

Abstract
The 2014 Ebola outbreak in West Africa is the largest documented for this virus. To examine the dynamics of this genome, we compare more than 100 currently available ebolavirus genomes to each other and to other viral genomes. Based on oligomer frequency analysis, the family Filoviridae forms a distinct group from all other sequenced viral genomes. All filovirus genomes sequenced to date encode proteins with similar functions and gene order, although there is considerable divergence in sequences between the three genera Ebolavirus, Cuevavirus and Marburgvirus within the family Filoviridae. Whereas all ebolavirus genomes are quite similar (multiple sequences of the same strain are often identical), variation is most common in the intergenic regions and within specific areas of the genes encoding the glycoprotein (GP), nucleoprotein (NP) and polymerase (L). We predict regions that could contain epitope-binding sites, which might be good vaccine targets. This information, combined with glycosylation sites and experimentally determined epitopes, can identify the most promising regions for the development of therapeutic strategies.
This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Keywords: Ebola, comparative genomics, viral genomes, epitope prediction, Ebola virus disease (EVD), Filovirus
Variation within Ebola genomes is most common in the intergenic regions and within specific areas of the genes encoding the glycoprotein (GP), nucleoprotein (NP) and polymerase (L); genomic conservation and epitope prediction, combined with glycosylation sites and experimentally determined epitopes, can identify the most promising regions for the development of therapeutic strategies.
Graphical Abstract Figure.
Variation within Ebola genomes is most common in the intergenic regions and within specific areas of the genes encoding the glycoprotein (GP), nucleoprotein (NP) and polymerase (L); genomic conservation and epitope prediction, combined with glycosylation sites and experimentally determined epitopes, can identify the most promising regions for the development of therapeutic strategies.
INTRODUCTION
The current 2014 Ebola virus disease (EVD) outbreak in West Africa is the largest in its four decades of history. The outbreak has dominated the news, inspiring the imaginations of many, often, unfortunately, in detrimental ways because of fear of the unknown. In this review, we examine the genomes of the Filoviridae family of viruses to which the genus Ebolavirus belongs, and compare the ebolavirus genomes to a set of roughly 4000 reference viral genomes, as well as to the genomes of other genera in the same virus family. We summarize how the different isolates of Ebola virus vary amongst the sequenced genomes currently available, and make some observations that will address basic questions about how changes in the viral genome might affect EVD virulence and immune responses, and efforts to defeat it.
‘So, what DO we know about this deadly viral scourge?’ Approximately 75% of emerging infectious diseases like Ebola are zoonoses that result from various anthropogenic, genetic, ecologic, socioeconomic and climatic factors (Gebreyes et al. 2014). The current Ebola epidemic in West Africa is a stark reminder of the role unknown animal reservoirs play in public health.
We know that in Central Africa, the location of all previous EVD outbreaks, several monkey species, chimpanzees, gorillas, baboons, duikers and fruit bats have been found to be infected with Ebola virus during trapping studies. Given their lack of overt disease while infected, there is good evidence that various species of bats, predominantly fruit bats, are significant natural reservoirs for ebolaviruses, marburgviruses and cuevaviruses (Olival and Hayman 2014) although the natural hosts for most ebolavirus species and variants are still unproven, as is still the case for the current outbreaks.
Transmission of EBOVs from bats and other zoonotic reservoirs to humans requires a hierarchy of enabling conditions that connect the redistribution of reservoir hosts, episodic viral infections within these hosts, random human exposure to infected blood or carcasses from these hosts and sufficient susceptibility of the new recipient human host (Plowright et al. 2015). Two hypotheses may explain 40 years of temporal and spatial pulses of Ebola outbreaks: episodic shedding from persistently infected reservoir hosts or transient epidemics that occur as the virus is transmitted among reservoir populations during animal migrations.
We know that EBOV's ability to jump from its natural reservoirs to humans and other animals is not new. From 1976, when an EBOV was first identified as the cause of two outbreaks of a viral hemorrhagic fever (VHF) that later was known as EVD, through 2013, the World Health Organization reported 1716 confirmed EVD cases, all occurring within documented outbreaks in tropical regions of Central Sub-Saharan Africa. However, EBOV probably infected humans many times prior to 1976, but was unrecognized with the tools available at the time among the many other such VHF diseases in the affected regions of Africa.
The 2014 outbreaks in West Africa and the Republic of the Congo are occurring at a time when we have the technology to rapidly and affordably sequence complete viral genomes in ways previously unimaginable. As technology has improved, blood samples and viral isolates collected from each of these outbreaks have been partially or completely sequenced, giving us an unprecedented opportunity to study the etiology of the virus related to changes in its genome.
HOW DIFFERENT IS EBOLA FROM OTHER VIRUSES?
We compared about 4000 complete virus genome sequences from the RefSeq database of viral sequences (downloaded 6 August 2014), and added additional available genomes from the family Filoviridae in GenBank. Figure 1 shows a dendrogram of all these viral genomes, which was based on 9-bp K-mer frequency analysis and was constructed using the feature frequency profile (FFP) method (Jun et al. 2010).
Figure 1.

A dendrogram of all viral genomes from RefSeq complemented with, in red, multiple genomes in the family Filoviridae extracted from GenBank. The dendrogram is constructed by the FFP method which is based on K-mers, with K set to 9, in the moderate range of optimal feature length based on analysis of cumulative relative entropy and relative sequence divergence as described previously in Wu et al. (2009).
One should be cautious about interpreting this figure. It is easy to assume that this is a ‘phylogenetic tree’, representing evolutionary distances between the viruses. However tempting, this dendrogram is NOT in general reflective of overall phylogeny. This tree contains viruses from all sequenced families, but there is not a single gene conserved amongst all viruses on which phylogeny could be based. So this figure is to be interpreted as roughly representing ‘genome distance’, based on similarity of (frequency of) 9-mers. Having said that, the deep branches that are observed for the virus families are as would be expected—most of the viral families are quite different from each other, and these often form clear clusters, including the family Filoviridae, shown in red. An interactive, zoomable version of this figure is available online (http://dtree.ornl.gov/ebola_ref_9mer.html).
HOW DIFFERENT IS THE GENUS EBOLAVIRUS FROM OTHER FILOVIRUSES?
The order Mononegavirales, family Filoviridae, genus Ebolavirus, has five species: Zaire ebolavirus (EBOV), Taï Forest ebolavirus (TAFV), Reston ebolavirus (RESTV), Sudan ebolavirus (SUDV) and Bundibugyo ebolavirus (BDBV). The taxonomy of the order Mononegavirales is described in detail in the Supplementary material.
In humans, EVD is caused by four viruses: EBOV, TAFV, SUDV and BDBV. The fifth ebolavirus, RESTV, is not known to cause disease in humans, but does cause EVD in non-human primates. EBOV is the most virulent of the viruses in humans and is responsible for the largest number of outbreaks and for the largest number of cases. Major EBOV outbreaks with 100 or more cases occurred in Yambuku, Zaire (now Democratic Republic of the Congo—DRC), in August 1976 (Pattyn et al. 1977); in Kikwit, Zaire, in 1995; in the Republic of the Congo (ROC—also known as Congo-Brazzaville) and Gabon, in 2001–02 (Nkoghe et al. 2005); in Lossi, ROC, in 2003; in Bamoukamba, DRC, in 2007; and in West Africa (Guinea, Sierra Leone and Liberia) in 2014–15. A smaller outbreak occurred in Ikanamongo, DRC, in 2014, concurrent with the West Africa outbreak.
SUDV was responsible for the first recorded case of EVD, during a major outbreak in Nzara Sudan in June 1976. The first international response, however, did not occur until two months later for the Yambuku outbreak, which gave the virus its name, taken from the nearby Ebola river. Another significant SUDV outbreak occurred in Gulu, Uganda, in 2000–01. BDBV has been responsible for two outbreaks, a major outbreak in Kabango, Uganda, in 2007 and a smaller outbreak in Isiro, DRC, in 2012.
The only known infection in humans by TAFV occurred in 1994 during an epizootic VHF event among western chimpanzees in the Taï National Park of Côte d'Ivoire. A scientist performing necropsies on infected chimpanzees contracted EVD but survived. Comprehensive descriptions of all known EVD outbreaks can be found in Corum (2014), Mylne et al. (2014) and CDC Outbreaks Chronology (2015).
