

Abstract
Recycling synaptic vesicles (SVs) transit through early endosomal sorting stations, which raises a fundamental question: are SVs sorted toward endolysosomal pathways? Here, we used snapin mutants as tools to assess how endolysosomal sorting and trafficking impact presynaptic activity in wild-type and snapin−/− neurons. Snapin acts as a dynein adaptor that mediates the retrograde transport of late endosomes (LEs) and interacts with dysbindin, a subunit of the endosomal sorting complex BLOC-1. Expressing dynein-binding defective snapin mutants induced SV accumulation at presynaptic terminals, mimicking the snapin−/− phenotype. Conversely, over-expressing snapin reduced SV pool size by enhancing SV trafficking to the endolysosomal pathway. Using a SV-targeted Ca2+ sensor, we demonstrate that snapin–dysbindin interaction regulates SV positional priming through BLOC-1/AP-3-dependent sorting. Our study reveals a bipartite regulation of presynaptic activity by endolysosomal trafficking and sorting: LE transport regulates SV pool size, and BLOC-1/AP-3-dependent sorting fine-tunes the Ca2+ sensitivity of SV release. Therefore, our study provides new mechanistic insights into the maintenance and regulation of SV pool size and synchronized SV fusion through snapin-mediated LE trafficking and endosomal sorting.
Keywords: BLOC-1, dynein motor, dysbindin, endosomal sorting and transport, snapin
Introduction
Synaptic vesicles (SVs) at presynaptic terminals are segregated into several functional pools: a limited recycling pool that undergoes dynamic exo- and endocytosis, and a large resting or reserve pool that is slowly recruited into the recycling pool under sustained synaptic activity (Rizzoli & Betz, 2005). SVs undergo the canonical SV cycle at active zones, where they dock to and fuse with the plasma membrane, before being retrieved for reuse (Kavalali, 2006). Priming SVs for Ca2+-dependent fusion requires a complex assembly of the SNARE membrane fusion machinery with regulatory proteins and the Ca2+ sensor synaptotagmin (Jahn & Scheller, 2006; Chapman, 2008). Proper regulation of SV pool size and Ca2+ sensitivity are critical for maintaining synaptic activity and synchronous neurotransmitter release.
Following endocytosis, recycling SVs can transit through early endosomal (EE) sorting stations (Hoopmann et al, 2010; Uytterhoeven et al, 2011; Korber et al, 2012). EEs mature into late endosomes (LEs), which are retrogradely transported toward the cell body to deliver internalized material to mature lysosomes for degradation. SVs can bud from EEs through an AP-3-dependent process and get back into the SV cycle (Faundez et al, 1998; Kokotos & Cousin, 2015). EEs thus represent crossroads between two pathways: endolysosomal trafficking and degradation (Route 1) and local AP-3-dependent SV sorting and recycling (Route 2). This raises a fundamental question as to how endosomal trafficking and sorting cooperate to maintain presynaptic function. To address this issue, it is crucial to apply molecular tools to dissect endosomal trafficking and sorting pathways at presynaptic terminals. Among the many regulatory components of SV exocytosis, snapin stands out as a unique protein with multivalent roles in intracellular organelle trafficking and sorting. Snapin was first identified as a SNAP25-binding protein enriched in synaptosomes and associated with SVs (Ilardi et al, 1999) and LEs (Cai et al, 2010). Transgene rescue experiments in snapin-deficient cortical neurons revealed a crucial role for snapin in synchronizing SV fusion by enhancing the efficacy of SV priming (Pan et al, 2009). snapin−/− mice display two striking phenotypes: (i) impaired priming of large dense-core vesicles in chromaffin cells (Tian et al, 2005) and (ii) desynchronized neurotransmitter release in neurons (Pan et al, 2009). We recently identified a new role for snapin as a molecular adaptor linking dynein motors to LEs, thus driving endosomal retrograde transport (Cai et al, 2010). Snapin-deficient neurons exhibit defective retrograde transport of LEs from axonal terminals to the soma, where mature acidic lysosomes are relatively enriched (Cai et al, 2010; Lee et al, 2011). Thus, snapin deficiency or disrupting snapin–dynein coupling impairs the delivery of endocytosed material to the endolysosomal system for degradation. This phenotype could be effectively rescued by reintroducing the snapin transgene but not a snapin-L99K mutant defective in dynein binding.
Several groups independently reported an interaction between snapin and dysbindin (Starcevic & Dell’Angelica, 2004; Talbot et al, 2006; Lee et al, 2012), the product of a susceptibility gene (DTNBP1) found among the most common genetic variations associated with schizophrenia (Straub et al, 2002). Similar to snapin deficiency, deletion of DTNBP1 in sandy mice reduces release probability and releasable pool size and slows glutamate release kinetics (Chen et al, 2008). Because the steady-state level of snapin in the hippocampus of sandy mice is significantly reduced (Feng et al, 2008), snapin deficiency might contribute to schizophrenia-like phenotypes. Importantly, dysbindin cooperates with snapin during the homeostatic regulation of neurotransmitter release (Dickman et al, 2012). Both snapin and dysbindin were co-purified as subunits of the biogenesis of lysosome-related organelle complex-1 (BLOC-1) (Starcevic & Dell’Angelica, 2004). BLOC-1 was shown to promote early endosome maturation in yeast (John Peter et al, 2013) and cargo sorting toward lysosome-related organelles such as melanosomes (Di Pietro et al, 2006; Setty et al, 2007). BLOC-1 interacts with the AP-3 complex (Di Pietro et al, 2006), influences sorting of SV cargoes (Salazar et al, 2006; Newell-Litwa et al, 2009), mediates SV budding from early endosomes (Faundez et al, 1998) and cargoes delivery to the synapse (Larimore et al, 2011). BLOC-1 deficiency perturbs AP-3 levels (Newell-Litwa et al, 2009, 2010), in turn affecting neurotransmitter release from hippocampal mossy fibers (Scheuber et al, 2006). BLOC-1 is thus involved in neurotransmission, although the mechanisms remain elusive.
Our central hypothesis is that coordinated endosomal transport and sorting influence SV recycling and presynaptic activity. As (i) an adaptor for dynein-mediated retrograde transport of endosomal cargoes (Cai et al, 2010) and (ii) a subunit of the BLOC-1 complex involved in endosomal sorting (Salazar et al, 2006; Newell-Litwa et al, 2009 Larimore et al, 2011), snapin represents an ideal tool to assess the importance of these two endolysosomal processes in the maintenance of presynaptic activity. In the current study, we tested our hypothesis by imaging live cortical neurons from WT and snapin−/− mice in combination with the expression of snapin mutants selectively impairing dynein-mediated LE trafficking or BLOC-1-dependent endosomal sorting. We demonstrate the regulation of SV pool size by endosomal transport and of the Ca2+ sensitivity of SV release by endosomal sorting.
Results
Snapin-deficient neurons display enlarged synapses retaining degradative organelles
Late endocytic trafficking maintains cellular homeostasis by delivering targeted material to lysosomes for degradation. Retrograde transport of LEs is driven by dynein from distal processes to the soma, where mature acidic lysosomes are mainly localized. To determine whether snapin-mediated and dynein-driven LE transport maintain presynaptic homeostasis, we first examined the ultrastructure of cultured cortical neurons at 14 days in vitro (DIV14) from snapin null mice (Fig1A and B). The average surface of presynaptic terminals was increased by 60% in snapin−/− neurons (0.61 ± 0.04 μm2) relative to wild-type (WT) neurons (0.38 ± 0.02 μm2, P < 0.0001). Snapin-deficient terminals contained ∼30% more SVs than WT ones (87 ± 5 and 68 ± 6 per terminal, respectively, P < 0.05). However, there was no significant difference in either active zone length (not shown) or the number of docked SVs per terminal (3.5 ± 0.2 in snapin+/+, 3.3 ± 0.2 in snapin−/−). Strikingly, a significant number of snapin−/− terminals retained late endocytic organelles—multi-vesicular bodies (MVBs), which were not readily observed in WT terminals. Accumulations of oversized MVBs were also found in neurites of snapin−/− neurons, consistent with the role of snapin as a motor adaptor in dynein-driven LE transport (Cai et al, 2010). Double-membraned autophagic-like vacuoles (AVs) were also present in snapin−/− terminals (0.52 ± 0.10 in snapin−/−; 0.05 ± 0.03 in snapin+/+, P < 0.0001), along with increased MVB-like organelles (0.17 ± 0.05 in snapin−/−; 0.02 ± 0.02 in snapin+/+, P < 0.0001). Altogether, snapin−/− terminals displayed a striking phenotype showing aberrant retention of degradative organelles involved in endolysosomal or autophagy processes: the total number of vacuoles, MVBs, and AV-like organelles per terminal was robustly increased (0.91 ± 0.16), while these structures were not readily found in WT terminals (0.08 ± 0.03, P < 0.0001). The increased number of AV-like structures in snapin−/− neurons reflects their impaired retrograde trafficking from terminals to the lysosomal system (Cai et al, 2010; Cheng et al, 2015).
Figure 1.

Snapin-deficient neurons display enlarged presynaptic terminals retaining various degradative organelles
- A, B Representative electron micrographs (A) and quantitative analysis (B) showing aberrant accumulation of degradative organelles within enlarged presynaptic terminals of snapin−/− cortical neurons at DIV14. Colored arrowheads indicate various types of degradative organelles: red, AVs; blue, endosomal vacuoles; and green, MVBs/LEs. All these organelles were not readily observed at WT presynaptic terminals.
- C, D Representative images (C) and quantitative analysis (D) showing enhanced intensity of synaptophysin in snapin−/− neurons. Cortical neurons at DIV14 were co-immunostained with antibodies against SV protein synaptophysin (green) and dendritic marker MAP2 (red).
- E Sequential immunoblots of synaptosomal fractions (Syn), cytosolic fractions (Cytosol), and total brain lysates (Total) showing elevated endolysosomal marker LAMP-1 and autophagy marker LC3-II (red boxes) in synapse-enriched preparations from snapin cKO mice at P40. Snapin is relatively enriched in synaptosomal fractions in WT mouse brains and is almost absent in synaptosomes from snapin cKO animals (green box). Note that synaptic protein levels (i.e., synaptotagmin) are not necessarily increased in synaptosomal preparations from adult snapin cKO mice, while EM and immunocytochemistry revealed increased SVs levels in snapin−/− presynaptic terminals in in vitro culture conditions. A possible explanation for this discrepancy is that oversized and dysfunctional terminals might be eliminated by microglia in adult snapin cKO animals to optimize network activity and survival.
Data information: Data were analyzed from the total number of electron micrographs (B) or neurons (D) indicated in parentheses (n) above bar graphs (B) or within bars (D) taken from three pairs of mice for each genotype and expressed as means ± s.e.m. with Student’s t-test (B) and Mann–Whitney U-test (D). P-values: *0.01–0.05; ***0.0001 to 0.001; **** < 0.0001. Scale bars: 200 nm (A) and 20 μm (C).
By immunostaining cortical neurons, we observed increased puncta size and mean intensity of the SV protein synaptophysin in snapin−/− neurons (Fig1C and D), consistent with the increased number of SVs and enlarged presynaptic terminals observed in electron micrographs. We further examined subcellular fractions from P40 mouse brains (Fig1E). Because homozygous snapin−/− mice die perinatally, we generated snapin conditional KO (cKO) mice using the Cre-Lox system. Mice carrying the floxed snapin gene were crossed with the Thy-1-Cre mice to delete snapin from cortical and hippocampal areas (Dewachter et al, 2002). Snapin is relatively enriched in synaptosomal fractions in WT mouse brains and is almost absent in synaptosomes from snapin cKO animals. Any residual signal is likely attributable to glial snapin. Levels of the lysosome-associated membrane protein-1 (LAMP-1), a marker for both late endosomes and lysosomes, and the lipidated form of the autophagy marker light chain-3 (LC3-II) are substantially increased in synaptosomes from adult snapin cKO mouse brains. Thus, biochemical data are consistent with our electron microscopy observations, highlighting the essential role of snapin in maintaining presynaptic homeostasis through endolysosomal trafficking.
