

Abstract
Clinical trials utilizing mesenchymal stem cells (MSCs) for severe vascular diseases have highlighted the need to effectively engraft cells and promote pro-angiogenic activity. A functional material accomplishing these two goals is an ideal solution as spatiotemporal and batch-to-batch variability in classical therapeutic delivery can be minimized, and tissue regeneration would begin rapidly at the implantation site. Gelatin may serve as a promising biomaterial due to its excellent biocompatibility, biodegradability, and non-immuno/antigenicity. However, the dissolution of gelatin at body temperature and quick enzymatic degradation in vivo have limited its use thus far. To overcome these challenges, an injectable, in situ crosslinkable gelatin was developed by conjugating enzymatically-crosslinkable hydroxyphenyl propionic acid (GHPA). When MSCs are cultured in 3D in vitro or injected in vivo in GHPA, spontaneous endothelial differentiation occurs, as evidenced by marked increases in endothlelial cell marker expressions (Flk1, Tie2, ANGPT1, vWF) in addition to forming an extensive perfusable vascular network after 2-week subcutaneous implantation. Additionally, favorable host macrophage response is achieved with GHPA as shown by decreased iNOS and increased MRC1 expression. These results indicate GHPA as a promising soluble factor-free cell delivery template which induces endothelial differentiation of MSCs with robust neovasculature formation and favorable host response.
Keywords: injectable biomaterials, in situ crosslinking, gelatin, mesenchymal stem cell, vasculogenesis
1. Introduction
Numerous material platforms have been funcionalized with biological molecules (e.g. vascular endothelial growth factor) to stimulate a pro-angiogenic response of embedded cells in order to effectively engineer functional new tissue.[1-3] In many of these studies, the pro-angiogenic activity is thought to rely exclusively on the biological molecules that are engrafted or delivered within these constructs, and the direct influence of the material itself has received little attention. Although these approaches have shown promise in vascularizing engineered constructs, they face significant challenges for stem cell delivery, because encapsulated stem cells show limited survival, vascular diffferentiation, and functional regeneration that are required for effective repair of vascular tissue.[4] Moreover, variations in the spatiotemporal release, batch-to-batch uniformity, and efficiency of presentation of biological molecules has hindered progress since these problems could result in heterogenous differentiation with unexpected side effects. Therefore, the principles for designing functional materials as a instructive cell delivery platform are evolving. While the traditional view considered the extracellular matrix (ECM) as a passive scaffold material mainly providing biomechanical support, it is now clear that ECM plays a central role in the outside-in signaling that influences the structure and functions of the cells with which it interacts.[5-7] Therefore, engineered ECM without the addition of extrinsic bioactive molecules represents an ideal functional material source that (1) can be specifically modified to engraft stem cells for maximal cell survival; (2) provide uniform functional and structural cues to cells in order to minimize spatiotemporal variations; and (3) instruct tissue regeneration as an all-in-one directive material platform that does not require release or presentation of additional biological molecules.
Mesenchymal stem cells (MSCs) are generated in culture from the adherent, non-hematopoietic population of the bone marrow (BM).[8] Due to their robust proliferation and survival, a single clinically-relevant dose (∼108) of therapeutic-grade MSCs can be obtained in 7 days from only a small starting volume (5 ml) of BM.[9] However, MSCs face the challenge of poor engraftment when delivered in vivo.[8,10] Moreover, the potential of MSCs to differentiate into endothelial cells has not been fully harnessed and remains unclear.[8,11] Published reports suggest that MSCs can contribute to endothelium, smooth muscle, or even myogenic tissue in cardiovascular repair at low levels; however, heterogeneous cellular responses have blurred the understanding of this process.[11, 12] Furthermore, clinical delivery of MSCs to diseased vascular tissue has proven unsuccessful, due primarily to ineffective retention of the cells at the site of implant, suggesting that an optimized, biocompatible delivery system for MSCs would be highly valuable. There have been several reports studying methods to differentiate MSCs into endothelial cells.[13-16] However, most studies employed in vitro experiments, thus would require pre-differentiation of MSCs prior to transplantation. Additionally, none of these approaches enables dynamic, in situ endothelial differentiation upon engraftment to a target site. Hence, a functional material that not only provides the mechanical support for the implanted stem cells but also serves as a guide for in situ stem cell differentiation while maintaining cell viability is highly desirable, but largely lacking to this date.
Gelatin, a form of denatured collagen, can be an ideal material source as it is known for its excellent biocompatibility and biodegradability, as well as adhesiveness for cell attachment, and absence of immuno/antigenicity.[17] However, the in vivo application of gelatin has been limited thus far due to its low upper critical solution temperature and quick enzymatic degradation, resulting in few experiments that have aimed to understand the functional impact of gelatin for stem cell delivery.[18] To address these issues, we have recently developed injectable, in situ-crosslinkable gelatin hydrogels.[19] Conjugation of hydroxyphenyl propionic acid to the free amines of gelatin (GHPA) enabled rapid H2O2- and horseradish peroxide (HRP)-mediated crosslinking. Such modification allowed the use of gelatin as an injectable thermostable hydrogel with tunable degradation resistance and mechanical properties for in vivo applications.
When encapsulated and cultured within GHPA hydrogels, MSCs showed high cell viability at 15 days with de novo expression of endothelial-specific markers in vitro, and formed perfusable vascular networks that resulted from MSCs undergoing endothelial differentiation in vivo. Our study indicates that GHPA hydrogels are an ideal platform for regenerating vascularized tissue from encapsulated MSCs in vivo, and display intrinsic properties that stimulate vascular induction. The GHPA hydrogels can be combined with established stem cell therapies to develop the next generation of clinically-applicable materials for treating severe vascular diseases and damage.
2. Results and Discussion
2.1. Gelatin-Hydroxyphenyl Propionic Acid (GHPA) Synthesis and Characterization
Injectable and sprayable hydrogels were successfully produced from hydroxyphenyl propionic acid-conjugated gelatin that underwent in situ oxidative crosslinking among the phenolic moieties catalyzed by HRP in the presence of H2O2 (Figure 1A and B). As seen in Figure 1B, two GHPA solutions were prepared in order to avoid premature gelation where one GHPA solution contains HRP with cells, and the other GHPA solution contains H2O2. HRP/cells- or H2O2-containing GHPA solutions are loaded into separate syringes, and the solutions can be injected or sprayed for in situ cross-linking towards mimally-invasive in vivo applications.[19] 1H NMR and UV-vis spectra of GHPA and unmodified gelatin in Figure 1C and D confirmed successful conjugation with 145.1 μmol HPA/g total polymer.
Figure 1.
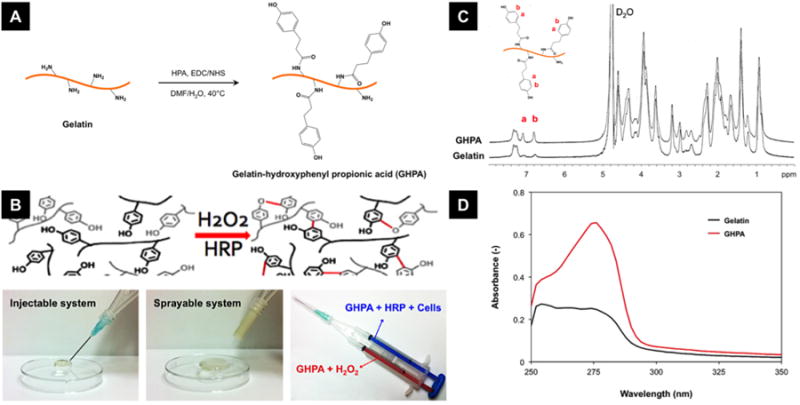
(A) Synthesis of gelatin-hydroxyphenyl propionic acid (GHPA). (B) Rapid gelation of GHPA by H2O2 and horseradish peroxidase (HRP)-catalyzed oxidative crosslinking. Bottom right image shows a dual-syringe system for cell-containing GHPA injections for in situ crosslinking, and this system can be used for injection or spraying. (C) 1H NMR and (D) UV-vis spectra of synthesized GHPA and unmodified gelatin.
