

Abstract
Advances in kidney cancer have occurred over the past decade, including the discovery of mutations in chromatin remodeling genes and genomic heterogeneity in clear cell renal cell carcinoma (ccRCC), altered metabolic patterns in ccRCC and papillary renal cell carcinoma and the approval of drugs for patients with ccRCC.
In the decade from 2004 to 2014 there have been remarkable improvements both in our understanding of the genetic basis of renal cell carcinoma (RCC) and in the treatment of patients with advanced kidney cancer. These developments include the approval of seven agents targeting the Von Hippel–Lindau/hypoxia-inducible factor (VHL/HIF) pathway for patients with advanced RCC by the FDA, and the identification of mutations in genes expressed by kidney tumours. These advances were built on the progress of the previous decade, which had seen the discovery of the VHL gene in VHL syndrome, the identification of mutations of the VHL gene in ccRCC and the delineation of the VHL pathway. Mutations in MET (proto-oncogene, receptor tyrosine kinase) and their involvement in hereditary type 1 papillary kidney cancer were also identified, and FLCN (folliculin) gene mutations were found to be the primary cause for the inherited form of chromophobe kidney cancer associated with Birt-Hogg-Dubé syndrome. Mutations in the Krebs cycle enzyme, fumarate hydratase (FH) gene were also discovered to be the cause of most cases of hereditary type 2 papillary kidney cancer associated with hereditary leiomyomatosis and renal cell carcinoma (HLRCC). The development of the understanding that kidney cancer is made up of a number of different types of cancer, each having a different histology, a different clinical course and genetic causes greatly aided progress in treating this disease.
At the start of this decade, only a single agent, interleukin-2 (IL-2), was approved for the treatment of patients with advanced kidney cancer. The discovery of the VHL gene in 1993, the identification of VHL gene mutations in a high percentage of clear cell renal tumours in 1994 and the delineation of the VHL/HIF pathway in the late 1990s provided the foundation for the development of targeted therapeutic agents for patients with this disease. The VHL protein functions as a component of a heteromeric protein complex that targets HIF for ubiquitin-mediated degradation. When the VHL gene is mutated, HIF accumulates and the transcription of a number of downstream genes, including the vascular endothelial growth factor (VEGF) and the platelet derived growth factor (PDGF), is increased, which drives tumour progression.
Several randomized, double-blind trials have since identified effective therapeutic agents for the treatment of kidney cancer, targeting VEGF and PDGF. The first targeted agent, sorafenib, which targets the VEGF and PDGF receptors, was approved for patients with advanced kidney cancer in December 2005. Sorafenib was found to have a median progression free survival (PFS) of 5.5 months for patients with kidney cancer, compared with 2.8 months for those given a placebo. In January 2006, sunitinib, which also targets the VEGF and PDGF receptors, was approved by the FDA. Treatment with sunitinib was associated with an 11 month PFS in patients with kidney cancer, compared with 5 months for patients treated with interferon (IFNα), a non-specific immune modulator that has been used for a number of years in patients with advanced kidney cancer. The mTOR (mammalian target of rapamycin) pathway is an important kidney cancer pathway that indirectly influences VEGF and PDGF expression by affecting HIF translation. In May 2007 temsirolimus, an agent that targets mTOR was approved for patients with advanced kidney cancer, after patients with poor prognostic factors and treated with temsirolimus had a 10.9 month survival compared with 7.3 months PFS for those treated with IFNα. Everolimus, another agent targeting the mTOR pathway, was approved in March 2009 for patients with RCC who had previously received sunitinib or sorafenib therapy. Those treated with everolimus demonstrated 4.0 months PFS compared with 1.9 months for patients in the placebo arm of the study. July 2009 saw the approval of bevacizumab, a monoclonal antibody based inhibitor of VEGF-A, in conjunction with IFNα, for patients with advanced RCC. Treatment with bevacizumab and IFNα increased PFS to a median of 10.2 months, compared with 5.4 months PFS in patients treated with IFNα alone. In October the same year, pazopanib, a VEGF Receptor (VEGFR) 1, 2 and 3 inhibitor, was approved to treat patients with advanced RCC. Pazopanib therapy was found to be associated with 9.2 months PFS compared with 4.2 months for patients given a placebo. Finally, in January 2012 axitinib, another VEGFR 1, 2 and 3 inhibitor was the seventh targeted therapeutic agent this decade approved by the FDA for the treatment of patients with kidney cancer. Patients treated with axitinib were found to have a median PFS of 6.7 months compared with 4.7 months for patient receiving sorafenib.1 These remarkable accomplishments provided physicians with, for the first time, a range of targeted agents approved for patients with advanced kidney cancer. While the response rates for treatment with agents such as sunitinib or pazopanib are reported to be in the 35% range, and there have been improvements in PFS and overall survival, few agents result in a complete response; most patients eventually progress and many die of their cancer. Why is it that a patients' cancer progresses through therapy? It could be because targeting of the VHL/HIF pathway with currently available therapies is inadequate, or because there are other genes associated with initiation or progression of kidney cancer that are not currently being targeted. At the moment there is no definitive answer to this question.
