

Abstract
Hydrogels are often employed as temporary platforms for cell proliferation and tissue organization in vitro. Researchers have incorporated photodegradable moieties into synthetic polymeric hydrogels as a means of achieving spatiotemporal control over material properties. In this study protein-based photodegradable hydrogels composed of methacrylated gelatin (GelMA) and a crosslinker containing o-nitrobenzyl ester groups have been developed. The hydrogels are able to degrade rapidly and specifically in response to UV light and can be photopatterned to a variety of shapes and dimensions in a one-step process. Micropatterned photodegradable hydrogels are shown to improve cell distribution, alignment and beating regularity of cultured neonatal rat cardiomyocytes. Overall this work introduces a new class of photodegradable hydrogel based on natural and biofunctional polymers as cell culture substrates for improving cellular organization and function.
Keywords: photodegradable hydrogels, photopatterning, Gelatin Methacrylate (GelMA), cardiomyocyte alignment, tissue engineering
1. Introduction
The application of hydrogels for soft tissue regeneration has been widely investigated, particularly in the design and manipulation of cell laden materials towards implantable tissues and devices for tissue repair.[1] Tissue development in the body is heavily influenced by the extracellular matrix (ECM), where the combination of physical properties and biochemical signals have all been demonstrated to play important roles in cellular development and differentiation. In tissue engineering research, the ability to manipulate the hydrogel properties during tissue formation to provide the necessary temporal cues for cells remains important yet elusive.[2] Developing hydrogels with physical properties which can be controlled over time would thus be a key achievement to assist in controlling and directing the development and differentiation of cells.[3] Furthermore, for in vivo applications, the hydrogel matrix should play a temporary role for supporting cells to form functional tissues but also be fully degradable or resorbable into harmless products after tissue formation.
The concept of photodegradable hydrogel networks with spatial-temporally tunable mechanical properties, introduced by the Anseth group[4] has opened up new ways of manipulating cell laden biomaterials. The photolabile o-nitrobenzyl ester moiety was incorporated into a polyethylene glycol (PEG) based hydrogel via radical polymerization, thus allowing the physical properties of the gel to be tuned with UV light. Other photosensitive gels utilizing alternative photodegradable moieties such as coumarins are concurrently being investigated with the aim of developing more bioinert degradation products.[5] Free radical polymerization is fast, requires very low amounts of initiators, can be initiated via a number of stimuli and has been extensively documented in tissue engineering applications. Alternatives to free radical polymerization include click reactions,[6] NHS coupling,[7] and disulfide fragmentation reactions.[8] Reversible additional fragmentation chain transfer (RAFT) polymerization[9] has also been used to synthesize photodegradable free radical gels providing a more homogenous network structure compared to uncontrolled free radical gels. Photodegradable hydrogels have been shown to influence cell signaling,[10] cell process extension and connectivity,[11] and cell attachment.[12] Recently photodegradable hydrogels were used as a platform to evaluate the mechanical dosing effect on human mesenchymal stem cell (hMSC) behavior,[13] and as a means to trigger the release of bioactive molecules or encapsulated compounds from the gel.[14]
The aforementioned photolabile hydrogel systems are in almost all cases based entirely on synthetic PEG polymer networks, and require conjugation of cell signaling peptides such as the arginyl-glycyl-aspartic acid (RGD) motif[15] for improving cellular attachment and growth. Such substrates may suffer from a loss of cell responsiveness over time as well as undergo limited cell-material interactions. As biomaterials used in regenerative medicine seek to recreate elements of the in vivo environment, much research has also focused on investigating hydrogels constituting ECM derived materials including collagen, fibrin, gelatin and elastin. These polymers are non-immunogenic and have intrinsic cell recognition sites facilitating cellular attachment, proliferation and differentiation.[16] A recent example by Yanagawa et al. crosslinked gelatin or PEG hydrogels with photodegradable multiarm PEG-NHS to enabled gel preparations with a 2 pot mixture.[7] However the NHS-chemistry is highly pH specific, requires specific buffer and salt concentrations as well as peptides or proteins present in the media or conjugated onto the polymer which can interfere with this crosslinking reaction.
In our study we incorporate photodegradable functionality into methacrylated gelatin (GelMA), which is made via the methacrylation of amine side groups of gelatin[17]. Gelatin itself is derived from collagen, is an inexpensive biopolymer and retains both matrix metalloproteinase (MMP) sensitive degradation sites as well as natural cell binding motifs such as the RGD sequence.[17] When crosslinked using free radical polymerization, GelMA forms a stable but flexible hydrogel matrix at 37°C, and it can support long term cell viability both as a two dimensional (2D) cell support layer or a three dimensional (3D) environment for their encapsulation.[18] In addition, GelMA is a versatile biopolymer that can be mixed with other materials to form copolymers or composites, including PEG,[19] hyaluronic acid, [20] and carbon nanotubes.[21] It has been shown that photopatterned GelMA hydrogels can assist in the formation of 3D endothelial cords[22]. Photopolymerization, via addition of photoinitiators such as Irgacure 2959 has been the preferred way of polymerizing GelMA due to a fast cure time, reproducibility and the ability to create a variety of patterns in the substrate[17]. However, the absorption wavelength with Irgacure 2959 overlaps significantly with the degradation wavelength of photodegradable groups and as such alternative methods of curing were utilized in this study.
It is well known that surface patterning of biomaterials influences cell organization and alignment[1]. Particularly for cardiac tissue engineering, patterned cues such as microablated grooves,[23] ridges, [24] nanofibres[25] or printed biomolecules such as fibronectin,[26] are necessary for aligning and the organization of cardiomyocytes (CM) on engineered substrates. Recently, we have shown that the use of micropatterned hydrogel substrates could significantly enhance the contractile activity of CMs[27]. Here, a facile method of patterning via photodegradable functionality is explored for the purpose of improving cell alignment.
In this work we have developed a photodegradable GelMA hydrogel which can be crosslinked using either redox initiators or photoinitiators, and combines the ability to accurately pattern and soften the hydrogel. Photodegradable GelMA gel is also made of gelatin, a biopolymer which can be digested by cells and degraded over a suitable time frame for biomedical applications. UV degradation provides a complementary and viable alternative to photopolymerization for patterning. Furthermore, UV degradation offers the benefit of allowing modifications and patterning after curing or even after cell seeding. We hypothesized that the photodegradability can be used to pattern the hydrogel to assist initial cellular organization, which when applied to CM cultures, has the potential to improve cell alignment and beating characteristics.