Mechanical properties were characterized without cells at 37°C in wet conditions. All test samples underwent complete gelation within 20 seconds and their storage moduli (G′) were measured at varying GHPA and H2O2 concentrations (Figure 2). Overall, crosslinked GHPA gels exhibited storage moduli ranging from ∼100 Pa to ∼2500 Pa which are typical of soft hydrogels and similar to native tissue.[20] As GHPA and/or hydrogen peroxide concentration (s) increased up to 7% and 0.01% (w/v) respectively, the storage modulus increased due to enhanced crosslinking. A previous study demonstrated a similar relationship wherein the resistance to degradation correlated directly with crosslinking density.[19] Three different formulations (arrows in Figure 2) were chosen for the cell experiments, each with different mechanical properties. Conditions with storage moduli < 500 Pa were excluded from the selection, as these soft gels were difficult to handle and degraded within a few days, even in in vitro cell cultures.
Figure 2.
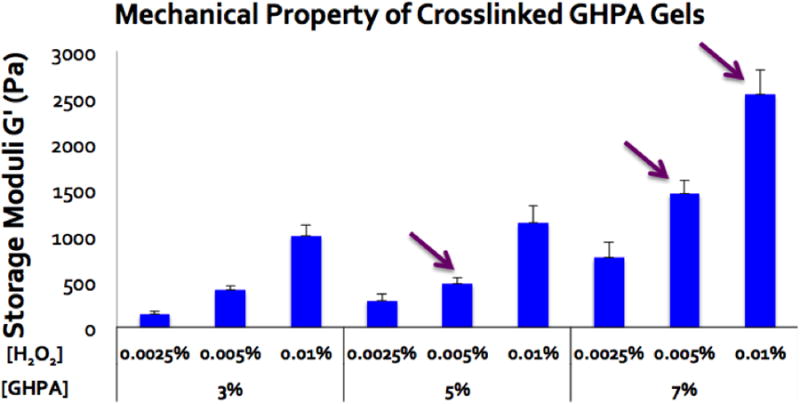
Storage moduli (G′) of crosslinked GHPA gels with varying concentrations of GHPA [%w/v] and H2O2 [%w/v] were measured using a rheometer with N=5 and error bars = +1 SEM. The compositions indicated with arrows were used for the following biological experiments.
2.2. In Vitro 3D Culture of Mesenchymal Stem Cells (MSCs) in GHPA
Using the dual-syringe system, GHPA solutions containing MSCs, HRP, and/or H2O2 were mixed upon injection and gelled in a 24-well plate of tissue culture polystyrene (TCPS) for 15 days of in vitro 3D culture. Reduction of resazurin was used as an indicator of live metabolic cells to measure viabiliity on days 1, 7, and 15 (Figure 3A).[21] Among the different GHPA compositions, there were no statistical differences in cell viability, although the condition with the highest GHPA and H2O2 contents (7%:0.01%) exhibited a slight decrease in viability. Hydrogen peroxide, a known cytotoxic agent, may account for the low viability at 1 and 7 days as well as the retarded growth in 7%:0.01% condition compared to other compositions.[22] In addition, inefficient diffusion of nutrients and wastes inherent in 3D static culture may also have negatively affected the cell viability.[23] Despite its shortcomings at the early culture period, viability of MSCs in GHPA gels greatly improved to around 80% for all GHPA conditions by day 15.
Figure 3.
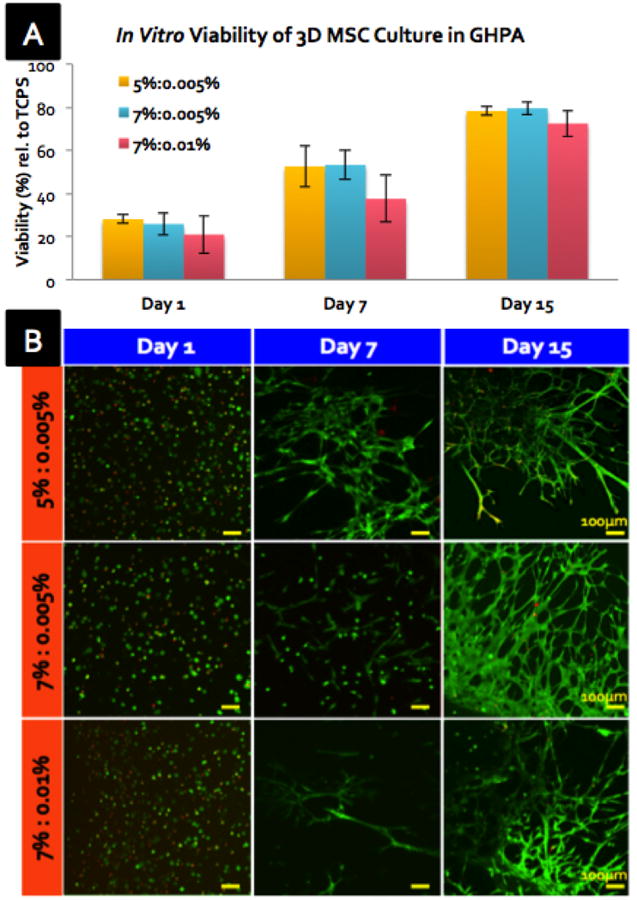
(A) In vitro cell viability of MSCs encapsulated in crosslinked GHPA gels on days 1, 7, and 15 compared to MSCs on tissue culture polystyrene (TCPS) by resazurin reduction with N=3 and error bars = ±1 SD. X%:Y% denotes X %w/v gelatin and Y %w/v H2O2. (B) Z-stacked confocal images of Live/Dead stained 3D MSC culture in GHPA on days 1, 7, and 15.
As a collagen-derived material, gelatin possesses numerous cell binding recognition sites with the RGD sequence being the most well-studied and prevalent site.[24] This is a crucial advantage of collagen- or gelatin-based materials over synthetics (ie. Poly (ethylene glycol) hydrogels) whose cell attachment relies critically on tethered molecules with reduced viability.[25] Most anchorage-dependent cells require attachment and spreading on a culture substrate for survival and proliferation, and poor cell attachment usually results in a rounded cell morphology and death due to anoikos.[26] The improved cell viability and healthy cell morphology upon 3D encapsulated culture were also evidenced in live/dead imaging over time (Figure 3B). MSCs appeared rounded yet viable at day 1; by day 7, most cells began extending through the gel, and by day 15 the cells formed highly-branched networks within the gel, while the top surface of GHPA gel was completely covered by a confluent cell monolayer (data not shown). Overall, crosslinked GHPA gels supported robust MSC viabilty and proliferation within and on the surface of the material, likely due to the favorable properties derived from collagen. Furthermore, changes in cell morphology and organization into unusual, highly-branched networks not only demonstrated active cell-material and cell-cell interactions, but led to question if MSCs began to differentiate within the GHPA gels.
2.3. In Vitro MSC Differentiation to Endothelial Lineage in GHPA
Since the organization of branched networks was observed in MSCs encapsulated in GHPA gels on day 15 in vitro (Figure 3B), we tested the ability of the material to promote MSC differentiation to specific lineages. An initial differentiation survey was performed by RT-PCR for several myogenic (MyoD, Myogenin), neuronal (NSE, Trk-A, NFL, NFM), and endothelial (VEGFA, CD31, Flk1, ANGPT1, ANGPT2, Tie1, Tie2, VE-cad, vWF) markers. Among the markers investigated, the expression of eight vascular-endothelial lineage markers was significnatly up-regulated, compared to the TCPS control (Figure 4A). Of note, MSCs in all conditions were positive for a neuronal marker NSE, and GHPA gels promoted very weak expression of Trk-A. However, no significant up-regulation in expression of neuronal markers in MSCs cultured in GHPA gels was observed relative to TCPS control, while the expression of eight vascular-endothelial markers were clearly up-regulated. Importantly, MSCs in all GHPA gels demonstrated de novo expression of critical, endothelial-specific markers, such as Tie2, ANGPT1, VE-Cadherin, and vWF, that are only observed in MSCs when stimulated with meticulous treatment with bioactive molecules or in co-culture with endothelial cells.[13,44]
Figure 4.