None of the viruses in the six EBOV outbreaks over the last 40 years have had clinical conditions unique enough to warrant designating any as new ‘strains’; therefore, they are called variants, using the following ICTV Virus nomenclature template: 〈virus name〉 / (〈strain〉) / 〈isolation host-suffix〉 / 〈country of sampling〉 / 〈year of sampling〉 / 〈genetic variant designation〉 - 〈isolate designation〉 (Kuhn et al. 2014).
The EBOV variant responsible for the 2014 West African outbreak has been named EBOV Makona (phonetic: mah-kaw’-nuh or muh-koh’-nuh) after the Makona River close to the border between Liberia, Guinea and Sierra Leone where the current outbreak originated (Kuhn et al. 2014) several thousand kilometers away from the DRC. In late 2014, EVD also broke out in the Boende District of the DRC. All cases were associated with one variant, for which the name ‘Lomela’ (law-me’-lah) has been proposed, after the Lomela River that runs through the DRC's Boende District (Kuhn et al. 2014).
As previously stated, filoviruses belong to the order Mononegavirales, which, as the name suggests, are non-segmented negative-sense, single-stranded RNA viruses that have inverse-complementary 3′ and 5′ termini (Pringle 2005). In addition to the genus Ebolavirus, the family Filoviridae contains the genus Marburgvirus, first described in Marburg, Germany, in 1967 (Siegert et al. 1967), and the genus Cuevavirus, first isolated from dead bats in Asturias, Spain, in 2002 (Negredo et al. 2011).
There is a strong similarity of genomic structure across the family Filoviridae, which points to a potential common origin. The presence of endogenous viral sequences in numerous mammalian genomes suggests that a common Ebola/Marburg/Cuevavirus-like ancestral virus existed between 32 and 53 million years ago (Belyi, Levine and Skalka 2010). The ebolaviruses and cuevaviruses diverged from marburgviruses at least 5 million years ago during the Miocene (Taylor et al. 2014).
As natural reservoirs for ebolaviruses, marburgviruses and cuevaviruses, infected bats are typically asymptomatic (although there have been occasional die-offs), suggesting stable host–virus relationships that have evolved over millions of years (Wynne and Wang 2013; Olival and Hayman 2014). The stability of this host–virus balance is illustrated by the relative similarity of EBOV sequences from the 1976 Yambuku outbreak to the sequences from the 2014 West Africa and DRC outbreaks; the genomes from the 1976 outbreak are, on average, 97% identical to the 2014 West Africa outbreak and 99% identical to the 2014 DRC outbreak.
To understand the global relationship between the three genera of the family Filoviridae, we constructed a maximum likelihood tree based on complete genomes of all 80 marburgvirus genomes, the only available complete cuevavirus genome, and 84 non-redundant ebolavirus genomes (Fig. 2). Descriptions of the ebolavirus and marburgvirus genome datasets are given in the Experimental Procedures section. As can be seen in Fig. 2, ebolaviruses separate into the five recognized species: Bundibugyo ebolavirus, Reston ebolavirus, Sudan ebolavirus, Taï Forest ebolavirus and Zaire ebolavirus.
Figure 2.

A maximum likelihood tree based on complete genomes of the three filovirus genera. The three genera Marburgvirus, Cuevavirus and Ebolavirus are separated and genus Ebolavirus is further split into species, indicated on the right. The tree was produced with PhyML (Guindon et al. 2010) with the GTR + I + G nucleotide substitution model to a multiple sequence alignment of complete genome sequences by MAFFT (Katoh and Standley 2013). The best substitution model was identified by jModelTest (Guindon and Gascuel 2003; Darriba et al. 2012) among a broad suite of evolutionary models based on BIC. The numeric values represent the number of members within the clades.
The single species of the genus Marburgvirus (Marburg marburgvirus) contains two groups, Marburg virus and Ravn virus, whose distinction is shown in the marburgvirus clade in Fig. 2. All three filovirus genera and all five of the ebolavirus species appear to be monophyletic. Genus Cuevavirus is more closely related to genus Ebolavirus than to genus Marburgvirus, in agreement with previous work (Carroll et al. 2013). The Reston genomes are grouped with Sudan genomes, and Taï Forest with Bundibugyo. The Zaire genomes share a common ancestor with a clade of the Taï Forest and Bundibugyo genomes first, and then with a clade of Reston and Sudan genomes.
Genomic structure of the EBOVs
All of the filovirus genomes sequenced to date are about 19 Kb in length and all encode seven predicted viral proteins, as shown in Fig. 3A; cuevavirus contains a potential extra open reading frame in the opposite direction, for which translation has not been verified, that overlaps for 29 bases with the VP24 gene.
Figure 3.

Gene organization of filovirus genomes and pan-core proteome analysis. (A) Gene organization of viruses from genera Marburgvirus, Ebolavirus and Cuevavirus. Conserved regions are indicated by color, with striped color for weak conservation. The furin cleavage site for glycoprotein GP is indicated by the inverted Y-shaped symbol. Gray blocks below the line indicate the position of conserved functional domains (predicated by InterProScan; Jones et al. 2014). (B) Venn diagram summarizing the results of the pan-core analysis based on protein sequence alignments with a cut-off based on the 50–50 rule (see the text). (C) Venn diagram of the protein functional domains resulting from pan-core analysis based on HMM.
The seven functional proteins encoded in EBOV are designated as NP (nucleoprotein), viral proteins (VP) VP24 (membrane-associated protein), VP30, VP35 (both polymerase matrix proteins), VP40 (matrix protein), L (RNA polymerase) and GP (glycoprotein) (Mühlberger 2007). Each of these genes encodes a single protein product, with the exception of GP, which also encodes sGP and ssGP, as a result of mRNA editing. VP40 is the primary EBOV matrix protein and regulates assembly and egress of infectious EBOV particles. VP40 assembles on the inner leaflet of the plasma membrane of human cells to regulate viral budding (Soni et al. 2013; Adu-Gyamfi et al. 2014).
In ebolaviruses and cuevaviruses, the GP gene encodes three proteins of different sizes: the full-length 676-residue surface glycoprotein GP1,2 (GP for short), which mediates virus–host cell attachment and fusion, the 364-residue pre-form of secreted glycoprotein (pre-sGP) and the 298-residue small secreted glycoprotein (ssGP) of unknown function. The various glycoproteins are produced from frameshifts as a result of mRNA editing of a polyadenosine site (Lee and Saphire 2009; Finn, Clements and Eddy 2011). Transcripts containing seven adenosine residues (without mRNA editing) encode pre-sGP, while addition of a single A residue at the RNA editing site results in GP, and addition of two A residues results in ssGP (Volchkov et al. 1995; Sanchez et al. 1996). Deep sequencing of mRNA in ebolavirus-infected Vero cells found approximately 70% of reads with 7 As, 25% with 8 As, 2% with 9 As and 2% with 10 As. Cell culture passaging studies have shown that GP with 8 As is the primary functional translation product (Shabman et al. 2014). Full-length GP protein is post-translationally cleaved by cellular proteases such as furin at position 511 to produce GP1 and GP2, which are covalently linked in the mature GP1,2 complex. Protein sGP, which can have immunogenic properties, is produced as pre-sGP (364 amino acid residues), which then undergoes post-translational proteolytic cleavage at position 324 by furin to yield soluble sGP.
Pan-core proteome analysis was performed using all 73 marburgvirus proteomes, the single cuevavirus proteome and 53 non-redundant ebolavirus proteomes, including only the largest GP (the 676-residue surface glycoprotein) for cuevaviruses and ebolaviruses. Descriptions of the ebolavirus and marburgvirus proteome datasets are given in the Experimental Procedures section. All proteins were clustered using USEARCH (Edgar 2010) with a cut-off of 50% minimum sequence identity over at least 50% of the longest sequence length. This resulted in 19 protein clusters, each of which corresponded to a different protein type. The results are summarized in the Venn diagram of Fig. 3B. All protein types of marburgviruses were found in separate clusters, while there were five singleton clusters unique to cuevaviruses and four clusters unique to ebolaviruses. The three clusters that were composed of proteins from both ebolaviruses and cuevaviruses contained VP35, VP24 and L. Thus, despite the conservation in gene organization observed with all filovirus genomes, only ebolaviruses and cuevaviruses share significant sequence similarity above the 50–50 cut-off. However, the underlying functional domains for all seven proteins are conserved across all the genomes from the family Filoviridae, when these domains were compared by Hidden Markov Model (HMM) analysis (Finn, Clements and Eddy 2011), as illustrated in Fig. 3C. The likely explanation for this difference is that viral genomes evolve relatively fast—hence, the protein sequences have varied substantially, to the point that by using standard cut-off values, the marburgvirus proteins appear non-conserved, despite their functional domain conservation.