Snapin mutants disturb LE retrograde transport in axons
Our previous study demonstrated that snapin serves as an adaptor that recruits dynein to LEs by binding to its intermediate chain (DIC) (Cai et al, 2010). Because snapin−/− embryos exhibit developmental defects, we assessed the role of snapin in axonal LE retrograde transport by expressing dominant-negative snapin mutants in WT neurons. First, to identify snapin mutants that disrupt dynein–snapin coupling, we tested the effect of already available mutations on DIC binding by pulling down His-tagged snapin using GST-DIC. The snapin-L99K mutation abolished its interaction with DIC (Fig2A). Interestingly, the snapin-S50D mutation also reduced snapin binding to DIC.
Figure 2.
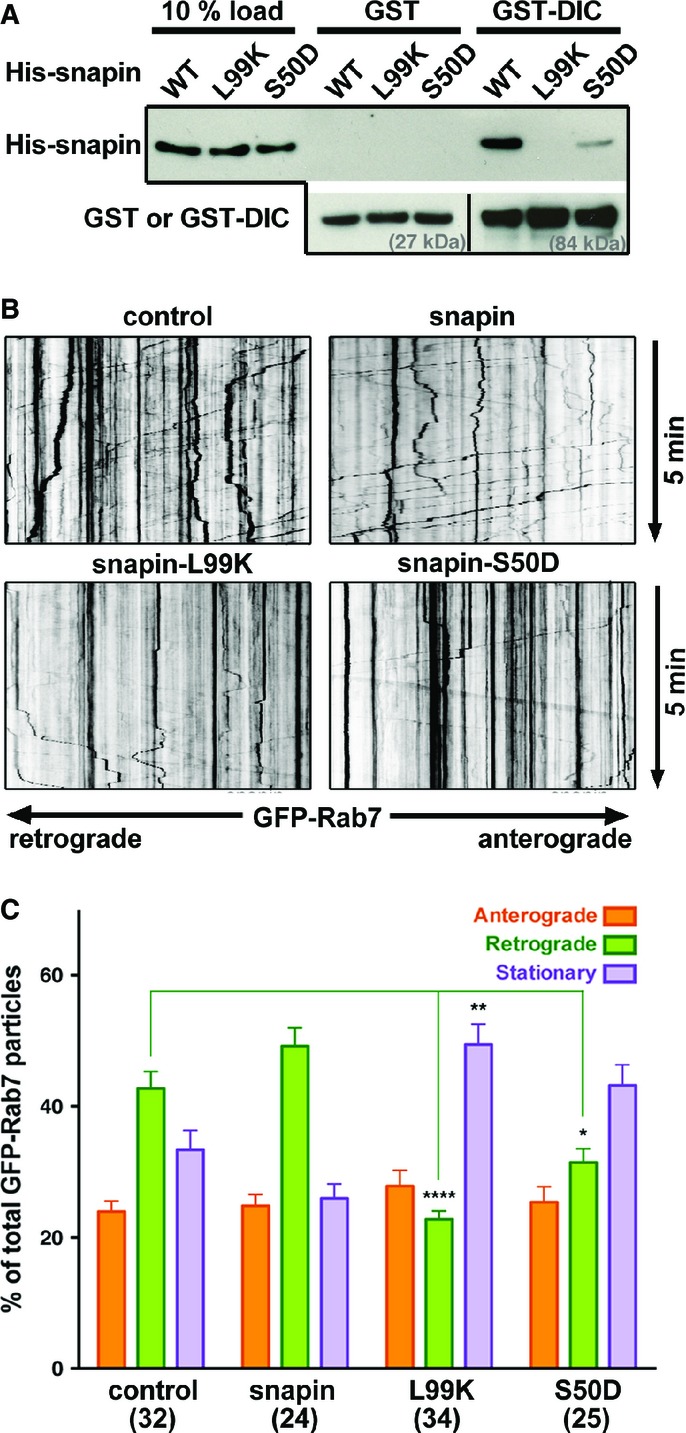
Snapin mutants disturb LE retrograde transport in axons
- A Representative GST-DIC pull-down illustrating distinct binding capacities of WT and mutant snapin with dynein intermediate chain (DIC). Note that the L99K mutation abolished snapin binding to DIC and the S50D mutation robustly decreased snapin–DIC coupling. The same membranes were sequentially blotted with anti-His and anti-GST antibodies. Lower panels demonstrate similar amount of GST or GST-tagged DIC used for the pull-down.
- B, C Representative kymographs (B) and quantitative analysis (C) showing the relative motility of axonal LEs during 5-min time-lapse imaging from cortical neurons at DIV14. Neurons were co-transfected with GFP-Rab7 and a pcDNA vector alone as a control or expressing WT or mutant snapin as indicated. Vertical lines correspond to stationary organelles; oblique traces reflect retrograde (leftward) or anterograde (rightward) transport. Motile or stationary organelles were normalized to the total number of organelles per axon segment selected for recording and averaged from the total number of axons indicated in parentheses. Retrograde transport of Rab7-labeled LEs was robustly inhibited by expressing snapin-L99K and reduced by expressing snapin-S50D. Data are means ± s.e.m., Kruskal–Wallis test with Dunn’s post hoc test. P-values: *0.01–0.05; **0.001 to 0.01; **** < 0.0001. (See also Supplementary Fig S1.)
We next verified the functional effects of these snapin mutations on axonal transport of LEs in WT cortical neurons at DIV14 co-transfected with GFP-Rab7 and a pcDNA vector alone as a control, or expressing WT or mutant snapin. Control cortical neurons displayed predominantly retrograde transport of LEs (Fig2B). While elevated WT snapin expression further enhanced retrograde motility, expressing the snapin-L99K mutant defective in DIC binding reduced retrograde motility from 42.7 ± 1.6% to 22.7 ± 2.4% (P < 0.0001) (Fig2C). Expressing the snapin-S50D mutant induced a smaller but significant decrease in LE retrograde motility (31.4 ± 2.4%, P < 0.05), consistent with its reduced binding to DIC (Fig2A). As a control, kinesin-driven anterograde transport of LEs was not affected by expressing snapin WT or mutants (Fig2C). Alternatively, we expressed DIC (108–268), the snapin-binding domain of DIC that competitively disrupts the endogenous snapin–DIC coupling (Cai et al, 2010). Expressing DIC (108–268) in WT neurons induced a phenotype similar to snapin-L99K expression: reduction in retrograde motility to 22.5 ± 2.0% (Supplementary Fig S1). These data further support the notion that reduced LE retrograde motility is due to impaired snapin–DIC coupling. Given that reduced retrograde motility was recorded during a short time-lapse imaging (5 min), LEs and SVs are expected to accumulate robustly at synaptic terminals and along neurites as observed in electron micrographs of snapin-deficient neurons (Fig1A and B).
Snapin-mediated LE transport influences the total SV pool size
Next, we tested whether snapin-mediated retrograde transport influences total SV pool size by examining presynaptic terminals labeled with HA-synaptophysin or GFP-bassoon, an active zone cytomatrix protein (Gundelfinger & Fejtova, 2012). Expressing WT snapin in WT cortical neurons induced a 24% decrease in the mean intensity of HA-synaptophysin (76.3 ± 5.8% of control, P < 0.001) (Fig3A and B), whereas expressing snapin-L99K and snapin-S50D mutants defective in DIC binding increased the mean intensity by ∼40% (139.2 ± 5.2% and 142.9 ± 11.3%, respectively, P < 0.001), mimicking the ultrastructural phenotype of snapin-deficient boutons (Fig1A and B). However, the mean intensity of the cytomatrix protein GFP-bassoon exhibited no significant change, suggesting that snapin selectively regulates SV levels at presynaptic boutons through dynein-driven SV cargo transport along the endolysosomal pathway. This assumption is further supported by the opposite effects of WT snapin and two DIC-binding defective mutants (snapin-L99K and snapin-S50D) on relative SV abundance at terminals (Fig3A) and the relative retrograde motility of LE cargoes along axons (Fig2B and C). To demonstrate the specificity of these effects, we reintroduced WT or mutant snapin constructs into snapin−/− cortical neurons at DIV3 and observed presynaptic terminals labeled with HA-synaptophysin at DIV14 (Fig3C and D). WT snapin fully rescued HA-synaptophysin signal intensity in snapin−/− boutons, while both DIC-binding-defective mutants failed to reverse the phenotype. These gene rescue experiments in snapin−/− neurons support our hypothesis that snapin regulates SV abundance at presynaptic terminals through the dynein-driven retrograde transport of SV components into the endolysosomal system.
Figure 3.

Snapin-mediated LE retrograde transport regulates total SV pool size
- A, B Representative images (A) and quantitative analysis (B) showing the impact of snapin and its mutants on the intensity of SV and active zone markers. WT cortical neurons co-transfected with HA-synaptophysin or GFP-bassoon and snapin constructs at DIV8, followed by immunostaining with antibodies against MAP2 and HA-tag at DIV14. Relative fluorescence intensities of HA-synaptophysin and GFP-bassoon were normalized to control neurons. Note that over-expressing WT snapin decreases, whereas the L99K and S50D mutants increase synaptophysin puncta intensities. As an internal control, expressing snapin or its mutant has no detectable effect on the intensity of GFP-bassoon.
- C, D Representative images (C) and quantitative analysis (D) showing the rescue of snapin-deficient boutons by snapin and its mutants. snapin−/− cortical neurons were co-transfected with HA-synaptophysin and snapin constructs as indicated at DIV3, and immunostained with antibodies against MAP2 and HA-tag at DIV14. Relative fluorescence intensities of HA-synaptophysin were normalized to snapin−/− neurons. WT snapin, but not the L99K or S50D mutants, rescues HA-synaptophysin-tagged SV fluorescence intensity to the wild-type level.
- E, F Sample kymographs (E) and quantitative analysis (F) illustrating the dynamic trafficking of recycling SV cargoes along axons of cortical neurons co-transfected with VGluT-pHluorin-mCherry and snapin or the L99K mutant. Active axons were selected based on the pHluorin response to 100 APs, and mobile VGluT-labeled SV cargoes were tracked through mCherry during 2.5-min dual-channel recordings. Note that over-expressing snapin enhances SV cargoes dynamics while the L99K mutant decreases trafficking.
- G Sample kymographs illustrating co-trafficking of Tomato-synaptophysin with GFP-LAMP-1-labeled LEs along axons from cortical neurons at DIV14. Axonal trafficking was monitored in neurons co-expressing the SV marker Tomato-synaptophysin and endolysosomal marker GFP-LAMP-1 during 5-min dual-channel time-lapse acquisitions. Arrowheads indicate retrogradely moving organelles containing both synaptophysin and LAMP-1.
- H Sample kymographs illustrating co-trafficking of VGluT-pHluorin with mApple-LAMP-1-labeled LEs along axons from cortical neurons at DIV14. VGlut-pHluorin fluorescence was elicited by 600 APs at 10 Hz in the presence of 1 μM bafilomycin to prevent SV re-acidification; axonal transport was monitored during 5-min dual-channel acquisitions. Arrowheads indicate retrogradely moving organelles containing both SV recycling material (VGluT-pHluorin) and LAMP-1.
Data information: Scale bars: 10 μm. The total number of images (B, D) or time-lapse videos (F) analyzed is indicated within bars. Data are means ± s.e.m., one-sample t-test, where each value is compared to the normalized control value (B, D); or ordinary one-way ANOVA with Dunnett’s post hoc test (F). P-values: *0.05–0.01; **0.001 to 0.01; *** < 0.001. Live imaging data were recorded during 2.5 (E) or 5 min (G, H). (See also Supplementary Fig S2.)