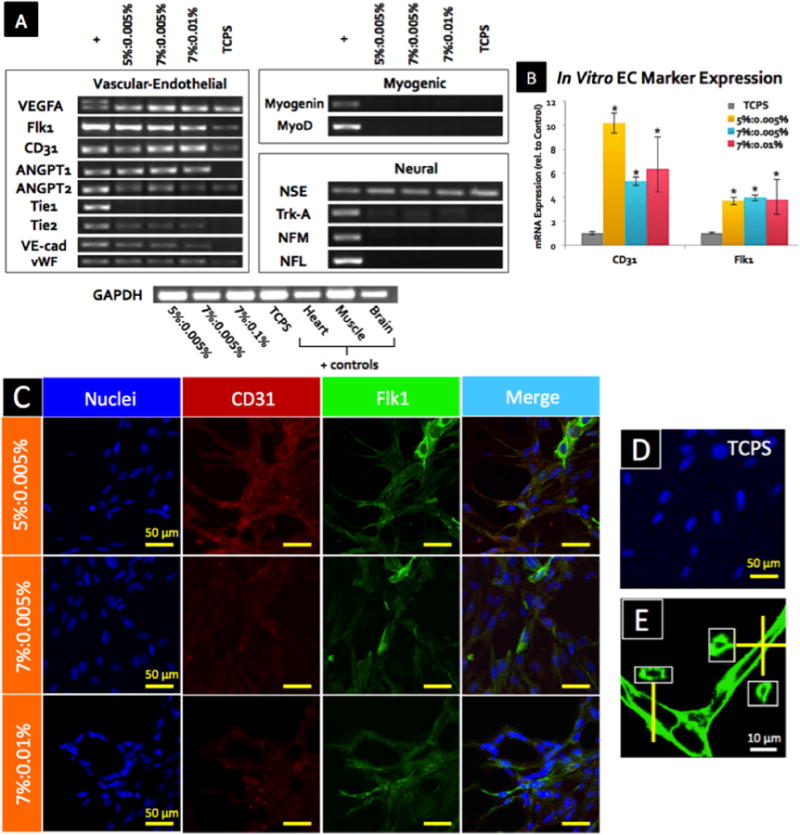
(A) Expression for vascular-endothelial, myogenic, and neural lineage markers in MSCs was determined from mRNA after 15 days of culture in GHPA gels by RT-PCR. + indicates positive controls from heart (vascular-endothelial), skeletal muscle (myogenic), and brain (neural) tissues, while TCPS indicates control MSCs cultured on on tissue culture poly styrene. (B) Expression for endothelial cell markers CD31 and Flk1 in MSCs was determined from mRNA after 15 days of culture in GHPA gels by qRT-PCR with N=3 and error bars = ±1 SEM. * indicates p<0.05 in comparison to the control MSCs on tissue culture plate. (C) CD31, Flk1 and nuclei were stained and imaged after 15 days of culture in GHPA gels, and a merged image for the TCPS control is shown in (D). Z-stacked confocal images are shown in (C). (E) F-actin staining of MSCs after 15 days of culture in 7%:0.005% condition showed clear lumen formation. Insets contain orthogonal views.
Hence, we decided to further characterize potential MSC differentiation into an endothelial lineage in vitro. First, quantitative RT-PCR (qRT-PCR) for CD31 and Flk1/VEGFR-2 showed remarkable up-regulation of CD31 (> 5 fold) and Flk1 (≈ 4 fold) expression in comparison to MSCs cultured on TCPS (p < 0.05) (Figure 4B). Subsequently, MSCs stained positive for Flk1 and CD31 in all crosslinked GHPA conditions (Figure 4C), compared to non-detectable levels of CD31 and Flk1 expression in TCPS control (Figure 4D). These results demonstrate the causal role of GHPA on in vitro MSC differentiation into the endothelial lineage at both the gene and protein levels. Furthermore, F-actin staining of MSCs in GHPA gel revealed clear lumen formation as shown in Figure 4E.
Previous studies employing similar gelatin materials have shown that gels < 600 Pa in stiffness promoted neuronal differentiation while stiff gels > 8000 Pa promoted myogenic differentiation of human mesenchymal stem cells in vitro with the use of mitomycin.[42,43] Taken together, our results suggest that GHPA gels with stiffness in the range of 600 ∼ 2500 Pa have potential to promote MSC differentiation towards an endothelial lineage in vitro.
Existing literature shows that cell binding to the RGD sequence on gelatin or denatured collagen involves integrin αvβ3, which is also found to cross-talk with Flk1, thereby promoting proliferation, migration and tubulogenesis of endothelial cells.[24, 27] Additionally, blocking of αvβ3 is proven to be an effective way to restrict angiogenesis as an anti-cancer therapy, signifying the necessity and importance of αvβ3 in angiogenesis.[28] Hence, prevalent RGD ligands on GHPA likely promoted endothelial differentiation and subsequent angiogenenesis by the MSCs via the αvβ3-Flk1 mechanism, which is in agreement with the previous works.[27, 29] Elucidating the exact mechanism of inducing endothelial differentiation and tubulogenesis from MSCs by crosslinked GHPA gels is a major subject of our next study.
2.4. In Vivo Delivery, Engraftment and Tracking of MSCs in GHPA Gels
In order to confirm the effect of GHPA gels on MSCs in vivo, Flk1-LacZ MSC-containing GHPA gels were injected into porous, non-biodegradable polyvinyl alcohol (PVA) sponges to allow for the tracking of delivered cells and GHPA as a target organ model.[30] Multiple gel-PVA sponge complexes were implanted into the ventral subcutaneous regions of wild type mice for 2 weeks (Figure 5A). Flk1, the murine analogue to human KDR/VEGFR2, is a well-established marker of vasculature and considered to be endothelial cell-specific. Therefore, Flk1-LacZ MSCs that begin expressing Flk1 in these experiments can be detected with β-galactosidase staining, and are indicative of endothelial differentiation. Flk1-LacZ transgenic MSCs were used to track and distinguish the implanted cells from host wild type cells, and provided a convenient reporter system for monitoring MSC differentiation into endothelial cells.
Figure 5.
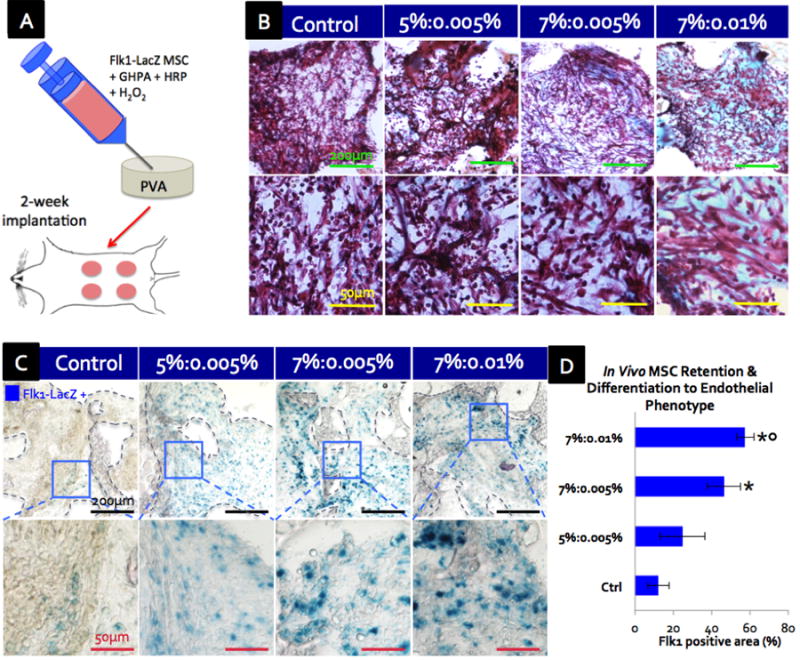
(A) Schematic of in vivo experiment where Flk1-LacZ MSCs-containing GHPA was injected into and crosslinked within a porous PVA scaffold for a murine ventral subcutaneous implantation. (B) Trichrome green staining of cross-sections of scaffolds at 2 weeks post implantation where cytoplasm is stained red, erythrocytes pink and collagen/GHPA gels blue/green. (C) β-galactosidase staining shows that delivered Flk1-LacZ transgenic MSCs were retained and became Flk1-LacZ+(blue) post 2-week implantation in crosslinked GHPA conditions. The boxes indicate Flk1-LacZ+ cell-containing areas. (D) Quantification of retained MSCs that differentiated into an endothelial phenotype (Flk1-LacZ+ cells) post 2-week implantation with error bars = ±1 SEM and N=4. Statistical significance with p < 0.05 is indicated with * in comparison to the control, and ○ in comparison to 5%:0.005%. (B-C) Top row images with scale bars = 200 μm, and bottom row images with scale bars = 50μm.
