

Abstract
An Arg345Trp (R345W) mutation in the last canonical calcium-binding epidermal growth factor (cbEGF) domain of fibulin-3 (F3) causes the rare macular dystrophy, Malattia Leventinese (ML). In cell culture studies, this mutation leads to inefficient F3 secretion and higher intracellular steady state levels, likely due to F3 disulfide bonding and/or protein folding problems. However, how the R345W mutation actually causes ML is still largely unknown. Herein we tested whether the introduction of analogous, ‘pseudo-pathogenic’ tryptophan mutations immediately after the bn cysteine (bn+1) in other cbEGF domains also caused protein folding/secretion challenges. We found that introduction of tryptophan mutations into each of the four other F3 canonical cbEGF domains caused a significant reduction in protein secretion ranging from 2.7 to 56% of wild-type (WT) F3 levels. Surprisingly, an R185W mutation in the first canonical cbEGF domain of F3 yielded the highest amount of secretion among the F3 tryptophan mutants, and its secretion defect could be rescued to near WT levels (95%) after growth temperature reduction. Interestingly, when similarly positioned tryptophan mutations were introduced into any of the canonical cbEGF domains of the highly homologous protein, fibulin-5 (F5), there was no effect on secretion. In an attempt to make F3 tolerant of tryptophan residues (like F5), we genetically engineered F3 to have a higher sequence homology with F5 by deleting three insert regions present in F3, but not F5. However, deletion of one or more of these regions did not have a beneficial effect on R345W F3 secretion. Overall, these results demonstrate that the introduction of tryptophan residues at the bn+1 position does not universally disrupt cbEGF domain folding and secretion, but that their effect is context dependent, and in this case, uniquely disrupt the folding of canonical cbEGF domains of F3, but not F5.
Keywords: Fibulin-3, Fibulin-5, Tryptophan mutations, Calcium binding epidermal growth factor domains, Malattia Leventinese, Protein misfolding
1. INTRODUCTION
Malattia Leventinese (ML), also known as Doyne Honeycomb Retinal Dystrophy, is a rare retinal disease which has striking phenotypic similarities to age-related macular degeneration (AMD) (Fu et al., 2007; Marmorstein et al., 2007; Stone et al., 1999). The primary similarity between ML and AMD is the formation of large extracellular deposits of proteins and lipids under the retinal pigment epithelial cell layer (Marmorstein, 2004). However, unlike AMD, these basal deposits form at a much earlier age in ML. In fact, drusen-like deposits have been detected in ML patients as young as twenty-two years of age (Michaelides et al., 2006). ML was found to be caused by a single R345W mutation in the secreted protein, fibulin-3 (F3) (Stone et al., 1999), yet exactly how this mutation ultimately leads to disease is still unknown. While no other mutations in F3 have been associated with retinal dysfunction or AMD, independent studies have found that drusen isolated from AMD patients, but not control patients, are surrounded by F3 (presumably WT F3) (Marmorstein et al., 2002; Sohn et al., 2014; Wyatt et al., 2013). Taken together, these results suggest that both WT and R345W F3 are associated with potentially pathogenic basal deposit formation. Thus, developing a deeper understanding of the molecular mechanisms that govern WT as well as R345W F3 folding and secretion will hopefully yield insights in to the etiology of ML and possibly AMD.
Cell culture studies have indicated that the R345W mutant is likely misfolded and inefficiently secreted from the endoplasmic reticulum (ER) (Hulleman et al., 2011; Marmorstein et al., 2002). Consistent with this hypothesis, over-expression of R345W F3 has been demonstrated to cause ER stress (Roybal et al., 2005), while reduction of ER stress through translational attenuation has been shown to partially rescue R345W F3 secretion (Hulleman et al., 2012). Prior to secretion of WT or R345W F3, each of the five canonical calcium binding epidermal growth factor (cbEGF) domains must first form three highly conserved disulfide bonds (Fig. 1A). The disulfide bond formation (and thus folding) of EGF domains proceeds via an established pathway wherein the formation of 1, 2 and 3 disulfide scrambled (non-native) intermediates are succeeded by the formation of native disulfide bond formation as seen in Fig. 1A (Chang et al., 2001; Chang et al., 1995). Consistent with the importance of proper disulfide bond formation prior to F3 secretion, elimination of any native disulfide bond within the 6th cbEGF domain (D6) of F3 all but prevent secretion of the full length protein (Hulleman et al., 2011).
Fig. 1.

Introduction of tryptophan residues into cbEGF domains of F3 have distinct effects on F3 secretion. (A) Schematic representation of domain 6 (D6) of F3, adapted from (Hulleman et al., 2011). dsb = disulfide bond (B) F3 domain organization and location of newly generated tryptophan residues, adapted from (Hulleman et al., 2011). (C) Conditioned media aliquots from transfected TREx-293 cells were taken 48 h after transfection and assayed for GLuc activity. (D) Western blot verification of GLuc activity levels shown in (C). (E) qPCR verification of similar F3 expression levels. mRNA was isolated from TREx-293 cells 48 h after transfection and the transcript levels of F3 relative to WT F3 were evaluated by qPCR. (F) Transfected TREx-293 cells were grown at 37°C followed by a media change and 24 h at 30°C. Conditioned media aliquots were assayed using the GLuc assay. n ≥ 3 independent experiments for panels C–F.