Retrograde trafficking of SV cargoes toward lysosomes is probably required for efficient degradation of SV components (Fernandes et al, 2014), in addition to degradation through the ubiquitin proteasome system (UPS) (Rizzoli, 2014). Thus, our findings raise the possibility that elevated snapin expression drives SV components toward the endolysosomal system. To test whether snapin influences the trafficking of recycling SV cargoes following exo- and endocytosis, we chose a well-established VGluT-mCherry-pHluorin probe (Kim & Ryan, 2010). We monitored VGluT-pHluorin-mCherry movements in neurons over-expressing snapin or snapin-L99K (Fig3E and F). Active synapses were selected based on the pHluorin response to 100 APs at 10 Hz, and VGluT-labeled SV components were tracked during a 2.5-min time-lapse imaging of mCherry. Elevated snapin expression increased the relative motility of VGluT-labeled SV cargoes along axons from 30.5 ± 2.6% to 42.3 ± 2.7% (P < 0.01), whereas expressing snapin-L99K mutant reduced their motility (21.8 ± 2.6%, P < 0.05). Thus, changes in the relative motility of the SV marker are inversely correlated with SV levels at presynaptic terminals (Fig3A and B).
Using the photoconvertible dendra-synaptophysin probe (Staras et al, 2010), we then verified whether snapin mediates local, inter-synaptic SV motility. First, we found that the photoconverted synaptophysin signal was significantly higher in snapin-deficient boutons (P < 0.05, Student’s t-test), consistent with enlarged terminals and increased SV number. However, while monitoring photoconverted dendra-synaptophysin trafficking in WT and snapin−/− neurons, we found no significant difference in the amount of SVs transferred to neighboring boutons (Supplementary Fig S2A and B). This suggests that snapin does not influence short-range actin-based movements of SVs from synapse to synapse, a dynamic SV local trafficking process described previously (Darcy et al, 2006; Staras et al, 2010). Because snapin mediates long-distance LE retrograde transport by recruiting the microtubule-based dynein to LEs, we further explored whether SV cargoes are transported through late endocytic organelles. Axonal trafficking was monitored in neurons co-expressing the SV marker Tomato-synaptophysin and endolysosomal marker GFP-LAMP-1 during 5-min dual-channel time-lapse acquisitions (Fig3G). Co-migration of synaptophysin-labeled SV cargoes and endolysosomal markers was readily detected in 5 out of 16 neurons over-expressing snapin. The low frequency of those co-migration events is likely due to limited recycling synaptophysin moving into the endolysosomal trafficking pathway in the absence of stimulation.
To confirm the entry of SV components into the endolysosomal pathway, we alternatively assessed the co-trafficking of recycling SV material with LEs using the activity reporter VGluT-pHluorin and mApple-LAMP-1. VGlut-pHluorin fluorescence was elicited by 600 APs at 10 Hz in the presence of 1 μM bafilomycin to prevent SV re-acidification and axonal transport was monitored during 5-min dual-channel acquisitions (Fig3H). Co-migration of those markers along axons could readily be observed. The relatively low fluorescence intensity of motile VGluT-pHluorin puncta co-migrating with mApple-LAMP-1 is consistent with the expected small amounts of recycling SV proteins entering the endolysosomal pathway at any given time, with respect to the total SV population at terminals. The rate of migration of VGluT-pHluorin is, however, higher than the trafficking of Tomato-synaptophysin, consistent with a previous observation that VGluT and synaptophysin are sorted through different mechanisms. AP-3 and BLOC-1 selectively influence VGluT, but not synaptophysin sorting (Newell-Litwa et al, 2009).
Alternatively, we labeled recycling SV cargoes during a 600-AP stimulation using an oyster-555-labeled antibody against the luminal domain of synaptotagmin, thus excluding possible caveats associated with over-expressed SV proteins. A significant amount of newly endocytosed SV components co-migrated with the LE marker GFP-Rab7 (Supplementary Fig S2C). Together, these time-lapse imaging data in live neurons consistently support our notion that dynein-driven axonal LE transport regulates SV pool size at presynaptic terminals. Disrupting snapin–DIC coupling by expressing dominant-negative DIC-binding defective snapin mutant impairs this transport, thus increasing SV levels at presynaptic terminals. Conversely, elevated WT snapin expression facilitates SV cargoes transport into the endolysosomal pathway, in turn reducing the SV pool size. We refer to SV components trafficking into the endolysosomal pathway as “Route 1” of endosomal regulation of presynaptic activity.
Altered SV exocytosis in snapin-deficient presynaptic terminals
To determine whether altered SV pool size impacts SV exocytosis, we visualized SV exocytosis at single boutons using VGluT-mCherry-pHluorin (Fig4A). We monitored pHluorin response to three different stimulation paradigms to compare releasable pool size and SV recycling between WT and snapin−/− presynaptic terminals (Fig4B and C). Previous studies using electrophysiology established that 30–50 action potentials (APs) at 20 Hz are necessary to deplete the readily releasable pool (RRP) (Rosenmund & Stevens, 1996; Murthy & Stevens, 1998; Moulder & Mennerick, 2005), and even stronger stimuli would be required according to a pHluorin imaging study (Ariel & Ryan, 2010). We reasoned that the response to a smaller, non-depleting stimulation (20 APs at 20 Hz) would reflect the probability of release (Fig4B, left panel). In snapin−/− boutons, the response to such stimulation was reduced by 27% (F/F0 peak amplitude = 1.72 ± 0.04 versus 2.37 ± 0.1 in WT boutons, P < 0.0001). Next, we applied a 10-s stimulation at 10 Hz to estimate the total releasable pool size (Fig4B, middle panel). A previous pHluorin study established that the response to such stimulation mainly relies on exocytosis, with little to no contribution from slow SV endocytosis and re-acidification (Kim & Ryan, 2009). We restricted our estimation to the total releasable pool size, since limited temporal resolution of imaging does not allow to distinguish the RRP within the total recycling pool (Ariel & Ryan, 2010). The pHluorin response to 100 APs at 10 Hz in snapin−/− terminals was reduced by 25% (F/F0 peak amplitude = 3.18 ± 0.07 versus 4.15 ± 0.08 in WT neurons, P < 0.0001), indicating a reduced total releasable pool size. Since the number of docked vesicles per synapse is not significantly changed in snapin−/− synaptic terminals (Fig1B), these results suggest a role for snapin at the priming step (Pan et al, 2009). In addition, we used a stimulation paradigm (1,500 APs at 5 Hz) established previously to assess SV recycling (Voglmaier et al, 2006). The pHluorin signal reached a peak 60 s after stimulation onset and then diminished slowly until stimulation ended, reflecting ongoing exocytosis, endocytosis, and re-acidification (Fig4B, right panel). The average pHluorin response was significantly decreased at snapin−/− synapses (F/F0 peak amplitude = 3.45 ± 0.14 versus 4.61 ± 0.19 in WT boutons, P < 0.0001).
Figure 4.
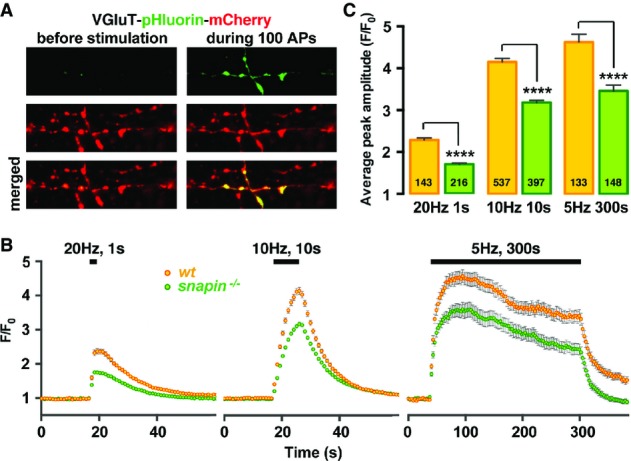
Altered SV exocytosis occurs at snapin-deficient presynaptic terminals
- A Sample pHluorin images from synapses expressing VGluT-pHluorin-mCherry before and during a 10-Hz, 10-s stimulation (100 APs) in cortical neurons at DIV14. Exposure of pHluorin to the alkaline extracellular media during exocytosis gives rise to green fluorescence emission.
- B, C Average pHluorin traces (B) and peak amplitudes (C) elicited by 20 (20 Hz, 1 s), 100 (10 Hz, 10 s), or 1,500 (5 Hz, 300 s) APs. Deleting snapin decreases total releasable pool size without affecting SVs recycling rate during a prolonged (300 s) stimulation. The total number of boutons analyzed is indicated within the columns. Data are means ± s.e.m., Student’s t-test. P-value: **** < 0.0001.
Snapin mutations differentially affect total releasable pool size and SV recycling
Snapin has three binding partners that could potentially influence presynaptic maintenance and activity: dynein, the BLOC-1 subunit dysbindin, and the SNARE protein SNAP25. Thus, snapin mutants with selective binding deficiencies could represent invaluable tools to assess the importance of related cellular processes in maintaining presynaptic activity. We first examined the relative binding capacities of snapin mutants by GST pull-down assays. As previously reported (Chheda et al, 2001; Navarro et al, 2012), snapin interacts with SNAP25 and the phosphomimetic S50D mutation enhances this interaction (Supplementary Fig S3A). Snapin also binds to dysbindin (Starcevic & Dell’Angelica, 2004; Lee et al, 2012), and this interaction is slightly increased by the S50D mutation under the same conditions. Moreover, the L99K mutation that abolishes snapin interaction with DIC (Fig2A; Cai et al, 2010) also prevented dysbindin binding to snapin, but did not impair SNAP25 binding. The differential binding capacities of snapin and its mutants to SNAP25, DIC and dysbindin were further confirmed by GST pull-down of HA-tagged snapin from HEK cells lysates (Supplementary Fig S3B). In that configuration, however, the S50D mutation increased SNAP25 binding, largely reduced DIC binding, and slightly reduced dysbindin binding, although the latter remained stronger than any of the interactions between WT or mutant snapin and either SNAP25 or DIC. We further characterized these three interactions by performing competitive binding assays, where His-snapin and GST-DIC were incubated in the presence of His-tagged dysbindin or SNAP25 (Supplementary Fig S3C and D). An equimolar amount of dysbindin reduced snapin binding to DIC while a fivefold molar excess was sufficient to abolish the interaction. No ternary complex was observed. In contrast, even a tenfold excess of SNAP25 failed to displace snapin from DIC, thus suggesting that dysbindin and DIC are snapin’s main binding partners under our in vitro conditions. These data also suggest that dysbindin and DIC share the same binding site on snapin. Thus, the DIC (108-268) interaction domain, which competes with DIC for binding to snapin and impairs LE retrograde transport (Supplementary Fig S1; Cai et al, 2010), will likely inhibit the snapin–dysbindin interaction and mimic the effect of snapin-L99K expression. Taken together with DIC-binding assays (Fig2A), these results suggest that the snapin-L99K mutation will affect both LE retrograde transport by disrupting snapin–DIC coupling and BLOC-1-mediated endosomal sorting by blocking snapin–dysbindin interaction (Supplementary Fig S3A and B), whereas the snapin-S50D mutation will selectively impair LE transport by reducing snapin–DIC coupling without impacting its binding to dysbindin. Neither the S50D nor the L99K mutation reduces SNAP25 binding. To assess the stability of secondary structures in these snapin mutants, we performed CD spectroscopy measurements by comparing WT snapin with snapin-L99K and snapin-S50D mutants. The alpha-helix profiles suggest these two mutants are properly folded (Supplementary Fig S3E). Thus, these snapin mutants are ideal molecular tools to assess the relative role of endosomal trafficking and sorting in maintaining presynaptic function.
We propose our working hypothesis where two snapin-mediated pathways influence presynaptic activity: dynein-driven endosomal transport shuttles SV components away from terminals (Route 1), and BLOC-1/AP-3-dependent endosomal sorting regulates SV exocytosis at synaptic terminals (Route 2). While the snapin-L99K mutation impairs both Routes 1 and 2, the S50D mutation selectively impairs LE retrograde transport (Route 1). Neither mutation negatively impacts binding between snapin and SNAP25, thus avoiding direct effects on SV fusion.