After 2 weeks of implantation, the scaffolds were harvested for analyses. Trichrome green staining was used to visualize newly-formed collagen or injected GHPA (green-light blue), cytoplasm of various cell types (purple-red), and erythrocytes (small pink rings due to the lack of nuclei) (Figure 5B). In all test conditions, there was robust leukocyte infiltration throughout the scaffolds, and groups of erythrocytes were often observed as well. However, there were two significant differences among the conditions: 1) more collagen and/or GHPA was present throughout the scaffolds in the conditions with higher GHPA and hydrogen peroxide contents, and sometimes lumps of the remaining GHPA were observed in crosslinked GHPA conditions (e.g. lower left corner in the upper image of 7%:0.01% condition in Figure 5B), and 2) crosslinked GHPA conditions exhibited extensive vascular networks throughout the scaffolds, with organized branches of cells extending a few hundred microns and containing erythrocytes which indicates functional, perfused vasculature, while the control condition lacks such structures (e.g. images in the lower panel of Figure 5B). Additionally, it was evident that there were no giant foreign body cells or fibrous capsule formation around the injected gels, indicating the non-inflammatory nature of GHPA. Hence, the conjugation of hydroxyphenyl propionic acid to geatin likely retains the non-immuno/antigenicity inherited from gelatin.
β-galactosidase staining revealed increasing numbers of the implanted Flk1-LacZ+ MSCs (blue) retained in crosslinked GHPA conditions at 2 weeks post injection, indicating differentiation of the implanted MSC into Flk1/VEGFR2+ endothelial lineage in vivo (Figure 5C and D). In particular 7%:0.01% and 7%:0.005% showed 4- and 3-fold increases in Flk1+ MSCs, respectively, compared to the control for which PVA scaffolds were loaded with a non-crosslinked GHPA solution containing Flk1-LacZ MSCs and HRP but without H2O2 for implantation. This result confirms that GHPA promotes MSC differentiation into an endothelial lineage in vivo as well as in vitro, and that crosslinking of the GHPA gel is necessary to drive this event.
2.5. In Vivo Angiogenesis and Endothelial Differentiation of MSCs Delivered in GHPA
In order to visualize the functional neovasculature in the implanted scaffolds, mice were perfused with heparinized saline containing fluorescent microbeads for micro-angiography before harvesting the scaffolds.[31] The resulting micro-angiograms from the surface and cross-sections of the scaffolds for each condition are shown in Figure 6A and quantified in Figure 6B. All angiograms presented are from the same mouse. Neovasculature in implanted scaffolds can be distinguished from the vasculature in the native host tissues around the implantation site in two important ways: 1) implanted scaffolds were not as profusely vascularized as the surrounding host tissues, and 2) neovasculature inside or on the surface of the scaffolds were irregular and tortuous in shape, while the vasculature in the neighboring host tissue exhibited well-organized vessel networks running straight and parallel to each other. Across all conditions, the surface of the implanted scaffold showed well-connected and higly-branched vasculature where smaller capillaries with diameters < 10 μm sprouted from larger arterioles that were 20-30 μm in diameters. The control scaffold also formed a considerable amount of neovasculature on its surface.
Figure 6.
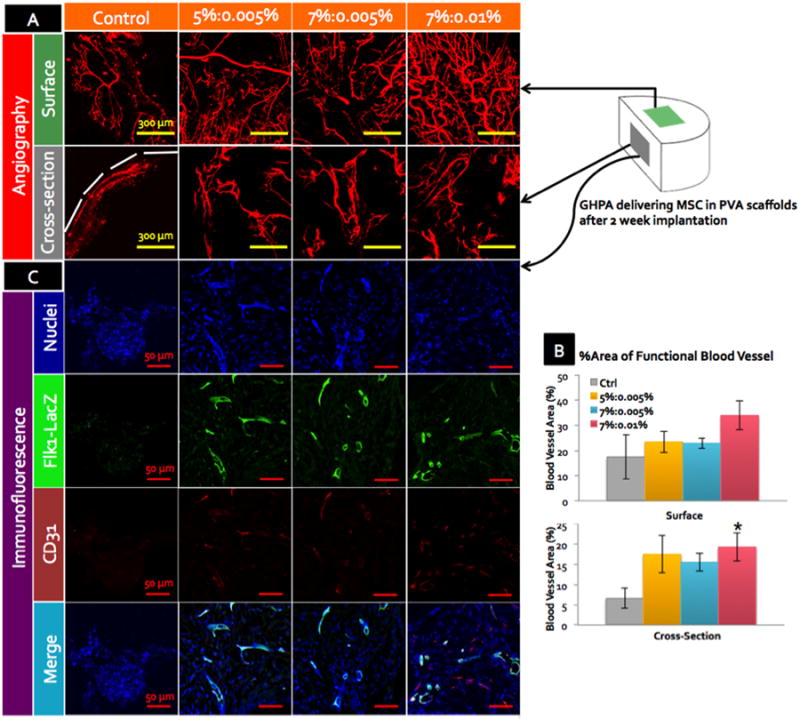
(A) Angiograms of the harvested scaffolds by perfusion with fluorescent microbeads at 2 weeks post implantation. Representative images from the outer surface and cross-sections are shown with scale bars = 300 μm. White dotted line marks the boundaries of the scaffolds. (B) % area of the functional blood vessels by angiograms in (A) with error bars = ±1 SEM and N=4. * indicates statistical significance with p < 0.05 in comparison to the control. (C) CD31, LacZ and nuclei staining of the cross-sections of the explanted scaffolds with scale bars = 50 μm. All images were acquired by confocal microscopy, and z-stacked.
Although there was no statistically significant differences among the crosslinked GHPA conditions, there appeared to be a trend indicating an increase in the blood vessel formation on the surface of the crosslinked GHPA conditions with the higher GHPA content and crosslinking. The crosslinked GHPA conditions, especially the 7%:0.01% condition showed a 100% increase in angiogenesis on the surface compared to the uncrosslinked control. On the other hand, the micro-angiograms from the cross-sections of the scaffolds revealed greater differences between the uncrosslinked control and crosslinked GHPA conditions: the control condition showed a limited degree of neovasculature at the perimeter of the scaffold while the crosslinked GHPA gels supported robust angiogenesis throughout the cross-sections. Quantification showed more than 200% increase in all three crosslinked GHPA conditions compared to the control. Understandably, there is less vasculature seen in the cross-sections than on the surfaces due to reduced access, and blood vessels exhibited even more tortuosity within the scaffold, likely due to the physical obstacles driven by the non-biodegradablity of PVA scaffold.