We crosschecked the existing functional domains of the eight proteins against the RefSeq virus protein database derived from about 4000 viruses. Most of the domains (based on InterPro functional domain identification; Hunter et al. 2009) are not found in other viruses. Two functional domains, IPR014023 and IPR026890, within the RNA-dependent RNA polymerase L gene were found in 90 other viruses.
HOW DIFFERENT ARE THE EBOLAVIRUSES IN THE CURRENT OUTBREAK FROM THOSE OF PREVIOUS EBOLA OUTBREAKS?
Using the same approach as described in Fig. 2, we constructed a maximum likelihood tree of 60 Zaire ebolavirus genomes (see Experimental Procedures section), including 34 sequences obtained from isolates of the 2014 West Africa outbreak, this time using the GTR + G nucleotide substitution model as the best model. We rooted our tree of species Zaire ebolavirus to the clade of the earliest recorded outbreak, which corresponds to the 1976 outbreaks (Fig. 4). The inference of the root is described in the Experimental Procedures section. This analysis resulted in clusters containing isolates from five distinct periods: 1976, 1994–96, 2002, 2007–08 and 2014, each representing a different unique outbreak. The tip labels of the tree describe, in order, features of accession, strain, country, host (only if non-human) and collection date according to metadata available from the European Bioinformatics Institute and the National Center for Biotechnology Information, confirmed by a literature survey.
Figure 4.

A maximum likelihood tree of 60 Zaire ebolavirus genomes. The tree was produced with the GTR + G model rooted by a clade of the 1976 outbreaks. All isolates were of human origin, with the exception of two isolates from the 1976 outbreak (AF499101 was mouse adapted, EU224440 was from a Guinea pig). The asterisk identifies the DRC 2014 isolate within a clade of 1994–1996 isolates from Gabon. The numbers on the major internal branches represent bootstrap support (%) out of 100 replicates. Abbreviations: DRC, Democratic Republic of Congo; GAB, Gabon; GIN, Guinea; SLE, Sierra Leone.
The Zaire ebolaviruses can be separated into clades, representing outbreaks in Central Africa (DRC and Gabon) and West Africa (Guinea, Sierra Leone and Liberia) showing distinct geographical and temporal clustering. Zaire ebolaviruses from DRC separate into three clades, corresponding to different time periods. One of these clades is grouped with a Gabon clade collected during the years 1994–96. The outbreak that occurred late in the summer of 2014 in the vicinity of Boende Town in DRC groups with a set of previous Gabon outbreak genomes isolated 20 years earlier, but not to the genomes of intervening outbreaks in that country. The 2014 DRC isolate is also clearly distinct from the large West African 2014 outbreak, as has been observed before (Maganga et al. 2014). The current West African 2014 outbreak, from which the majority of sequences are derived, is well documented to have originated in Guinea and subsequently spread to Sierra Leone and Liberia. From there, travelers involuntarily exported the virus to additional countries as single cases or as patients were relocated for care. The 2014 West African group forms a distinct clade, instead of being nested within the previous Zaire ebolavirus outbreaks, which suggests that the current outbreak in the West Africa is caused by a divergent lineage of Zaire ebolaviruses (Dudas and Rambaut 2014).
So far, we have focused on complete genomes to study the Ebola outbreak, which mostly correspond to human isolates. However, many Ebola studies published before the 2014 outbreaks used mainly partial genomes obtained by sequencing gene PCR products (Wittmann et al. 2007). To be as comprehensive as possible in our study, we included a set of 21 full or partial sequences encoding GP deposited by Wittmann et al. (2007). We also included sequences encoding NP, among which were 12 partial genes of viruses isolated from wild apes (i.e. EU051639 and EU051639), each covering about 65% of the complete CDS of the NP gene. After alignment, the complete sequences were shortened so that only matching regions remained. The resulting maximum likelihood trees for GP and NP are available in Fig. S1A and B (Supporting Information). Both trees show branching patterns very similar to the one shown in Fig. 4. The additional NP proteins from Gorilla isolates (collected in Gabon between 2001 and 2005) form a monophyletic group with the other human isolates collected during this period. The inclusion of additional GP sequences results in a more complex picture of molecular evolution of Zaire ebolaviruses. The isolates from animals clustered together with 100% bootstrap support, but show a distinct evolutionary distance unlike the current outbreaks of human isolates. Both the NP and the GP analysis grouped a 2002 outbreak of strain Ilembe in Gabon with a clade of animal isolates from the same country. The Gabon outbreaks fall into two clades; one is placed in such a way that the tree topology agreed with the temporal pattern of Zaire ebolaviruses, but the other clade, including the animal isolates (Gorilla, Chimpanzee) from between 2001 and 2005, is placed between two clades of 2007–08 DRC outbreaks and the current outbreak in West Africa, violating a temporal arrangement with the bootstrap support of 66%. Inclusion of GP coding genes from zoonotic viruses in the analysis might imply a transmission route through a potential connection between the current human infection in Guinea and Sierra Leon and an infection in animals in Gabon more than 10 years ago, even though at first this seems unlikely because of the geographical distance between the two outbreaks. However, a long-distance migration of infected bats might explain the anomaly.
IS THE CURRENT OUTBREAK STRAIN OF EBOLAVIRUSES ‘RAPIDLY EVOLVING’?
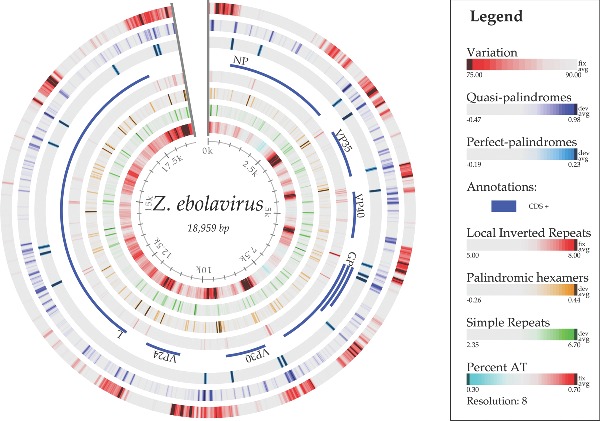
Figure 5 maps the sequence conservation along the chromosome of one reference ebolavirus genome, comparing it to all of the other genomes sequenced to date. As can be seen from the outer ring, the intergenic regions and parts of the GP encoding region have much less conservation (that is, a higher mutational frequency) than the regions encoding the internal viroid proteins. This finding is consistent with the structurally constrained relationship among proteins that are integrally involved in fitting and working functionally together, and it predicts this patchwork of mutability. The membrane-protruding glycoprotein GP is less constrained, and there is even a potential advantage to external antigen/epitope variability for escaping host defenses and gaining new cellular receptor tropisms, allowing better adaptations to new hosts.
Figure 5.

Atlas of the genome of ebolavirus KJ660347, showing, from the outer ring inwards, variations within 84 other ebolavirus genomes, structural cruciforms and palindromes (van Noort et al. 2003), the coding sequences, local inverted repeats, palindromic hexamers, simple repeats and AT content. The conservation percentage (%) is defined as the number of genomes with the same letter on a multiple sequence alignment normalized to range from 0 to 100% for each site along the chromosome of Ebola KJ660347.