Next, we examined how these sorting/trafficking routes impact presynaptic activity by expressing snapin mutants in cortical neurons. Expressing WT snapin induced a slight reduction of the F/F0 pHluorin response to 20 APs, from 2.42 ± 0.04 to 2.12 ± 0.04 (P < 0.0001) (Fig5A and B, left), reminiscent of the smaller excitatory postsynaptic currents (EPSCs) recorded from hippocampal neurons over-expressing snapin (Thakur et al, 2004). In contrast, expressing snapin-S50D, a mutant displaying enhanced binding to dysbindin and SNAP25, potentiated the pHluorin response (F/F0 =2.83 ± 0.07, P < 0.0001), consistent with the previous findings that expressing this mutant increased Pr (Thakur et al, 2004). Conversely, expressing snapin-L99K, a dominant-negative mutant defective in both DIC and dysbindin binding, inhibited the F/F0 pHluorin response by 20% (1.99 ± 0.04, P < 0.0001). We further showed that expressing snapin or snapin-L99K reduced the size of the releasable pool, illustrated by a decreased F/F0 pHluorin amplitude from 4.27 ± 0.08 to 3.48 ± 0.07 (P < 0.001), and 3.35 ± 0.06 (P < 0.001), respectively, in response to 100 APs (Fig5A and B, middle). However, such a reduction was not detected in neurons expressing snapin-S50D (4.3 ± 0.12). Since S50D mutation impairs LE transport by disrupting snapin–DIC coupling without reducing SNAP25 or dysbindin binding, this mutation likely turns off the effect of LE trafficking on releasable pool size. Therefore, the smaller releasable pool observed after WT snapin over-expression likely results from increased trafficking of recycling SV cargoes into the endolysosomal system. In addition, the lack of effect of snapin-S50D on releasable pool size argues against an unspecific effect arising from snapin overexpression and, in particular, excludes the titration of SNAP25 or dysbindin as a potential explanation for impaired release.
Figure 5.
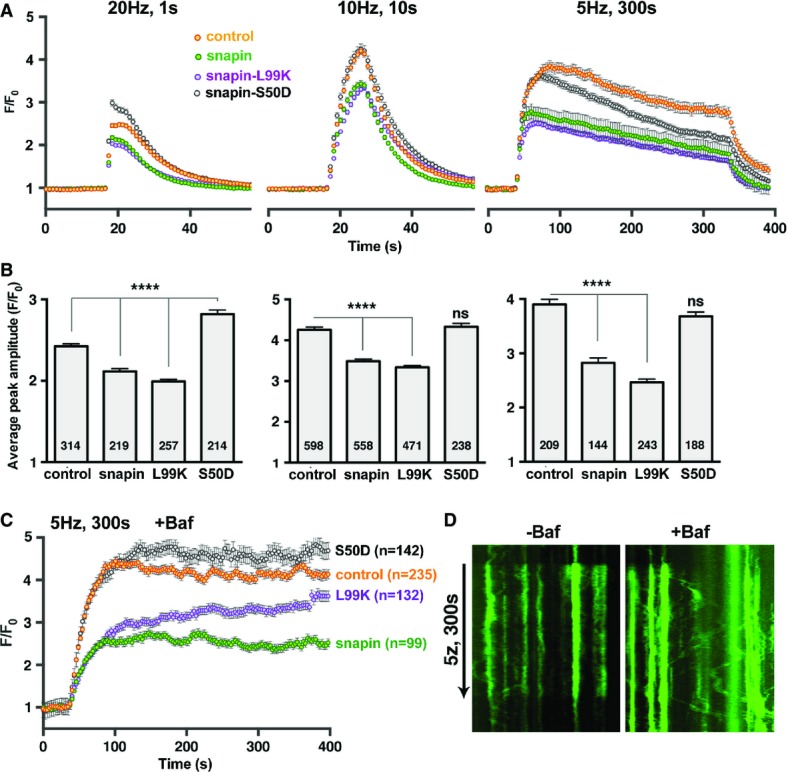
Snapin mutations differentially affect presynaptic activity
- A, B Average pHluorin traces (A) and corresponding peak amplitudes (B) elicited by 20 (left), 100 (middle), or 1,500 (right) APs from cortical neurons at DIV14. Note that over-expressing snapin or snapin-L99K decreases releasable pool size without affecting the recycling rate. Conversely, expressing snapin-S50D accelerates decay during a prolonged stimulation without affecting the releasable pool size. Data are means ± s.e.m., ANOVA with Dunnett’s post hoc test, where each value is compared to the control. The total number of boutons imaged is indicated within the columns. P-value: **** < 0.0001.
- C, D Average pHluorin traces elicited by 1,500 APs in the presence of 1 μM of the V-ATPase inhibitor bafilomycin (C) and sample kymographs illustrating pHluorin responses from control cells in the absence (left) or presence (right) of 1 μM bafilomycin (D). Note that in the presence of bafilomycin, trafficking of the pHluorin signal out of boutons becomes readily detectable. “n” indicates the total number of boutons imaged and averaged. Error bars represent s.e.m. (See also Supplementary Figs S3 and S4.)
We speculate that the inhibition by snapin-L99K results from defective dysbindin/BLOC-1-dependent sorting rather than reduced LE transport, because snapin-S50D, which also reduces LE transport, does not affect the releasable pool size. Inhibitory effects were more pronounced with a lower stimulation frequency (1,500 APs at 5 Hz): the F/F0 response was reduced by 28% in neurons over-expressing snapin (P < 0.0001) and by 37% in neurons expressing snapin-L99K (P < 0.0001) (Fig5A and B, right). Importantly, expressing DIC (108–268) mimicked the effect of snapin-L99K on pHluorin responses under all stimulation conditions, further arguing against unspecific inhibition due to over-expression of the mutant (Supplementary Fig S4). In addition, snapin-S50D accelerated the signal decay, suggesting either faster endocytosis/re-acidification, or faster depression of release due to higher Pr and/or slower SV recruitment; the latter possibility is more likely because pHluorin responses to prolonged stimulations in 6 mM CaCl2 also displayed such a fast decay (Fig6E).
Figure 6.

A snapin mutant defective in dysbindin binding impairs SV positional priming
- A Schematic illustration of Ca2+ imaging using the high-sensitivity green fluorescent Ca2+ sensor GECO. The synaptophysin-GECO chimera selectively reports SV exposure to activity-triggered Ca2+ influx, while soluble GECO serves as a reporter for global Ca2+ levels within terminals.
- B Representative images (left) and average traces of Ca2+ imaging and SV cycling (right) during a 100-AP stimulation. Dual-channel time-lapse imaging of synaptophysin-GECO and pH-sensitive VGluT-mOrange demonstrates that the newly developed probe is targeted to active synapses. Average traces illustrate the relative timing of Ca2+ influx with exo- and endocytosis. Data are means ± s.e.m. from 60 boutons. Scale bar: 5 μm.
- C, D Average synaptophysin-GECO or GECO traces (C) and peak values (D) recorded from cortical neurons at DIV14 in response to 100 APs. Neurons were co-transfected with either synaptophysin-GECO or GECO and snapin or its mutants, as indicated. Note that SVs experience higher Ca2+ levels in terminals over-expressing snapin and lower Ca2+ levels in the ones over-expressing snapin-L99K. The global Ca2+ waveform monitored through soluble GECO was unchanged.
- E, F Average pHluorin traces (E) and peak amplitudes (F) elicited by 20 (left), 100 (middle), or 1,500 (right) APs in the presence of 6 mM extracellular Ca2+. Elevating extracellular Ca2+ levels rescues release in synapses expressing snapin-L99K that impairs positional priming, but fails to rescue snapin-over-expressing boutons where the release defect is attributable to reduced SV pool size by enhancing SV transport into the endolysosomal pathway.
Data information: Data are means ± s.e.m., ANOVA with Dunnett’s post hoc test, where each value was compared to the control. P-values: **** < 0.0001. The total number of boutons analyzed is indicated within columns. (See also Supplementary Fig S5.)
To provide mechanistic insights into snapin-mediated regulation of the SV cycle, we applied the V-ATPase inhibitor bafilomycin during the 1,500-AP stimulation (Fig5C and D). When SV re-acidification is blocked by bafilomycin, the pHluorin signal mainly reflects accumulated exocytosis, thus displaying a biphasic pHluorin response: the initial rapid rise in fluorescence is followed by a slow continued increase due to SV recruitment from the reserve pool. Surprisingly, in control neurons and neurons expressing WT snapin or snapin-S50D, the F/F0 pHluorin response quickly reached a steady-state 60 s after stimulation onset (Fig5C). Inhibiting re-acidification further revealed extensive pHluorin movements out of presynaptic boutons during prolonged stimulations, a phenotype not readily observed in the absence of bafilomycin (Fig5D). The recruitment of SVs from the resting pool into the recycling pool could therefore be counterbalanced by pHluorin-labeled SV trafficking out of terminals (Route 1), thus explaining the flat appearance of the F/F0 pHluorin response during prolonged stimulations. In contrast, in boutons expressing snapin-L99K, there was a biphasic fluorescence increase: after the initial fast rise, the F/F0 pHluorin signal kept increasing steadily at a slower rate (Fig5C). One explanation is that accumulating Ca2+ at presynaptic boutons during the 5-min stimulation may facilitate the release of SVs with reduced Ca2+ sensitivity or impaired positioning to the Ca2+ entry sites. Taken together, the pHluorin results suggest that over-expressing WT or snapin-L99K reduces the releasable pool size through two different mechanisms: WT snapin acts by increasing SV components trafficking into the endolysosomal pathway (Route 1) while snapin-L99K likely acts by impairing BLOC-1-dependent SV sorting from endosomes (Route 2).
A snapin mutant defective in dysbindin/BLOC-1 binding impairs SV positional priming
While both snapin-S50D and snapin-L99K mutants induced SV accumulation at presynaptic boutons (Fig3A and B) by disrupting the retrograde transport of SV cargoes along the endolysosomal pathway (Fig2B and C), only snapin-L99K reduced the releasable pool size (Fig5A–C). In view of the biochemical data (Supplementary Fig S3A), this raises the possibility that dysbindin/BLOC-1-dependent function (Route 2) is required for maintaining the releasable pool size. Our previous study revealed desynchronized EPSCs in snapin−/− neuronal cultures (Pan et al, 2009), reminiscent of manipulations that decrease the physical coupling of SVs with voltage-dependent Ca2+ channels (Mochida et al, 1996). Such deficits can be readily rescued by raising the extracellular Ca2+ concentration [Ca2+]ext (Pan et al, 2009). Therefore, snapin represents an attractive candidate in facilitating the positional priming of SVs toward Ca2+ entry sites (Wadel et al, 2007), thus influencing the Ca2+ sensitivity of neurotransmitter release.
Determination of SV positioning with respect to Ca2+ channels was previously reported from GABAergic synapses (Bucurenciu et al, 2008). Based on the differential effects of fast and slow Ca2+ chelators, the study revealed SV positioning within 10–20 nm from Ca2+ channels, a distance beyond the resolution of current fluorescence imaging techniques. To circumvent that limitation, we assessed the exposure of SVs to activity-triggered Ca2+ influx at presynaptic terminals. We constructed a SV-targeted Ca2+ sensor by linking a green-genetically encoded calcium indicator for optical imaging (G-GECO, Zhao et al, 2011) to the cytosolic C-terminal domain of synaptophysin (Fig6A). As evidenced by its co-localization with VGluT-mOrange (Li et al, 2011), synaptophysin-GECO was targeted to active synapses in cortical neurons and exhibited a rapid fluorescence increase in response to activity-triggered Ca2+ influx during a 100-AP stimulation (Fig6B). Over-expressing WT snapin increased the peak amplitude of the Ca2+ transient detected by synaptophysin-GECO from 3.30 ± 0.06 in control neurons to 3.76 ± 0.11 (P < 0.0001), although the Ca2+ waveform sensed by the SV remained globally equivalent to the control. In contrast, expressing snapin-L99K substantially reduced the peak amplitude of the synaptophysin-GECO response to 2.55 ± 0.06 (P < 0.0001) (Fig6C and D). Expressing WT snapin or snapin-L99K did not perturb the global Ca2+ transient measured by the cytosolic GECO probe (peak F/F0 values: control, 2.94 ± 0.07; snapin, 2.90 ± 0.06; snapin-L99K, 3.07 ± 0.07) (Fig6C and D). In contrast, snapin-S50D, which retains dysbindin-binding capacity but displays reduced binding to dynein DIC, had only a minor effect on the peak amplitudes of both cytosolic GECO and synaptophysin-GECO responses (3.17 ± 0.06 and 3.11 ± 0.06, respectively, Supplementary Fig S5A and B). This suggests that the snapin–dynein coupling is unlikely required for the positional priming of SVs. Expressing DIC (108–268) reduced synaptophysin-GECO response to the same extent as snapin-L99K (2.30 ± 0.05, P < 0.0001) without affecting the GECO signal (2.93 ± 0.06), thus further supporting biochemical competition data (Supplementary Fig S3C) showing that DIC and dysbindin compete with each other to bind snapin. Expressing DIC (108–268) would impair SV positional priming by disrupting snapin coupling with dysbindin, thus disturbing BLOC-1- and AP-3-dependent sorting mechanisms.