Interestingly, there also appeared to be a positive correlation between the amount of neovasculature and the degree of GHPA content and crosslinking (Figure 6B). This implies that the stability of GHPA gels in vivo is a crucial factor in promoting angiogenesis, as uncrosslinked gelatin is known to degrade rapidly in vivo by matrix metalloproteases.[32] Additionally, the tubulogenic effect observed in in vitro experiments was lost in non-crosslinked GHPA control condition in vivo, while the condition containing the most GHPA with the highest level of crosslinking showed the highest degree of angiogenesis among the test groups.
Finally, simultaneous staining of LacZ and CD31 confirmed that Flk1-LacZ+ MSCs were incorporated into the blood vessels (Figure 6C). Immunostaining for LacZ yielded similar results to the β-galactosidase staining in Figure 5C, with only few weakly Flk1-LacZ+ MSCs detected in the control condition. For the crosslinked GHPA conditions, numerous cells stained positive for LacZ, however, LacZ expression was the strongest at and co-localized with the CD31+ blood vessels, indicating that the delivered MSCs indeed differentiated into endothelial cells and formed blood vessels in vivo.
Taken together, the angiograms and co-staining of LacZ and CD31 showed branched, perfused neovasculature formation throughout the implanted scaffolds for crosslinked GHPA conditions with clear co-localization between delivered Flk1-LacZ+ MSCs and several CD31+ blood vessels, confirming MSC differentiation into endothelial cells with the aid of crosslinked GHPA in vivo as well as in vitro.
2.6. In Vivo Gene Expression in GHPA Gels Delivering MSCs
The gene expression from the harvested scaffolds was analyzed by qRT-PCR. Several markers (Flk1, VE-cadherin, CD31, vWF) for angiogenesis were analyzed (Figure 7A). All angiogenic markers surveyed were significantly up-regulated in all crosslinked GHPA conditions compared to the uncrosslinked control. For Flk1 expression, crosslinked GHPA conditions showed approximately 1-, 2-, 3-fold increases in expression for 5%:0.005%, 7%:0.005%, and 7%:0.01%, respectively. For VE-cadherin expression, crosslinked GHPA conditions showed approximately 1-, 4-, and 12-fold increases in expression for 5%:0.005%, 7%:0.005%, and 7%:0.01%, respectively. In a similar trend, CD31 expression showed 1-, 1-, and 3-fold increases for 5%:0.005%, 7%:0.005%, and 7%:0.01%, respectively. There was a clear positive correlation between the angiogenic marker expression and the GHPA content/crosslinking density, and these results are in agreement with the angiograms and CD31 staining. For vWF expression, however, all crosslinked GHPA conditions had 60% increase in comparison to the control condition. Collectively, these results demonstrate that overall there were significant increases in the expression of angiogenesis markers in the crosslinked GHPA conditions, and that such increases were even more pronounced in conditions with higher amounts of GHPA and crosslinking.
Figure 7.
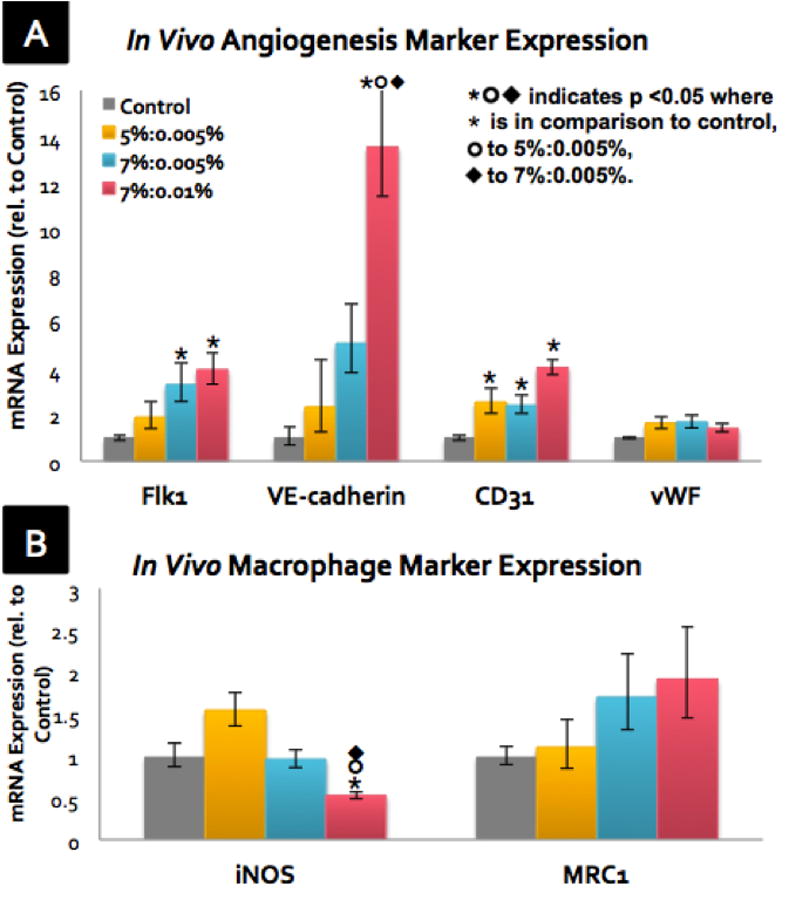
After 2-week subcutaneous implantation, explanted scaffolds were assayed for gene expression of (A) angiogenesis/endothelial cell markers and (B) macrophage markers by qRT-PCR with N=4 and error bars = ±1 SEM.
The expression of two markers (iNOS and MRC1) that represent the host macrophage response to the implants were also measured (Figure 7B). iNOS expression is associated with a classically-activated/inflammatory macrophage phenotype, while MRC1 expression is regarded as a marker for an alternatively-activated/reparative macrophage phenotype.[33] For iNOS expression, the 5%:0.005% showed a 50% increase compared to the control; however, iNOS expression for 7%:0.005% did not change, and 7%:0.01% showed a 50% decrease in comparison to the control. For MRC1, there was again a GHPA/crosslinking-dependent trend of increasing expression, with 7%:0.01% condition having the highest level of MRC1 expression at 1.9-fold that of the control. These results indicate that the 7%:0.01% condition invoked a favorable response from the host macrophages with reduced inflammation and increased a reparative macrophage phenotype, and this group also demonstrated the highest degree of vascularization and endothelial marker expression, all of which may forecast better long-term integration with the host tissues and functional vascularity. It is also possible that such positive interactions between crosslinked GHPA and host immune cells may have contributed to the increased angiogenesis seen in the crosslinked GHPA conditions since angiogenesis and inflammation are known to be coupled, interdependent processes
3. Conclusion
In this study, injectable and in situ crosslinkable gelatin demonstrated excellent biocompatiblity, tunable mechanical properties, and a marked pro-angiogenic effect by promoting endothelial differentiation of MSCs, resulting in robust neovasculature formation throughout the implants, as well as favorable macrophage responses. Previous studies have shown MSC differentiation into endothelial cells using soluble factors such as VEGF and/or bFGF in vitro and/or in vivo.[1-3] In constrast, currently there is only one other study showing differentiation of adipose tissue-derived MSCs into endothelial cells by encapsulating MSCs in PEGylated fibrin hydrogels.[34] In contrast to our study, MSCs in PEGylated fibrin hydrogels did not show increase in Flk1 expression, which implies that there may be multiple mechanisms responsible for endothelial differentiation of MSCs. Nevertheless, our GHPA can be considered as a unprecedented injectable biomaterial platform that is equipped with advanced functions to direct endothelial differentiation of BM-derived MSCs both in vitro and in vivo via purely material-driven signaling pathways. Such biomaterial-driven stem cell differentiation would be preferred to soluble factor-mediated differentiation due to the reproducibility, relatively economic production cost, reduced spatiotemporal variations commonly observed in soluble factor treatments, minimized side effects, and the physical and instructive support provided for tissue regeneration at the target site. The results are also highly significant as this is one of the first studies to use a purely gelatin-based material in the form of an injectable hydrogel for vasculogenic delivery of stem cells in the fields of tissue engineering and regenerative medicine, which has been almost impossible to-date.