2. Results and Discussion
2.1. Degradation assessment and gel characterization
The aim of this work was to create a photopatternable cell culture substrate using GelMA with a high degree of methacrylation (~80% methacrylation) as the base hydrogel. The methacrylate groups of the GelMA were crosslinked with a photodegradable (PD) PEG crosslinker using a redox initiated or visible light photoinitiated free radical polymerization process (Figure 1A). The PD crosslinker consists of a PEG chain (Mw ~3000 gmol−1) linked to a photodegradable o-nitrobenzyl ester modified with a terminal methacrylate group on each end, to enable crosslinking (via methacrylate groups) and to impart photodegradability respectively. The photodegradable moiety is based on that reported previously by Kloxin and colleagues[4].
Figure 1.

(A) Schematic representation of a photodegradable GelMA hydrogel before and after irradiation: methacrylate groups (represented by light blue branches) were attached to gelatin’s pendant NH2 groups (represented by black dashes). GelMA was crosslinked with a bifunctional PEG based crosslinker containing photodegradable groups via radical polymerization (schematic shows photodegradable gel crosslinked with photodegradable dimethacrylate). The hydrogel can be degraded by exposure to UV light. (B) UV-visible spectroscopy of the photodegradable crosslinker at 0.025% (w/v) showing the decrease in intensity of absorbance peaks of the photodegradable moiety in response to UV irradiation at 365 nm, 15 mW cm−2. (C) Percentage mass loss profile showing the decrease in dry weight of 5% GelMA hydrogels with 5% photolabile o-NBE group with increasing UV exposure time at 0, 10 or 20 mW cm−2. Physical properties of the hydrogels investigated in this study with (D) representative rheology curve of 6% (w/v) hydrogel with 50% (w/w) photodegradable group. UV irradiation began from 17 mins at 365 nm, 20 mW cm−2. (E) Normalized degradation curves of samples from 33% to 66% photodegradable groups were compared and (F) comparison of hydrogel swelling with increasing quantities of photodegradable groups. Error bars show standard deviation, ***p< 0.0001, n=5. (G) Photorheology measurements of elastic modulus of hydrogels cured with LAP photoinitiator. Gels were exposed to visible light wavelengths of 400–500 nm for 5 min for gelation, then left to stabilize and degraded with UV light at 365nm, 20 mW cm−2.
GelMA hydrogels with different compositions of PD crosslinker: 0, 33, 50 and 67% (w/w) to GelMA were compared throughout the study (hereafter referred as the 33% PD, 50% PD and 67% PD, respectively). The total solids content of GelMA and crosslinker in the buffer during curing was kept constant at 6% (w/v). A critical amount of GelMA is required to retain cell attaching properties of the hydrogel, whilst the presence of PD groups is also necessary to impart photodegradability. The network structure of the hydrogel can be is disrupted by UV/Vis light in the range of 270 – 400 nm which induces the cleavage of the nitrobenzyl group, yielding a nitroso ketone and carboxylic acid group as outlined in Figure 1A. UV light in this range can thus be used to induce a photodegradation process. The UV-Vis degradation response of GelMA and the PEG crosslinker over time is shown in Figure 1B. The spectra for the PEG crosslinker showed two absorption peaks at around 305 and 356 nm. Following UV exposure at 365 nm for up to 5 min the absorbance of both peaks moved towards a higher wavelength, indicating that the photodegradation products may cause a slight attenuation of the incident light at 365 nm. The GelMA solution was slightly turbid, which may account for the background absorption in the UV-Vis spectra, but this was insignificant above 300 nm. When hydrogel samples were degraded at different intensities of UV light, shown in Figure 1C, there was greater mass loss with both increasing irradiation time and intensity of the PD samples compared to GelMA control. This is to be expected and indicates sensitivity of the hydrogel in response to the UV dose.
The photodegradability of the hydrogel was assessed via photorheology measurements using a parallel plate rheometer fitted with a quartz glass bottom plate through which a UV beam can irradiate (365 nm, 20 mW cm−2). After redox initiated curing (Figure 1C), the storage modulus was in the range of 600–3000 Pa which is comparable to GelMA materials reported in literature.[17]
When UV light was then illuminated upon the gel, there was a rapid drop in the storage modulus to half its value after 6 min. With further degradation, the G′ plateaued at around 15–20% of G′0 (Figure 1D), showing a similar trend to the gels from Kloxin et al[12]. The gels were not completely liquefied due to non-photodegradable GelMA-GelMA crosslinked sites in the network formed either during curing or possibly from radicals generated during UV irradiation. The rate and extent of degradation increased with increasing photodegradable group concentration as expected, with the 67% PD gels being 14% softer than the 33% PD gels after 5 min (Figure 1E).
The swelling ratios of redox initiated hydrogels were compared across compositions. The hydrogel made up of 100% PD PEG showed the greatest swelling ratio, compared to one made using 100% GelMA. Interestingly, when the two polymers were combined, the swelling properties of the GelMA hydrogels appeared to be dominated more by the GelMA than the PD crosslinker. At the percentage weight solids tested there was a slight increase in swelling ratio with increasing PD concentration, but this was not statistically significant (Figure 1F). This may be the result of two effects. As the PD crosslinker content increases, GelMA-to-GelMA crosslinks are replaced with PEG-GelMA crosslinks, which could produce the overall effect of increasing the distance between the junctions in the network. However, more significantly, a higher degree of methacrylation from GelMA may create a more highly constrained network. This may have restricted the overall extent of swelling of the PD hydrogels, even with the incorporation of the linear and more hydrophilic PEG crosslinker.
Free radical polymerization offers the advantage of having multiple means of triggering gelation. In addition to the redox initiated polymerization, visible light polymerization using lithium acylphosphinate[8] (LAP) photoinitiator provides a complementary means of creating photocurable and photoadaptable hydrogels. LAP was selected as it has an absorption tail above 400 nm and therefore does not overlap with the o-nitrobenzyl moieties in the PD crosslinker. Photopolymerization using visible light (400–500 nm) successfully crosslinked the hydrogels and photodegradation was subsequently achieved using UV light (365 nm), (Figure 1G). As with redox initiated hydrogels, the 67% PD sample showed faster rate and extent of degradation than the 50% and 33% PD hydrogels. Following photocuring, a drop in the storage modulus was observed even after photoirradiation was stopped but eventually stabilized. This effect may be due to chain rearrangement or stress relaxation effects by the hydrogel after being in a non-equilibrium state during curing, but further investigations may be required to understand this effect.
Free radical polymerization can take place across a large range of conditions and is not tightly constrained by factors such as pH, temperature, solvents, presence of chemically or biologically reactive groups. Redox initiated polymerization provided a means for creating PD hydrogels that can be gelled with cells in situ allows cells to be studied in a 3D matrix environment and this has proven feasible with our photodegradable GelMA system to demonstrate the proliferation and expansion of encapsulated cells (Figure S6). At the same time, the flexibility of curing and releasing on demand using different wavelengths of light imparts great utility for hydrogels for use as a tissue engineering platform both to study cell-material interactions and to create organized tissues. Therefore, both the LAP and the redox initiated crosslinking further provide complementary means towards creating hydrogels quickly and efficiently depend on the intended application.
