30°C with high aeration (225 rpm) in 18-mm culture tubes. Growth was measured as the optical density at 600 nm (OD600) using a Spec20D+ spectrophotometer (Thermo Scientific) or a Tecan M200Pro microplate reader (Tecan Trading AG, Switzerland). All growth experiments were performed in biological triplicate, and where appropriate statistical significance was calculated using analysis of variance (ANOVA) (17) with the GraphPad Prism, version 6, software package (GraphPad, La Jolla, CA). When required, kanamycin was used at a concentration of 50 μg/ml. All chemicals were purchased from Fisher Scientific.
Construction of C. japonicus mutants.
Mutants of C. japonicus were made through triparental mating as previously described (11) with the following modifications. The plasmid pK18mobsacB (18) was used for deletion mutant generation as follows. A 500-bp region upstream of the start codon of the gene to be deleted and a 500-bp region downstream of the stop codon of the gene to be deleted were assembled into a single 1-kb construct and cloned into the pK18mobsacB vector. These up- and downstream regions were synthesized at GeneWIZ (South Plainfield, NJ) and cloned into the EcoRI and XbaI sites of pK18mobsacB using the assembly method of Gibson et al. (19). The vector was transformed into chemically competent E. coli S17 λpir (20) and selected for with kanamycin. The deletion-containing plasmid was introduced into C. japonicus through conjugation using the S17 λpir strain as the donor with an additional DH5α helper strain containing the plasmid pRK2013 (21) (Table 1). Mating was performed on MOPS defined medium plates containing glucose and supplemented with a mating mix (100 μM proline, 100 μM arginine, and 10 μM thiamine). A single colony of the S17 λpir donor containing the pK18mobsacB deletion plasmid, a single colony of the DH5α strain containing the helper plasmid, and a single colony of the C. japonicus recipient strain were streaked within a 2-cm by 2-cm common area and incubated overnight at 30°C. All of the growth within the 2-cm by 2-cm shared region was collected, resuspended in 200 μl of 8.5% sterile saline, and serially plated onto MOPS defined medium plates supplemented with kanamycin (selection 1A) and grown for 48 h at 30°C. Twenty well-isolated colonies were picked and restreaked on MOPS defined medium plates containing kanamycin (selection 1B) and grown for 48 h at 30°C. Finally, the 20 isolated colonies from selection 1B were then streaked on MOPS defined medium plates containing 10% sucrose (selection 2) and grown for 48 h at 30°C. To determine the total number of potential C. japonicus recipients, CFU counting was performed in biological triplicate. Conjugations were performed as described above, and the growth within the 2-cm by 2-cm region was removed and serially diluted with 8.5% sterile saline and then plated on MOPS defined medium plates without mating mix, thereby allowing only C. japonicus to grow. Finally, C. japonicus colonies were enumerated to determine the efficiency of the conjugation.
TABLE 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Genotype | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Laboratory collection |
| E. coli S17 λpir | Tpr Smr recA thi pro hsdR hsdM+ RP4-2-Tc::Mu::Km Tn7 λpir | Laboratory collection |
| C. japonicus Ueda107 | Wild type | Laboratory collection |
| C. japonicus gspD KO | Ueda107 gspD::pK18gspDKO kan+ | 11 |
| C. japonicus Δgsp | Ueda107 Δgsp | This study |
| C. japonicus Δcel5B | Ueda107 Δcel5Ba | This study |
| C. japonicus Δcel6A | Ueda107 Δcel6Ab | This study |
| C. japonicus Δcbp2E | Ueda107 Δcbp2Ec | This study |
| C. japonicus Δcbp2D | Ueda107 Δcbp2Dd | This study |
| C. japonicus Δcel5B Δcel6A | Ueda107 Δcel5B Δcel6A | This study |
| Plasmids | ||
| pRK2013 | ColE1 RK2-Mob+ RK2-Tra+; Kmr | 21 |
| pK18mobsacB | pMB1 ori mob+ sacB+; Kmr | 18 |
| pK18gspDKO | Contains 500-bp internal gspD cloned into plasmid pK18mobsacB; Kmr | 11 |