Another important advantage of gelatin-based material is its non-immuno/antigenicity in vivo, as the harsh gelatin extraction process is thought to remove known antigens existing on intact 3D collagen fibrils.[35] Injections of unmodified gelatin into several animals has also failed to produce antibodies.[36] Similarly, past studies involving crosslinked gelatin substrates showed negligible inflammation and no sign of scarring or fibrous capsule formation when implanted, and these results are in agreement with the data presented here.[37-39]
Only a small number of studies have investigated angiogenesis using thermally-or chemically-crosslinked, pre-fabricated solid gelatin scaffolds, and these studies often used clinically-available hemostatic agent Gelfoam®. These studies showed significant angiogenesis in the implants, even when implanted alone.[37, 38] Interestingly, it was also shown that crosslinked gelatin scaffolds significantly improved angiogenesis as compared to collagen scaffolds prepared in a similar method.[37] Our study is also in support of the in vivo pro-angiogenic effect of crosslinked gelatin, however, our results are convoluted by the inclusion of highly trophic MSCs.
Due to the ease of isolation and high capacity for ex vivo expansions, BM-derived MSCs represent a highly desirable candidate cell type for in vivo regeneration of vascularized host tissue. However, results from multiple clinical trials demonstrated that the promise of BM-derived cell therapy has fallen short due to two major obstacles: poor long-term engraftment of cells within the ischemic, hostile wound environment, and poor vascularization. Therefore, our study has focused on overcoming the major hurdles of utilizing easily-obtained BM-MSCs for vascular applications by optimizing a minimally-invasive, targeted delivery strategy, ensuring long-lasting survival and retention of implanted cells post delivery, and promoting angio/vasculogenesis in vivo. In order to strive towards clinical translation, the angiogenic effects of crosslinked gelatin material alone and especially in comparisons to other materials need to be investigated further. In addition, because of the short history of using gelatin-based materials in tissue engineering applications, the exact mechanisms for improved angiogenesis by GHPA and 3D gelatin-cell interactions remain to be elucidated. Further studies are required to better understand the apparent and numerous advantages of GHPA and its optimal applications in specific biomedical fields.
4. Experimental Section
Materials
Gelatin (type A from porcine skin, >300 Bloom), 3-(4-hydroxyphenyl) propionic acid (HPA), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxy-succinimide (NHS), peroxidase from horseradish (HRP type VI, 250–330 U/mg solid), hydrogen peroxide (H2O2) were obtained from Sigma Aldrich (St. Louis, MO, USA). Dimethylformamide (DMF) was obtained from Junsei (Tokyo, Japan). Dulbecco's modified Eagle medium (DMEM), penicillin–streptomycin (P/S), fetal bovine serum (FBS), Dulbecco's phosphate buffered saline (DPBS) and trypsin–EDTA were purchased from Gibco BRL (Grand Island, NY, USA). All chemicals and solvents were used as received.
Synthesis and chemical characterization of Gelatin-Hydroxyphenyl Propionic Acid (GHPA)
Synthesis of GHPA has been described previously.[19] Briefly, hydroxyphenyl propionic acid (HPA) was first activated with 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS) in a co-solvent of water and DMF (volume ratio of 3: 2). The activated HPA solution was then added to the pre-heated gelatin solution and stirred at 40°C for 24 hours. The resulting solution was transferred into a dialysis bag (MWCO. 3.5 kDa), dialyzed against deionized water for 3 days, filtered, and lyophilized to obtain the GHPA conjugates (Figure 1A). GHPA was characterized by 1H NMR spectroscopy (AS400, OXFORD instruments, UK), and the phenolic contents of the conjugates were measured quantitatively at 275 nm using a UV visible spectrophotometer (V-750 UV/vis/NIR, Jasco, Japan).
Characterization of Elastic/Storage Moduli (G′) of GHPA
GHPA was dissolved in DMEM media at 3-7% (wt) and divided into two aliquots; one was mixed with horseradish peroxidase (HRP) at the final concentration of 2.5 μg/ml, while the other aliquot was mixed with H2O2 at the final concentrations of 0.0025-0.01% (w/v). Solutions were loaded onto separate syringes, and a dual-syringe applicator were used to evenly eject the two solutions, ensuring proper mixing and gelling (Figure 1B). Storage moduli (G′) was measured in a parallel plate setting on a TA Instrument RA2000 rheometer in oscillation mode with a frequency of 1 Hz and 0.1% strain at 37°C.
In Vitro 3D Culture of MSC in GHPA Gels
Wild type murine mesenchymal stem cells (MSCs, GIBCO) or Flk1-LacZ transgenic murine MSCs were used (Jackson Laboratories). GHPA and H2O2 were dissolved in DMEM media at various % (w/v) as indicated, while a constant concentration of 2.5 μg/ml HRP was used in all conditions. Cells were added to the GHPA+HRP solution at the final concentration of 106 cells/ml. The same number of cells was seeded on tissue culture plate without GHPA gel to serve as a control. After GHPA gelled on the well plate, DMEM supplemented with 10% FBS and 1% penicillin-streptomycin was added and media was changed every day over 15 days.
Cell Viability Assay
Cell viability was measured at days 1, 7, and 15 post culture using 5μM resazurin (Sigma). After 4 hours incubation of resazurin with cells, test culture media were transferred to a new 96-well plate for fluorescence readout at 590 nm using a plate reader (M1000, Tecan, Mannedorf, Switzerland). On the same days, cells were also incubated in media containing 1μM calcein AM (Invitrogen) and 1μg/ml propidium iodide (Sigma) for 15 minutes and then imaged by a Zeiss 710 confocal laser scanning microscope for identification of live/dead cells.
MSC Delivery in GHPA Gels on Polyvinyl Alcohol (PVA) Scaffolds In Vivo
All animal procedures were approved and performed in accordance with Vanderbilt Institutional Animal Care and Use Committee. With heterozygous Flk1-LacZ transgenic murine MSCs whose Flk1 was partially replaced by promoter-less LacZ, Flk1+ cells can then be detected by beta-galactosidase staining. GHPA and H2O2 were dissolved in DMEM media at various % (w/v) as described above, while a constant concentration of 2.5μg/ml HRP was used in all conditions. Flk1-LacZ MSC (5×105)-containing GHPA gel solutions in a total volume of 60 μl were loaded on porous 6 mm-diameter PVA scaffolds (Medtronics). As a control, porous PVA scaffolds loaded with non-crosslinked GHPA + HRP gel solution containing Flk1-LacZ MSCs were implanted. The gel-scaffold complexes were then subcutaneously implanted aseptically on the ventral side of 5-month-old female C57/bl6 mice (Jackson Lab) for 2 weeks, and the procedure was previously described (Figure 5A).[40] Briefly, mice were anesthesized with 1.5 L/min oxygen and 1.5% isoflurane on a warm water blanket, and shaved. A small 1.5 cm longitudinal incision was made on the ventral side, and four different gel-scaffold complexes were inserted into individual subcutaneous pockets. The skin incision was closed with sutures.
Characterization of Implanted Scaffolds
At 2 weeks post implantation, mice were perfused under heavy, near-lethal level of anesthesia with 4% isoflurane in 2 L/min oxygen. First, PBS containing 0.1 mg/ml heparin sulfate was injected into the left ventricle to exsanguinate via the cut inferior vena cava. Then mice were perfused with PBS containing fluorescent microbeads (Invitrogen) for micro-angiography.[31] Scaffolds were subsequently harvested and analyzed for mRNA expression by qRT-PCR, β-galactosidase activity by x-gal staining, angiogenesis by micro-angiography and CD31 staining, and the presence of remaining GHPA gel and general histological analysis by trichrome staining.