It has been suggested (Wittmann et al. 2007) that RNA negative strand viruses may undergo recombination of genetic material, noting that contradictory evolutionary tree relationships among the strains derived from specific sites in ebolavirus proteins can show different relationships. We examined this hypothesis, focusing on a region of surface protein GP, which is known to evolve more rapidly than the other proteins. Figure 6 indicates, in the context of a multiple sequence alignment, that a mosaic pattern exists in GP—that is, sequences in different regions of the protein cluster differently than the full-length protein. The block surrounding the GP furin cleavage site highlights the similarity of this region in TAFV GP to that of the Zaire ebolavirus, while the region from residue 380–425 shows a much higher similarity to the SUDV. The general mosaic structure of this alignment is not consistent with a model in which evolution is happening with equal frequency along the length of the protein. Different parts of the protein are evolving at divergent rates—perhaps due to the selective pressure variability along the protein sequence—which in some cases could provide selective immune system advantage.
Figure 6.

Multiple sequence alignment of a portion of the ebolavirus glycoprotein (GP) from four species of ebolavirus genomes, showing identities between the Taï Forest genome (gi|208436395) and three others (numbered using the Zaire ebolavirus genome of gi|208436395). Identities between Taï Forest and others are shaded in gray. At each position of the alignment, the genome with the highest identity to Taï Forest GP is shown above the alignment by color and the first letter of the genome type: Z = Zaire, gi|667853009 (green), S = Sudan, gi|165940954 (cyan), and R = Reston, gi|253317719 (yellow). The highest similarity at each position was determined by the largest number of identities in a five-residue window centered at each location, with dashes indicating a tie or an undetermined result. Dashes between blocks of the same letter are colored by the surrounding color.
Based on palindrome predictions (van Noort et al. 2003), there are many loci on the chromosome that contain quasi- and perfect palindromes, as shown in Fig. 5. Some of these are located at the beginning and end of genes. Palindromes can form hairpin loop structures in the RNA transcript that might be similar to the initiation step of microRNA (miRNA) biogenesis found in the host. The virus hairpin transcript might be cleaved off to become pre-miRNA and eventually become mature miRNA via the host Drosha and Dicer proteins, respectively. The mature virus miRNA could thus play an inhibition role on expression of host genes.
Indeed, Liang et al. (2014) have recently identified two pre-miRNAs derived from the EBOV genome and conserved among different EBOV strains. These pre-miRNAs were processed through the host's cellular Dicer processing machinery into mature miRNAs. Liang et al. have also predicted potential target genes for the regulatory function of these miRNAs and their possible immunomodulatory functions in EVD. EBOV may produce these miRNAs in abundance early in an infection, which could serve as non-invasive early biomarkers for the diagnosis and prognosis of EBOV infection and as therapeutic targets for Ebola viral infection treatments.
From the host side of miRNA involvement in viral pathogenicity, Sheng et al. (2014) recently showed that human umbilical vein endothelial cells that overexpress EBOV GP undergo cell detachment and rounding; GP expression caused at least 18 different host cell miRNAs to be differentially expressed. Knock-down experiments with specific miRNA inhibitors showed that three of the induced host miRNAs were essential for mediating the EBOV GP-mediated cell damage.
HOW SIMILAR ARE PHYLOGENETIC TREES CONSTRUCTED FROM DIFFERENT FEATURES (DNAs, PROTEINS) OF EBOLAVIRUSES?
The DNA sequence variations observed in Fig. 5 indicate different degrees of variation at different loci of the EBOV chromosome. We investigated these differences by comparing topology of phylogenetic trees constructed from different features. A minimum dataset consisting of 53 non-redundant Ebola genomes was used for the tree comparison. We constructed maximum likelihood trees based on whole genomes, coding genes, intergenic regions and individual proteins, which, in total, led to 23 different alignment-based trees. The substitution models identified as the best model for each feature are listed in Table 1 in the Experimental Procedures section. We constructed three more trees by applying BIONJ (Gascuel 1997) to distance matrices of whole genomes generated by alignment-free approaches—the FFP method with K = 11, Repeating Pattern Distance (RPD) (described in Experimental Procedures section) and Normalized Compression Distance (NCD) (Rosa et al. 2008; Ito, Zeugmann and Zhan 2010). Figure 7 is an image plot of branch score distances (Kuhner and Felsenstein 1994) between the various trees, where the branch score distance is defined as the sum of the squares of the differences between each branch's lengths in the two trees, if it is found in both trees. If a branch is found only in one tree, then a branch of length 0 is considered to exist in the other tree. The NCD approach resulted in the tree topology most distant from maximum likelihood trees. Except for the intergenic region between VP35 and VP40, all intergenic regions showed tree topology relatively different from trees of coding genes and proteins. Again, the glycoprotein tree showed the highest variability among individual protein trees. The glycoprotein nucleotide tree and GP coding sequence tree best captured the topology of an alignment-based whole genome tree, implying that GP may be indicative of molecular evolution of ebolavirus genomes.
Table 1.
A summary of substitution models used for the ebolavirus trees comparison in Fig. 7.
| Feature | Substitution | Feature | Substitution |
|---|---|---|---|
| Whole genome | GTR + I + G | VP30 protein | JTT + G |
| NP coding gene | GTR + I + G | VP24 protein | JTT + G |
| VP35 coding gene | HKY + I + G | L protein | FLU + I + G + F |
| VP40 coding gene | HKY + G | Intergenic: - NP | HKY + G |
| GP coding gene | GTR + I + G | Intergenic: NP-VP35 | HKY + I |
| VP30 coding gene | HKY + I + G | Intergenic: VP35-VP40 | HKY + I |
| VP24 coding gene | GTR + G | Intergenic: VP40-GP | HKY + I |
| L coding gene | GTR + I + G | Intergenic: GP-VP30 | HKY + G |
| NP protein | JTT + G | Intergenic: VP30-VP24 | HKY + G |
| VP35 protein | JTT + G | Intergenic: VP24-L | HKY + G |
| VP40 protein | JTT + G | Intergenic: L- | HKY + G |
| GP protein | FLU + G |
Figure 7.
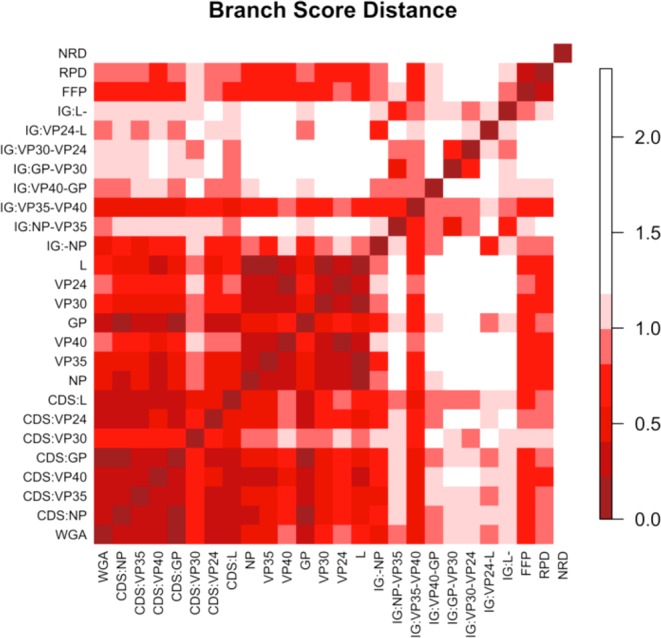
Ebolavirus trees comparison. This is an image plot of branch score distances between alignment-based trees constructed by different features including whole genome alignment (WGA), coding gene sequences (CDS), intergenic (IG) sequences, protein sequences and three alignment-free-based whole genome trees by FFP, NRD and RPD.
HOW CAN BETTER VACCINES BE DEVELOPED FOR EVD?
The virulence of Ebola is extreme, at the heart of which is a multi-layered evasion of the host's immune system (Wong et al. 2014). The evasion strategies include the downregulation of type I interferon production, the masking of viral epitopes and the abundant expression of secreted sGP, which overloads the host's adaptive humoral immunity (Wang, Liu and Dai 2014). By these mechanisms, the specific and non-specific host antiviral immune responses that normally limit viral replication are ineffective, so that a rapid increase in viral load can quickly result in death. Four of the proteins EBOV produces have been shown to interact with the host in ways that counteract the host's immune response. VP35 is capable of capping dsRNA and interacts with IRF7 (interferon regulatory factor 7) to prevent detection of the virus by immune cells; VP24 interferes with the production of interferon (IFN) and with IFN signaling in infected cells; GP1,2 protein has shown anti-tetherin activity and the ability to hide cell-surface proteins (Audet and Kobinger 2014). The main role of soluble sGP is still unclear, but it is reportedly capable of subverting the anti-GP1,2 antibody response. In addition, as pointed out before, pre-miRNAs can have immunomodulatory functions.