The R345W mutation in F3 occurs immediately adjacent to a cysteine reside that forms disulfide bond 2 (dsb2) within D6 of F3 (Fig. 1A, B). We have demonstrated previously that the R345W mutation, as well as other aromatic substitutions (and proline) at the 345th position partially disrupt dsb2 formation, and thus affect the folding of D6 and the eventual secretion of F3 (Hulleman et al., 2011). However, it remains unknown whether effects of the R345W mutation on F3 secretion are specific to D6 of F3, or if the presence of a tryptophan residue (in an identical position) in other F3 cbEGF domains would have similar detrimental effects on F3 secretion. In the experiments described herein, we explored this possibility within the context of F3, and, additionally, we tested the effects of tryptophan introduction more broadly in another highly homologous fibulin protein, fibulin-5 (F5). Our results demonstrate that the effects of tryptophan residues at the bn+1 position are context-dependent. Not only are tryptophan residues differentially tolerated within the cbEGF domains of F3, reducing the secretion of full length F3 by 44 – 97%, but surprisingly, they have no apparent effect on F5 secretion efficiency.
2. METHODS
2.1 Plasmid generation
Plasmids encoding for WT F3 eGLuc2, R345W F3 eGLuc2 and WT F5 eGLuc2 were generated previously and are described in (Hulleman et al., 2012). Tryptophan mutants and deletion constructs were generated using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, NEB, Ipswich, MA). The pEGFP-N1 vector (Clontech, Mountain View, CA) was used as a control plamid in some experiments. All constructs were verified by sequencing.
2.2 Cell culture
TREx-293 cells (Life Technologies, Carlsbad, CA) were grown in DMEM (Corning, Corning, NY) supplemented with 1% penicillin/streptomycin (Corning), glutamine (Corning) and 10% FBS (Omega Scientific, Tarzana, CA) and maintained at 37°C, 5% CO2 unless otherwise noted. While TREx-293 cells allow for doxycycline/tetracycline-regulated expression in a ‘TET-ON’ manner, this feature was not used for any of the experiments described herein; all constructs were constitutively expressed. TREx-293 cells were transfected with pcDNA constructs encoding for the eGLuc2 fusion proteins using X-tremeGENE9 (Roche, Picataquay, NJ) as described previously using 250–500 ng of DNA per well of a 24 well plate (Hulleman et al., 2012).
2.3 GLuc assay
For GLuc assays, an identical protocol to that described in (Hulleman et al., 2011) was used. Briefly, 50 µL of conditioned media was removed from the cultures and reacted with 10 µL GLuc buffer and 50 nL GLuc substrate from the NEB BioLux kit (NEB). The luminescence was read immediately using a BioTek Synergy 2 plate reader (BioTek, Winooski, VT). For growth temperature reduction experiments, cells were transfected for 48 h, followed by a media change and 24 h at 30°C.
2.4 Western blotting
For all western blotting experiments, media containing 2% FBS was used to minimize gel distortions caused by excess bovine serum albumin. Conditioned media aliquots from transfected TREx-293 cells were denatured using reducing Laemlli buffer and run on Tris-Gly SDS-PAGE gels. Proteins were transferred to a nitrocellulose membrane using a G2 Blotter (Thermo Scientific, Waltham, MA) blocked in LI-COR blocking buffer (LI-COR, Lincoln, NE) and probed with a rabbit anti-GLuc antibody (1:2000, NEB) followed by a goat anti-rabbit IRDye 680RD secondary antibody (1:10,000–1:15,000, LI-COR). Blots were imaged on a LI-COR Odyssey Fc Imager (LI-COR).
2.5 Quantitative PCR (qPCR)
Messenger RNA (mRNA) was extracted from transfected TREx-293 cells using a Promega Wizard RNA kit (Promega, Madison, WI). Equal amounts of RNA (10–100 ng) were then reverse transcribed into cDNA using a 5 Prime reverse transcriptase kit (5 Prime, Gaithersburg, MD). The cDNA was diluted 4–5 fold, and mixed with qPCR primers and 5 Prime master mix (5 Prime). SybrGreen amplification was detected on a Quant Studio 6 using a 60°C/30 sec annealing temperature/duration. Primer sequences can be found in a previous publication (Hulleman et al., 2013).
2.7 Sequence homology
To compare fibulin protein sequence similarity, a constraint-based alignment tool (COBALT, http://www.st-va.ncbi.nlm.nih.gov/tools/cobalt, (Papadopoulos and Agarwala, 2007)) was used.