| pK18Δgsp | Contains 500 bp upstream of the gspC start codon and 500 bp downstream of the stop codon of gspK cloned into pK18mobsacB; Kmr | This study |
| pK18Δcel5B | Contains 500 bp upstream and downstream of cel5B cloned into pK18mobsacB; Kmr | This study |
| pK18Δcel6A | Contains 500 bp upstream and downstream of cel6A cloned into pK18mobsacB; Kmr | This study |
| pK18Δcbp2E | Contains 500 bp upstream and downstream of cbp2E cloned into pK18mobsacB; Kmr | This study |
| pK18Δcbp2D | Contains 500 bp upstream and downstream of cbp2D cloned into pK18mobsacB; Kmr | This study |
| Primers | ||
| Δgsp CONF (5′) | 5′-ACGGGTAGATGGTGT-3′ | This study |
| Δgsp CONF (3′) | 5′-TAATTTCCAGCCCCAG-3′ | This study |
| Δgsp INT (5′) | 5′-TGCCGATCT TTCTGT-3′ | This study |
| Δgsp INT (3′) | 5′-GATGACTCGTAGCGAT-3′ | This study |
| Δcel5B CONF (5′) | 5′-ATCTTCACGGATCACG-3′ | This study |
| Δcel5B CONF (3′) | 5′-TCCATAATGTCGATCTCG-3′ | This study |
| Δcel5B INT (5′) | 5′-AGTATTACAACGCCCA-3′ | This study |
| Δcel5B INT (3′) | 5′-GCCATTGGCATTGACT-3′ | This study |
| Δcel6A CONF (5′) | 5′-GTTGGAGTGTTAGCTG-3′ | This study |
| Δcel6A CONF (3′) | 5′-TGCGGTTGTGGTAGA-3′ | This study |
| Δcel6A INT (5′) | 5′-ATCAAGCAGCTCTGTTC-3′ | This study |
| Δcel6A INT (3′) | 5′-CACCGATACGATCCAT-3′ | This study |
| Δcbp2E CONF (5′) | 5′-ATTGATGCAGGACCTG-3′ | This study |
| Δcbp2E CONF (3′) | 5′-GTGTCCAATTGCGTAG-3′ | This study |
| Δcbp2E INT (5′) | 5′-TACAGCCTCTGCGAT-3′ | This study |
| Δcbp2E INT (3′) | 5′-GCTGATGTTATTCGCCA-3′ | This study |
| Δcbp2D CONF (5′) | 5′-CAGACGGTTGAATTTGGA-3′ | This study |
| Δcbp2D CONF (3′) | 5′-TCTTCGATTTCACGAATC-3′ | This study |
| Δcbp2D INT (5′) | 5′-TAACCTGATCCTGCAC-3′ | This study |
| Δcbp2D INT (3′) | 5′-GATGATGAGCTGCCT-3′ | This study |
BioCyc accession number CJA_2983.
BioCyc accession number CJA_2473.
BioCyc accession number CJA_2615.
BioCyc accession number CJA_2616.
Confirmation of gene deletion mutants.
PCR confirmation of C. japonicus mutants was performed as follows. Primers (confirmation set [CONF]) were designed ∼750 bp from either the start or stop codon of the gene to be deleted (Table 1). As the gene deletion constructs were designed to remove the sequence between the start and stop codons, these confirmation primers would result in a 1.5-kb PCR product for a gene deletion strain. Additionally, primers were designed for a 500-bp internal portion of the gene to be deleted (internal set [INT]). These primers would yield no PCR product in a correctly constructed deletion strain but would generate a 500-bp PCR product using wild-type C. japonicus DNA as the template. The PCR program used was as follows: 95°C for 1 min, followed by 29 cycles of 95°C for 30 s, 45°C for 30 s, and 72°C for 1 to 2 min, with a final step at 72°C for 10 min and a hold at 4°C.
The strains that produced a 1.5 CONF PCR product and did not yield a 500-bp INT PCR product were tested for antibiotic sensitivity. Those that tested Kans were then saved as permanent glycerol stocks.
Cellulase secretion assay.
Cellulase secretion was determined using the CMC-Congo red staining method (22). Briefly, 10 μl of overnight cultures of wild-type and mutant strains was spotted onto MOPS–0.5% CMC plates supplemented with 0.5% glucose and grown statically at 30°C for 24 h. Plates were then stained with 0.1% aqueous Congo red solution for 10 min. After removal of excess stain, the plates were then destained with 1 M NaCl for 10 min before air drying overnight at ambient temperature. Cellulase secretion-deficient mutants were identified by a diminished zone of CMC hydrolysis compared to that of the wild type.
Strain genotypes, plasmids, and primers used in this study are found in Table 1.
Accession numbers.
Data for C. japonicus Δcel5B, Δcel6A, Δcbp2E, and Δcbp2D have been deposited in the BioCyc database (http://biocyc.org) under accession numbers CJA_2983, CJA_2473, CJA_2615, and CJA_2616, respectively.