The best long-term approach for dealing with EVD is the development of reliable and broad vaccines that increase the proportion of resistant individuals in key populations at risk, either to prevent future outbreaks or to ensure they are more easily containable. However, little is currently known about human antibody targets for Zaire ebolaviruses and about whether the observed ongoing genetic drift will affect their roles as immunogenic targets. The goal of recent research efforts is to produce more effective vaccines, those that produce copious amounts of potent virus-inactivating antibodies and a persistent long-term broad-spectrum immunity to the current strain of species Zaire ebolavirus. Ideally, this immunity would extend to other ebolaviruses. To help achieve this goal, we need to be able to better predict viral epitopes to use as vaccine materials that will result in the production of desirable human immune responses.
Microarray analysis recently identified antigens for NP, VP40 and GP from isolates that represented the six known species of filoviruses (Kamata et al. 2014). This work was extended with antibody responses in rhesus monkeys vaccinated with virus-like particles bearing these epitopes, followed by a challenge with MARV or EBOV. These findings showed an increase in immunoglobulin G (IgG) as a result of the immunization, though the antibody response that resulted from a rechallenge was far more extensive. Moreover, cross-reactivity was observed between antibodies raised against NP and VP40 of the five Ebolavirus species, but antibodies against GP were strain specific (Kamata et al. 2014).
Recovery of infected EVD patients is associated with an efficient EBOV-specific humoral IgG response, whereas fatal outcome is apparently the result of insufficient immune responses. Becquart et al. (2014) identified specific B-cell epitopes in EVD patient sera for the four EBOV proteins GP, NP, VP40 and VP35. They also tested EBOV IgG-positive sera from asymptomatic individuals (EBOV sero-positive with no memory of an infection) and from symptomatic survivors, comparing sera from the early humoral response (7 days after the end of symptoms) with the late memory phase (7–12 years post-infection). Surprisingly, they found that serum from asymptomatic individuals more strongly reacted to VP40 than to GP, NP or VP35, suggesting that a vaccine made with VP40 might be more protective than vaccines from the other viral proteins. In support of this hypothesis, anti-EBOV IgG from these asymptomatic patients targeted three regions of VP40 that were reported to play a crucial role in virus assembly and budding. In contrast, serum from the early humoral response of survivors of three EBOV outbreaks reacted mainly with GP peptides. These observations, and the fact that GP appears to evolve faster than the other genes of EBOV, suggest that GP may not be the only suitable target for vaccine development; among the other EBOV proteins, at least VP40 should be considered a potential vaccine candidate.
A closer look on GP epitopes: humoral responses and experimentally verified B-cell epitopes
The surface glycoprotein GP1,2 is usually considered the target of choice for antibody production and vaccine research, since it is the only viral protein exposed on the surface of the virion; GP1,2 is also the most immunogenic of the EBOV antigens, as determined by DNA vaccine studies that expressed various EBOV antigens (de La Vega et al. 2015). Recombinant GP1,2 as a vaccine component induces a broad immune response, both at the cellular and humoral level. However, heavy glycosylation of GP1,2 shields the virus through epitope masking (reviewed by de La Vega et al. 2015), thus counteracting the host immune response. Soluble sGP, which is more abundant than GP, forms a disulfide-linked 110 kDa homodimer, whose role was recently reviewed (de La Vega et al. 2015). sGP is antigenetic, as antibodies reacting with sGP have been observed in the sera of human EVD survivors; moreover, sGP was able to inhibit the virus-specific neutralizing activity of some GP antisera (de La Vega et al. 2015). Indeed, when vaccines based on both GP1,2 and sGP were tested in a mouse model, the elicited antibodies cross-reacted between the two proteins, which is not surprising since they share a common N-terminus. It has been hypothesized that sGP plays a role in controlling host humoral immune responses by adsorbing antibodies elicited against GP1,2 (de La Vega et al. 2015).
ZMAb, the highly experimental drug, which has been used to treat several care workers stricken with EVD, consists of three mouse antibodies, 1H3, 2G4 and 4G7, whose GP epitope-binding properties have been reported by Audet et al. (2014). The antibodies 2G4 and 4G7 were shown to cross-inhibit each other in vitro and probably recognize the same epitope, as they both selected for the same escape mutation at amino acid position 508 of GP. The 1H3 antibody selected an escape mutant at amino acid 273 (Audet et al. 2014).
Serological evidence from surviving patients reveals information on B-cell responses to epitopes that are active during infection. So far, the sparse available information indicates that neutralizing antibodies found in the blood of surviving victims react to four proteins: NP, VP30, VP40 and GP (Sobarzo et al. 2013). We focus here on GP, since vaccine development has tended to concentrate on this protein. To investigate the potential for vaccine therapeutic approaches, we combined experimentally mapped B-cell epitope data for GP from a number of experimental studies (Wilson et al. 2000; Shahhosseini et al. 2007; Takada et al. 2007; Lee et al. 2008a,b; Bale et al. 2012; Becquart et al. 2014; Lennemann et al. 2014; Qiu et al. 2014; Wang, Liu and Dai 2014). Certain sites, such as in the center of the mucin domain near amino acid 400 and around amino acids 480–500, just prior to the furin site, are identified in four or more independent studies and are also consistent with sera-recognized epitopes in Ebola patients.
Figure 9 shows a diversity of epitope locations identified in various studies, perhaps reflecting differences in the structure and sequence of GP from various Ebola strains, as well as differences in the hosts in which the studies were done. In most cases, animal studies were used, but some data from human patients are included. The most consistently recognized region of GP is in the middle of the protein, around residues 390–412, where four experimental studies find epitopes and two Ebola patients had mapped epitopes. These amino acid residues lie in uncleaved GP or in GP1 in the center of the mucin domain. This region is disordered in crystal structures but is positioned where it is accessible outside the Ebola membrane (Lee et al. 2008a). Additionally, a region from about residues 480–500 seems to be strongly recognized in several studies. This region is just prior to the furin cleavage site that cleaves the long GP precursor into GP1 and GP2. Portions of the GP glycan cap region (from about residue 227–313) are recognized in only one study, but was found in two patients (Becquart et al. 2014).
Figure 9.

Position of 10 predicted MHC class I (red) and 10 class II (blue) epitopes in six ebolavirus proteins, and the allelic variation detected in the 53 non-redundant proteomes. Sequence variation that destroys a predicted epitope is shown in red, while all variants shown in green were equally strong or only marginally less strong, compared to the sequences shown in black. Gray blocks above the proteins indicate the position of experimentally proven B-cell epitopes, after Becquart et al. (2014).
Predicted T-cell epitopes
In addition to humoral immunity, mediated by B cells, another essential arm of the host adaptive immune response is cellular immunity, mediated by T cells. T cells interact with small peptide fragments displayed on the surface of cells in complex with Major Histocompatibility Complexes, MHC (human leukocyte antigens, HLA in humans). Generally speaking, two main types of T cells exist; cytotoxic T cells (CTL) and helper T cells. Likewise, two types of MHC molecules exists; class I that presents peptides to CTLs, and class II that presents peptides to helper T cells. MHC class I molecules present peptides derived from inside the cell, and MHC class II molecules present peptides derived from endocytosed proteins. As the target of both types of T cells is a peptide bound to an MHC molecule, binding to MHC is a necessary prerequisite for a peptide to induce a T-cell response. While factors other than MHC binding can dictate whether a peptide will end up being an epitope (i.e. be able to induce a T-cell response), several studies have demonstrated that MHC binding is the single most selective step in the T-cell antigen presentation process (Yewdell and Bennink 1999; Stranzl et al. 2010; Trolle and Nielsen 2014).