3. RESULTS
3.1 Introduction of ‘pseudo-pathogenic’ tryptophan residues in F3 cbEGF domains have unique effects on F3 secretion
The pathogenic R345W mutation in F3 occurs immediately adjacent to a cysteine residue (the bn cysteine) involved in the formation of dsb2 in within D6 (Fig 1A). We designated this position as the bn+1 residue within the cbEGF domain. We have shown previously that the presence of a tryptophan, phenyalanine, tyrosine or proline at the bn+1 residue in D6 causes secretion defects in F3 and a reduced ability to properly form dsb2 within D6 (Hulleman et al., 2011). Due to the similarities in disulfide bond formation across canonical cbEGF domains (Chang et al., 2001; Chang et al., 1995; Chang et al., 2002), as well as a generalized similarity in the final fold of cbEGF domains, we hypothesized that placing a tryptophan residue at the bn+1 position of other cbEGF domains in F3 (Fig. 1B) would have a similar effect as the R345W mutation and cause inefficient F3 secretion. Surprisingly, introduction of tryptophan residues into the separate cbEGF domains of F3 were differentially tolerated (Fig. 1C). The tryptophan mutants, H225W, A265W, and Q306W which correspond to mutations in domains 3, 4 and 5 (D3, D4, and D5), respectively, reduced F3 secretion by 92–97% relative to WT (Fig. 1C). In contrast, mutation of an arginine residue at position 185 in domain 2 (D2), had a substantially smaller effect on F3 secretion, reducing secreted F3 levels by 44% (Fig. 1C). These results were verified by measuring the amount of secreted F3 in the conditioned media in parallel experiments using western blotting (Fig. 1D). Furthermore, the differences in the amounts of F3 secretion were not simply due to transcriptional changes or transfection efficiency differences (Fig. 1E).
Previously we have demonstrated that the secretion defects in R345W F3 can be partially rescued by reducing the temperature at which the transfected cells are grown (Hulleman et al., 2011). We used a similar strategy to determine if the secretion of the newly engineered tryptophan mutants could also be partially or fully rescued to WT levels. Interestingly, the secretion of the majority of new tryptophan mutants could be rescued by reduction of growth temperatures to 30°C for 24 h (Fig. 1F). While R185W F3 secretion was raised to near-WT F3 levels (95% of WT), secreted levels of H225W and Q306W were elevated 3.2 and 3.6 fold, respectively, to 39% and 36% of WT levels (Fig. 1F). In contrast, secretion of A265W F3 was virtually unaltered by reduced growth temperatures (4% of WT F3 levels at 37°C vs. 3% of WT F3 levels at 30°C, Fig. 1F). The inability of the A265W mutant to be rescued by growth temperature reduction may indicate that the protein folding difficulties this mutation causes are distinct from the other tryptophan mutants. A similar inability to be rescued by reduced growth temperatures was observed with cysteine-to-alanine mutations which completely prevented native disulfide bond formation (Hulleman et al., 2011).
3.2 Tryptophan introduction into cbEGF domains of F5 does not alter protein secretion
To determine how broadly tryptophan mutations at the bn+1 position affected the folding and secretion of proteins containing typical cbEGF domains, we introduced five new tryptophan mutations into a protein with high homology to F3, F5 (Fig. 2A). We then quantified the amount of secreted F5 in the conditioned media from transfected TREx-293 cells. Surprisingly, all of the tryptophan mutations that we generated (N139W, Q178W, V218W, Q259W and N300W) were well tolerated and had no discernible effect on F5 secretion (Fig. 2B). These results were also verified by western blotting (Fig. 2C). These results suggest that the simple presence of a tryptophan at the bn+1 position is not universally disruptive of the folding of cbEGF domains, but rather, that the effect of the tryptophan on protein secretion is context-dependent; factors such as which domain is mutated as well as what protein is mutated ultimately dictate whether a tryptophan residue is tolerated.
Fig. 2.

Tryptophan mutations have no effect on F5 secretion. (A) F5 domain organization and location of newly generated tryptophan residues, adapted from (Hulleman et al., 2012). (B) Conditioned media aliquots from transfected TREx-293 cells were taken 48 h after transfection and assayed for GLuc activity. (C) Western blot verification of GLuc activity levels shown in (B). n ≥ 3 independent experiments for all panels.
3.3 F3 and F5 share similar domain organization and sequence homology
Both F3 and F5 are comprised of a single atypical cbEGF domain with an insertion, followed by a series of five canonical cbEGF domains and a C-terminal fibulin-type domain (Fig. 1B, Fig. 2A). We aligned the protein sequences of F3 and F5 to identify regions of similarity as well as discrepant areas. We found that F3 shares a 44.6% (220/493 residues) sequence homology with F5 (Sup. Fig. 1). The region with the highest amount of discrepant residues is located within the first atypical cbEGF domain (Sup. Fig. 1). This region in F3 contains three insertions which are not present in F5. These regions correspond to amino acids 22–24 (insert 1), amino acids 92–115 (insert 2) and amino acids 133–152 (insert 3) in F3 (Sup. Fig. 1). We rationalized that removing one or more of the F3 insertions would potentially make F3 fold and behave more similar to F5, i.e., not affected by tryptophan introduction. Thus, we generated WT and R345W F3 variants lacking F3 residues 22–24 (Δ1), lacking residues 92–115 (Δ2), lacking residues 133–152 (Δ3), lacking residues 92–115, 133–152 (Δ2,3), or lacking residues 22–24, 92–115, 133–152 (Δ1,2,3). Δ1,2,3 F3 increased the sequence homology to WT F5 to 49.5% (Sup. Fig. 2, 220/444 residues).