Exciting new insights into the genetic basis of kidney cancer came from reports identifying mutations in several chromatin remodeling, histone modifying and SWItch/sucrose non-fermentable (SNF) nucleosome remodeling complex genes in ccRCC.2, 3 When the Cancer Genome Atlas Research Network performed a comprehensive molecular characterization of genetic changes associated with ccRCC and evaluated more than 400 tumours using different genomic platforms, 19 significantly mutated genes were identified. After the VHL gene, the most commonly mutated genes were the chromosome 3 chromatin remodeling genes—PBRM1, (protein polybromo-1), SETD2, (histone-lysine N-methyltransferase) and BAP1, (ubiquitin carboxyl-terminal hydrolase), and mutations of BAP1 were found to be correlated with poor survival. Furthermore, mutations were identified in KDM5C (Lysine-specific demethylase 5C), TP53 (cellular tumor antigen p53), and the phosphoinositide 3-kinase / RAC-alpha serine/threonine-protein kinase (PI3K/AKT) pathway genes, PTEN (phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase), MTOR, and PIK3CA (phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform). It was also demonstrated that the PI3K/AKT has an important role in tumour progression. Integrative analysis showed that high grade, high-stage, low-survival clear cell tumours demonstrated evidence of a Warburg-like metabolic shift, involving downregulation of genes involved in the Krebs cycle, decreased 5′ adenosine monophosphate-activated protein kinase (AMPK) and increased fatty acid synthesis.4 These findings, which are consistent with the isotopomer spectral analysis of VHL -/- ccRCC and FH -/- type 2 papillary RCC, reveal that a dependence on reductive glutamine metabolism for lipid biosynthesis is important in kidney cancer and could provide the foundation for the development of novel forms of therapy targeting the metabolic basis of renal cell carcinoma.5, 6
In order to further understand the genetic basis of ccRCC, Gerlinger and colleagues reported the use of multiregion exome sequencing (M-seq) to study the genomic architecture and evolution of ccRCC. In a study with major implications for our understanding of tumour evolution and the effect of targeted therapeutics, these investigators found extensive genomic heterogeneity in CCRCC. ‘Truncal’ mutations of the VHL gene were found in every segment of each sample, while driver mutations in the chromatin remodeling genes SETD2, BAP1 and KDM5C, as well as TP53 and genes in the PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase) -mTOR pathway (MTOR, PIK3CA, PTEN and TSC2[tuberin]), were found only in some segments of the tumours, these were labeled ‘branch’ mutations. Of all the driver mutations, 75% were subclonal, and intratumoral heterogeneity increased with the number of biopsies analysed.7 These findings raise profound questions about the most effective way to detect driver mutations in ccRCC, and which gene pathways to target. Should M-seq be performed on primary tumours and/or metastatic sites in order to identify common driver mutations that are associated with the initiation and progression of kidney cancer? Do both the truncal VHL/HIF and branching driver gene pathways need to be targeted, singularly or together, to develop an effective form of therapy for ccRCC? These critical questions will need to be answered in the next decade in kidney cancer research.