RESULTS
Evaluation of the markerless in-frame deletion system for C. japonicus.
Previous mutational analyses in C. japonicus used techniques that heavily borrowed from E. coli protocols (20) and, while functional, were time-consuming and inefficient. By optimizing for growth and recovery of the recipient bacterium (C. japonicus), we have significantly accelerated the mutant generation process and greatly improved the success rate of generating targeted mutations. The first optimization was choice of mating medium and a change from LB to MOPS defined medium. Previous work indicated that C. japonicus grew poorly in LB but well in MOPS defined medium (10). In order to allow the donor and helper strains to grow (S17 λpir and DH5α, respectively), the MOPS defined medium was supplemented as these E. coli strains are amino acid auxotrophs (Table 1). Second, we standardized the mating area (Fig. 1A). By using a 2-cm by 2-cm area, we ensured a more thorough outgrowth and mixing of the three strains. Previous filter mating techniques used set volumes of conjugation strains, and while these were successful, the conjugation efficiency appeared to be low (23). Finally, during the selection process, we were able to remove the step involving supplementation with valine, which was previously used for counterselection (11). Using MOPS defined medium, we are now able to use E. coli auxotrophs to prevent growth. In this manner, we allowed for more robust growth of the recipient C. japonicus strain during mating and streamlined the selection process. Due to the standardization of the mating protocol, the total number of potential recipients was approximately the same when the various mutant strains were generated (Table 2).
FIG 1.
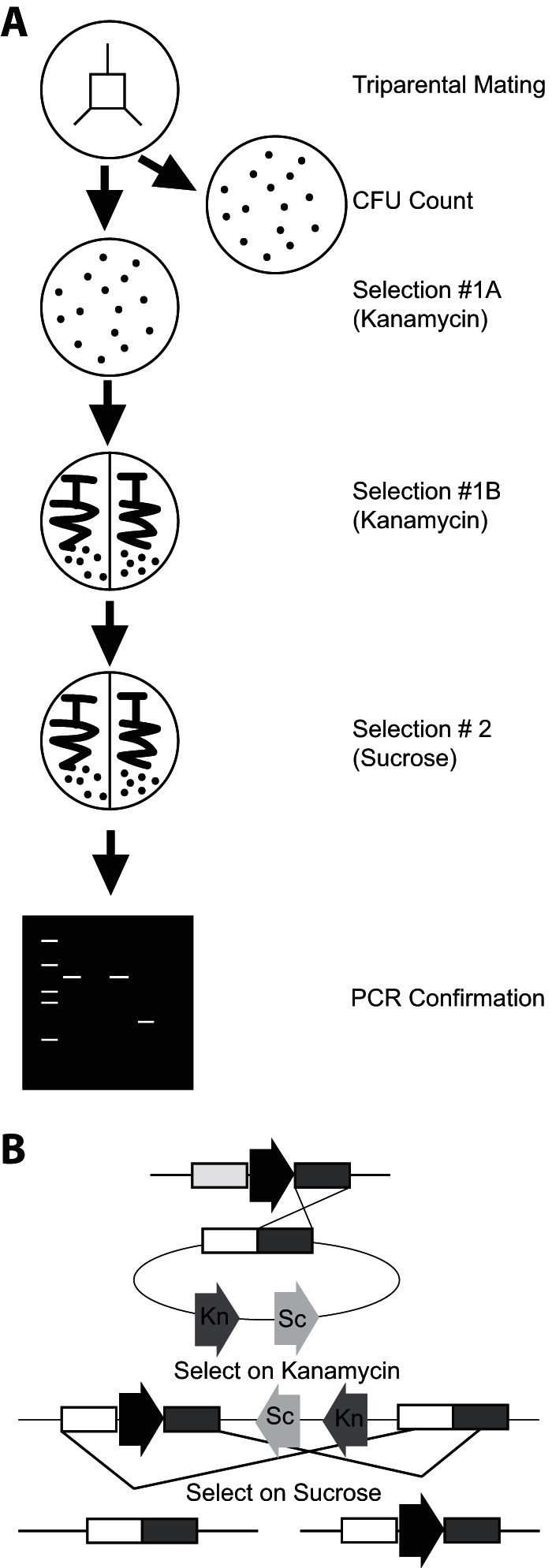
Flowchart for C. japonicus mutation generation. (A) Bacterial conjugations and selections were carried out as described in the text. Selection 1A separates potential recombinants from the donor strains, while selection 1B further isolates potential mutants. The second selection (2) is performed to generate the deletion. PCR screening confirms gene deletion. (B) Integration of the deletion-generating vector. The black arrow represents the gene to be deleted on the chromosome, while the kanamycin (Kn) and sucrose (Sc) arrows represent kanamycin resistance and sucrose sensitivity genes on the plasmid, respectively. The first recombination under kanamycin selection forces integration of the plasmid into the chromosome. The figure depicts the plasmid integrating at the 3′ end, but it is equally possible for it to integrate at the 5′ end. A second recombination event, under sucrose counterselection, forces removal of the plasmid and results in one of two outcomes: reversion to wild type or generation of the deletion.