Previously we noted that WT F5 was secreted more efficiently than WT F3 (Fig. 3A, (Hulleman et al., 2012)). We hypothesized that deletion of one or more of the insertions in F3 would increase F3 secretion kinetics and make F3 tolerant of tryptophan residues. Indeed, individual removal of residues 92–115 (Δ2) and 133–153 (Δ3) increased F3 secretion by 24 and 54%, respectively (Fig. 3B). However, combination of these deletions (Δ2,3) was not additive nor synergistic in favoring F3 secretion (Fig. 3B). Furthermore, removal of residues 22–26 (Δ1) or the combination of all three deletions (Δ1,2,3) was in fact detrimental to F3 secretion (Fig. 3B). These results were verified at the western blotting level (Fig. 3C). In parallel, we generated a series of deletions constructs of R345W F3. Surprisingly, the effects of the deletions on R345W F3 secretion were distinct from those observed with F3 (cf Fig. 3B vs. 3D). Removal of the insertion regions had very little effect on R345W F3 secretion (Fig. 3D). These results were also verified at the protein level (Fig. 3E). The differences in how F3 and R345W F3 tolerate the deletions suggest that the two variants proceed down different folding pathways and/or have different intramolecular contacts during the folding process.
Fig. 3.

Deletion of one or more insert regions in F3 does not increase F3 secretion to F5 levels or make it tolerant of tryptophan residues. (A) WT F5 is secreted more efficiently from TREx-293 cells than WT F3 under identical conditions. Absolute luciferase activity was measured 48 h after initial transfection. (B) Secretion of F3 deletion variants does not reach F5 levels. Single, double or triple deletion constructs were transfected into TREx-293 cells and 48 h afterward the amount of eGLuc2 was assayed in the conditioned media. F5 secretion levels 48 h after transfection are shown by a dotted line. (C) Western blot confirmation of (B). (D) Deletion variants do not have an increased tolerance for the pathogenic R345W mutation. Single, double or triple deletion constructs were generated in the R345W background and transfected into TREx-293 cells. 48 h afterward, the amount of eGLuc2 was assayed in the conditioned media. (E) Western blot confirmation of (D). n ≥ 3 independent experiments for all panels.
3.4 Deletion of residues 22–26 (Δ1) combined with growth temperature reduction normalizes R345W F3 secretion to WT levels
Even though deletion of one or more of the insertion regions in F3 had no beneficial effect in rescuing R345W secretion defects, we still explored how the deletion variants responded to growth temperature reduction. While nearly 69% (relative to WT F3) of R345W F3 was secreted after growth temperature reduction (Fig. 4A), compared to 8% of R345W F3 secreted relative to WT F3 at 37°C (Fig. 1C), 105% of Δ1 R345W F3 was secreted relative to Δ1 F3 (Fig. 4A, B) after 24 h at 30°C. While this result is due to the combination of enhanced R345W F3 secretion at reduced growth temperature in addition to the disruptive effect the Δ1 deletion has on WT F3, it is the first time that we have been able to rescue R345W F3 to WT levels.
Fig. 4.

The combination of growth temperature reduction and deletion of residues 22–26 allows for R345W to be secreted at WT levels. (A) Transfected TREx-293 cells were grown at 37°C followed by a media change and 24 h at 30°C. Conditioned media aliquots were assayed using the GLuc assay. Data are presented as % of respective WT F3 variant, i.e., full length R345W F3 vs. full length WT F3; Δ1 R345W F3 vs. Δ1 F3, etc. (B) Western blot verification of GLuc results in (A). n ≥ 3 independent experiments for all variants, * = p < 0.01 when compared to full length R345W F3.
4. DISCUSSION
This study was the first to explore the consequences of introducing ‘pseudo-pathogenic’ tryptophan mutations (based on the R345W F3 mutation) into canonical cbEGF domains of F3 and F5. We found that tryptophan mutations at the bn+1 position in F3 cbEGF domains are apparently destabilizing and can reduce F3 secretion by up to 97%. However, tryptophan mutations placed in identical positions in F5 cbEGF domains had no effect on F5 secretion. Removal of three insertion regions in F3 to increase its similarity with F5 did not result in better tolerance of the R345W mutation. Overall, these results are surprising given that F3 and F5 share high sequence homology, have a nearly identical domain organization, do not have any substantive differences in aromatic amino acid content (Sup. Table 1), and neither F3 nor F5 have any naturally occurring aromatic residues at the bn+1 position (Hulleman et al., 2011). Based on these observations, one would predict that tryptophan residues would be universally disfavored in the bn+1 position of any cbEGF domain, which is not the case.