We also assessed the global Ca2+ transient at snapin−/− synapses with the cytosolic GECO sensor in response to 100 APs. The average amplitude of activity-triggered Ca2+ influx was similar to WT neurons, although the rising and decay time courses were delayed (not shown), suggesting that synaptic levels of voltage-dependent calcium channels are not significantly reduced in snapin-deficient boutons. This conclusion is supported by our previous electrophysiological study showing that Ca2+ currents from snapin-deficient synapses are similar to wild-type Ca2+ currents (Pan et al, 2009).
We then verified the specificity of these effects by reintroducing snapin and its mutants into snapin-deficient neurons (Supplementary Fig S5C and D). snapin−/− boutons displayed severely decreased GECO-synaptophysin responses to 100-AP stimulations (2.09 ± 0.06) with slowed-down kinetics. While reintroducing WT or mutant snapin could rescue the timing of GECO-synaptophysin responses, only WT snapin and snapin-S50D, which retains dysbindin-binding capacity, could rescue the peak amplitude (2.87 ± 0.10 and 3.15 ± 0.11, respectively). The dysbindin-binding defective mutant snapin-L99K failed to rescue the defective GECO-synaptophysin responses in snapin−/− boutons. Together, these data support our hypothesis that snapin regulates SV positioning with respect to Ca2+ entry sites via a dysbindin/BLOC-1-dependent sorting mechanism (Route 2).
Next, we raised [Ca2+]ext from 1.2 to 6 mM to increase the electrochemical driving force for Ca2+ entry and yield larger presynaptic Ca2+ domains. If our hypothesis is correct, enhanced Ca2+ entry should alleviate defective positional priming in neurons expressing snapin-L99K but should not rescue the size of the releasable pool in neurons over-expressing WT snapin, which reduces the total SV pool by enhancing LE transport. Indeed, when pHluorin responses were recorded in 6 mM CaCl2, the amplitudes of the F/F0 traces from neurons expressing snapin-L99K were fully rescued to control levels in all stimulation paradigms (Fig6E and F), while the amplitudes of the F/F0 traces in neurons over-expressing snapin remained smaller when compared to control neurons [2.54 ± 0.07 for controls versus 1.99 ± 0.04 for snapin in response to 20 APs (left); 3.63 ± 0.08 for controls versus 2.56 ± 0.06 for snapin in response to 100 APs (middle); 3.75 ± 0.1 for controls versus 2.05 ± 0.05 for snapin in response to 1,500 APs (right, P < 0.0001 in all cases)]. In addition, defective pHluorin responses from neurons over-expressing the snapin-binding domain of DIC (108–268), which competes with dysbindin to bind snapin (Supplementary Fig S3B), could also be rescued under those conditions (Supplementary Fig S5E and F). These results confirm that: (i) the mechanisms leading to an inhibition of release by snapin or snapin-L99K over-expression are distinct and (ii) snapin plays a role in SV positional priming by its interaction with dysbindin/BLOC-1, likely through AP-3-dependent sorting (Route 2).
Emerging lines of evidence show that different recycling pathways generate SVs with variable molecular identities (Voglmaier & Edwards, 2007; Hua et al, 2011; Morgan et al, 2013). For instance, the loss of AP-3 function is known to reduce the presynaptic level of VAMP7 (the tetanus toxin-insensitive VAMP or Ti-VAMP, Scheuber et al, 2006), which is a v-SNARE preferentially associated with the reserve pool of SVs (Hua et al, 2011; Ramirez & Kavalali, 2012) and a marker of the AP-3 pathway (Salazar et al, 2006; Newell-Litwa et al, 2009, 2010). BLOC-1 deficiency perturbs AP-3 levels and function (Newell-Litwa et al, 2009, 2010) and could affect SV recycling from the EE route. To understand how perturbing snapin–dysbindin interaction by expressing snapin-L99K affects SV positioning with respect to Ca2+ entry sites, we examined the composition of SVs immunopurified from snapin cKO adult mouse brains using magnetic beads coated with an antibody against synaptophysin. Sequential immunoblotting analysis demonstrated an enriched SV fraction, which was devoid of mitochondrial (TOM20) or cytosolic (GAPDH) markers (Fig7A). While the amount of synaptotagmin or VAMP2 in purified SVs from snapin cKO mouse brains did not differ from WT, the signal intensity for Ti-VAMP was reduced on average to 50 ± 10% of WT (Fig7B), suggesting a defective AP-3-dependent SV recycling pathway. Interestingly, Rab3a was also robustly reduced to 39 ± 10% of WT levels. Rab3a is a SV-associated GTPase that interacts with the active zone protein RIM (Rab3-interacting molecule) (Wang et al, 1997, 2001; Gracheva et al, 2008). RIM plays an essential role in tethering voltage-dependent Ca2+ channels to release sites (Han et al, 2011; Kaeser et al, 2011). Reduced Rab3a levels in SVs purified from snapin-deficient mouse brains might thus provide a molecular clue as to how snapin mutant defective in dysbindin/BLOC-1 binding impairs SV positional priming (Fig6) and how deleting snapin impairs the synchronization of neurotransmitter release (Pan et al, 2009).
Figure 7.
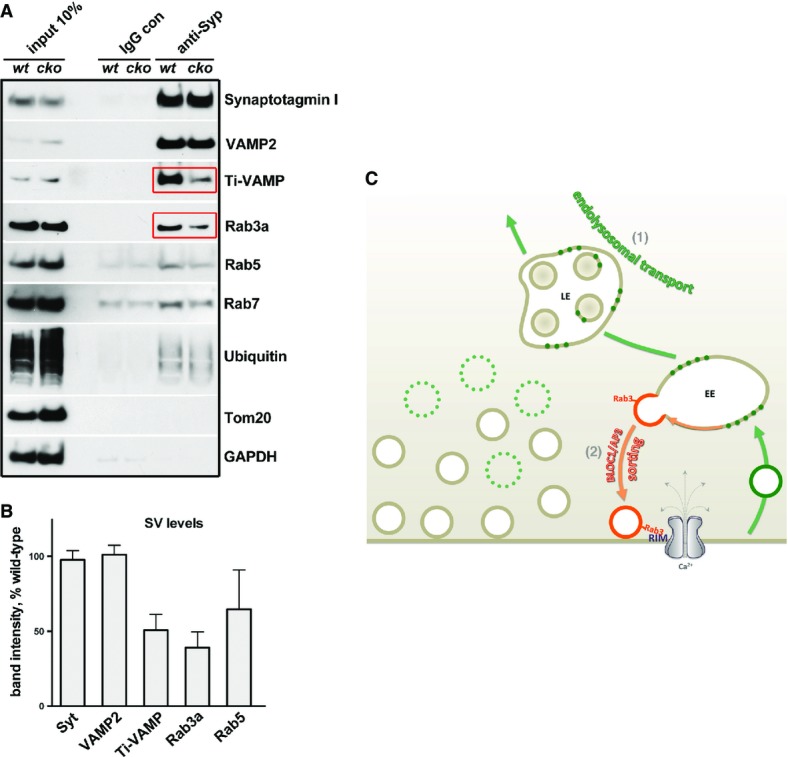
Deleting snapin affects SV molecular identity with BLOC-1/AP-3 inhibition
- A, B SVs were immuno-isolated from synaptosomal preparations of wild-type and snapin cKO (flox/flox; thy/Cre) adult mouse brains using magnetic beads coated with an antibody against synaptophysin. Sequential immunoblotting was conducted on the same membrane after stripping between each antibody application (A). Quantification of protein intensities from three independent experiments (B). Note the reduced amounts of Rab3a and the AP-3-dependent cargo Ti-VAMP in purified SVs from snapin cKO mouse brains (red boxes). Data are means ± s.e.m.
- C Model illustrating the bipartite regulation of synaptic activity by the endolysosomal sorting and trafficking pathways. Late endosomal transport regulates SV pool size by shuttling SV components into the endolysosomal pathway (Route 1), while BLOC-1/dysbindin/AP-3-dependent endosomal sorting regulates SV composition, and consequently positional priming and release probability (Route 2). By balancing these two dynamic pathways, snapin coordinates the releasable pool size and Ca2+ sensitivity of neurotransmitter release.
Discussion
Snapin was previously shown to influence the synchronization of SV fusion (Pan et al, 2009), regulate the retrograde transport of LEs by linking the microtubule-based motor dynein to LEs (Cai et al, 2010), and function as a BLOC-1 subunit (Starcevic & Dell’Angelica, 2004). In the current study, we used our snapin mouse model in combination with dominant-negative snapin mutants specifically impairing LE transport or BLOC-1 endosomal sorting function. We reveal that the endolysosomal pathway exerts a bipartite regulation of presynaptic activity (Fig7C). First, LE transport influences the total SV pool size by shuttling SV components along the endolysosomal pathway (Route 1). Second, BLOC-1-dependent endosomal sorting determines SV composition and positional priming via AP-3-dependent recycling (Route 2). By balancing these two dynamic pathways, snapin coordinates the releasable pool size and Ca2+ sensitivity of neurotransmitter release. Thus, our study provides new mechanistic insights into how presynaptic terminals can maintain proper SV pool size by regulating LE retrograde transport and fine-tune the Ca2+ sensitivity of evoked exocytosis through EE sorting.
Endolysosomal trafficking serves as a new pathway regulating total SV pool size
Snapin plays a key role in dynein-driven retrograde transport and degradation of targeted or internalized material through the endolysosomal system (Cai et al, 2010). The current study provides multiple lines of evidence consistently supporting an essential role for axonal LE transport in maintaining the size of presynaptic SV pools. First, ultrastructural analysis of snapin-deficient neurons displayed striking phenotypes: enlarged presynaptic terminals retaining various degradative organelles from the endolysosomal or autophagy pathways, containing 30% more SVs than WT ones. Those degradative organelles were not readily found in WT terminals. Secondly, immunostaining analysis of cortical neurons confirmed increased puncta size and intensity of the SV marker synaptophysin in snapin−/− neurons. Third, subcellular fractionation showed increased levels of endolysosomal (LAMP-1) and autophagic (LC3-II) markers in snapin−/− synaptosomes. Fourth, we further demonstrated that over-expressing WT snapin induced a 24% decrease in the mean intensity of synaptophysin, whereas expressing dynein-binding defective snapin mutants increased the mean intensity by ∼40%, thus mimicking the ultrastructural features of snapin-deficient boutons. In addition, we tested whether snapin influences trafficking of SV cargoes by monitoring VGluT-pHluorin-mCherry movements along axons after synaptic stimulation. Elevated snapin expression increased the mobility of VGluT-labeled SV cargoes along activated axons, whereas expressing the dynein-binding defective snapin-L99K mutant reduced their trafficking. Finally, we revealed co-migration of SV cargoes with LEs in live cortical neurons and confirmed that elevated snapin expression enhanced such co-migration. Thus, our findings consistently suggest that the endolysosomal system regulates SV pool size through snapin-mediated and dynein-driven LE transport. Alterations of this endolysosomal trafficking (Route 1) will influence SV releasable pool size.