TABLE 2.
Conjugation frequencies and mutation generation ratesa
| Mutant | No. of potential recipients (× 108 CFU)b | No. of potential recombinants (CFU)c | Mutant generation efficiency (%)d |
|---|---|---|---|
| Negative controle | 2.0 ± 0.8 | 3 ± 2 | NAf |
| gspD KO strain | 31.0 ± 20.0 | 67 ± 18 | 17 ± 3 |
| Δgsp strain | 23.0 ± 4.2 | 121 ± 14 | 23 ± 3 |
| Δcel5B strain | 5.5 ± 1.1 | 431 ± 73 | 32 ± 3 |
| Δcel6A strain | 2.5 ± 0.8 | 29 ± 10 | 13 ± 7 |
| Δcel5B Δcel6A strain | 6.3 ± 3.7 | 513 ± 189 | 27 ± 3 |
| Δcbp2E strain | 3.2 ± 1.3 | 63 ± 18g | 22 ± 10 |
| Δcbp2D strain | 6.2 ± 2.1 | 405 ± 76 | 50 ± 33 |
Experiments were performed in biological triplicate, and standard deviations were calculated using the GraphPad Prism, version 6, software package.
Potential number of C. japonicus recipients was determined through CFU counting from the conjugation plate as outlined in Fig. 1.
Potential recombinants were determined by counting colonies present after selection 1A.
The efficiency of mutation generation was determined by the number of PCR-confirmed deletion mutants divided by the number of colonies screened (n = 20).
The number of colonies that arose when an S17 strain with no plasmid was used as a donor in the triparental mating; used to enumerate spontaneous Kanr C. japonicus colonies.
NA, not applicable.
Duplicate experiment.
With mating conditions standardized, we next enumerated the number of C. japonicus colonies that arose from selection 1. These colonies represent both authentic mutations and spontaneous kanamycin-resistant isolates. We termed these colonies potential recombinants. Negative-control experiments using an S17 λpir donor strain with no plasmid indicated that the number of spontaneous kanamycin-resistant colonies was low (3 ± 2 per conjugation). From the selection 1A plate, 20 potential recombinants were taken and restreaked for isolation onto MOPS-glucose plates supplemented with kanamycin (selection 1B). This step ensures removal of any residual donor or helper E. coli cells mixed with the recipient C. japonicus mutants. Finally, the 20 potential recombinants underwent sucrose counterselection to resolve the plasmid and generate the deletion (selection 2). Utilizing the sacB gene present on the pK18mobsacB plasmid, we selected for a second recombination event that excised the plasmid from the chromosome (24). The second recombination event can occur in either the 5′ or the 3′ region of homology on the integrated deletion plasmid, reverting to the wild-type genotype or creating the desired gene deletion (Fig. 1B). The total time from triparental mating to completion of selection 2 was 7 days.
At this point, the putative deletion mutants were screened via PCR. The confirmation primer set (CONF) was designed to anneal 750 bp up- and downstream of the start and stop codons of the gene to be deleted. As these primers anneal to regions of the genome that are not duplicated on the deletion-generating plasmid, a 1.5-kb PCR product will be formed only by amplification of a chromosomal deletion. A second set of primers internal to the gene to be deleted (INT) was used as an additional confirmation of the deletion. A correct gene deletion would not yield a PCR product using the INT primer set but would generate a PCR product using wild-type chromosomal DNA. We were able to confirm via PCR the deletion of the entire gsp operon, as well as the cellulase genes cel5B, cel6A, cbp2E, and cbp2D (Fig. 2). As expected, we found that all of the PCR-confirmed deletion mutants were kanamycin sensitive. The deletion frequency varied from 13% ± 7% to 50% ± 33%. As a comparison, we also regenerated a gsp vector integration mutant using a previously published protocol (11) and found that the success rate using the in-frame deletion protocol was similar (23% ± 3% for the deletion protocol versus 17% ± 3% for the vector integration protocol). Table 2 summarizes the success rates for the construction of the C. japonicus gene deletion mutants.