T-cell epitopes were predicted for both MHC class I and MHC class II MHC molecules for the ebolavirus proteome using the software described in the Experimental Procedures section. Since the immune response of the human population in West Africa and in the United States (US) is likely/known to differ (caused by variations in the MHC), this was accounted for by factoring in allele frequency data for Caucasians, Hispanics, African Americans, Asians and Native Americans for US population. A total of 3000 potential epitopes were predicted for class I binding and 4500 for class II. From these epitopes, selections were made for each population (US and West Africa), 10 peptides with strong predicted MHC class I binding properties, and 10 peptides with strong predicted MHC class II binding properties, that in combination could be expected to produce an immune response in both populations under study, covering all the ebolavirus variants included in this analysis. The selection for class I contained seven strong epitopes in common between the two populations (four in protein L, and one each in GP, NP and VP40), and three that differed in each of these human populations. Of the 13 different epitopes selected for class I, 7 were found in the L protein and none were detected for VP24. For MHC class II peptide selection, there were six predicted strong epitopes in common between the West African and US populations (four in NP, and one each in VP24 and GP). Two more epitopes (one in protein L and one in GP/sGP) were shifted by one amino acid only between the West African and US populations; these pairs were also interpreted to be conserved epitopes for the two populations. Population-specific epitopes were identified for VP24 and L (West African only) and for VP40 and GP (US only). That produced 12 different class II epitopes, in which NP was slightly overrepresented (4 out of 12 epitopes). The data are summarized in Tables S1 and S2 (Supporting Information).
Next, we mapped the epitopes along the respective ebolavirus protein sequences in Fig. 8. This revealed that for NP three of the four class II epitopes that were predicted to be recognized in the combined West African and US populations were overlapping. This region positioned at amino acid 199–217 thus seems to bear a strong signal for MHC class II, in both populations under study. This same region was also recognized as an MHC class I epitope (for US population only).
Figure 8.

Experimentally verified B-cell epitopes for Ebola GP protein based on selected studies (Wilson et al. 2000; Shahhosseini et al. 2007; Takada et al. 2007; Lee et al. 2008a,b; Bale et al. 2012; Becquart et al. 2014; Qiu et al. 2014; Wang et al. 2014), represented by colored bars in the GP schematic. Glycan sites are also indicated. Some mapped data, where the epitope was not well localized within 50 amino acids, were omitted.
Figure 8 also displays the observed variability in protein alleles for the location of the epitopes. Our model predicts that all the observed variant epitopes within NP maintain a similar HLA-binding profile (data not shown). A similar mapping experiment identified that the three predicted epitopes listed for VP40 (two for class I and one for class II) were also overlapping, at position 153–180. However, at least one variant of ebolavirus VP40 exists with an alternative amino acid sequence that abolishes binding to HLA (Fig. 9B). The three epitopes identified for sGP (secreted spike glycoprotein) are distributed at three different locations, and although none of them are 100% conserved in the GP protein sequences of various ebolavirus strains, all analyzed variants share a similar HLA-binding profile (data not shown). For protein VP24, two epitopes are separated by 20 amino acids, while for protein L, most class I epitopes are located between amino acid 526 and 563. This region also contains an overlapping epitope for MHC class I and class II (Fig. 9E). For the predicted epitope in L located closest to the carboxyl end of the protein, an alternative protein sequence exists that was predicted to have lost its HLA-binding potential (data not shown). Finally, two epitopes were mapped next to each other for VP35, of which the MHC class I epitope is completely conserved, while variations in class II epitope maintain a similar HLA class II binding profile (data not shown).
Theoretical prediction of epitopes that could serve as vaccine candidates should take available information on sequence variation between virus strains into account, as well as variation in population responses. Our predictions identified differences between the US and West African populations, but also found epitopes that are strongly conserved and recognized in both populations, which could present optimal vaccine candidates. Experimental evidence for T-cell epitopes that are actually recognized by the host is only available from mouse models. Three predicted MHC class I epitopes for GP from ebolaviruses (two for SUDV and two for Zaire ebolavirus) were able to induce strong IFN-γ responses in mice (Wu et al. 2012). A vaccine trial in mice with an epitope derived from NP, expressed in murine cytomegalovirus, resulted in long-term expression of CD8+ T cells; the epitope in question was located between amino acid 43 and 54; that region was not flagged in our analysis (Tsuda et al. 2011).
CONCLUSIONS
Ebolavirus genomes provide clues as to the relationship with each other and reflect information that can be used to trace back their likely geographical and temporal locations. In our opinion, one of the most reliable methods for detection of Ebolavirus is from its genome sequence. Novel sequencing methods can detect the presence of ebolavirus within a few hours, which will allow for rapid characterization of the virus. Further, with hundreds of genomes, it is possible to measure the sequence variability, knowledge useful in the development of approaches to help prevent the spread and recurrence of the outbreak.
EXPERIMENTAL PROCEDURES
Datasets
Ebolavirus genomes
On 28 October 2014, we downloaded all ebolavirus genomes available from GenBank excluding all sequences with ‘from Patent’ in the description and also removing all sequences less than 200 bases in length. This resulted in 149 complete genomes which included genomes representing the May 2014 outbreaks in Sierra Leone (Gire et al. 2014) and genomes representing the July 2014 outbreak in DRC (Maganga et al. 2014). Based on a criterion of 100% sequence identify for matches that span a pair of genome sequences to the extent that all coding sequences in their entirety were included in the match using NUCmer program from MUMmer (Kurtz et al. 2004), we reduced a dataset of 149 ebolavirus genomes down to 84 ebolavirus genomes (60 Zaire, 10 Sudan, 8 Reston, 5 Bundibugyo, 1 Taï Forest), We modified two genome sequences (KM034563, L11365) to estimate missing segments using nearest neighbor sequences. The GenBank sequence KM034563 contains four runs of N's of length 94, 45, 43 and 51. However, the four regions containing these runs were so highly conserved (100% identity) in closely related species that we felt justified in replacing the N's with the most likely bases. L11365, a Zaire sequence from 1976, is complete up through the CDS for the first 54 amino acids of the final L-protein, and differs from its nearest neighbor at 10 known locations, 4 SNPs and 6 indels. Because the L-protein was 100% identical elsewhere in L11365's four nearest neighbors, we added this sequence into the L11365.
Ebolavirus proteomes
We reduced the 84 non-redundant Ebola genomes to 53 non-redundant Ebola proteomes (34 Zaire, 7 Sudan, 8 Reston, 3 Bundibugyo, 1 Taï Forest) based on the 100% amino acid sequence identity excluding the current DRC outbreak, KM519951 due to the lack of annotation of its mRNA in GenBank. Note that the sequences filtered out had the same metadata (isolate, collection date and country) as the one kept when they belonged to the same cluster by 100% sequence identity. The resulting Ebola datasets represented outbreaks in nine different countries, Guinea (GIN), Sierra Leone (SLE) and Côte d'Ivoire (COT) in West Africa, Democratic Republic of Congo (DRC, formerly Zaire), Gabon (GAB), Uganda (UGA), Sudan in Central Africa, Philippines (PHI) and United States (USA) between 1976 and 2014.
Marburgvirus genomes and proteomes
On 28 October 2014, we downloaded all marburgvirus genomes available from GenBank. This resulted in a dataset of 80 complete genomes, 73 of which had CDS features specified from which complete proteomes could be determined.
Rooting phylogenetic trees
It is well known that rooting a tree of Zaire ebolaviruses using any distantly related ebolavirus is problematic in the sense that the branching pattern of Zaire ebolaviruses differs depending on the deriving features; for example, using coding gene sequences versus intergenic sequences changes the molecular evolution story of the current outbreaks (Dudas and Rambaut 2014). With different datasets of Zaire ebolaviruses, the root-to-tip regression analysis showed much better correlation between genetic divergence and isolation date when the trees were rooted with the 1976 outbreak (Carroll et al. 2013; Dudas and Rambaut 2014; Gire et al. 2014). Thus, we rooted the Zaire ebolavirus tree in Fig. 4 and the two trees in Fig. S1 (Supporting Information) to the clade of the earliest recorded outbreak in 1976.