2.2 Substrate patterning, characterization and cardiac fibroblast alignment
Having demonstrated the photodegradability of the hydrogels, the materials were then patterned, characterized and prepared for cell culture. Hydrogels for the following studies were all prepared using redox initiated free radical polymerization unless otherwise specified. With the use of various photomasks a variety of patterns and geometries were created in the hydrogel (Figure 2A–C). For the purposes of cell alignment, striped masks of 20 μm microchannels separated with 20 μm spacing were used as they have been reported to be optimal for CM alignment[26] and specifically because this spacing correlates to the average functional intercapillary distance in rat hearts[28]. An irradiation time of 5 min at 320–500 nm, 20 mW cm−2 was used to degrade the gel and form well-defined micro-size patterns after optimization of the relevant parameters. The degradation time chosen was selected to ensure a good pattern was obtained without overly degrading and weakening the gel. Current commonly utilized techniques for photopatterning such fine patterns either use a photoinitator and photomask to create striped blocks of gels,[19, 22] or a prepatterned polydimethylsiloxane (PDMS) mold over the polymer solution as a micromolding technique.[29] The first technique does not produce a base layer of polymer between channels, and the second requires the preparation of many PDMS molds as well as requiring careful attention to the demolding process so as to not damage the gels. In contrast, the photodegradable method uses light to etch the pattern within the gel and is a simpler process overall.
Figure 2.
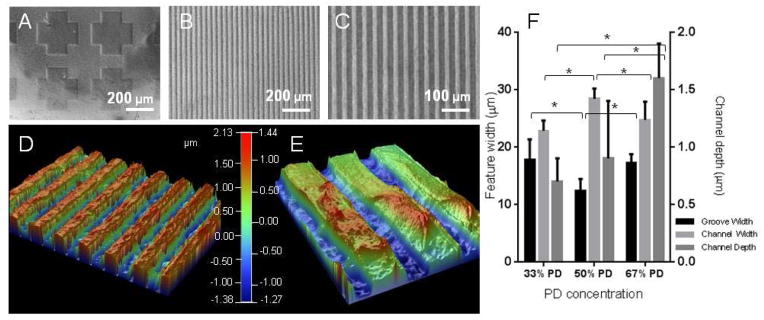
Visual characterisation of the UV patterned hydrogels with brightfield micrographs of the PD hydrogel etched with a cross shaped pattern (A), or hydrogel patterned with striped grooves of 20 μm and channels of 20 μm at x 10 (B) and x 20 (C) magnification. The hydrogel patterns were plated with copper and visualised with optical profilometry, exemplified in (D) and (E) at different magnifications of the 67% PD hydrogel. Quantification of the groove width (bottom of channel), channel width (top of channel) and channel depth (step height between the two) are shown in (F). The groove and channel widths were similar across compositions, but channel depth increased with PD concentration. * denotes statistical significance with p < 0.05, n = 6, Error bars denote standard deviation.
The patterned hydrogels were visualized and quantified for optical profilometry by first applying a plated copper thin film on the sample using a modified version of a previously reported protocol, [30] (Figure 2D–F). The widths of the channels (top section) and grooves (bottom section) created on the substrates were measured as well as the channel depth across the three concentrations. The widths of the patterns were similar to the width of the photomask used (20 μm) although the 50% PD sample showed a lower groove width of 12 ± 2 μm. The channel widths measured (22 – 28 μm) were consistently higher than that of the groove widths (12 – 17 μm). This is likely due to the edges of the channels having a lower crosslink density than the bulk of the material, resulting in local swelling and increase in the channel width and reduction in the groove width correspondingly. Interestingly the channel depth increased with increasing PD concentration, reaching 1.6 ± 0.3 μm for the 67% PD concentration and 0.6 ± 0.2 μm for the lowest concentration. Although it is has been reported that PD groups and their degradation products may increase light attenuation,[31] the concentration of PD groups used in this study resulted in accelerated degradation of the hydrogel leading to the formation of deeper channels.
2.3 Cell alignment and immunocytochemical analysis
Cardiac tissue is comprised of two major cell types- cardiac fibroblasts and CMs. Cardiac fibroblasts are integral to creating and maintaining the desirable environment of a normal heart including the structural, biochemical, mechanical and electrical properties.[32] CMs are the contractile cells of the heart and exist in a 3D network with endothelial cells, vascular smooth cells and fibroblasts and require high directionality and gap junctions between cells to maintain strong and regular muscle contractions[33]. Our previous work has shown GelMA to be a suitable base matrix material for CM culture.[34] Incorporation of the PD crosslinker into the hydrogel showed no adverse effects to cultured L929 fibroblasts as tested by live/dead staining (Figure S7).
Neonatal CMs were seeded on unpatterned and patterned GelMA and monitored over 7 days. Cardiomyocytes were round when seeded, but a majority of cells do spread and attached onto the substrate shortly after seeding across all sample compositions, demonstrating the PD GelMA substrate is suitable for CM attachment and spreading. Cytoskeleton staining with phalloidin was carried out to study cell spreading. On unpatterned samples there were more rounded cells organized in small clumps initially (Figure 3A–C), while on patterned gels the majority of cells were spread and elongated (Figure 3D–F), with only a small proportion of individual rounded cells being scattered across the patterned samples. The spread cells usually have an elongated, oval shaped nuclei (Figure 3E, insert). The orientation of the major axis of the nuclei was measured with respect to the channel directions to determine the cell orientation.
Figure 3.

CM culture and nuclei alignment quantification. Fluorescent images of CMs fixed and stained with phalloidin (F-actin filaments, green) and DAPI (nuclei, blue) on unpatterned (A–C) and patterned (D–F) 67% PD hydrogels showed increased spreading and elongation from day 1 through to day 7. Insert images in D–F show the elongation of nuclei towards the channel directions over time. The orientation of the nuclei with respect to channel direction were quantified for the three timepoints respectively. The orientation of nuclei were equally distributed for unpatterned samples but showed an increase in orientation towards channel directions over time. *p< 0.05, ** p < 0.01 and *** p < 0.001, n =3, error bars denote standard deviation..