FIG 2.
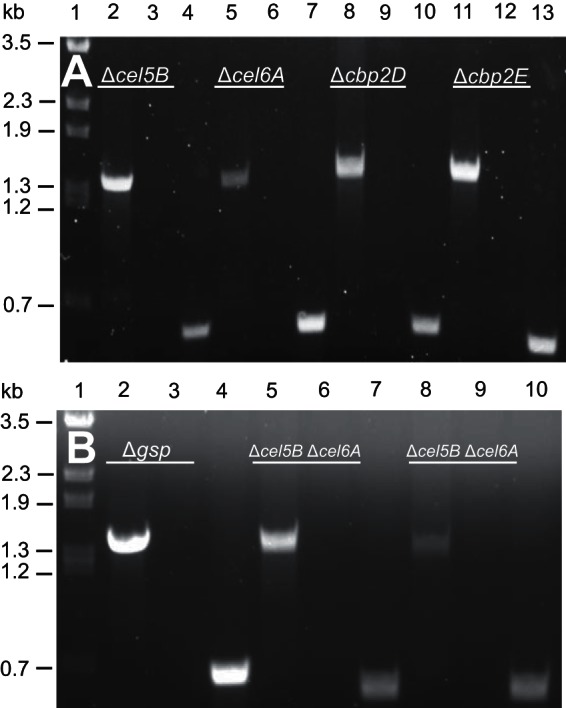
PCR confirmation of C. japonicus deletion mutants. All primer pairs (confirmation, CONF; internal, INT) specific to the gene deletion being confirmed are listed in Table 1. (A) Lane 1, λ BstEII ladder; lane 2, Δcel5B mutant using cel5B CONF primers; lane 3, Δcel5B mutant using cel5B INT primers; lane 4, wild-type using cel5B INT primers; lane 5, Δcel6A mutant using cel6A CONF primers; lane 6, Δcel6A mutant using cel6A INT primers; lane 7, wild type using cel6A INT primers; lane 8, Δcbp2D mutant using cbp2D CONF primers; lane 9, Δcbp2D mutant using cbp2D INT primers; lane 10, wild type using cbp2D INT primers; lane 11, Δcbp2E mutant using cbp2E CONF primers; lane 12, Δcbp2E mutant using cbp2E INT primers; lane 13, wild type using cbp2E INT primers. (B) Lane 1, λ BstEII ladder; lane 2, Δgsp mutant using gsp CONF primers; lane 3, Δgsp mutant using gsp INT primers; lane 4, wild type using gsp INT primers; lane 5, Δcel5B Δcel6A mutant using cel5B CONF primers; lane 6, Δcel5B Δcel6A mutant using cel5B INT primers; lane 7, wild type using cel5B INT primers; lane 8, Δcel5B Δcel6A mutant using cel6A CONF primers; lane 9, Δcel5B Δcel6A mutant using cel6A INT primers; lane 10, wild type using cel6A INT primers.
Multiple targeted gene deletions in a C. japonicus strain facilitate functional characterization of the cellulose degradation apparatus.
Previous mutational analysis in C. japonicus used either random transposon insertions (23) or a targeted gene disruption method using vector integration (11). While both have been valuable in functionally characterizing the genes critical for lignocellulose degradation in C. japonicus, advancement of in vivo analyses requires the ability to make multiple mutations in a strain. A previous report indicated that the cel5B and cel6A genes were important for efficient cellulose degradation (12). We created a Δcel5B Δcel6A double mutant and found that it displayed a growth defect more severe than that with either of the single mutants (Fig. 3C). The growth rate of the Δcel5B single deletion mutant when it was grown on cellulose was 0.15 generations per hour (gen/h), that of the Δcel6A single deletion mutant was 0.09 gen/h, and that of the Δcel5B Δcel6A double deletion mutant was 0.07 gen/h. Additionally, the Δcel5B Δcel6A double deletion mutant had a lag phase of 48 h that was absent from either single mutant or the wild type. All strains grew similarly on glucose, and as previously described, mutants lacking cel5B were unable to grow using soluble cellulose (11, 12) (Fig. 3A and B). These data represent the construction and analysis of the first directed double mutant in C. japonicus.
FIG 3.