Phylogeny construction
We investigated the molecular evolution of ebolaviruses based on many different features, from complete genomes to individual proteins. The substitution models were identified based on Bayesian Information Criterion (BIC) using jModelTest (Darriba et al. 2012) for DNA sequences and ProtTest (Darriba et al. 2011) for amino acid sequences. Table 1 lists the substitution models used for the ebolavirus trees comparison presented in Fig. 4. The GP and L Protein identified FLU models for influenza proteins as the best model.
Repeating pattern distance
The RPD method examines the distances between short sequence motifs such as three or four nucleotide-long sequences (e.g. GAC or GTAC). If several distances between such motifs and their order are shared between organisms, this is used to provide evidence of an evolutionary relationship and the quantitative similarity can be used to score evolutionary distance.
Epitope selection
A set of epitopes with optimal coverage of both the different Ebola strains and the HLA alleles prevalent in the populations of interest (US and West Africa) was selected as follows.
HLA allele selection
HLA allele frequency data were obtained from the allele frequency net database (Gonzalez-Galarza et al. 2011). A set of relevant HLA alleles specific for the US and West Africa populations was chosen such that there was 95% population coverage at each of the three HLA-A, HLA-B and HLA-DRB1 loci. For the US, this led to the selection of 27 HLA-A, 58 HLA-B and 33 HLA-DRB1 alleles. For West Africa, the numbers were 19, 31 and 20 (these allele sets are available in Table S2, Supporting Information). For the HLA-DP and HLA-DQ loci, the allele combinations recommended by the Immune Epitope Database were used (Kim et al. 2012).
T-cell epitope selection
T-cell epitopes were predicted in the 53 ebolavirus genomes that were non-redundant at the protein level. HLA alleles were used as targets for the epitope predictions. HLA class I-restricted 9-mer epitopes were predicted using NetMHCcons1.1 (Karosiene et al. 2012) and NetChop3.1 C-terminal peptide processing algorithm (Nielsen et al. 2005). HLA class II-restricted 15-mer epitopes were predicted using NetMHCIIpan3.0 (Karosiene et al. 2013). Epitope selection used both weak and strong binding thresholds, default settings for HLA-I and proteasomal cleavage and IEDB recommendations for HLA-II:
Weak—HLA-I: Peptides with a C-terminal cleavage score greater than 0.5 and either a predicted binding affinity of less than 500 nM or a rank score ≤2%. HLA-II: Peptides with rank scores ≤ 10%.
Strong—HLA-I: Peptides with a predicted binding affinity of less than 50 nM or a rank score ≤0.5%. HLA-II: Peptides with rank scores ≤2%.
The strong threshold produces the best binders for vaccine candidates. However, when studying the natural immune response, it is well recognized that a strong threshold is often overly restrictive and filters out the virus-inactivating epitopes one may be looking for (Stranzl et al. 2010; Paul et al. 2013). Tables of the top predicted epitopes are presented in the Supplemental Material.
Candidate epitope selection
Each amino acid position in the ebolavirus proteome was given an epitope score based on the number of epitopes overlapping at that given position. The scores were weighted by allele frequency, giving epitopes bound by prevalent MHC molecules a larger contribution to the final epitope score. Separate scores were calculated for MHC class I and class II.
Final epitope selection
Final epitope selections were based on calculated population coverage for each candidate epitope using the PopCover method (Buggert et al. 2012), as described by Schubert, Lund and Nielsen (2013). Epitope tables can be found in the Table S1 (Supporting Information).
Acknowledgments
This work would not be possible without the thousands of sequenced viral genomes deposited in GenBank and made publicly available.
SUPPLEMENTARY DATA
FUNDING
Funding was provided by internal funds of Oak Ridge National Laboratory (ORNL), managed by UT-Battelle, LLC for the U.S. Department of Energy under Contract No. DE-AC05–00OR22725. The Open Access funding for this paper was provided by the Oak Ridge National Laboratory.
Conflict of interest. None declared.
REFERENCES
- Adu-Gyamfi E, Soni SP, Jee CS, et al. A loop region in the N-terminal domain of Ebola virus VP40 is important in viral assembly, budding, and egress. Viruses. 2014;6:3837–54. doi: 10.3390/v6103837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet J, Kobinger G. Immune evasion in Ebolavirus infections. Viral Immunol. 2014;28:10–8. doi: 10.1089/vim.2014.0066. [DOI] [PubMed] [Google Scholar]
- Audet J, Wong G, Wang H, et al. Molecular characterization of the monoclonal antibodies composing ZMAb: a protective cocktail against Ebola virus. Sci Rep. 2014;4:6881. doi: 10.1038/srep06881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S, Dias JM, Fusco ML, et al. Structural basis for differential neutralization of Ebolaviruses. Viruses. 2012;4:447–70. doi: 10.3390/v4040447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becquart P, Mahlakoiv T, Nkoghe D, et al. Identification of continuous human B-cell epitopes in the VP35, VP40, nucleoprotein and glycoprotein of Ebola virus. PloS One. 2014;9:e96360. doi: 10.1371/journal.pone.0096360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyi VA, Levine AJ, Skalka AM. Unexpected inheritance: multiple integrations of ancient bornavirus and Ebolavirus/Marburgvirus sequences in vertebrate genomes. PloS Pathog. 2010;6:e1001030. doi: 10.1371/journal.ppat.1001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggert M, Norström MM, Czarnecki C, et al. Characterization of HIV-specific CD4+ T cell responses against peptides selected with broad population and pathogen coverage. PLoS One. 2012;7:e39874. doi: 10.1371/journal.pone.0039874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SA, Towner JS, Sealy TK, et al. Molecular evolution of viruses of the family filoviridae based on 97 whole-genome sequences. J Virol. 2013;87:2608–16. doi: 10.1128/JVI.03118-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC Outbreaks Chronology. Ebola Virus Disease. Centers for Disease Control and Prevention; 2015. http://www.cdc.gov/vhf/ebola/outbreaks/history/chronology.html (17 June 2015, date last accessed) [Google Scholar]
- Corum J. The New York Times. 2014. A history of Ebola in 24 outbreaks. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, et al. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, et al. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Vega M-A, Wong G, Kobinger GP, et al. The multiple roles of sGP in Ebola pathogenesis. Viral Immunol. 2015;28:3–9. doi: 10.1089/vim.2014.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas G, Rambaut A. Phylogenetic analysis of Guinea 2014 EBOV Ebolavirus outbreak. PLoS Curr. 2014;6 doi: 10.1371/currents.outbreaks.84eefe5ce43ec9dc0bf0670f7b8b417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–95. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- Gebreyes W, Dupouy-Camet J, Newport M, et al. The global one health paradigm: challenges and opportunities for tackling infectious diseases at the human, animal, and environment interface in low-resource settings. PLoS Negl Trop D. 2014;8:e3257. doi: 10.1371/journal.pntd.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–72. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Galarza FF, Christmas S, Middleton D, et al. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011;39:D913–9. doi: 10.1093/nar/gkq1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hunter S, Apweiler R, Attwood TK, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Zeugmann T, Zhan JJ. Clustering the normalized compression distance for influenza virus data. In: Elomaa T, Mannila H, Orponen P, editors. Algorithms and Applications. Berlin: Springer; 2010. pp. 130–46. [Google Scholar]