The exact reason(s) for why tryptophan mutations are disfavored in F3 cbEGF domains, while tolerated in F5 cbEGF domains are unclear. One possibility is that since F5 is secreted more efficiently than F3 (Fig. 3A, (Hulleman et al., 2012)), it spends less time accessing the ER quality control machinery which regulate whether a malfolded protein can escape the cell (reviewed in (Araki and Nagata, 2011)). Thus, subtle changes in F5 folding may not be detected by the cell and F5 secretion is not efficiently retarded or prevented. Consistent with this possibility, a C217R mutation, which prevents dsb2 formation within D4 of F5, does not completely prevent F5 secretion (Claus et al., 2008; Hulleman et al., 2012; Lotery et al., 2006). Furthermore, a naturally occurring R351W mutation in F5, which was identified in an AMD patient (Stone et al., 2004), was found previously to not significantly affect F5 secretion (Lotery et al., 2006), yielding similar results to the tryptophan mutants we presented herein.
A second possibility as to why tryptophan mutations are disfavored in F3 cbEGF domains, but tolerated in F5 domains could be due to differences in protein folding kinetics. In contrast to F5, F3 folds more slowly, and it is more likely to engage the ER quality control machinery (chaperones, lectins and protein disulfide isomerases) for a longer period. As such, F3 is likely subjected to stricter quality control mechanisms and scrutiny. Evidence supporting this idea arise from the observation that cysteine-to-alanine mutations in D6 of F3 (which also eliminates dsb2 formation) completely prevent F3 secretion and render it incapable of enhanced secretion at reduced growth temperatures (Hulleman et al., 2011).
Overall amino acid composition differences between F3 and F5 could represent another possibility as to why F3 and F5 are affected differently by tryptophan residues. While both F3 and F5 share a relatively high sequence homology, over half of the residues (52.4%) within the typical cbEGF domains are not conserved between F3 and F5 (Sup. Fig. 3). In accordance with the variability of residues in cbEGF domains, among the tryptophan mutants generated herein, only one pair (Q306W in F3 and Q259W in F3) had the same native reside at the bn+1 position (glutamine). While these two mutations altered the same native amino acid to a tryptophan residue, they had drastically different effects on fibulin secretion. Overall, there are no clear trends in amino acid composition differences between F3 and F5 cbEGF domains that could explain why the two proteins tolerate tryptophan residues so differently (i.e., a predominance of negatively charged residues in F3 while a lack of them in F5 cbEGF domains). Thus, in order to better understand the reasons behind the difference in tryptophan mutation tolerance, a more in-depth biochemical and mutational analysis must be performed.
Our work was based the assumption that a tryptophan mutation which was placed in the same location within a cbEGF domain would have similar effects on protein folding and secretion. However, in reality, we now know that we must also take into account potential differences in tertiary contacts (i.e. interaction of tryptophan residues with residues of other cbEGF domains, not just within the mutated domain). These interactions/contacts could also affect protein secretion and provide yet another reason for the discrepancies between the tryptophan mutants of F3 and F5. Such contacts are difficult to predict a priori without a structure of the full length protein, but are nonetheless worth exploring.
We rationalized that increasing F3’s sequence homology to F5 may be a way to make F3 behave more like F5 (e.g. have faster secretion kinetics and be insensitive to tryptophan residues). While individual removal of insertion 2 and 3 was beneficial in enhancing WT F3 secretion, deleting the first insertion, the second and third insertions together, or deleting all three insertion regions together did not benefit WT F3 secretion. These results suggest that the insert regions are not vestigial and that they can regulate WT F3 secretion efficiency. Residues 22–26 appear to be quite important for efficient WT F3 secretion, as deletion this region resulted in a 40% reduction in WT F3 secretion. In contrast to WT F3, individual (or combinatorial) deletion of any insert region of F3, did not substantially alter R345W F3 secretion. In fact, deletion of residues 22–26 of R345W F3 in combination with temperature reduction resulted in a similar amount of Δ1 R345W F3 and Δ1 F3 secretion. Overall, these results suggest that the R345W mutation may prevent the folding of F3 and lock the protein in a conformation whereby it is not benefited by removal of insert 2 or 3, like in WT F3. We speculate that cbEGF domains fold first by forming intradomain native disulfide bonds, followed by forming interdomain contacts prior to F3 secretion. Furthermore, we speculate that interdomain contacts between D1 and D6 are required for efficient F3 secretion. Prevention of such contacts, as would be predicted in the case of Δ1 F3 or R345W F3, significantly reduces F3 secretion. Therefore, it appears that the R345W variant is largely unaffected by deletion of insert regions in F3 because it is locked in a folding state that precedes the interdomain contact formation required for efficient secretion.
According to our results, introduction of a tryptophan residue at the bn+1 position in D3, D4 or D5 of F3 would be predicted to have the same pathogenic effect as R345W, assuming that secretion-associated defects are linked to ML disease progression. However, mutation of the bn+1 residues in D3, D4 or D5 to a tryptophan residue would require at least two base pair changes (Sup. Table 2), which is an unlikely biological event. Furthermore, our results demonstrate that the presence of a tryptophan residue at the bn+1 position does not always lead to dramatic protein folding problems. The R185W mutant still retained 56% of WT F3 secretion levels, and its secretion could be rescued to near 100% of WT levels after growth temperature reduction. Given that F3 folding and secretion is affected by numerous engineered mutations (described herein and in (Hulleman et al., 2011)), we speculate that co- or post-translational changes to F3 such as N-linked glycosylation (Hulleman and Kelly, 2014) or oxidation may significantly alter WT and/or R345W F3 secretion and intracellular steady state levels. Additional testing of the mutants described herein as well as those described previously (Hulleman et al., 2011) in emerging cell culture or mouse models (i.e. F3 knockout mice (McLaughlin et al., 2007)) could yield beneficial insight into the underlying causes of ML.