The degree of cell alignment was quantified based on the orientation of the elongated nuclei compiled from multiple images, with each image containing 300–1500 cells. The analysis of nuclei orientation showed that with unpatterned samples (Figure 3 B, D, F, Figure S8) there was no preference for nuclei orientation across the compositions from day 1 to 7. This was in contrast to cells cultured on patterned samples (Figure 3 A, C, E) where a majority of cell nuclei showed a preference for alignment towards the channel directions. Alignment on day 7 of culture decreased compared to day 3, which was likely from a combination of lack of available space for cell spreading, possible cell proliferation and multiple layers of cells being present. A high cell seeding density was selected to achieve a confluent basal monolayer of cells on the substrate, however this also means that any cells present over the basal layer may have obscured the effect of topographical cues from the substrate. Statistical analysis by two-way ANOVA showed a significance difference in orientation over time between all comparisons of timepoints for 33% and 50% PD samples, and a significant difference between day 1 and 3, and between day 1 and 7 for the 67% PD sample with an overall p value <0.0001. No significant difference was found across time points with unpatterned samples. Within a particular timepoint hydrogels with increased PD concentration (and therefore increased channel depth) did in some cases have a significant effect on improving nuclei alignment. Overall, the trend of cell alignment over time was consistent to results using an elastin substrate from a previous study.[27] Topographic cues with grooves ranging from 0.2 to 50 μm have been reported to be sufficient to serve as contact guidance cues for CM[35] and cardiac fibroblasts[36]. High degrees of cell alignment were observed when patterned substrates, particularly the 50% and 67% PD were cultured with both CM (Figure 3, S8) and cardiac fibroblasts (Figure S9). Both the 50% and 67% PD samples showed higher degrees of alignment compared to the 33% and unpatterned samples. This is most likely due to the increased channel depth of the 50% and 67% PD samples as shown in Figure 3F.
In addition to studying the orientation of the cell nuclei, the conformation of cultured CM on the patterned and unpatterned substrates was also visualized by immunostaining of the cytoskeleton with phalloidin (binding to F-actin, Figure 3) and myofilaments with troponin I (Figure 4 A–F), which is also a specific marker of CM. Cells were more evenly distributed across the patterned hydrogels but remained in large cell clumps on the unpatterned samples. Staining of F-actin fibers revealed filaments preferentially aligned along the patterned channels. Extensive bridging and contact between channels was also evident (Figure 3F, 4 D–F inserts), where cells connected two adjacent channels, presumably to ensure strong communications between channels for signal conduction purposes. On unpatterned surfaces the filaments were disorganized with filaments oriented in multiple directions.
Figure 4.

Fluorescent micrographs comparing the effects of PD concentration and patterning on CM alignment and distribution. Figure A–F show epifluoresence images of day 7 CM fixed and stained with DAPI (blue) which stains the nuclei, and troponin I antibody (green) present in CM. Inserts in images D–F show higher magnification regions obtained via confocal microscopy showing cell-cell contact between adjacent channels. Figures G–L show confocal images of day 7 CM fixed and stained with DAPI (blue), connexin 43 (red) and sarcomeric α-actinin (green) which stains for the striated Z-lines of myotubes in cardiac muscle. Image plane is focused on cells at the top of the channels.
On day 7 of culture, CM-seeded substrates were fixed and stained with antibodies to assess the expression of cardiac markers. It was found that PD hydrogels supported the development of CM phenotypes with positive staining for troponin I, sarcomeric α-actinin and connexin 43 (Figure 4 G–L). There were no major differences in phenotype across the compositions of unpatterned and patterned samples.
Confocal microscopy confirmed the CMs were elongated with well-defined striated sarcomeric structures representing typical cardiac cross-striated patterns as stained by sarcomeric α-actinin and troponin I. Actinin in cells without contractile phenotype would not otherwise be stained by sarcomeric α-actinin. The cross-striation sarcomeres were clearly defined on the hydrogels. Quantification of the overall sarcomere direction showed that sarcomeres were preferentially aligned on patterned compared to unpatterned samples (Figure S10). On the unpatterned samples there was no preferential cross-striation orientation due to cells spread over different orientations.
In all cases a large number of gap junctions were found in proximity to cell clusters as shown by connexin 43. The presence of gap junctions is crucial to allow for spreading of action potentials between cardiac cells and enable depolarization of heart muscle. The gap junctions appeared to be well organized along the patterns, although this was harder to visualize compared to the unpatterned samples due to their distribution across different focal planes when imaging patterned samples. For similar reasons the density of connexin appeared higher on unpatterned samples.
2.4 Beating characterization
Spontaneous beating was observed on samples seeded with CMs as early as 2–3 days after seeding. Examples of beating patterns at each sample composition are shown in Figure 5A–B. On unpatterned samples, the cells were generally organized in clusters, (Figure 4A–C) resulting in beating rhythms that were eccentric in their frequency and regularity (Supplementary Video S11). The patterned samples, in comparison, showed significant improvement in both beating regularity and the beats per minute (BPM) due to the channels promoting a more even distribution of CM across the substrate and between channels. This improvement was found to correlate with increasing PD concentration and channel depth, and this was reasonable given the improvements in cell alignment. The contractions were also uniform and consistent in direction across the substrate (Supplementary Video S12). The beat homogeneity between patterned and unpatterned hydrogels were characterized by quantifying the average beat period length. The F tests also showed a significant difference in variance between patterned and unpatterned samples indicating more regular beating periods on patterned samples. At present we believe that this is due to the presence of patterned channels organizing and distributing cells more uniformly across the hydrogel. Contractile activity of the cells were maintained for at least 7 days, and there appears to be a general decrease in beating activity as culture time increases as reported previously[27]. This could be due to a combination of cell degradation and remodeling of the hydrogel material as well as continual proliferation of cardiac fibroblasts on the samples which may act to dampen contractile activity.
Figure 5.

CM Beating characterization. Beating characteristics of day 4 CMs seeded on unpatterned (A) and patterned substrates (B) across different compositions are represented by monitoring pixel intensity in regions of interests over a period of 30 s. The average time between beats and beat homogeneity are compared (C) between patterned and unpatterned substrates. * denotes statistical significance from t-tests comparing means between patterned and unpatterned conditions, p< 0.05. D. Representation of CM beating on patterned 67% PD substrate on day 6 in response to an externally applied electric field at 1 and 2 Hz. The measured BPM of CM was quantified on patterned substrates at 0.5, 1.0 and 2.0 volts at input frequency of 1 Hz (E) and 2 Hz (F), error bars indicate standard deviation. As input voltage increased the BPM of CMs increased towards the desired frequency.
Electrical stimulation by input of a square wave was able to override and control the beating frequency of CMs on the substrate (Figure 5D). A relatively low threshold voltage of 2 V was sufficient to conform CM BPM to the input signal (Figure 5E and F) compared to the previous study with elastin based substrates[27] (~3–8 V), and this could be due to the mechanical properties of the hydrogel materials in this study, which are about an order of magnitude lower in modulus. Since the minimal threshold voltage was low there was no discernable difference across the three sample compositions. Increasing applied voltage from 0.5 to 2 V in all cases was sufficient for obtaining controlled beating of CM seeded on the samples.