SV degradation may also proceed through the autophagy pathway (Hernandez et al, 2012). However, ultrastructural analysis of snapin−/− neurons did not reveal SV-like organelles engulfed within double-membraned AVs, suggesting that autophagy may not be a predominant pathway in SV turnover. Little is known about how SVs and their components are recycled or degraded. It is thought that the UPS plays a major role in removing misfolded or damaged presynaptic proteins and SV components (Wheeler et al, 2002; Speese et al, 2003; Willeumier et al, 2006; Yao et al, 2007), thus regulating synaptic strength. Interestingly, aberrant accumulation of SVs or SV material at snapin−/− boutons was not accompanied by enhanced ubiquitination levels in either synaptosomes (Fig1E) or immunopurified SVs from snapin cKO mouse brains (Fig7A). This further consolidates our findings that SV components can be transported along the endolysosomal trafficking route, likely traveling from presynaptic terminals to the soma, where mature lysosomes are mainly localized (Cai et al, 2010; Lee et al, 2011).
Unexpectedly, blocking LE retrograde transport by expressing the dynein-binding defective snapin-S50D mutant in WT neurons had no impact on the releasable pool size. Even when both dynein-driven LE transport and BLOC-1/dysbindin-dependent sorting were impaired by expressing snapin-L99K, SV exocytosis could be effectively rescued by increasing [Ca2+]ext. Thus, synaptic activity is relatively impervious to perturbations of LE transport, possibly due to the sizeable population of SVs at terminals, where only a small subset of SVs are actively recycled (Denker et al, 2011; Marra et al, 2012; also see Rose et al, 2013). SVs from the large resting pool are mobilized to the recycling pool only during prolonged stimulations (Rizzoli & Betz, 2005; Denker and Rizzoli, 2010) or following pharmacological inhibition of CDK5 (Kim & Ryan, 2010; Marra et al, 2012). Therefore, SVs that accumulate during LE transport inhibition likely belong to the resting pool. This assumption is based on the previous report that the resting pool consists of “mature” SVs that do not actively recycle (Kamin et al, 2010). In contrast, enhancing LE retrograde transport by over-expressing snapin profoundly impacted synaptic activity: total SV pool size in presynaptic terminals was decreased, and the releasable pool size was accordingly reduced, in agreement with a previous report (Thakur et al, 2004). We speculate that snapin over-expression facilitates BLOC-1-mediated sorting and AP-3-dependent SV recycling through EE intermediates (Di Pietro et al, 2006; Setty et al, 2007), from where SV components would be more efficiently shuttled back to the cell body due to faster endosome maturation (John Peter et al, 2013) and enhanced LE transport (Cai et al, 2010). It is noteworthy that snapin, but not dysbindin, was recently identified as a member of the newly characterized BLOC-1-related complex (BORC) in non-neuronal Hela cells (Pu et al, 2015). BORC enables microtubule-based lysosome movements toward the cell periphery. It remains to be determined whether BORC also regulates dysbindin-independent and dynein–snapin-mediated retrograde transport of LEs in axons.
BLOC-1/dysbindin-dependent sorting contributes to the Ca2+ sensitivity of neurotransmitter release
Tight structural and functional coupling between SVs and Ca2+ channels at release sites is essential for fast synchronous release (Wadel et al, 2007; Bucurenciu et al, 2008; Young & Neher, 2009). EPSCs recorded from snapin−/− neurons are strongly desynchronized (Pan et al, 2009), similar to the response recorded from superior cervical ganglion neurons while disrupting the association between SVs and Ca2+ channels (Mochida et al, 1996). In addition, over-expressing snapin accelerates exocytosis at low [Ca2+]i in chromaffin cells (Schmidt et al, 2013). These studies raise a fundamental mechanistic question as to how snapin regulates Ca2+ sensitivity of neurotransmitter release. In the current study, we assessed the role of snapin in SV positional priming by using two different snapin mutants and by generating a SV-targeted Ca2+-sensing probe, synaptophysin-GECO. Over-expressing snapin-L99K, defective in binding to both dynein DIC and dysbindin, revealed a striking phenotype: SVs were exposed to a lower amount of Ca2+ during trains of 100 APs (Fig6). The resulting defect in pHluorin response could be effectively reversed by raising [Ca2+]ext to 6 mM. However, this phenotype was not readily observed in neurons expressing snapin-S50D, a mutant defective in DIC-binding but retaining dysbindin-binding capacity. Based on these findings, we propose that SVs recycled through the BLOC-1/AP-3-dependent sorting pathway are more likely to be localized in close proximity to Ca2+ entry sites. This conclusion is supported by our findings that immuno-isolated SVs from adult snapin cKO mouse brains have substantially lower levels of the AP-3-dependent cargo Ti-VAMP and the Rab3a GTPase. Rab3a, by tethering SVs to the active zone protein RIM, plays a key role in recruiting SVs in close proximity to Ca2+ channels (see reviews by Gundelfinger & Fejtova, 2012; Sudhof, 2013). Interestingly, dysbindin−/− hippocampal neurons exhibit slower kinetics and smaller RRP size (Chen et al, 2008) and Drosophila dysbindin−/− neuromuscular junctions display reduced Ca2+ sensitivity of neurotransmitter release (Dickman et al, 2012). The BLOC-1 subunit dysbindin was identified as a central mediator of the presynaptic homeostatic plasticity that follows postsynaptic glutamatergic receptors inhibition (Dickman & Davis, 2009), functioning in concert with snapin in flies (Dickman et al, 2012). Interestingly, it was recently proposed that this homeostatic phenomenon involves a tightening of SV coupling to Ca2+ influx (Müller et al, 2015), and both Rab3 and dysbindin have been suggested to influence SV positional priming during such presynaptic homeostasis (Müller et al, 2011). Thus, our data and reports from others consistently indicate that BLOC-1-dependent processes contribute to the Ca2+ sensitivity of SV exocytosis, by regulating the SV-Ca2+ channels coupling. In addition, NCAM-deficient synapses were shown to rely mainly on the AP-3-dependent pathway for SV recycling and release neurotransmitters with enhanced Ca2+ sensitivity (Polo-Parada et al, 2001), also suggesting that AP-3-dependent sorting influences the Ca2+ sensitivity of SV exocytosis.
We therefore suggest that snapin mediates SV endosomal sorting through BLOC-1, driving the trafficking of SVs through the AP-3-dependent route (Route 2). This pathway would generate SVs enriched in Rab3a, which is required for positional priming and high release probability. Abolishing snapin–dysbindin coupling likely perturbs BLOC-1 function and the targeting of SVs to Ca2+ entry sites, thus impairing the synchronization of release.
Snapin coordinates endolysosomal transport and SV endosomal recycling
Remarkably, the snapin-S50D mutation, which mimics snapin phosphorylation by PKA (Chheda et al, 2001), has opposite effects on its interactions with three binding partners: reduced interaction with dynein and enhanced interaction with dysbindin and SNAP25. This mutation concomitantly inhibits endolysosomal transport by impairing snapin–dynein coupling (Fig2B and C) and increases the probability of release (Thakur et al, 2004). It has long been known that PKA enhances transmitter release (Chavez-Noriega & Stevens, 1994; Capogna et al, 1995) and enhances the intrinsic Ca2+ sensitivity of the fast-releasing SVs (i.e. “positionally” primed ones) at the calyx of Held (Yao & Sakaba, 2010). Although the phosphorylated targets were not identified in the latter study, our current findings highlight snapin as a candidate substrate for PKA-mediated regulation of fast SV exocytosis.
The phosphorylation of snapin by PKA could constitute a molecular switch between its LE transport function mediating SV turnover (Route 1) and BLOC-1/SNARE/-related functions driving SV positional priming and fusion (Route 2) (Fig7C). We propose that the balance between the two pathways is critical to fine-tune presynaptic strength. Increasing LE transport by non-phosphorylated snapin will increase SV turnover through the endolysosomal pathway (Route 1), thus diverting SV components off the presynaptic endosomal recycling pathway (Route 2) and also decreasing the releasable pool size. In contrast, phosphorylated snapin will reduce LE transport and SV turnover (Route 1), in turn maintaining releasable pool size by favoring AP-3-dependent recycling (Route 2).
The presence of endosomal intermediates in the SV cycle has been a matter of controversy since it was proposed by Heuser and Reese (1973). An alternative pathway bypassing endosome-sorting stations was suggested (Murthy & Stevens, 1998), where SVs are retrieved through a single endocytotic step involving AP-2 and clathrin (Takei et al, 1996; Granseth et al, 2006). It was proposed that AP-2-dependent recycling replaces the AP-3-dependent mechanism during development (Shetty et al, 2013). However, both pathways coexist in vivo (Korber et al, 2012). Endosomes were confirmed as sorting intermediates for SV recycling (Hoopmann et al, 2010; Uytterhoeven et al, 2011; Watanabe et al, 2014), and SVs contain various endosomal proteins as a result (Takamori et al, 2006; Morgan et al, 2013). A recent study showed that clathrin-dependent endocytosis is essential for release sites clearance rather than vesicles reuse (Hua et al, 2013). Consistently, two studies indicated that SV recycling during moderate stimulation does not involve clathrin coat assembly (Watanabe et al, 2013; Schikorski, 2014), and a third study revealed that clathrin and AP-2 are not essential for SV recycling (Kononenko et al, 2014). In particular, a newly described “ultrafast” endocytosis mechanism retrieves small endosome-like structures from the plasma membrane in response to single APs (Watanabe et al, 2013), implying that additional sorting mechanisms would be necessary to maintain appropriately sized releasable SVs.
In summary, our study reveals for the first time that endoly-sosomal processes regulate SV pool size and Ca2+ sensitivity of neurotransmitter release (Fig7C). Retrograde transport of SV cargoes through the endolysosomal pathway (Route 1) influences the SV releasable pool size, while BLOC-1/dysbindin/AP-3-dependent endosomal sorting (Route 2) regulates the positional priming of SVs. Our findings thus provide new mechanistic insights into the fine coordination of releasable pool size and synchronization of SV fusion.
Materials and Methods
Animal care and use were carried out in accordance with NIH guidelines and approved by the NIH and NINDS/NIDCD Animal Care and Use Committee.
Cortical neuron culture and transfection
Mouse cortical neuron cultures were prepared from cortical tissues of E18–19 mouse embryos using the papain method as described previously (Cai et al, 2010). Cells were plated onto 10-day-old glial feeder layers sitting on polyornithine- and Matrigel (BD Biosciences)-coated coverslips and grown overnight in plating medium: 5% FBS (Thermo Scientific), insulin (30 μg/ml, Sigma), GlutaMAX (Gibco), and B27 (Gibco) in Neurobasal (Invitrogen). Following a half media change at DIV2 using neuronal feed (1 × B27 and GlutaMAX in Neurobasal), one-fourth media change was performed every 3 days. Unless otherwise noted, neurons were transfected at DIV8 using a modified calcium phosphate method and imaged on DIV14–15.
Immunocytochemistry
Neurons were fixed at DIV10–12 with a solution containing 4% formaldehyde (Polyscience Inc.) and 4% sucrose (Sigma) in 1× PBS for 30 min at room temperature and then permeabilized with 0.1% Triton X-100 for 10 min. Cells were washed 3 times with PBS, incubated for 1 h in a PBS-based blocking buffer containing 2% BSA (Sigma) and 5% goat serum, then incubated with primary anti-bodies in blocking buffer overnight at 4°C. After three 15-min PBS washes, secondary antibodies (Alexa 488 or 546 conjugated) were applied for 1 h. Antibodies used for immunocytochemistry are as follows: monoclonal against MAP2 (BD Pharmingen, 1:1,000), polyclonal against synaptophysin (Santa Cruz H93, 1:200), and secondary antibodies (Molecular Probes, 1:400). Confocal images were obtained using an Olympus Fluoview FV1000 confocal microscope with a 1.45 NA 63× objective with sequential-acquisition setting. For fluorescence quantification, images were acquired using the same settings below saturation at a resolution of 1,600 × 1,600 pixels.