Supplementary Material
Sup. Fig. 1. F3 and F5 share high sequence homology. The protein sequences of WT F3 and WT F5 were aligned using COBALT. Residues which are present in only one of the two protein sequences are shown in grey. Residues identical across all sequences are shown as red letters, whereas non-consensus sequences are shown in blue. Residues mutated in this manuscript are underlined.
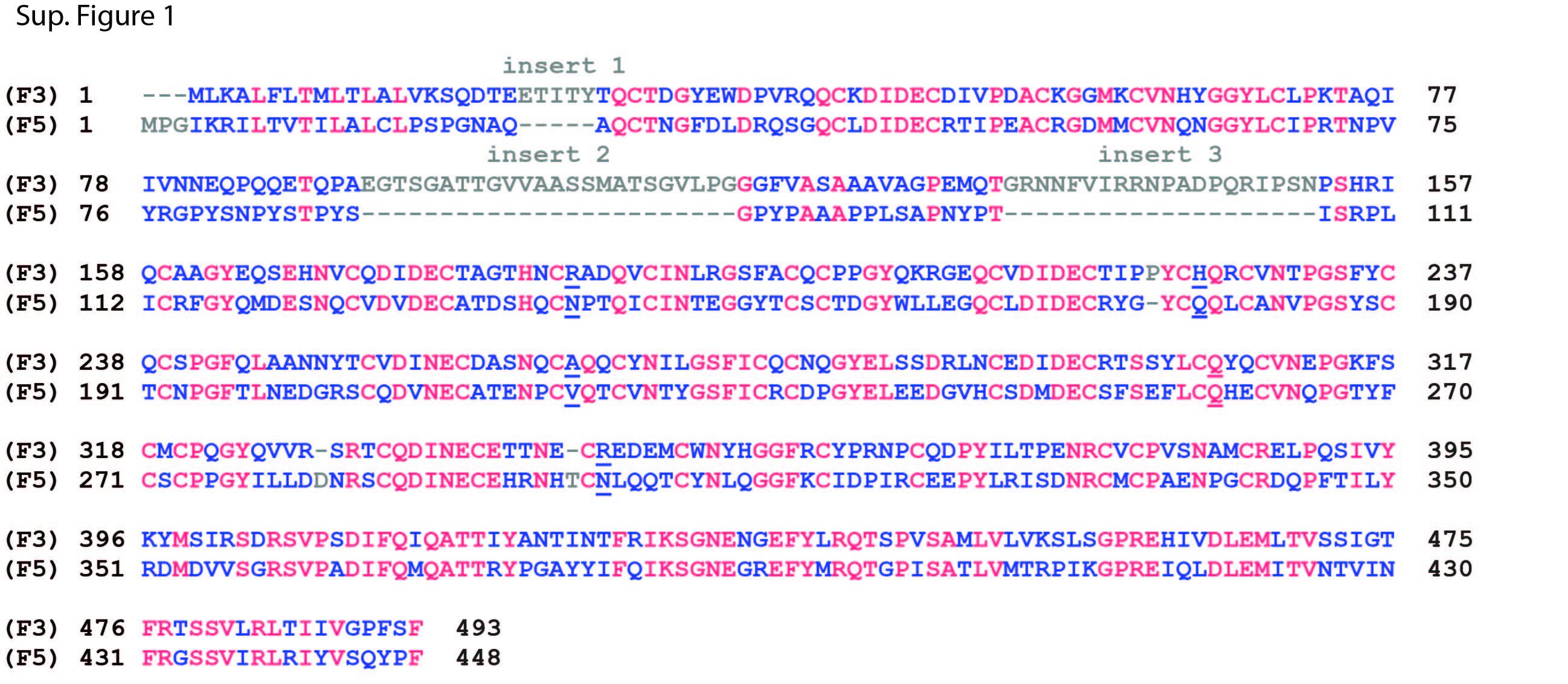{kind=link}
Sup. Fig. 2. The protein sequences of F3 Δ1–3 and full length F5 were aligned using COBALT. Residues which are present in only one of the two protein sequences are shown as grey letters. Residues present, but not identical in all three sequences are shown as blue letters. Residues identical across all sequences are shown as red letters. Overall, 220 residues were identical across both proteins, yielding a 49.5% sequence homology.