Gene Expression Analysis via Quantitative Polymerase Chain Reaction (qRT-PCR)
Samples were homogenized in Trizol (Invitrogen), and RNA was collected using RNeasy kit (Qiagen). RNA concentration and 260/280 ratios were measured on a TECAN M1000 plate reader. RNA was treated with DNAse to eliminate genomic contamination, and reverse-transcribed using High Capacity cDNA Synthesis Kit (ABiosystems). SYBR Green PCR mix (Biorad) was used for quantitative PCR. Each sample containing at least 40 ng cDNA and 500 nM of each primer with annealing temperature at 55°C was run in technical triplicates, followed by melting curve analysis. Raw data were analyzed using CFX Manager (Biorad), and biological replicates from different animals were combined.[41] GAPDH expression was used as a reference gene, where the GAPDH expression level divides each gene expression level for normalization. This relative gene expression to GAPDH is then normalized to that of the control condition. Primers used in this study are listed in Table 1.
Table 1.
List of RT-PCR primers used in this study.
| Genes | Forward Primer | Reverse Primer | Accession |
|---|---|---|---|
| ANGPT1 | TCACTCAGTGGCTGCAAAAACTTG | CTAGCAGTTGTATTTCAAGTCGGG | NM_001286062.1 |
| ANGPT2 | CACAGTGGCTGATGAAGCTGG | GTCGTCTGGTTTAGTACTTGGGC | NM_007426.4 |
| CD31 | TCCCTGGGAGGTCGTCCAT | GAACAAGGCAGCGGGGTTTA | NM_008816 |
| Flk1 | GAGAGCAAGGCGCTGCTAGC | GACAGAGGCGATGAATGGTG | NM_010612 |
| GAPDH | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG | NM_001289726 |
| iNOS | CCAAGCCCTCACCTACTTCC | CTCTGAGGGCTGACACAAGG | NM_010927 |
| MRC-1 | TTGTGGTGAGCTGAAAGGTG | GTGGATTGTCTTGTGG | NM_008625 |
| MyoD | AGGCTCTGCTGCGCGACC | TGCAGTCGATCTCTCAAAGCACC | NM_010866.2 |
| Myogenin | CCAGGAGATCATTTGCTCG | TTCTGGACATCAGGACAGCC | NM_031189.2 |
| NFL | CCAGGAAGAGCAGACAGAGGT | GTTGGGAATAGGGCTCAATCT | NM_010910.1 |
| NFM | ACCAGGACACCATCCAGCAG | GCTGTCGGTGTGTGTACAGAGG | NM_008691.2 |
| NSE | AGCGTTACTTAGGCAAAGGTGT | AGATACCTGAGCTGATGAGGGC | NM_013509.2 |
| Tie1 | ACCCACTACCAGCTGGATGT | ATCGTGTGCTAGCATTGAGG | NM_011587.2 |
| Tie2 | GCCTTAATGAACCAGCACCAAG | CCTTATAGCCTGTCCTCGAAC | NM_001290549.1 |
| Trk-A | GCAGCCACCGTGAAGAAAT | GCACCAATGATGCTGCTCCA | NM_001033124.1 |
| VE-cadherin | TCCTCTGCATCCTCACCATCACA | TAAGTGACCAACTGCTCGTGAAT | NM_009868 |
| VEGFA | ATGCGGATCAAACCTCACCA | CCGCTCTGAACAAGGCTCAC | NM_001110267.1 |
| vWF | GCTTGAACTGTTTGACGGAGAGG | TGACCCAGCAGCAGGATGAC | NM_011708 |
Tissue Preparation for Immunohistochemistry
Samples were fixed in 4% paraformaldehyde (PFA) for 24 hours at 4 °C, washed with PBS, and immersed in 5%-30% sucrose solution until samples sank. Samples were then embedded in Optimal Cutting Temperature compound (TissueTek) and frozen in acetone and dry ice bath. 5 μm-thick sections were obtained by cryosectioning.
Trichrome Green Staining
Trichrome green staining for implanted GHPA gel cryosections was performed by the Vanderbilt Research Histology Core.
β-Galactosidase Staining
Sample cryosections as well as positive and negative controls were fixed with 4% PFA for 10 min at room temperature, washed with PBS, and incubated at 37°C for 2 days in a solution containing the following: 27 mM NaH2PO4, 73 mM Na2HPO4, 2 mM MgCl2, 2 mM EGTA, 1 μg/ml NP40, 5 mM K4[Fe(CN)6], 5 mM K3[Fe(CN)6], and 1 mg/ml x-gal (all chemicals from Sigma-Aldrich). Slides were then washed with dH2O and mounted for imaging.
Immunostaining
Samples were fixed with 4% PFA for 10 min at room temperature, washed with PBS, blocked with 10% goat serum and 0.1% Triton-X100 overnight at 4°C, washed with PBS, and incubated with 1:100 goat anti-mouse CD31 antibody (sc-1505, Santa Cruz Biotechnology), 1:100 rabbit anti-LacZ antibody (ab616, Abcam), 1:100 rabbit anti-Flk1 antibody (sc-504, Santa Cruz Biotechnology) for 2 hours, followed by incubation with 1:1500 IR680LT-conjugated anti-rat antibody (92668029, Licor) and ReadyProbes® AlexaFluor 488-conjugated anti-rabbit antibody (R37116, Invitrogen). Sections were then counter-stained with DAPI and mounted for imaging.
Imaging
Bright-field microscopy for β-galactosidase and trichrome green stain was performed on a Nikon Eclipse Ti microscope, and fluorescence images for immunostaining and micro-angiography were acquired using a Zeiss 710 confocal laser microscope. ImageJ (National Institutes of Health, USA) was used for all image preparation and analysis, including z-stacking fluorescence images and quantification.
Statistical Analysis
Results are presented as means ± standard deviation (SD) or standard error mean (SEM) as indicated. Comparisons among different conditions were performed via ANOVA, followed by Tukey's HSD test in Prism 6 (Graphpad). For all statistics, p < 0.05 was considered statistically significant, and such significance is indicated where appropriate.
Acknowledgments
This study was supported by NIH HL091465, NIH 1UH2 TR000491, NSF DMR 1006558, and NSF CAREER CBET 1056046. The authors would also like to acknowledge the use of resources at the Vanderbilt Institute of Nanoscale Science and Engineering (VINSE), a facility renovated under NSF ARI-R2 DMR-0963361, and the Vanderbilt Institue for Integrative Biosystems Research and Education (VIIBRE) Automated Biosystems Core at Vanderbilt University, which was established by NIH grant 1S10RR027485-01. Confocal images were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). 1H-NMR was conducted in the Small Molecule NMR Facility Core, and the Translational Pathology Shared Resources Core assisted in the preparation and staining of histological specimens.
Contributor Information
Sue Hyun Lee, Dept. of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235 USA; Center for Stem Cell Biology, Vanderbilt University Medical Center, Nashville, TN, 37235 USA.
Yunki Lee, Dept. of Molecular Science & Technology, Ajou University, Suwon 443-749 South Korea.
Dr. Young Wook Chun, Dept. of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235 USA; Center for Stem Cell Biology, Vanderbilt University Medical Center, Nashville, TN, 37235 USA
Spencer W. Crowder, Dept. of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235 USA; Center for Stem Cell Biology, Vanderbilt University Medical Center, Nashville, TN, 37235 USA
Prof. Pampee P. Young, Dept. of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN 37235 USA
Prof. Ki Dong Park, Dept. of Molecular Science & Technology, Ajou University, Suwon 443-749 South Korea
Prof. Hak-Joon Sung, Email: hak-joon.sung@vanderbilt.edu, Dept. of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235 USA; Center for Stem Cell Biology, Vanderbilt University Medical Center, Nashville, TN, 37235 USA.