Growth analyses of C. japonicus deletion mutants. Wild-type and mutant strains of C. japonicus were grown in MOPS minimal medium supplemented with 0.25% glucose (A), 0.5% CMC (B), or 0.5% filter paper (C and D) as the sole carbon source. All experiments were performed in biological triplicate. Error bars represent standard deviations. Open circles, wild type; filled diamonds, Δgsp strain;, open squares, Δcel5B strain; open inverted triangles, Δcel6A strain; filled triangles, Δcel5B Δcel6A strain; filled circles, Δcbp2E strain; open diamonds, Δcbp2D strain. For the sake of clarity, the filter paper growth experimental data are shown as two panels, but since the data are part of one experiment, the control strain data (wild type and Δgsp strain) are the same in both panels. Growth rates were calculated between the 24- and 58-h time points, depending on when a given strain was exponentially growing, and used a minimum of 8 h of growth data from the filter paper experiments for the calculation.
Deletion of entire operons or single genes within operons elucidates critical genes for cellulose degradation in C. japonicus.
A gspD vector integration mutant (gspD knockout [gspD KO] strain) was previously shown to be cellulase secretion deficient and was unable to grow on cellulose as a sole carbon source (11). Growth analysis with a strain that had a deletion of the entire 9.4-kb gsp operon recapitulated both the growth defect on cellulose (Fig. 3B and C) and the secretion-deficient phenotype (Fig. 4). Despite the large size of the Δgsp deletion, the number of potential recombinants and overall mutation generation efficiency were similar to those of the single gene deletions generated (Table 2).
FIG 4.

Visualization of the C. japonicus gsp cellulase secretion defect via Congo red staining. Representative spot test for cellulase secretion shown for 10 Δgsp mutants taken from selection 1A. A diminished zone of clearance compared to that of the wild type (WT) identified secretion-deficient mutants. The gspD KO strain was used as a known secretion-deficient control. This plate shows four Δgsp secretion-deficient mutants (1, 2, 5, and 8). Subsequent PCR screening indicated that these four putative mutants had the entire gsp operon deleted.
While the elimination of an entire operon will ensure that a complete biological process is removed from a cell, there are times when elimination of a single gene within an operon is advantageous to determine its contribution to a biological process. Previous work showed that the cbp2ED operon played a critical role in cellulose degradation (12). However, at the time, there were no genetic techniques to separate the individual contributions of the genes in the operon. Using our in-frame deletion system, we deleted individually the cbp2E and cbp2D genes and assessed their roles in cellulose degradation. While it appears that both contribute to cellulose degradation, the Δcbp2E mutant was slightly, albeit significantly, more impaired for growth on cellulose. The growth rate of the Δcbp2E mutant was 0.09 gen/h, and that of the Δcbp2D mutant was 0.06 gen/h. These rates were accelerated compared to the reported growth rate of 0.01 gen/h for the cbp2ED insertion mutant when it was grown on filter paper as a sole carbon source (12). As expected, there was no growth defect when these mutant strains were grown in glucose or soluble cellulose (CMC) medium, as was previously shown (12) (Fig. 3A and B).
DISCUSSION
Better understanding of lignocellulose degradation will impact diverse fields of study from carbon cycle science to human health and sustainable energy (25–27). While there are currently several model organisms used to study lignocellulose degradation, genetic tools that allow rapid and efficient generation of mutants are needed to accelerate in vivo studies of recalcitrant polysaccharide depolymerization. To this end, we have provided an optimized protocol for targeted in-frame deletions in Cellvibrio japonicus and shown its utility in further characterizing genes important for cellulose degradation.
The initial report of targeted gene disruption in C. japonicus indicated that both lysogenic broth and M9 defined medium were poor for growing C. japonicus and that MOPS defined medium allowed superior growth (10). According to Neidhardt et al., MOPS is a more complete minimal medium as it has iron, copper, manganese, cobalt, molybdenum, boron, and zinc, which are missing in the M9 recipe (14). The use of MOPS minimal medium rather than M9 minimal medium reduces incubation times of C. japonicus from 4 to 5 days to 24 to 48 h on agar plates. The improvement in C. japonicus recipient cell growth on MOPS medium was also leveraged to improve the selections against the donor and helper conjugation strains. The supplements supplied on the conjugation plate allowed all donor and recipient strains to have robust growth on a MOPS-glucose plate, but removal of the mating mix from selection plates efficiently prevented E. coli growth.
The reported mutant-making procedure is superior due to the rate at which mutations can be generated. Mutants created using the previous vector integration protocol, from conjugation to PCR confirmation, took 2 weeks. With our described method, a minimum of 6 days has been eliminated from the mutation generation time due to using optimal growth conditions (i.e., MOPS medium). The entire procedure for C. japonicus shown in Fig. 1A can be performed in 8 days, from conjugation to PCR confirmation. This improvement in speed does not impact the efficiency of successful mutant generation, as indicated by the data shown in Table 2.