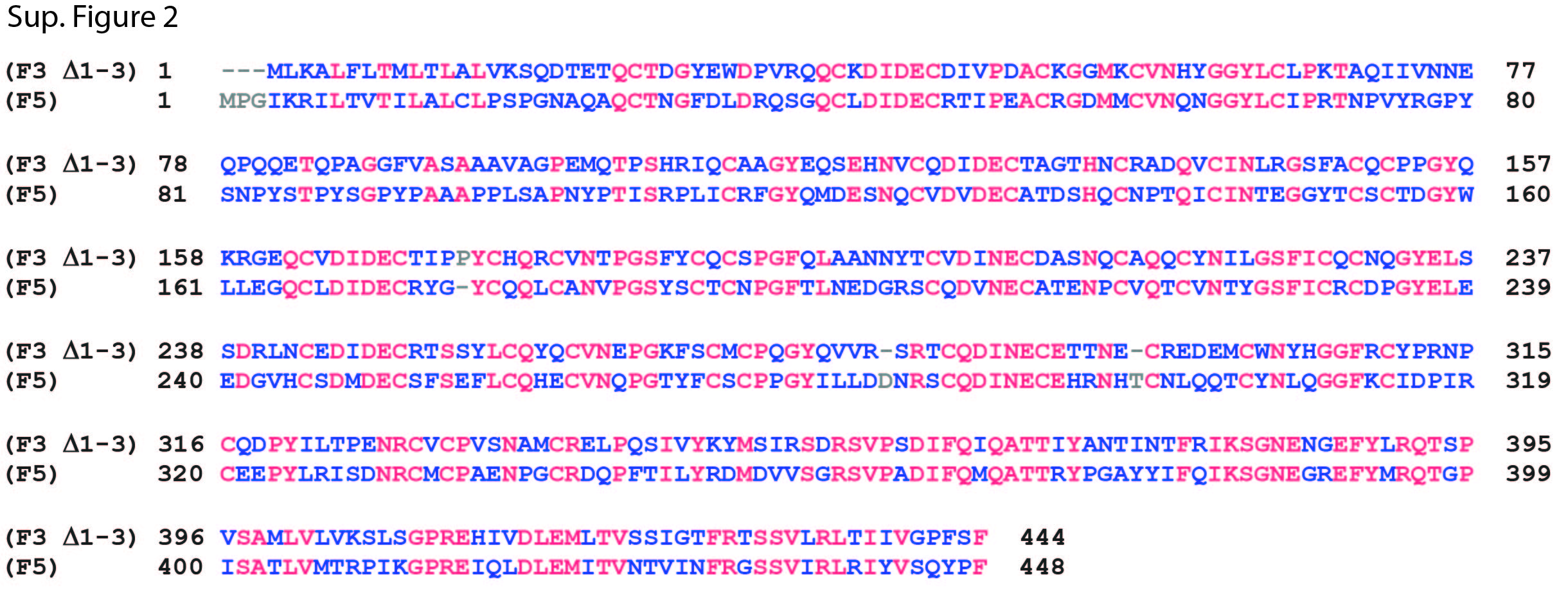{kind=link}
Sup. Fig. 3. The protein sequences of D2–D6 of F3 and F5 were aligned using COBALT. Residues which are present in only one of the two protein sequences are shown as grey letters. Residues present, but not identical in all three sequences are shown as blue letters. Residues identical across all sequences are shown as red letters. On average, D2–D6 are 52.4% dissimilar.
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was funded in part by an endowment from the Roger and Dorothy Hirl Research Fund, a National Eye Institute Visual Science Core Grant (EY020799), and an unrestricted grant from Research to Prevent Blindness.
Abbreviations
- AMD
age-related macular degeneration
- cbEGF
calcium-binding epidermal growth factor
- CMV
cytomegalovirus
- ER
endoplasmic reticulum
- F3
fibulin-3
- F5
fibulin-5
- ML
Malattia Leventinese
- qPCR
quantitative polymerase chain reaction
REFERENCES
- Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harbor perspectives in biology. 2011;3:a007526. doi: 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Li L, Lai PH. A major kinetic trap for the oxidative folding of human epidermal growth factor. The Journal of biological chemistry. 2001;276:4845–4852. doi: 10.1074/jbc.M005160200. [DOI] [PubMed] [Google Scholar]
- Chang JY, Schindler P, Ramseier U, Lai PH. The disulfide folding pathway of human epidermal growth factor. The Journal of biological chemistry. 1995;270:9207–9216. doi: 10.1074/jbc.270.16.9207. [DOI] [PubMed] [Google Scholar]
- Chang YJ, Wu HL, Hamaguchi N, Hsu YC, Lin SW. Identification of functionally important residues of the epidermal growth factor-2 domain of factor IX by alanine-scanning mutagenesis. Residues Asn(89)-Gly(93) are critical for binding factor VIIIa. The Journal of biological chemistry. 2002;277:25393–25399. doi: 10.1074/jbc.M105432200. [DOI] [PubMed] [Google Scholar]
- Claus S, Fischer J, Megarbane H, Megarbane A, Jobard F, Debret R, Peyrol S, Saker S, Devillers M, Sommer P, Damour O. A p.C217R mutation in fibulin-5 from cutis laxa patients is associated with incomplete extracellular matrix formation in a skin equivalent model. J Invest Dermatol. 2008;128:1442–1450. doi: 10.1038/sj.jid.5701211. [DOI] [PubMed] [Google Scholar]
- Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E, Stone EM, Zhang K, Pierce EA. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Human molecular genetics. 2007;16:2411–2422. doi: 10.1093/hmg/ddm198. [DOI] [PubMed] [Google Scholar]
- Hulleman JD, Balch WE, Kelly JW. Translational attenuation differentially alters the fate of disease-associated fibulin proteins. FASEB J. 2012;26:4548–4560. doi: 10.1096/fj.11-202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman JD, Brown SJ, Rosen H, Kelly JW. A high-throughput cell-based Gaussia luciferase reporter assay for identifying modulators of fibulin-3 secretion. J Biomol Screen. 2013;18:647–658. doi: 10.1177/1087057112469405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman JD, Kaushal S, Balch WE, Kelly JW. Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol Biol Cell. 2011;22:4765–4775. doi: 10.1091/mbc.E11-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman JD, Kelly JW. Genetic ablation of N-linked glycosylation reveals two key folding pathways for R345W fibulin-3, a secreted protein associated with retinal degeneration. FASEB J. 2014 doi: 10.1096/fj.14-255414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotery AJ, Baas D, Ridley C, Jones RP, Klaver CC, Stone E, Nakamura T, Luff A, Griffiths H, Wang T, Bergen AA, Trump D. Reduced secretion of fibulin 5 in age-related macular degeneration and cutis laxa. Human Mutation. 2006;27:568–574. doi: 10.1002/humu.20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein L. Association of EFEMP1 with malattia leventinese and age-related macular degeneration: a mini-review. Ophthalmic genetics. 