Although we have seen the development of a number of novel therapies for patients with advanced clear cell kidney cancer over the past decade, we still have no effective treatment for patients with advanced papillary kidney cancer. Significant insight into papillary kidney cancer has come from studies of the hereditary form of type 2 papillary renal carcinoma that is found in patients with HLRCC, and is caused by mutation of the FH gene. FH-deficient kidney cancer cells, which exhibit impaired oxidative phosphorylation, undergo a Warburg-like metabolic shift to aerobic glycolysis with decreased AMPK (5′ adenosine monophosphate-activated protein kinase) and increased mTOR and HIF1α levels.8 Studies by Adam et al.9 and Ooi et al.10 showed a striking upregulation of the antioxidant signaling pathways in both FH-deficient kidney cancer, as well as sporadic (non-inherited) papillary kidney cancer. Elevated fumarate in FH-deficient RCC targets and inactivates the electrophile sensor, KEAP1 (Kelch-like ECH-associated protein 1), which results in NRF2 (Nuclear factor erythroid 2-related factor 2) accumulation and upregulation of antioxidant response element (ARE)-controlled genes, such as NAD(P) H quinone oxidoreductase (NQO1) and the glutathione S-transferases (GST's). These ARE-controlled genes are critical for the survival of cancers, which are characterized by high levels of oxidative stress, such as hereditary and sporadic type 2 papillary kidney cancer.9, 10 This critical pathway provides a number of opportunities for the development of targeted therapeutic approaches for patients with advanced papillary RCC.
Overall, this past decade has seen a number of historic advances towards the understanding and treatment of RCC, with the FDA approving seven drugs targeting the VHL/HIF pathway for patients with advanced kidney cancer. While the sobering finding of extensive genomic heterogeneity poses a significant therapeutic challenge, intense efforts are underway to translate the discoveries of chromatin remodeling gene mutations into new therapeutic approaches for patients with advanced RCC. The finding of a Warburg-like metabolic shift and the dependence of the KEAP1-NRF2 antioxidant pathway in both clear cell and papillary RCC provides very promising opportunities for the development of therapies targeting the metabolic basis of kidney cancer.
Figure 1. Implications of Genomic Heterogeneity.
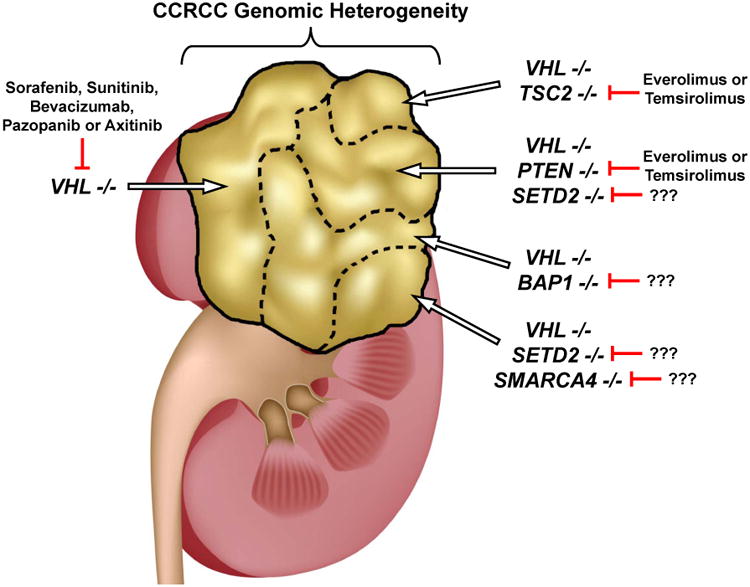
The finding of genomic heterogeneity of clear cell renal cell carcinoma (CCRCC) has profound implications, not only for our understanding of the critical factors associated with the initiation and evolution of this disease, but also for the development of effective targeted therapeutic strategies.7 This diagram shows genomic heterogeneity that can be found within different regions of a CCRCC. The ‘truncal’ VHL gene is mutated in all regions, and could be targeted with with agents such as sunitinib, pazopanib or axitinib. The ‘branch’ mutations, present in selected regions of the tumour, can occur in genes for which potential targeted therapies exist, or in genes for which no current targeted treatment exists, such as SETD2, BAP1 or SMARCA4. Thus, the most effective strategy may involve combinatorial approaches targeting the ‘truncal’ VHL-HIF pathway, or a more global strategy that includes targeting ‘branch’ chromatin remodeling gene pathways.
References
- 1.Jonasch E, Motzer RJ. Ten years of progress in renal cell carcinoma. J Natl Compr Canc Netw. 2012;10:690–693. doi: 10.6004/jnccn.2012.0071. [DOI] [PubMed] [Google Scholar]
- 2.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullen AR, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong WH, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–327. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adam J, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ooi A, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]