References
- 1.Zieris A, Prokoph S, Levental KR, Welzel PB, Grimmer M, Freudenberg U, Werner C. Biomaterials. 2010;31:7985. doi: 10.1016/j.biomaterials.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 2.Chiu LL, Radisic M. Biomaterials. 2010;31:226. doi: 10.1016/j.biomaterials.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 3.Moon JJ, Saik JE, Poche RA, Leslie-Barbick JE, Lee SH, Smith AA, Dickinson ME, West JL. Biomaterials. 2010;31:3840. doi: 10.1016/j.biomaterials.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawson E, Mapili G, Erickson K, Taqvi S, Roy K. Advanced drug delivery reviews. 2008;60:215. doi: 10.1016/j.addr.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 5.Juliano RL, Haskill S. The Journal of cell biology. 1993;120:577. doi: 10.1083/jcb.120.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleinman HK, Philp D, Hoffman MP. Current Opinion in Biotechnology. 2003;14:526. doi: 10.1016/j.copbio.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Badylak SF, Freytes DO, Gilbert TW. Acta biomaterialia. 2009;5:1. doi: 10.1016/j.actbio.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 8.Alfaro MP, Saraswati S, Young PP. Vitamins and hormones. 2011;87:39. doi: 10.1016/B978-0-12-386015-6.00023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfaro MP, Young PP. Cell transplantation. 2012;21:1065. doi: 10.3727/096368911X612477. [DOI] [PubMed] [Google Scholar]
- 10.Saraswati S, Deskins DL, Holt GE, Young PP. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2012;20:185. doi: 10.1111/j.1524-475X.2012.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner J, Kean T, Young R, Dennis JE, Caplan AI. Curr Opin Biotechnol. 2009;20:531. doi: 10.1016/j.copbio.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Cao Y, Sun Z, Liao L, Meng Y, Han Q, Zhao RC. Biochemical and biophysical research communications. 2005;332:370. doi: 10.1016/j.bbrc.2005.04.135. [DOI] [PubMed] [Google Scholar]; Silva GV, Litovsky S, Assad JA, Sousa AL, Martin BJ, Vela D, Coulter SC, Lin J, Ober J, Vaughn WK, Branco RV, Oliveira EM, He R, Geng YJ, Willerson JT, Perin EC. Circulation. 2005;111:150. doi: 10.1161/01.CIR.0000151812.86142.45. [DOI] [PubMed] [Google Scholar]
- 13.Oswald J, Werner C. Stem Cells. 2004;22:377. doi: 10.1634/stemcells.22-3-377. [DOI] [PubMed] [Google Scholar]
- 14.Liu JW, Dunoyer-Geindre S, Serre-Beinier V, Mai G, Lambert JF, Fish RJ, Pernod G, Buehler L, Bounameaux H, Kruithof EK. Journal of thrombosis and haemostasis: JTH. 2007;5:826. doi: 10.1111/j.1538-7836.2007.02381.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang CH, Wang TM, Young TH, Lai YK, Yen ML. Biomaterials. 2013;34:4223. doi: 10.1016/j.biomaterials.2013.02.062. [DOI] [PubMed] [Google Scholar]
- 16.Portalska KJ, Groen N, Krenning G, Georgi N, Mentink A, Harmsen MC, van Blitterswijk C, de Boer J. Tissue engineering Part A. 2013;19:2318. doi: 10.1089/ten.TEA.2012.0646. [DOI] [PubMed] [Google Scholar]
- 17.Gorgieva S, Kokol V. In: Biomaterials Applications for Nanomedicine. R P, editor. Vol. 17 InTech; 2011. [Google Scholar]
- 18.Bohidar HB, Gupta A. Biomacromolecules. 2005;6:1623. doi: 10.1021/bm0492430. [DOI] [PubMed] [Google Scholar]
- 19.Lee Y, Bae JW, Oh DH, Park KM, Chun YW, Sung HJ, Park KD. Journal of Materials Chemistry B. 2013;1:2407. doi: 10.1039/c3tb00578j. [DOI] [PubMed] [Google Scholar]
- 20.Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Adv Mater. 2010;22:3484. doi: 10.1002/adma.200904179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Brien J, Wilson I, Orton T, Pognan F. European journal of biochemistry/FEBS. 2000;267:5421. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 22.Burdon RH. Free radical biology & medicine. 1995;18:775. doi: 10.1016/0891-5849(94)00198-s. [DOI] [PubMed] [Google Scholar]
- 23.Griffith CK, Miller C, Sainson RC, Calvert JW, Jeon NL, Hughes CC, George SC. Tissue engineering. 2005;11:257. doi: 10.1089/ten.2005.11.257. [DOI] [PubMed] [Google Scholar]
- 24.Davis GE. Biochemical and biophysical research communications. 1992;182:1025. doi: 10.1016/0006-291x(92)91834-d. [DOI] [PubMed] [Google Scholar]
- 25.Benoit DS, Durney AR, Anseth KS. Biomaterials. 2007;28:66. doi: 10.1016/j.biomaterials.2006.08.033. [DOI] [PubMed] [Google Scholar]; Hern DL, Hubbell JA. Journal of biomedical materials research Part B, Applied biomaterials. 1997;39:266. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 26.Ingber DE. Proc Natl Acad Sci USA. 1990;87:3579. doi: 10.1073/pnas.87.9.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks PC, Clark RAF, Cheresh DA. Science. 1994;264:569. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 28.Ruegg C, Alghisi GC. In: Angiogenesis Inhibition. PM S, HJ S, editors. Springer; 2010. [Google Scholar]
- 29.Somanath PR, Malinin NL, Byzova TV. Angiogenesis. 2009;12:177. doi: 10.1007/s10456-009-9141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan GS, Mak TW. Journal of immunology. 1999;162:3022. [PubMed] [Google Scholar]
- 31.Zachman AL, Crowder SW, Ortiz O, Zienkiewicz KJ, Bronikowski CM, Yu SS, Giorgio TD, Guelcher SA, Kohn J, Sung HJ. Tissue engineering Part A. 2013;19:437. doi: 10.1089/ten.tea.2012.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agren MS. British Journal of Dermatology. 1994;131:634. doi: 10.1111/j.1365-2133.1994.tb04974.x. [DOI] [PubMed] [Google Scholar]
- 33.Sica A, Mantovani A. The Journal of clinical investigation. 2012;122:787. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang G, Drinnan CT, Geuss LR, Suggs LJ. Acta biomaterialia. 2010;6:3395. doi: 10.1016/j.actbio.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 35.Lynn AK, Yannas IV, Bonfield W. Journal of biomedical materials research Part B, Applied biomaterials. 2004;71:343. doi: 10.1002/jbm.b.30096. [DOI] [PubMed] [Google Scholar]
- 36.Starin WA. the journal of infectious diseases. 1918;23:139. [Google Scholar]
- 37.Dreesmann L, Ahlers M, Schlosshauer B. Biomaterials. 2007;28:5536. doi: 10.1016/j.biomaterials.2007.08.040. [DOI] [PubMed] [Google Scholar]
- 38.Ribatti D, Nico B, Vacca A, Presta M. Nature protocols. 2006;1:85. doi: 10.1038/nprot.2006.13. [DOI] [PubMed] [Google Scholar]
- 39.Ponticiello MS, Barry FP. Journal of biomedical materials research Part B, Applied biomaterials. 2000;52:246. doi: 10.1002/1097-4636(200011)52:2<246::aid-jbm2>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 40.Deskins DL, Ardestani S, Young PP. Journal of visualized experiments: JoVE. 2012 doi: 10.3791/3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willems E, Leyns L, Vandesompele J. Analytical biochemistry. 2008;379:127. doi: 10.1016/j.ab.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 42.Wang LS, Du C, Chung JE, Kurisawa M. Acta biomaterialia. 2012;8:1826. doi: 10.1016/j.actbio.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Wang LS, Chung JE, Chan PP, Kurisawa M. Biomaterials. 2010;31:1148. doi: 10.1016/j.biomaterials.2009.10.042. [DOI] [PubMed] [Google Scholar]
- 44.Menge T, Gerber M, Wataha K, Reid W, Guha S, Cox CS, Jr, Dash P, Reitz MS, Jr, Khakoo AY, Pati S. Stem cells and development. 2013;22:148. doi: 10.1089/scd.2012.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]