- Jones P, Binns D, Chang HY, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun SR, Sims GE, Wu GHA, et al. Whole-proteome phylogeny of prokaryotes by feature frequency profiles: an alignment-free method with optimal feature resolution. P Natl Acad Sci USA. 2010;107:133–8. doi: 10.1073/pnas.0913033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T, Natesan M, Warfield K, et al. Determination of specific antibody responses to the six species of ebola and marburg viruses by multiplexed protein microarrays. Clin Vaccine immunol. 2014;21:1605–12. doi: 10.1128/CVI.00484-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karosiene E, Lundegaard C, Lund O, et al. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64:177–86. doi: 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- Karosiene E, Rasmussen M, Blicher T, et al. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65:711–24. doi: 10.1007/s00251-013-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Ponomarenko J, Zhu ZY, et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40:W525–30. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn JH, Andersen KG, Baize S, et al. Nomenclature- and database-compatible names for the two Ebola virus variants that emerged in Guinea and the Democratic Republic of the Congo in 2014. Viruses. 2014;6:4760–99. doi: 10.3390/v6114760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner MK, Felsenstein J. Simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol Biol Evol. 1994;11:459–68. doi: 10.1093/oxfordjournals.molbev.a040126. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, et al. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008a;454:177–82. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Kuehne A, Abelson DM, et al. Complex of a protective antibody with its Ebola virus GP peptide epitope: unusual features of a V lambda(x) light chain. J Mol Biol. 2008b;375:202–16. doi: 10.1016/j.jmb.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009;4:621–35. doi: 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennemann NJ, Rhein BA, Ndungo E, et al. Comprehensive functional analysis of N-Linked glycans on Ebola virus GP1. MBio. 2014;5:e00862–13. doi: 10.1128/mBio.00862-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang HW, Zhou Z, Zhang SY, et al. Identification of Ebola virus microRNAs and their putative pathological function. Sci China Life Sci. 2014;57:973–81. doi: 10.1007/s11427-014-4759-2. [DOI] [PubMed] [Google Scholar]
- Maganga GD, Kapetshi J, Berthet N, et al. Ebola virus disease in the Democratic Republic of Congo. New Engl J Med. 2014;371:2083–91. doi: 10.1056/NEJMoa1411099. [DOI] [PubMed] [Google Scholar]
- Mühlberger E. Filovirus replication and transcription. Future Virol. 2007;2:205–15. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylne A, Brady OJ, Huang Z, et al. A comprehensive database of the geographic spread of past human Ebola outbreaks. Sci Data. 2014;1:140042. doi: 10.1038/sdata.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negredo A, Palacios G, Vazquez-Moron S, et al. Discovery of an ebolavirus-like filovirus in Europe. PloS Pathog. 2011;7:e1002304. doi: 10.1371/journal.ppat.1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Lund O, et al. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- Nkoghe D, Formenty P, Leroy EM, et al. Multiple Ebola virus haemorrhagic fever outbreaks in Gabon, from October 2001 to April 2002. Bull Soc Pathol Exot. 2005;98:224–9. [PubMed] [Google Scholar]
- Olival KJ, Hayman DT. Filoviruses in bats: current knowledge and future directions. Viruses. 2014;6:1759–88. doi: 10.3390/v6041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn S, van der Groen G, Jacob W, et al. Isolation of Marburg-like virus from a case of haemorrhagic fever in Zaire. Lancet. 1977;1:573–4. doi: 10.1016/s0140-6736(77)92002-5. [DOI] [PubMed] [Google Scholar]
- Paul S, Weiskopf D, Angelo MA, et al. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. Immunol. 2013;191:5831–9. doi: 10.4049/jimmunol.1302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowright RK, Eby P, Hudson P, et al. Ecological dynamics of emerging bat virus spillover. Proc Biol Sci. 2015;282:20142124. doi: 10.1098/rspb.2014.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle CR. Order mononegavirales. In: Fauquet CM, Mayo MA, Maniloff J, et al., editors. Virus Taxonomy: Eighth Report of the International Comittee on Taxonomy of Viruse. San Diego: Elsevier Academic Press; 2005. pp. 609–66. [Google Scholar]
- Qiu XG, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514:47–53. doi: 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa M, Rizzo R, Urso A, et al. Comparison of genomic sequences clustering using normalized compression distance and evolutionary distance. In: Ignac Lovrek, Robert J. Howlett, Lakhmi C. Jain., editors. Knowledge-Based Intelligent Information and Engineering Systems. Berlin: Springer; 2008. pp. 740–6. [Google Scholar]
- Sanchez A, Trappier SG, Mahy BWJ, et al. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. P Natl Acad Sci USA. 1996;93:3602–7. doi: 10.1073/pnas.93.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert B, Lund O, Nielsen M. Evaluation of peptide selection approaches for epitope-based vaccine design. Tissue Antigens. 2013;82:243–51. doi: 10.1111/tan.12199. [DOI] [PubMed] [Google Scholar]
- Shabman RS, Jabado OJ, Mire CE, et al. Deep sequencing identifies noncanonical editing of Ebola and Marburg virus RNAs in infected cells. MBio. 2014;5:e02011. doi: 10.1128/mBio.02011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahhosseini S, Das D, Qiu X, et al. Production and characterization of monoclonal antibodies against different epitopes of Ebola virus antigens. J Virol Methods. 2007;143:29–37. doi: 10.1016/j.jviromet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Sheng MM, Zhong Y, Chen Y, et al. Hsa-miR-1246, hsa-miR-320a and hsa-miR-196b-5p inhibitors can reduce the cytotoxicity of Ebola virus glycoprotein in vitro. Sci China Life Sci. 2014;57:959–72. doi: 10.1007/s11427-014-4742-y. [DOI] [PubMed] [Google Scholar]
- Siegert R, Shu HL, Slenczka W, et al. Zur Ätiologie einer unbekannten, von Affen ausgegangenen menschlichen Infektionskrankheit. Deut Med Wochenschr. 1967;92:2341–3. doi: 10.1055/s-0028-1106144. [DOI] [PubMed] [Google Scholar]
- Sobarzo A, Groseth A, Dolnik O, et al. Profile and persistence of the virus-specific neutralizing humoral immune response in human survivors of Sudan Ebolavirus (Gulu) J Infect Dis. 2013;208:299–309. doi: 10.1093/infdis/jit162. [DOI] [PubMed] [Google Scholar]
- Soni SP, Adu-Gyamfi E, Yong SS, et al. The Ebola virus matrix protein deeply penetrates the plasma membrane: an important step in viral egress. Biophys J. 2013;104:1940–9. doi: 10.1016/j.bpj.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranzl T, Larsen MV, Lundegaard C, et al. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357–68. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A, Ebihara H, Feldmann H, et al. Epitopes required for antibody-dependent enhancement of Ebola virus infection. J Infect Dis. 2007;196:S347–56. doi: 10.1086/520581. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Ballinger MJ, Zhan JJ, et al. Evidence that ebolaviruses and cuevaviruses have been diverging from marburgviruses since the Miocene. PeerJ. 2014;2:e556. doi: 10.7717/peerj.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolle T, Nielsen M. NetTepi: an integrated method for the prediction of T cell epitopes. Immunogenetics. 2014;66:449–56. doi: 10.1007/s00251-014-0779-0. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Caposio P, Parkins CJ, et al. A replicating cytomegalovirus-based vaccine encoding a single Ebola virus nucleoprotein CTL epitope confers protection against Ebola virus. Plos Neglect Trop D. 2011;5:e1275. doi: 10.1371/journal.pntd.0001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort V, Worning P, Ussery DW, et al. Strand misalignments lead to quasipalindrome correction. Trends Genet. 2003;19:365–9. doi: 10.1016/s0168-9525(03)00136-7. [DOI] [PubMed] [Google Scholar]
- Volchkov VE, Becker S, Volchkova VA, et al. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology. 1995;214:421–30. doi: 10.1006/viro.1995.0052. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu Z, Dai Q. A highly immunogenic fragment derived from Zaire Ebola virus glycoprotein elicits effective neutralizing antibody. Virus Res. 2014;189:254–61. doi: 10.1016/j.virusres.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Wilson JA, Hevey M, Bakken R, et al. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–6. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- Wittmann TJ, Biek R, Hassanin A, et al. Isolates of Zaire ebolavirus from wild apes reveal genetic lineage and recombinants. P Natl Acad Sci USA. 2007;104:17123–7. doi: 10.1073/pnas.0704076104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G, Kobinger GP, Qiu X. Characterization of host immune responses in Ebola virus infections. Expert Rev Clin Immunol. 2014;10:781–90. doi: 10.1586/1744666X.2014.908705. [DOI] [PubMed] [Google Scholar]
- Wu GA, Jun SR, Sims GE, et al. Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. P Natl Acad Sci USA. 2009;106:12826–31. doi: 10.1073/pnas.0905115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SP, Yu T, Song XH, et al. Prediction and identification of mouse cytotoxic T lymphocyte epitopes in Ebola virus glycoproteins. Virol J. 2012;9:111. doi: 10.1186/1743-422X-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne JW, Wang LF. Bats and viruses: friend or foe? PloS Pathog. 2013;9:e1003651. doi: 10.1371/journal.ppat.1003651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]