Our findings indicate that PD GelMA hydrogels of all compositions enabled CM to adhere directly to the hydrogel and did not require additional steps for protein adhesion or attachment. PD GelMA hydrogels were suitable substrates for CM culture and furthermore the 20 μm patterns created from photodegradation influenced the orientation of CMs in agreement with previous studies[27, 35] to resemble tissue microstructure of the native rat heart. The arrangement of localized gap junctions along channels and orientation of contractile units enabled spontaneous, coordinated and more regular beating. Photodegradable GelMA is a suitable biomaterial for single step patterning of cells and it is expected that the inherent photodegradability can be further applied to the hydrogel with cells in situ similar to reported studies with valvular interstitial cells[12, 37] to further influence cellular behaviors by modulation of factors such as material stiffness and local patterning.
3. Conclusion
In summary, the engineering of a novel photodegradable hydrogel derived from a natural polymer, gelatin, was demonstrated. Photodegradable GelMA was shown to be sensitive to UV irradiation, which in turn enabled a facile technique of photopatterning with precision down to the micrometer scale in a one step process. The photodegradable GelMA material was found to facilitate attachment, proliferation and elongation of both neonatal rat cardiac fibroblasts and CM. Hydrogels with greater PD content had deeper patterned channels, which in turn led to better alignment of cell nuclei along the channel directions.
Micropatterned GelMA samples generated by spatially controlled photodegradation resulted in high degrees of alignment of cardiac fibroblasts and CMs mimicking the native myocardium. The phenotype, viability and contractility of CMs on photodegradable GelMA were confirmed by both immunostaining and observation of more regular beating rhythms. The photodegradable GelMA hydrogels thus show promise as a dynamic cell culture substrates with tunable material and topographical properties which are important for achieving improved cell-material interactions. These novel photodegradable hydrogel materials are expected to find applications in tissue engineering and regenerative medicine, including cardiovascular tissue engineering.
4. Experimental Section
GelMA preparation
GelMA with a high degree of methacrylation was synthesized as reported previously.[20] Briefly, methacrylate anhydride (8% (v/v), MA, Sigma) was added to a 10% (w/v) solution of porcine gelatin type A (Sigma) in Dulbecco’s Phosphate Buffer Saline (DPBS; Invitrogen). The resulting emulsion was continuously stirred at 50 °C for 3 hours, then diluted and dialyzed in distilled water at 40 °C for 7 days in 12–14 kDa cutoff tubing (Spectrum Labs). The solution was then filtered with a 0.2 μm filter, frozen at 80 °C and lyophilized. The methacrylation of the solution was confirmed by 1H NMR analysis as previously described to be ~80%.[20].
Photodegradable PEG crosslinker synthesis (PD group)
The crosslinker incorporating an o-nitrobenzyl ester was synthesized in house, with details in the supplementary information. Following a published method[38], 4-(4-(1-(methacryloyloxy)ethyl)-2-methoxy-5-nitrophenoxy)butanoic acid was synthesized from a solution of 4-(4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy)butanoic acid) and triethylamine and either methacryloyl chloride (for dimethacrylate crosslinkers) or acryloy chloride (for diacrylate crosslinkers). The purified product was reacted with oxalyl chloride under nitrogen, then redissolved in dry dichloromethane (DCM) and added to a solution of PEG-3000 and triethylamine overnight under nitrogen to form the PD crosslinker. The crosslinker was purified by precipitation into cold diethyl ether to remove excess 4-(4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy)butanoyl chloride.
UV-Vis Characterization
A solution of the photodegradable PEG crosslinker was dissolved in Milli-Q water at 0.025% (w/v). Aliquots of 1 mL solution were pipetted in a 12-well cell culture plate and exposed to UV light (EXFO, Canada) at 365 nm, 15 mW cm−2 for times of 1, 2, and 5 mins. Samples were then collected in a 1cm quartz glass UV cell and scanned in a Cary-300Bio UV-Vis spectrometer (Agilent Technologies, USA) to assess degradation behavior.
Hydrogel fabrication
Stock solutions of ammonium persulfate (APS, Sigma) (20 % (w/v) in DPBS), N,N,N′,N′-tetramethylethylenediamine (TEMED (10 % (w/v) in DPBS, Sigma), GelMA (10% (w/v) in DPBS) and PD crosslinker (20 % (w/v) in DPBS) were prepared. GelMA solutions were heated above 40 °C to homogenize and ensure solubility. To prepare gel samples, corresponding amounts of stock solutions were combined to obtain final w/v concentrations as shown in Table 1. For brevity, the hydrogel compositions were named according to the amount of PEG crosslinker i.e.: 0% PD, 33% PD, 50% PD and 67% PD.
Table 1.
Compositions of hydrogels used for experiments.
| Hydrogel composition | GelMA concentration (%w/v) | Photo-crosslinker concentration (%w/v) | Total macromer concentration (%w/v) |
|---|---|---|---|
| 0% PD | 6 | 0 | 6 |
| 33% PD | 4 | 2 | 6 |
| 50% PD | 3 | 3 | 6 |
| 67% PD | 2 | 4 | 6 |
| 100% PD | 0 | 6 | 6 |
To initiate redox free radical polymerization, 3% (v/v) of the APS and TEMED solutions were added to the prepolymer, then solutions were cast rapidly in droplets of 8 μL onto polystyrene spacers with a step height of 150 μm. Each drop was then covered with a 3-(trimethoxysilyl)propyl methacrylate (TMSPMA, Sigma) coated glass slide square of 0.8 by 0.8 mm to allow covalent attachment between the gel and the backing slide during the curing process. The gel typically cured within 5–10 min. The spacers were wetted with PBS to release the gels, and the gels were washed/soaked in PBS water either for photopatterning or cell culture.
Photopatterning
UV light was delivered by an OmniCure S2000 unit equipped with a 320 – 500 nm filter at an intensity of 20 mW cm−2. For patterning, laser-printed polymer photomasks consisting of 20 μm channels and 20 μm spacing were placed over the gel and exposed to UV irradiation for 300 s. Patterning was also carried out with a cross shaped photomask for visualization purposes only. After patterning the samples were rinsed twice in PBS and allowed to reach equilibrium swelling overnight.
Swelling ratio measurement
Samples for swelling ratio measurements were made following steps described above with redox initiated free radical polymerization, but with a thickness of 1mm. Gels were swollen in DPBS for 24 hours and their wet mass were recorded. Gel samples were then vacuum dried using SpeedVac systems (Savant). The dry mass was then recorded, and the swelling ratio, defined as wet mass/dry mass, was calculated. For UV degradation studies, 67% PD samples were exposed to 0, 180, 300 and 600 s of UV light at 365 nm and 10 or 20 mW cm−2 respectively. Samples were left swollen overnight and their wet mass was recorded, followed by drying as described.
