

Abstract
Bone grafting procedures are performed to treat wounds incurred during wartime trauma, accidents, and tumor resections. Endogenous mechanisms of repair are often insufficient to ensure integration between host and donor bone and subsequent restoration of function. We investigated the role that bone marrow-derived cells play in bone regeneration and sought to increase their contributions by functionalizing bone allografts with bioactive lipid coatings. Polymer-coated allografts were used to locally deliver the immunomodulatory small molecule FTY720 in tibial defects created in rat bone marrow chimeras containing genetically-labeled bone marrow for monitoring cell origin and fate. Donor bone marrow contributed significantly to both myeloid and osteogenic cells in remodeling tissue surrounding allografts. FTY720 coatings altered the phenotype of immune cells two weeks post-injury, which was associated with increased vascularization and bone formation surrounding allografts. Consequently, degradable polymer coating strategies that deliver small molecule growth factors such as FTY720 represent a novel therapeutic strategy for harnessing endogenous bone marrow-derived progenitors and enhancing healing in load-bearing bone defects.
Keywords: Phospholipid, Progenitor cells, Bone marrow, Immunomodulation, Bone graft
1. Introduction
Bone grafting is the second most common transplant operation after blood, with more than 500,000 bone grafting procedures in the United States annually and 2.2 million worldwide [1]. Autograft usage is the gold standard for bone graft transplant due to its osteoinductive, osteoconductive, and osteogenic properties [1–3]. However, autografting procedures may lead to donor site complications in as many as 30% of patients and is often insufficient for the treatment of massive bone loss such as that suffered during acute trauma and limb salvage surgeries [3]. Allograft, or transplantation tissue harvested from a donor, is a viable alternative to autograft and constitutes approximately one-third of all bone grafting procedures performed in the United States [4]. While use of allografts eliminates the risk of donor site morbidity, the risk of disease transmission from donor to patient necessitates the need for extensive sterilization procedures that often minimize the osteoinductive and osteogenic properties [3,4]. New strategies are needed to restore the lost therapeutic properties of allografts to enable adequate integration with host bone and ensure restoration of function.
Endogenous cell populations represent a powerful and adaptable tool for improving the host regenerative response after injury. Typical stages of bone healing include hematoma formation at the fracture site, which leads to the infiltration of myeloid cells (primarily neutrophils and monocytes) and the development of granulation tissue and new blood vessels. Mesenchymal stem cells (MSCs) then migrate into the fracture site and begin proliferating and differentiating into osteoblasts, chondrocytes, and fibroblasts that eventually form a callus, ending with growth and remodeling of new bone [5]. Both myeloid cells and mesenchymal cells can migrate from adjacent bone marrow and may infiltrate the injury site from the blood in response to damaged endothelium and chemotactic cues. The precise origin of MSCs and osteoblastic progenitor cells is not well understood despite evidence that the blood, bone marrow, and adjacent tissue compartments may serve as sources for mesenchymal cells [6]. MSCs serve as both direct and indirect contributors to bone repair, directly differentiating into bone-forming cells and secreting immunomodulatory factors that condition the injury microenvironment and participate in reciprocal signaling with inflammatory cells [5]. Bone marrow-derived monocytes and macrophages appear to play multi-faced roles in bone repair, regulating the acute inflammatory response [7], contributing to the pool of myeloid-derived pre-osteoclasts [8], and secreting factors that enable intramembranous ossification [9]. Osteal macrophages or “osteomacs” are present in bone both homeo-statically and after injury and their depletion completely eliminates the endosteal bone-forming osteoblast lining [10]. Moreover, the desired phenotype of macrophages, whether pro-inflammatory “M1-like” or anti-inflammatory “M2-like”, and the dynamics of their persistence in tissue after initial injury is likely important, but not well understood.
Regulating the relative contributions of endogenous therapeutic cells from distinct compartments is likely critical to improving biomaterial-based therapies for promoting bone regeneration. Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid growth factor that plays physiological roles in immune cell trafficking [11,12], vascular network formation and maturation [13,14], and osteogenic activities [15] and thus represents a likely therapeutic target for regulating cell populations within the injury niche. FTY720, a structural analog of sphingosine that activates signaling through four of the five known S1P receptors, has previously been shown to improve endothelial integrity, promote osteogenesis, and inhibit osteoclastogenesis [15–17]. Previous studies in our lab have demonstrated that biomaterial-based delivery of FTY720 induces angiocrine signaling within the endothelium to recruit pro-regenerative cell types such as anti-inflammatory monocytes and pro-angiogenic M2 macrophages [18,19] that facilitate neovascularization and bone formation [20–22]. Encapsulation within poly(lactic-co-glycolic acid) (PLGA) enables sustained release and signaling of FTY720 for at least 3 days in vivo and generates gradients that spatially localize pro-regenerative responses to peri-implant tissue [12]. S1P signaling also regulates reciprocal signaling between osteoblasts and osteoclasts [15], as well as induces platelet-derived growth factor (PDGF)-BB secretion by monocytic pre-osteoclasts that promotes coupling between angiogenesis and osteogenesis [8].