2004;25:219–226. doi: 10.1080/13816810490498305. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, McLaughlin PJ, Peachey NS, Sasaki T, Marmorstein AD. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Human molecular genetics. 2007;16:2423–2432. doi: 10.1093/hmg/ddm199. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, Traboulsi E, Marmorstein AD. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13067–13072. doi: 10.1073/pnas.202491599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PJ, Bakall B, Choi J, Liu Z, Sasaki T, Davis EC, Marmorstein AD, Marmorstein LY. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Human molecular genetics. 2007;16:3059–3070. doi: 10.1093/hmg/ddm264. [DOI] [PubMed] [Google Scholar]
- Michaelides M, Jenkins SA, Brantley MA, Jr, Andrews RM, Waseem N, Luong V, Gregory-Evans K, Bhattacharya SS, Fitzke FW, Webster AR. Maculopathy due to the R345W substitution in fibulin-3: distinct clinical features, disease variability, and extent of retinal dysfunction. Investigative ophthalmology & visual science. 2006;47:3085–3097. doi: 10.1167/iovs.05-1600. [DOI] [PubMed] [Google Scholar]
- Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23:1073–1079. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- Roybal CN, Marmorstein LY, Vander Jagt DL, Abcouwer SF. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Investigative ophthalmology & visual science. 2005;46:3973–3979. doi: 10.1167/iovs.05-0070. [DOI] [PubMed] [Google Scholar]
- Sohn EH, Wang K, Thompson S, Riker MJ, Hoffmann JM, Stone EM, Mullins RF. Comparison of Drusen and Modifying Genes in Autosomal Dominant Radial Drusen and Age-Related Macular Degeneration. Retina. 2014 doi: 10.1097/IAE.0000000000000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, Eastman CG, Casavant TL, Sheffield VC. Missense variations in the fibulin 5 gene and age-related macular degeneration. The New England journal of medicine. 2004;351:346–353. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH, Vandenburgh K, Cousin P, Nishimura D, Swiderski RE, Silvestri G, Mackey DA, Hageman GS, Bird AC, Sheffield VC, Schorderet DF. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nature genetics. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- Wyatt MK, Tsai JY, Mishra S, Campos M, Jaworski C, Fariss RN, Bernstein SL, Wistow G. Interaction of complement factor h and fibulin3 in age-related macular degeneration. PLoS One. 2013;8:e68088. doi: 10.1371/journal.pone.0068088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sup. Fig. 1. F3 and F5 share high sequence homology. The protein sequences of WT F3 and WT F5 were aligned using COBALT. Residues which are present in only one of the two protein sequences are shown in grey. Residues identical across all sequences are shown as red letters, whereas non-consensus sequences are shown in blue. Residues mutated in this manuscript are underlined.
Sup. Fig. 2. The protein sequences of F3 Δ1–3 and full length F5 were aligned using COBALT. Residues which are present in only one of the two protein sequences are shown as grey letters. Residues present, but not identical in all three sequences are shown as blue letters. Residues identical across all sequences are shown as red letters. Overall, 220 residues were identical across both proteins, yielding a 49.5% sequence homology.
Sup. Fig. 3. The protein sequences of D2–D6 of F3 and F5 were aligned using COBALT. Residues which are present in only one of the two protein sequences are shown as grey letters. Residues present, but not identical in all three sequences are shown as blue letters. Residues identical across all sequences are shown as red letters. On average, D2–D6 are 52.4% dissimilar.