This new method of mutant generation has three major advantages for physiological analyses of C. japonicus. The first is the ability to delete kilobase-long regions of the genome, thereby eliminating entire operons. As proof in concept, we successfully deleted the entire 9.4-kb locus that encodes the type II secretion system. The entirety of this chromosomal section was precisely and efficiently removed with no chance of reversion or antibiotic pressure required after deletion. The deletion mutant behaved identically to the vector integration mutant derived from insertion of a suicide plasmid in the gspD gene (Fig. 4). A second beneficial feature is the ability to delete genes within operons without altering frame or leaving a scar sequence, an advantage over Flp/Flp recognition target (FRT) and Cre/LoxP systems that leave small recognition sites in the chromosome (28, 29). When the Δcbp2E and Δcbp2D mutants were grown on cellulose, we were able to observe different phenotypes that had not been discerned previously. Therefore, this technique makes it possible to see individual contributions of genes in operons. The third and perhaps greatest advancement of the described deletion system is the ability to make multiple deletions in the same strain of C. japonicus. Multiple deletions allow the synergy of lignocellulose-degrading enzymes to be observed in an in vivo context. This is especially important in working with complex enzyme systems, like that for cellulose degradation by C. japonicus, which contains 12 cellulases and four β-glucosidases (9). We sequentially deleted the cel5B and cel6A genes, which are at chromosomally distant locations, in the same C. japonicus strain and assessed growth on cellulose (Fig. 3). The growth defect of the double mutant is more severe than that of either single mutant, giving physiological evidence for these enzymes working in concert to degrade cellulose. There is only one predicted cellobiohydrolase (cel6A) in C. japonicus, so, as expected, the deletion of this gene prevents the bacterium from degrading cellulose effectively. Interestingly, there are eight predicted GH5 endoglucanases, and only the cel5B gene has been demonstrated to be critical for cellulose utilization, particularly for noncrystalline cellulose. Our data presented here indicate that the cel5B gene product is also important for crystalline cellulose degradation. These in vivo data corroborate previous biochemical studies on the enzyme synergy between the Cel5B and Cel6A proteins (12). For the complete degradation of cellulose, there is a typical requirement for endoglucanase, cellobiohydrolase, and β-glucosidase activity (30). We are currently investigating the four β-glucosidase genes of C. japonicus to determine those that have critical roles in cellulose degradation.
Due to the iterative nature of the deletion system described, making strains with several genes deleted can be accomplished and will further allow for the dissection of complex phenotypes. For example, many lignocellulose-degrading bacteria encode several genes that have been bioinformatically described as functionally redundant but have not been tested experimentally. Additionally, the physiological effects of enzyme synergy can be assessed, as was done in this report with the Δcel5B Δcel6A double deletion mutant. Current studies by our group are examining these “redundant” genes for lignocellulose degradation in C. japonicus.
In summary, we have established a rapid, efficient, and precise method of markerless gene deletion in Cellvibrio japonicus. This method will be an invaluable tool that will greatly facilitate the study of complex enzyme systems in a physiologically relevant manner to provide a better understanding of lignocellulose degradation. Additional work aims to apply this protocol to delete only portions of genes or to precisely place point mutations in genes for structure/function studies. Another application under investigation is introduction of nonnative genes into exact locations for heterologous expression.
ACKNOWLEDGMENTS
Start-up funds from the College of Natural and Mathematical Sciences at the University of Maryland—Baltimore County supported this work. Additional support to C.E.N. came from a U.S. Department of Education Graduate Assistantship in Areas of National Need (GAANN) and the Meyerhoff Graduate Fellows Program by an NIGMS Initiative for Maximizing Student Development grant (2 R25-GM55036).
We declare that we have no conflicts of interest.