Rheology Assessment
Photorheology studies were carried out on a strain controlled rheometer (Anton Paar Physica series) with the same setup as previously reported[9] using a gap size of 0.08 mm, a frequency of 1 Hz and constant strain of 0.2%. The UV source used was an OmniCure S2000 UV/Vis spot curing system (365 nm, 20 mW cm−2). For redox initiated free radical polymerization, solutions containing the initiators and prepolymers were mixed and immediately placed on the rheometer. The solution was allowed to gel and stabilize before the light was switched on to monitor degradation characteristics. For photoinitiated free radical polymerization, compositions of hydrogels as shown in Table 1 were dissolved in a 1% (w/v) solution of LAP in Milli-Q water. Aliquots of ~150 μL of the polymer solution were pipetted on the rheometer and the top plate was lowered. After allowing the solution to settle for 2 min, the OmniCure lamp with the aforementioned intensity was switched on for 5 min using the 400–500 nm filter. The light was then switched off for 13 min, then the filter was changed back to 365 nm and switched on to induce photodegradation.
Optical profilometry visualization
A copper layer was deposited onto patterned redox initiated hydrogels using a modified electroless process[30]. Gels were then imaged using a Veeko Optical Profilometer (Veeco NT1100, NY, US). Images of gels were then used to quantify the channel width, depth and height (Supporting Information, Methods).
Cell culture
For cell culture, all solutions for hydrogel preparation were prepared with sterile DPBS as the solvent in a sterile biosafety cabinet, and solutions were filtered with a 0.2 μm syringe filter. After photopatterning, substrates were transferred into 48 well plates after rinsing with sterile DPBS, where they were rinsed twice more with sterilizing solution consisting of sterile DPBS with 2% (v/v) Penicillin-Streptomycin. The samples were then soaked overnight in cell culture media comprising of DPBS with 1% (v/v) Penicillin Streptomycin and 1% (v/v) Glutamine.
CMs and cardiac fibroblasts were obtained from two day old neonatal Sprague-Dawley rats and extraction carried out as previous reported[27] (Supporting Information, Methods). Hydrogels were rinsed once with fresh culture media prior to seeding. CMs were seeded at 220,000 cells per 48 well, and kept at 37 °C with 5% CO2. Media was changed after one day, from day 3 onwards 50% of the media was removed and refreshed with new media every day.
Electrical Stimulation
An electrical pulse generator delivered biphasic square waves of 1 or 2 Hz to a sample placed in a custom built well between two carbon rod electrodes by using the same setup in our previous study[27]. The voltage was gradually increased from 0 to 10 V to induce synchronous beating of cells in accordance to input electrical function.
Staining
At days 1, 3 and 7, the cell seeded substrates were rinsed with DPBS, fixed and stained with rhodamine-phalloidin (Alexa-Flour 594, Invitrogen) and 4′,6-diamidino-2-phenylindole (DAPI; Sigma) to visualize the nuclei (Supporting Information, Methods). Samples were imaged on a Nikon fluorescence microscope. For immunocytochemical analysis samples were blocked and connexin43 and either sarcomeric alpha-actinin or troponin I (Abcam) at 1:200 dilution ratio in DPBS overnight at 4 °C (Supporting information, Methods). Stained samples were also imaged with an inverted laser scanning confocal microscope (Leica SP5 XMP, Germany) with a x40 water immersion objective.
Video imaging and quantification
For quantifying contractile properties of CMs were imaged with both images and movies taken with a video camera (Sony XCD-X710) attached to a fluorescence microscope (Nikon, Eclipse TE 2000U, Japan) at 10× magnification. Video sequences were digitized at 15 frames per second. Videos were analyzed by extracting the frames and rebuilding the sequence via Metafluor software (Universal Imaging), then assessing the light intensity variation across a selected region of interest over time as a representation of beating regularity. The periods of beats across different conditions were measured and quantified in order to compare beating regularity and homogeneity.
Nuclei Alignment analysis
Following a previously reported method[27], fluorescent images were of DAPI stained nuclei were acquired using an inverted fluorescence microscope (Nikon Eclipse Ti-5) and processed using ImageJ to convert to a binary image and distinguish individual nuclei which were fitted as an ellipse. For alignment analysis, the angle between the major elliptical axis of the elongated nuclei and the pattern direction (the vertical axis) was quantified as the angle of deviation and tallied in bins of 10 degrees across the whole image. Nuclei from at least 3 images per sample per condition were quantified. As images were taken at x10 objective magnification, each image has on average 300–1500 cells.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 software package. One-way ANOVA followed by Tukey’s post hoc test was used to characterize statistical difference between mean ± standard deviation (SD) of measurements for swelling ratio (Figure 1D) and channel features (Figure 2F). Two-way ANOVA with Tukey’s post hoc test was used to analyze differences in nuclei orientation (Figure 3G–I). Unpairted T-test with Welch’s correction were used to assess significance between mean and variance of average time between beats for patterned and unpatterned CMs (Figure 5C), and variances were compared with F tests. Significance level was deemed at p < 0.05.
Supplementary Material
Figure S1. Reaction scheme for synthesis of photodegradable methacrylate unit.
Figure S2. Reaction scheme for incorporation of photodegradable methacrylate into PEG.
Figure S3. 1H NMR spectroscopic analysis of the PEG-photodegradable dimethacrylate.
Figure S4. Reaction scheme for synthesis of photodegradable acrylate unit.
Figure S5. 1H NMR spectroscopic analysis of the PEG-photodegradable diacrylate
Figure S6. Brightfield images showing the proliferation and spreading of L929 mouse fibroblasts cells within in redox-initiated 50%PD GelMA hydrogels from zero (A), 4 (B), 6 (C) and 14 (D) days. Scale = 20 μm.
Figure S7. Brightfield and fluoresence images showing the quantity of live (green) versus dead (red) cells of L929 mouse fibroblasts cells on GelMA and PD hydrogels after 3 days. Scale = 40 μm.
Figure S8. Nuclei orientation quantification from DAPI stained images of CM seeded on patterned and unpatterned hydrogels. Cell orientation was calculated and grouped at 10° deviations from the patterned direction.
Figure S9. Cardiac fibroblasts cultured on 67% PD unpatterned substrates showing improved no preference to cell alignment on day 1 (A), day 3 (C) and day 7 (E) compared to patterned samples (B,D,F) at the same timepoints. (B) Quantification of neonatal rat cardiac fibroblast cell alignment across 7 days. Samples (i) to (iii) are for unpatterned samples from day 1, 3 and 7 respective, and samples (iv) to (vi) are patterned samples from the same timepoints. Cell orientation was calculated and grouped at 10° deviations from the patterned direction. Scale = 100 μm.