Electron microscopy
Cultured cortical neurons at DIV14 were fixed at room temperature with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 N Na+ cacodylate buffer and then stored at 4°C overnight. The samples were then treated with osmium tetroxide, en bloc mordanted with uranyl acetate, dehydrated through a series of graded ethanol washes, and embedded in epoxy resins. Thin sections were stained with uranyl acetate and lead citrate (EM Facility, National Institute of Neurological Disorders and Stroke, National Institutes of Health). Quantitative analysis was based on previous descriptions (Pan et al, 2009). Briefly, the EM thin sections were examined on a JEOL (Akishima, Japan) 1200 EX electron microscope, and digital images were captured with a CCD camera system (XR-100; Advanced Microscopy Techniques, Danvers, MA). Both symmetrical and asymmetrical SV-filled presynaptic boutons were imaged at 30,000× magnification and then analyzed by ImageJ 10.2 (NIH) with the same scaling system. Only SVs immediately adjacent to the presynaptic membrane were considered docked at the active zone (i.e., the portion of membrane apposed closely to a postsynaptic element). For some presynaptic elements not attaching to a postsynaptic element, docked SVs were not counted in those boutons. Total SVs were counted within presynaptic boutons from the AZ to the edge of the terminal. The surface area of the presynaptic terminal was traced by outlining synaptic terminal structure using the region selection tool (Schikorski and Stevens, 1997) and measured using ImageJ.
Live-cell imaging
Neurons were transferred to Tyrode’s solution containing (in mM) 10 HEPES, 10 glucose, 145 NaCl, 3 KCl, 1.3 CaCl2, and 1.3 MgCl2, pH 7.4 and ∼305 mOsm supplemented with 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μM 2-amino-5-phosphonopentanoic acid (AP5). Where indicated, 1 μM bafilomycin A (Sigma) was added to the imaging media. For the live labeling of recycling SVs, an Oyster-555-labeled antibody directed against the luminal domain of rat synaptotagmin (Synaptic Systems) was added at a 1/100 dilution. During all experiments, the temperature was maintained at 37°C using a stage-mounted incubator. Time-lapse images were acquired with an Olympus Fluoview FV1000 confocal microscope using a 1.45 NA 63× objective and a CCD camera (Hamamatsu) using either a 488-nm excitation in TIRF mode at ∼1 Hz (single-channel imaging of pHluorin, GECO, GFP-Rab7 axonal transport) or a mercury lamp source and a filter-wheel setup alternating GFP and Texas Red filter sets (Olympus) at ∼0.5 Hz (all dual-channel imaging). Data were obtained from a minimum of three independent transfections, analyzed with custom-written macros and open-source plug-ins in ImageJ.
Subcellular fractionation
Synaptosomes were isolated by centrifugation on sucrose gradients as described previously (Talbot et al, 2006). Two to three mouse brains were homogenized immediately after decapitation using a Teflon-glass tube in buffer A (mM): 320 sucrose, 1 NaHCO3, 1 MgCl2, 0.5 CaCl2, 1 PMSF, plus protease inhibitor mix (complete, Roche). After 15 min of 1,400 × g centrifugation at 4°C to clear cell debris, supernatants were pooled as total extract and centrifuged for 10 min at 13,800 × g. The resulting supernatants were recentrifuged for 25 min at 15,000 × g and concentrated in a Speed Vac (Savant) to be used as the cytosolic fraction. Pellets were resuspended in buffer A minus MgCl2 and CaCl2 (buffer B) and distributed equally on top of gradients (6 tubes) consisting in 0.9-ml layers of (top to bottom) 0.8 M, 1 M, and 1.2 M sucrose containing 1 mM of NaHCO3. These gradients were centrifuged for 2 h at 82,500 × g in a Beckman-Coulter centrifuge using a SW50Ti rotor, and ∼600 μl of crude synaptosomes was retrieved from the interface between the 1.2 and 1 M layers. The retrieved fractions were pooled and washed with 14 ml buffer B and then run for 30 min at 32,800 × g in a Sorvall RC-5 centrifuge. The resulting pellet was resuspended in TBS supplemented with protease inhibitors and used as the synaptosomal fraction.
Immuno-isolation of synaptic vesicles
Dynabeads coated with sheep anti-mouse IgG (Life Technologies) were washed once with an isolation buffer consisting of PBS with 2 mM EDTA and 0.1% BSA, and then 50-μl beads/tube were incubated with 3 μg of a monoclonal anti-synaptophysin antibody (Synaptic Systems) or a control mouse IgG overnight at room temperature. After three washes with the homogenization buffer HB (mM: 320 sucrose, 5 HEPES, 1 MgCl2, 1 PMSF, plus protease inhibitor mix), beads were incubated with a crude SV preparation consisting in a combination of SVs directly released during homogenization and the ones released from osmotically ruptured synaptosomes (Ahmed et al, 2013). Two to three mouse brains were used per immuno-isolation. Brain homogenates were centrifuged for 10 min at 1,000 × g to remove all cell debris. The supernatant (S1) was re-run for 15 min at 15,000 × g. While the supernatant (S2) containing directly released SVs was kept on ice, the pellet (P2) was resuspended in 9 ml ice-cold water and homogenized using the Teflon-glass tube to lyse synaptosomal membranes. Fifty microliters of 1 M HEPES (pH 7.4) and protease inhibitors were added immediately, and the homogenate was centrifuged for 15 min at 17,000 × g. The supernatant containing osmotically released SVs (LS1) was combined with the S2, and the resulting crude SV preparation was incubated with the antibody-coated Dynabeads for 3 h at room temperature. After three washes with HB buffer, samples were processed for SDS–PAGE and Western blotting.
Protein purification and pull-down assay
GST- and His-tagged proteins were purified according to standard procedures. Briefly, bacteria pellets were resuspended in TBS supplemented with 1 mM PMSF and a protease inhibitor mix (Roche), and cells were passed through a high-pressure homogenizer to release the recombinant proteins. After adding 20 μg/ml of DNase and RNase, the homogenates were incubated for 20 min at room temperature. One percent protein-grade Triton X-100 (Calbiochem) was then added and lysates were run for 1 h at 14,000 × g. Clarified lysates were incubated for 1 h with either glutathione sepharose (GE Healthcare) or Ni-NTA agarose (Qiagen) beads (GST- and His-tagged proteins, respectively). Beads were then loaded onto glass econo-columns (Bio-Rad) and washed extensively with either TBS (GST-tagged proteins) or MCAC buffer (500 mM NaCl, 20 mM Tris pH 8, 10% glycerol) with added PMSF and protease inhibitors. Note that the first washes were carried out at high ionic strength (1 M NaCl added to either buffer), to remove proteins from RNA and DNA fragments. Elution was carried out with either 15 mM glutathione or 500 mM imidazole, and eluates were dialyzed overnight in TBS pH 7.4 containing 10% glycerol. For the pull-down experiments, 70 pmoles of GST-tagged proteins was immobilized on glutathione sepharose beads, washed, and incubated for 1 h at room temperature with either an equimolar amount of the relevant His-tagged proteins in TBS supplemented with 0.04% Triton TX-100 and 1 mM PMSF, or supernatant from HEK cells transfected with HA-tagged snapin constructs using lipofectamine (lysed using TBS supplemented with 0.4% Triton TX-100 and protease inhibitors under agitation). After four washes, samples were processed for SDS–PAGE and Western blotting.
Western blotting
Samples were loaded onto Novex NuPAGE 4–12% Bis-Tris gels (Life Technologies) and then transferred onto nitrocellulose membranes using standard methods. Antibodies used were as follows: polyclonal against synaptotagmin, VAMP2, and Rab3a were kind gifts from Pr M. Takahashi; Snapin (Synaptic Systems); Ti-VAMP, GADPH, Rab7, and Tom20 (Santa Cruz); LC3 I/II (Novus); LAMP-1 (Abcam); Rab5 (Stressgen); Ubiquitin (Dako); monoclonal anti-ubiquitin (Santa Cruz); and T7 tag (Novagen).
Circular dichroism spectroscopy
Circular dichroism spectra were recorded in TBS pH 7.4 in the presence of 0.5 mM dithiothreitol using a JASCO J-815 spectropolarimeter equipped with thermally controlled cuvette holder. Spectra were recorded from protein samples at 0.5 mg/ml in 0.5-mm quartz cuvettes, from 200 nm to 240 nm with 1-nm step resolution and 4-s integration time.
Acknowledgments
The authors would like to thank the following people for their help: Timothy Ryan for the kind gift of VGluT-mCherry-pHluorin and VGluT-mOrange plasmids; Kevin Staras for dendra-synaptophysin; Wei Li for dysbindin antibody and constructs; Susan Cheng and Virginia Crocker at the NINDS facility for electron microscopy; D. Krepkiy in the Swartz laboratory for help with CD spectra acquisitions; members of the Sheng laboratory for helpful discussions; and Devera Schoenberg for proof editing. The work was supported by the Intramural Research Program of NINDS, NIH ZIA HS003029, and ZIA NS002946 (Z-H. Sheng).