REFERENCES
- 1.Lynd LR, Weimer PJ, Pretorius IS. 2002. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577. doi: 10.1128/MMBR.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson DB. 2011. Microbial diversity of cellulose hydrolysis. Curr Opin Microbiol 14:259–263. doi: 10.1016/j.mib.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Bokinsky G, Peralta-Yahya PP, George A, Holmes BM, Steen EJ, Dietrich J, Lee TS, Tullman-Ercek D, Voigt CA, Simmons BA, Keasling JD. 2011. Synthesis of three advanced biofuels from ionic liquid-pretreated switchgrass using engineered Escherichia coli. Proc Natl Acad Sci U S A 108:19949–19954. doi: 10.1073/pnas.1106958108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Brink J, de Vries RP. 2011. Fungal enzyme sets for plant polysaccharide degradation. Appl Microbiol Biotechnol 91:1477–1492. doi: 10.1007/s00253-011-3473-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilbert HJ, Stalbrand H, Brumer H. 2008. How the walls come crumbling down: recent structural biochemistry of plant polysaccharide degradation. Curr Opin Plant Biol 11:338–348. doi: 10.1016/j.pbi.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Rogowski A, Zhao L, MG H, Avci U, Knox JP, Gilber HJ. 2014. Understanding how complex molecular architecture of mannan-degrading hydrolysis contributes to plant cell wall degradation. J Biol Chem 289:2002–2012. doi: 10.1074/jbc.M113.527770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cartmell A, McKee LS, Pena MJ, Larsbrink J, Brumer H, Kaneko S, Ichinose H, Lewis RJ, Vikso-Nielsen A, Gilbert HJ, Marles-Wright J. 2011. The structure and function of an arabinan-specific alpha-1,2-arabinofuranosidase identified from screening the activities of bacterial GH43 glycoside hydrolases. J Biol Chem 286:15483–15495. doi: 10.1074/jbc.M110.215962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naas AE, Mackenzie AK, Mravec J, Schuckel J, Willats WG, Eijsink VG, Pope PB. 2014. Do rumen Bacteroidetes utilize an alternative mechanism for cellulose degradation? mBio 5(4):e01401-14. doi: 10.1128/mBio.01401-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBoy RT, Mongodin EF, Fouts DE, Tailford LE, Khouri H, Emerson JB, Mohamoud Y, Watkins K, Henrissat B, Gilbert HJ, Nelson KE. 2008. Insights into plant cell wall degradation from the genome sequence of the soil bacterium Cellvibrio japonicus. J Bacteriol 190:5455–5463. doi: 10.1128/JB.01701-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardner JG, Keating DH. 2012. Genetic and functional genomic approaches for the study of plant cell wall degradation in Cellvibrio japonicus. Methods Enzymol 510:331–347. doi: 10.1016/B978-0-12-415931-0.00018-5. [DOI] [PubMed] [Google Scholar]
- 11.Gardner JG, Keating DH. 2010. Requirement of the type II secretion system for utilization of cellulosic substrates by Cellvibrio japonicus. Appl Environ Microbiol 76:5079–5087. doi: 10.1128/AEM.00454-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardner JG, Crouch L, Labourel A, Forsberg Z, Bukhman YV, Vaaje-Kolstad G, Gilbert HJ, Keating DH. 2014. Systems biology defines the biological significance of redox-active proteins during cellulose degradation in an aerobic bacterium. Mol Microbiol 94:1121–1133. doi: 10.1111/mmi.12821. [DOI] [PubMed] [Google Scholar]
- 13.Reyrat JM, Pelicic V, Gicquel B, Rappuoli R. 1998. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect Immun 66:4011–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neidhardt FC, Block PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertani G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol 186:595–600. doi: 10.1128/JB.186.3.595-600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranyi J, Pin C. 1999. Estimating bacterial growth parameters by means of detection times. Appl Environ Microbiol 65:732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schafer A, Tauch A, Jager W, Kalinowski J, Theirbach G, Puhler A. 1994. Small mobilization multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 19.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 20.Miller JH. 1975. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 21.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1684–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teather R, Wood P. 1982. Use of Congo red-polysaccharide interaction in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl Environ Microbiol 43:777–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beylot MH, Emami K, McKie VA, Gilbert HJ, Pell G. 2001. Pseudomonas cellulosa expresses a single membrane-bound glycoside hydrolase family 51 arabinofuranosidase. Biochem J 358:599–605. doi: 10.1042/0264-6021:3580599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donnenberg MS, Kaper JB. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun 59:4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, Creagh AL, Haynes CA, Kelly AG, Cederholm SN, Davies GJ, Martens EC, Brumer H. 2014. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506:498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solomon BD. 2010. Biofuels and sustainability. Ann N Y Acad Sci 1185:119–134. doi: 10.1111/j.1749-6632.2009.05279.x. [DOI] [PubMed] [Google Scholar]
- 27.Wilson DB. 2012. Processive and nonprocessive cellulases for biofuel production—lessons from bacterial genomes and structural analysis. Appl Microbiol Biotechnol 93:497–502. doi: 10.1007/s00253-011-3701-9. [DOI] [PubMed] [Google Scholar]
- 28.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 29.Marx CJ, Lidstrom ME. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33:1062–1067. [DOI] [PubMed] [Google Scholar]
- 30.Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG. 2012. Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45. doi: 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]