Figure S10. Quantification of sarcomere alignment based on sarcomeric alpha actinin staining (in Figure 4G–L) showing the average sarcomere direction, where zero degrees denotes horizontal aligned and 90° vertical alignment (channel direction). A random sample is expected to have an alignment of 45°. Error bars denote standard error of the mean. Overall the patterned samples showed a preference of sarcomeres towards the horizontal direction. *** denotes statistical significance determined by unpaired T-tests with Welch’s correction, p< 0.0001.
Video S11. Video showing beating of CM on unpatterned 67% PD hydrogels at day 4.
Video S12. Video showing beating of CM on patterned 67% PD hydrogels at day 4.
Acknowledgments
K.M.T. acknowledges support from the CRC for Polymers (top-up scholarship), CSIRO, the Australian-American Fulbright Association, BHP-Billiton and the Australian Postgraduate Award. J.S.F acknowledges funding from the Australian Research Council (Discovery grant DP 1093848). A. K. acknowledges funding from the National Science Foundation Career Award (DMR 0847287), the Office of Naval Research Young Investigator Award, the National Institutes of Health (HL092836, DE019014, EB012597, AR057837, DE021468, HL099073, EB008392) and the German Heart Foundation (S/04/12). The authors wish to thank Ben Fairbanks for his gift of LAP photoinitiator as well as helpful discussions with the manuscript.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Kelly M.C. Tsang, Department of Materials Engineering, Wellington Road, Monash University, Clayton, VIC 3800, Australia. CSIRO Manufacturing Flagship, Bayview Avenue, Clayton, VIC 3168, Australia. CRC for Polymers, 8 Redwood Drive, Notting Hill, VIC 3168, Australia
Dr. Nasim Annabi, Biomaterials Innovation Research Center, Division of Biomedical Engineering, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, 02139, MA, USA. Harvard-Massachusetts Institute of Technology Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge 02139, MA, USA. Wyss Institute for Biologically Inspired Engineering, Harvard University, 02115, MA, USA
Dr. Francesca Ercole, Department of Materials Engineering, Wellington Road, Monash University, Clayton, VIC 3800, Australia
Dr. Kun Zhou, Department of Materials Engineering, Wellington Road, Monash University, Clayton, VIC 3800, Australia
Daniel Karst, Biomaterials Innovation Research Center, Division of Biomedical Engineering, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, 02139, MA, USA. Harvard-Massachusetts Institute of Technology Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge 02139, MA, USA.
Fanyi Li, Department of Materials Engineering, Wellington Road, Monash University, Clayton, VIC 3800, Australia.
Dr. John M. Haynes, Faculty of Pharmacy and Pharmaceutical Sciences; Drug Discovery Biology, Department of Pharmaceutical Biology, Monash University, Parkville, VIC 3052, Australia
Dr. Richard A. Evans, CSIRO Manufacturing Flagship, Bayview Avenue, Clayton, VIC 3168, Australia. CRC for Polymers, 8 Redwood Drive, Notting Hill, VIC 3168, Australia
Dr. Helmut Thissen, CSIRO Manufacturing Flagship, Bayview Avenue, Clayton, VIC 3168, Australia. CRC for Polymers, 8 Redwood Drive, Notting Hill, VIC 3168, Australia
Prof. Ali Khademhosseini, Email: alik@rics.bwh.harvard.edu, Biomaterials Innovation Research Center, Division of Biomedical Engineering, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, 02139, MA, USA. Harvard-Massachusetts Institute of Technology Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge 02139, MA, USA. Wyss Institute for Biologically Inspired Engineering, Harvard University, 02115, MA, USA. Department of Maxillofacial Biomedical Engineering and Institute of Oral Biology, School of Dentistry, Kyung Hee University, Seoul 130-701, Republic of Korea. Department of Physics, King Abdulaziz University, Jeddah 21569, Saudi Arabia
A/Prof John S. Forsythe, Email: john.forsythe@monash.edu, Department of Materials Engineering, Wellington Road, Monash University, Clayton, VIC 3800, Australia.
References
- 1.Rice JJ, Martino MM, De Laporte L, Tortelli F, Briquez PS, Hubbell JA. Adv Healthcare Mater. 2013;2:57. doi: 10.1002/adhm.201200197. [DOI] [PubMed] [Google Scholar]
- 2.Rehmann MS, Kloxin AM. Soft Matter. 2013;9:6737. doi: 10.1039/c3sm50217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeForest CA, Anseth KS. Annu Rev Chem Biomol Eng. 2012;3:421. doi: 10.1146/annurev-chembioeng-062011-080945. [DOI] [PubMed] [Google Scholar]
- 4.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azagarsamy MA, McKinnon DD, Alge DL, Anseth KS. ACS Macro Lett. 2014;3:515. doi: 10.1021/mz500230p. [DOI] [PubMed] [Google Scholar]
- 6.Ki CS, Shih H, Lin CC. Polymer. 2013;54:2115. doi: 10.1016/j.polymer.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanagawa F, Sugiura S, Takagi T, Sumaru K, Camci-Unal G, Patel A, Khademhosseini A, Kanamori T. Adv Healthcare Mater. 2014:n/a. doi: 10.1002/adhm.201400180. [DOI] [PubMed]
- 8.Fairbanks BD, Singh SP, Bowman CN, Anseth KS. Macromolecules. 2011;44:2444. doi: 10.1021/ma200202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ercole F, Thissen H, Tsang K, Evans RA, Forsythe JS. Macromolecules. 2012;45:8387. [Google Scholar]
- 10.Kloxin AM, Tibbitt MW, Kasko AM, Fairbairn JA, Anseth KS. Adv Mater. 2010;22:61. doi: 10.1002/adma.200900917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kloxin AM, Lewis KJR, DeForest CA, Seedorf G, Tibbitt MW, Balasubramaniam V, Anseth KS. Integr Biol. 2012;4:1540. doi: 10.1039/c2ib20212c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kloxin AM, Benton JA, Anseth KS. Biomaterials. 2010;31:1. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang C, Tibbitt MW, Basta L, Anseth KS. Nat Mater. 2014;13:645. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shenoy R, Tibbitt MW, Anseth KS, Bowman CN. Chem Mater. 2013;25:761. doi: 10.1021/cm303913f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kloxin AM, Tibbitt MW, Anseth KS. Nat Protocols. 2010;5:1867. doi: 10.1038/nprot.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sreejit P, Verma RS. Stem Cell Rev Rep. 2013;9:158. doi: 10.1007/s12015-013-9427-6. [DOI] [PubMed] [Google Scholar]