Author contributions
JDG designed and conducted the experiments and data analysis. Z-HS is a senior author who designed the project. JDG and Z-HS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures
Supplementary Figure Legends
Review Process File
References
- Ahmed S, Holt M, Riedel D, Jahn R. Small-scale isolation of synaptic vesicles from mammalian brain. Nat Protoc. 2013;8:998–1009. doi: 10.1038/nprot.2013.053. [DOI] [PubMed] [Google Scholar]
- Ariel P, Ryan TA. Optical mapping of release properties in synapses. Front Neural Circuits. 2010;4:18. doi: 10.3389/fncir.2010.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron. 2008;57:536–545. doi: 10.1016/j.neuron.2007.12.026. [DOI] [PubMed] [Google Scholar]
- Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68:73–86. doi: 10.1016/j.neuron.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, Gahwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Feng YQ, Hao CJ, Guo XL, He X, Zhou ZY, Guo N, Huang HP, Xiong W, Zheng H, Zuo PL, Zhang CX, Li W, Zhou Z. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J Cell Biol. 2008;181:791–801. doi: 10.1083/jcb.200711021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X-T, Zhou B, Mei-Yao Lin M-Y, Qian Cai Q, Sheng Z-H. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209:377–386. doi: 10.1083/jcb.201412046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chheda MG, Ashery U, Thakur P, Rettig J, Sheng ZH. Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat Cell Biol. 2001;3:331–338. doi: 10.1038/35070000. [DOI] [PubMed] [Google Scholar]
- Darcy KJ, Staras K, Collinson LM, Goda Y. Constitutive sharing of recycling synaptic vesicles between presynaptic boutons. Nat Neurosci. 2006;9:315–321. doi: 10.1038/nn1640. [DOI] [PubMed] [Google Scholar]
- Denker A, Bethani I, Krohnert K, Korber C, Horstmann H, Wilhelm BG, Barysch SV, Kuner T, Neher E, Rizzoli SO. A small pool of vesicles maintains synaptic activity in vivo. Proc Natl Acad Sci USA. 2011;108:17177–17182. doi: 10.1073/pnas.1112688108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A, Rizzoli SO. Synaptic vesicle pools: an update. Front Synaptic Neurosci. 2010;2:135. doi: 10.3389/fnsyn.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002;22:3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro SM, Falcon-Perez JM, Tenza D, Setty SR, Marks MS, Raposo G, Dell’Angelica EC. BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol Biol Cell. 2006;17:4027–4038. doi: 10.1091/mbc.E06-05-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Tong A, Davis GW. Snapin is critical for presynaptic homeostatic plasticity. J Neurosci. 2012;32:8716–8724. doi: 10.1523/JNEUROSCI.5465-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93:423–432. doi: 10.1016/s0092-8674(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Feng YQ, Zhou ZY, He X, Wang H, Guo XL, Hao CJ, Guo Y, Zhen XC, Li W. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res. 2008;106:218–228. doi: 10.1016/j.schres.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Fernandes AC, Uytterhoeven V, Kuenen S, Wang YC, Slabbaert JR, Swerts J, Kasprowicz J, Aerts S, Verstreken P. Reduced synaptic vesicle protein degradation at lysosomes curbs TBC1D24/sky-induced neurodegeneration. J Cell Biol. 2014;207:453–462. doi: 10.1083/jcb.201406026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Hadwiger G, Nonet ML, Richmond JE. Direct interactions between C. elegans RAB-3 and Rim provide a mechanism to target vesicles to the presynaptic density. Neurosci Lett. 2008;444:137–142. doi: 10.1016/j.neulet.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Gundelfinger ED, Fejtova A. Molecular organization and plasticity of the cytomatrix at the active zone. Curr Opin Neurobiol. 2012;22:423–430. doi: 10.1016/j.conb.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Sudhof TC, Schneggenburger R. RIM determines Ca(2)+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D, Torres CA, Setlik W, Cebrian C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, Gershon M, Sulzer D. Regulation of presynaptic neurotransmission by macroautophagy. Neuron. 2012;74:277–284. doi: 10.1016/j.neuron.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann P, Punge A, Barysch SV, Westphal V, Buckers J, Opazo F, Bethani I, Lauterbach MA, Hell SW, Rizzoli SO. Endosomal sorting of readily releasable synaptic vesicles. Proc Natl Acad Sci USA. 2010;107:19055–19060. doi: 10.1073/pnas.1007037107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, Edwards RH. v-SNARE composition distinguishes synaptic vesicle pools. Neuron. 2011;71:474–487. doi: 10.1016/j.neuron.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Woehler A, Kahms M, Haucke V, Neher E, Klingauf J. Blocking endocytosis enhances short-term synaptic depression under conditions of normal availability of vesicles. Neuron. 2013;80:343–349. doi: 10.1016/j.neuron.2013.08.010. [DOI] [PubMed] [Google Scholar]
- Ilardi JM, Mochida S, Sheng ZH. Snapin: a SNARE-associated protein implicated in synaptic transmission. Nat Neurosci. 1999;2:119–124. doi: 10.1038/5673. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- John Peter AT, Lachmann J, Rana M, Bunge M, Cabrera M, Ungermann C. The BLOC-1 complex promotes endosomal maturation by recruiting the Rab5 GTPase-activating protein Msb3. J Cell Biol. 2013;201:97–111. doi: 10.1083/jcb.201210038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Sudhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamin D, Lauterbach MA, Westphal V, Keller J, Schonle A, Hell SW, Rizzoli SO. High- and low-mobility stages in the synaptic vesicle cycle. Biophys J. 2010;99:675–684. doi: 10.1016/j.bpj.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. Synaptic vesicle reuse and its implications. Neuroscientist. 2006;12:57–66. doi: 10.1177/1073858405281852. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. Synaptic vesicle recycling at CNS synapses without AP-2. J Neurosci. 2009;29:3865–3874. doi: 10.1523/JNEUROSCI.5639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokotos AC, Cousin MA. Synaptic vesicle generation from central nerve terminal endosomes. Traffic. 2015;16:229–240. doi: 10.1111/tra.12235. [DOI] [PubMed] [Google Scholar]
- Kononenko NL, Puchkov D, Classen GA, Walter AM, Pechstein A, Sawade L, Kaempf N, Trimbuch T, Lorenz D, Rosenmund C, Maritzen T, Haucke V. Clathrin/AP-2 mediate synaptic vesicle reformation from endosome-like vacuoles but are not essential for membrane retrieval at central synapses. Neuron. 2014;82:981–988. doi: 10.1016/j.neuron.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Korber C, Horstmann H, Satzler K, Kuner T. Endocytic structures and synaptic vesicle recycling at a central synapse in awake rats. Traffic. 2012;13:1601–1611. doi: 10.1111/tra.12007. [DOI] [PubMed] [Google Scholar]
- Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, Craige B, Lee JD, Talbot K, Pare JF, Smith Y, Faundez V. The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell. 2011;22:4854–4867. doi: 10.1091/mbc.E11-07-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Nemecek D, Schindler C, Smith WJ, Ghirlando R, Steven AC, Bonifacino JS, Hurley JH. Assembly and architecture of biogenesis of lysosome-related organelles complex-1 (BLOC-1) J Biol Chem. 2012;287:5882–5890. doi: 10.1074/jbc.M111.325746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Foss SM, Dobryy YL, Park CK, Hires SA, Shaner NC, Tsien RY, Osborne LC, Voglmaier SM. Concurrent imaging of synaptic vesicle recycling and calcium dynamics. Front Mol Neurosci. 2011;4:34. doi: 10.3389/fnmol.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra V, Burden JJ, Thorpe JR, Smith IT, Smith SL, Hausser M, Branco T, Staras K. A preferentially segregated recycling vesicle pool of limited size supports neurotransmission in native central synapses. Neuron. 2012;76:579–589. doi: 10.1016/j.neuron.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2 + channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Morgan JR, Comstra HS, Cohen M, Faundez V. Presynaptic membrane retrieval and endosome biology: defining molecularly heterogeneous synaptic vesicles. Cold Spring Harb Perspect Biol. 2013;5:a016915. doi: 10.1101/cshperspect.a016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Pym EC, Tong A, Davis GW. Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron. 2011;69:749–762. doi: 10.1016/j.neuron.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Genç Ö, Davis GW. RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron. 2015;85:1056–1069. doi: 10.1016/j.neuron.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Navarro A, Encinar JA, Lopez-Mendez B, Aguado-Llera D, Prieto J, Gomez J, Martinez-Cruz LA, Millet O, Gonzalez-Ros JM, Fernandez-Ballester G, Neira JL, Ferrer-Montiel A. Mutation of Ser-50 and Cys-66 in Snapin modulates protein structure and stability. Biochemistry. 2012;51:3470–3484. doi: 10.1021/bi201574t. [DOI] [PubMed] [Google Scholar]
- Newell-Litwa K, Salazar G, Smith Y, Faundez V. Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell. 2009;20:1441–1453. doi: 10.1091/mbc.E08-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Chintala S, Jenkins S, Pare JF, McGaha L, Smith Y, Faundez V. Hermansky-Pudlak protein complexes, AP-3 and BLOC-1, differentially regulate presynaptic composition in the striatum and hippocampus. J Neurosci. 2010;30:820–831. doi: 10.1523/JNEUROSCI.3400-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PY, Tian JH, Sheng ZH. Snapin facilitates the synchronization of synaptic vesicle fusion. Neuron. 2009;61:412–424. doi: 10.1016/j.neuron.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/s0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- Pu J, Schindler C, Jia R, Jarnik M, Backlund P, Bonifacino JS. BORC, a Multisubunit Complex that Regulates Lysosome Positioning. Dev Cell. 2015;33:176–188. doi: 10.1016/j.devcel.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET. The role of non-canonical SNAREs in synaptic vesicle recycling. Cell Logist. 2012;2:20–27. doi: 10.4161/cl.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO. Synaptic vesicle recycling: steps and principles. EMBO J. 2014;33:788–822. doi: 10.1002/embj.201386357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T, Schoenenberger P, Jezek K, Oertner TG. Developmental refinement of vesicle cycling at Schaffer collateral synapses. Neuron. 2013;77:1109–1121. doi: 10.1016/j.neuron.2013.01.021. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Salazar G, Craige B, Styers ML, Newell-Litwa KA, Doucette MM, Wainer BH, Falcon-Perez JM, Dell’Angelica EC, Peden AA, Werner E, Faundez V. BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol Biol Cell. 2006;17:4014–4026. doi: 10.1091/mbc.E06-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuber A, Rudge R, Danglot L, Raposo G, Binz T, Poncer JC, Galli T. Loss of AP-3 function affects spontaneous and evoked release at hippocampal mossy fiber synapses. Proc Natl Acad Sci USA. 2006;103:16562–16567. doi: 10.1073/pnas.0603511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T. Readily releasable vesicles recycle at the active zone of hippocampal synapses. Proc Natl Acad Sci USA. 2014;111:5415–5420. doi: 10.1073/pnas.1321541111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Schirra C, Matti U, Stevens DR, Rettig J. Snapin accelerates exocytosis at low intracellular calcium concentration in mouse chromaffin cells. Cell Calcium. 2013;54:105–110. doi: 10.1016/j.ceca.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Setty SR, Tenza D, Truschel ST, Chou E, Sviderskaya EV, Theos AC, Lamoreux ML, Di Pietro SM, Starcevic M, Bennett DC, Dell’Angelica EC, Raposo G, Marks MS. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol Biol Cell. 2007;18:768–780. doi: 10.1091/mbc.E06-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Sytnyk V, Leshchyns’ka I, Puchkov D, Haucke V, Schachner M. The neural cell adhesion molecule promotes maturation of the presynaptic endocytotic machinery by switching synaptic vesicle recycling from adaptor protein 3 (AP-3)- to AP-2-dependent mechanisms. J Neurosci. 2013;33:16828–16845. doi: 10.1523/JNEUROSCI.2192-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol. 2003;13:899–910. doi: 10.1016/s0960-9822(03)00338-5. [DOI] [PubMed] [Google Scholar]
- Staras K, Branco T, Burden JJ, Pozo K, Darcy K, Marra V, Ratnayaka A, Goda Y. A vesicle superpool spans multiple presynaptic terminals in hippocampal neurons. Neuron. 2010;66:37–44. doi: 10.1016/j.neuron.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starcevic M, Dell’Angelica EC. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1) J Biol Chem. 2004;279:28393–28401. doi: 10.1074/jbc.M402513200. [DOI] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O’Neill FA, Walsh D, Kendler KS. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71:337–348. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Cho DS, Ong WY, Benson MA, Han LY, Kazi HA, Kamins J, Hahn CG, Blake DJ, Arnold SE. Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet. 2006;15:3041–3054. doi: 10.1093/hmg/ddl246. [DOI] [PubMed] [Google Scholar]
- Thakur P, Stevens DR, Sheng ZH, Rettig J. Effects of PKA-mediated phosphorylation of Snapin on synaptic transmission in cultured hippocampal neurons. J Neurosci. 2004;24:6476–6481. doi: 10.1523/JNEUROSCI.0590-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian JH, Wu ZX, Unzicker M, Lu L, Cai Q, Li C, Schirra C, Matti U, Stevens D, Deng C, Rettig J, Sheng ZH. The role of Snapin in neurosecretion: snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J Neurosci. 2005;25:10546–10555. doi: 10.1523/JNEUROSCI.3275-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell. 2011;145:117–132. doi: 10.1016/j.cell.2011.02.039. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Edwards RH. Do different endocytic pathways make different synaptic vesicles? Curr Opin Neurobiol. 2007;17:374–380. doi: 10.1016/j.conb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- Wang X, Hu B, Zimmermann B, Kilimann MW. Rim1 and rabphilin-3 bind Rab3-GTP by composite determinants partially related through N-terminal alpha -helix motifs. J Biol Chem. 2001;276:32480–32488. doi: 10.1074/jbc.M103337200. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Rost BR, Camacho-Perez M, Davis MW, Sohl-Kielczynski B, Rosenmund C, Jorgensen EM. Ultrafast endocytosis at mouse hippocampal synapses. Nature. 2013;504:242–247. doi: 10.1038/nature12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Trimbuch T, Camacho-Perez M, Rost BR, Brokowski B, Sohl-Kielczynski B, Felies A, Davis MW, Rosenmund C, Jorgensen EM. Clathrin regenerates synaptic vesicles from endosomes. Nature. 2014;515:228–233. doi: 10.1038/nature13846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TC, Chin LS, Li Y, Roudabush FL, Li L. Regulation of synaptophysin degradation by mammalian homologues of seven in absentia. J Biol Chem. 2002;277:10273–10282. doi: 10.1074/jbc.M107857200. [DOI] [PubMed] [Google Scholar]
- Willeumier K, Pulst SM, Schweizer FE. Proteasome inhibition triggers activity-dependent increase in the size of the recycling vesicle pool in cultured hippocampal neurons. J Neurosci. 2006;26:11333–11341. doi: 10.1523/JNEUROSCI.1684-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Sakaba T. cAMP modulates intracellular Ca2+ sensitivity of fast-releasing synaptic vesicles at the calyx of Held synapse. J Neurophysiol. 2010;104:3250–3260. doi: 10.1152/jn.00685.2010. [DOI] [PubMed] [Google Scholar]
- Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron. 2009;63:482–496. doi: 10.1016/j.neuron.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca(2)(+) indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figure Legends
Review Process File