- 17.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Biomaterials. 2010;31:5536. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aubin H, Nichol JW, Hutson CB, Bae H, Sieminski AL, Cropek DM, Akhyari P, Khademhosseini A. Biomaterials. 2010;31:6941. doi: 10.1016/j.biomaterials.2010.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutson CB, Nichol JW, Aubin H, Bae H, Yamanlar S, Al-Haque S, Koshy ST, Khademhosseini A. Tissue Eng Part A. 2011;17:1713. doi: 10.1089/ten.tea.2010.0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camci-Unal G, Cuttica D, Annabi N, Demarchi D, Khademhosseini A. Biomacromolecules. 2013;14:1085. doi: 10.1021/bm3019856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahadian S, Ramón-Azcón J, Estili M, Liang X, Ostrovidov S, Shiku H, Ramalingam M, Nakajima K, Sakka Y, Bae H, Matsue T, Khademhosseini A. Sci Rep. 2014;4 doi: 10.1038/srep04271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikkhah M, Eshak N, Zorlutuna P, Annabi N, Castello M, Kim K, Dolatshahi-Pirouz A, Edalat F, Bae H, Yang Y, Khademhosseini A. Biomaterials. 2012;33:9009. doi: 10.1016/j.biomaterials.2012.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillemette MD, Park H, Hsiao JC, Jain SR, Larson BL, Langer R, Freed LE. Macromol Biosci. 2010;10:1330. doi: 10.1002/mabi.201000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heidi Au TH, Cui B, Chu ZE, Veres T, Radisic M. Lab Chip. 2009;9:564. doi: 10.1039/b810034a. [DOI] [PubMed] [Google Scholar]
- 25.Kharaziha M, Nikkhah M, Shin SR, Annabi N, Masoumi N, Gaharwar AK, Camci-Unal G, Khademhosseini A. Biomaterials. 2013;34:6355. doi: 10.1016/j.biomaterials.2013.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feinberg AW, Feigel A, Shevkoplyas SS, Sheehy S, Whitesides GM, Parker KK. Science. 2007;317:1366. doi: 10.1126/science.1146885. [DOI] [PubMed] [Google Scholar]
- 27.Annabi N, Tsang K, Mithieux SM, Nikkhah M, Ameri A, Khademhosseini A, Weiss AS. Adv Funct Mater. 2013;23:4950. doi: 10.1002/adfm.201300570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korecky B, Hai CM, Rakusan K. Can J Physiol Pharmacol. 1982;60:23. doi: 10.1139/y82-003. [DOI] [PubMed] [Google Scholar]
- 29.McCain ML, Agarwal A, Nesmith HW, Nesmith AP, Parker KK. Biomaterials. 2014;35:5462. doi: 10.1016/j.biomaterials.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao YC, Kao ZK. ACS Appl Mater Interfaces. 2012;4:5109. doi: 10.1021/am301654j. [DOI] [PubMed] [Google Scholar]
- 31.Wong DY, Griffin DR, Reed J, Kasko AM. Macromolecules. 2010;43:2824. [Google Scholar]
- 32.Souders CA, Bowers SL, Baudino TA. Circ Res. 2009;105:1164. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen MS, Nygaard Axelsen L, Sorgen PL, Verma V, Delmar M, Holstein-Rathlou N-H. Compr Physiol. John Wiley & Sons, Inc; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin SR, Jung SM, Zalabany M, Kim K, Zorlutuna P, Kim Sb, Nikkhah M, Khabiry M, Azize M, Kong J, Wan K-t, Palacios T, Dokmeci MR, Bae H, Tang X, Khademhosseini A. ACS Nano. 2013;7:2369. doi: 10.1021/nn305559j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Au HTH, Cheng I, Chowdhury MF, Radisic M. Biomaterials. 2007;28:4277. doi: 10.1016/j.biomaterials.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Haque S, Miklas JW, Feric N, Chiu LLY, Chen WLK, Simmons CA, Radisic M. Macromol Biosci. 2012;12:1342. doi: 10.1002/mabi.201200042. [DOI] [PubMed] [Google Scholar]
- 37.Kirschner CM, Alge DL, Gould ST, Anseth KS. Adv Healthcare Mater. 2014;3:649. doi: 10.1002/adhm.201300288. [DOI] [PubMed] [Google Scholar]
- 38.Holmes CP. J Org Chem. 1997;62:2370. doi: 10.1021/jo961602x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reaction scheme for synthesis of photodegradable methacrylate unit.
Figure S2. Reaction scheme for incorporation of photodegradable methacrylate into PEG.
Figure S3. 1H NMR spectroscopic analysis of the PEG-photodegradable dimethacrylate.
Figure S4. Reaction scheme for synthesis of photodegradable acrylate unit.
Figure S5. 1H NMR spectroscopic analysis of the PEG-photodegradable diacrylate
Figure S6. Brightfield images showing the proliferation and spreading of L929 mouse fibroblasts cells within in redox-initiated 50%PD GelMA hydrogels from zero (A), 4 (B), 6 (C) and 14 (D) days. Scale = 20 μm.
Figure S7. Brightfield and fluoresence images showing the quantity of live (green) versus dead (red) cells of L929 mouse fibroblasts cells on GelMA and PD hydrogels after 3 days. Scale = 40 μm.
Figure S8. Nuclei orientation quantification from DAPI stained images of CM seeded on patterned and unpatterned hydrogels. Cell orientation was calculated and grouped at 10° deviations from the patterned direction.
Figure S9. Cardiac fibroblasts cultured on 67% PD unpatterned substrates showing improved no preference to cell alignment on day 1 (A), day 3 (C) and day 7 (E) compared to patterned samples (B,D,F) at the same timepoints. (B) Quantification of neonatal rat cardiac fibroblast cell alignment across 7 days. Samples (i) to (iii) are for unpatterned samples from day 1, 3 and 7 respective, and samples (iv) to (vi) are patterned samples from the same timepoints. Cell orientation was calculated and grouped at 10° deviations from the patterned direction. Scale = 100 μm.
Figure S10. Quantification of sarcomere alignment based on sarcomeric alpha actinin staining (in Figure 4G–L) showing the average sarcomere direction, where zero degrees denotes horizontal aligned and 90° vertical alignment (channel direction). A random sample is expected to have an alignment of 45°. Error bars denote standard error of the mean. Overall the patterned samples showed a preference of sarcomeres towards the horizontal direction. *** denotes statistical significance determined by unpaired T-tests with Welch’s correction, p< 0.0001.
Video S11. Video showing beating of CM on unpatterned 67% PD hydrogels at day 4.
Video S12. Video showing beating of CM on patterned 67% PD hydrogels at day 4.