Given that successful bone requires precise coordination between multiple cell types, we designed a series of studies to assess the ability of FTY720-coated allografts to direct the in situ fate of marrow-derived cells in a rat critical-sized segmental tibial defect model. We used a rat bone marrow chimera model that is derived from transplantation of bone marrow cells harvested from transgenic eGFP-expressing animals into lethally irradiated wild type (WT) animals. The use of rat bone marrow chimeras is particularly advantageous in that GFP+ hematopoietic cells are stably retained in vivo and easily monitored using fluorescence microscopy without the need for histochemical staining or in situ hybridization [23]. Consequently, the eGFP rat chimera has been used for investigating the multipotentiality and migration of bone marrow-derived cells in a number of different models, including tendon repair, bladder regeneration, and adipose tissue [23–25]. However, bone repair, in which the rat model offers a significantly better representation of human physiology than parallel mouse models, has not been investigated in rat bone marrow chimeras. Consequently, we investigated the relative contributions of the blood and bone marrow to bone repair using transgenic-based labeling of hematopoietic cells and assessed the fate of cells infiltrating in response to volumetric bone loss.
2. Materials and methods
2.1. Chimera creation
33 8-week old female eGFP−/− Sprague Dawley (SD) rats received transgenic bone marrow transplantation from 11 Sprague Dawley-eGFP (SD-eGFP) male rats (Rat Resource & Research Center, University of Missouri, Columbia, MO). The host rats were irradiated at 10 grays using a Sheperd Mark 1 irradiator. Within a two-hour window immediately post-irradiation, each animal received an intravenous tail injection of 500,000 bone marrow cells suspended in 1 mL of serum-free media. Host rats received equal portions of bone marrow aspirate from each donor SD-eGFP rat. The rats were given the antibiotic Enrofloxacin (trade name Baytril, Bayer Corporation, Germany) for two weeks post-bone marrow transplant and carefully monitored for 21 days; weight loss of greater than 10% resulted in the animal being euthanized. At 6 weeks post-bone marrow transplant, 30 chimeric rats were randomly assigned to experimental groups in the tibial defect study.
2.2. Assessment of rat chimerism
Blood was collected from WT, eGFP, and chimeric rats via the tail vein. Red blood cells were lysed using Ammonium Chloride Solution (Stem Cell Technologies). The cells were then stained with CD11b, CD29, CD45, and CD90 (Abcam) antibodies for 1 h at 4 °C. After 1 h, the cells were fixed with 2% PFA solution for 30 min and then re-suspended in 10% fetal bovine serum (FBS), and stored in 4°C prior to analysis by flow cytometry.
2.3. Allograft coating
Tibial bone was harvested from 3-month old SD male rats and cut length-wise into 4 mm-long segments for use as allografts in tibial defects. The grafts were stripped of soft tissue, cleaned with detergent, hydrogen peroxide and ethanol sonication washes and then allowed to dry [26]. The cleaned allografts were divided into 3 groups: allograft alone, graft with polymer coating, graft with FTY720-loaded polymer coating. The polymer coating used 50:50 poly(lactic-co-glycolic acid) (PLGA, 69.85 kDa, Lakeshore Bio-materials) dissolved in methylene chloride at a polymer to solvent ratio of 1:12 (wt:wt). For the drug-loaded group, FTY720 (343.9 Da, Cayman Chemical) was agitated until dissolved in the polymer solution at 1:200 drug to polymer ratio (wt:wt). The allograft segments were vortexed in the FTY720-polymer solution for 10 min. To dry the coating, allografts were stored at 4 °C overnight and then lyophilized for 24 h to remove the remaining solvent. We have previously demonstrated that this coating technique enables sustained delivery of FTY720 for at least two weeks in vitro [27] and that the drug remains bioactive [22].
2.4. Rat tibial defect surgery
All animal surgeries were performed in accordance with an approved protocol from the University of Virginia Animal Care and Use Committee. 30 chimeric rats were randomly assigned to three different experimental groups (n = 10): allograft alone, graft with polymer coating, graft with FTY720-loaded polymer coating. Rats were anesthetized with isoflurane gas prior to and during surgery. Prior to surgery, the left hind limb was shaved and sterilized with betadine and 70% ethanol. A small incision was made longitudinally over the midshaft of the tibia. Subperiosteal dissection was made after the subcutaneous tissue was dissected over the anterior aspect of the tibia. Once the bone is exposed, a rotating saw (Dremel 400XPR) was used to make a 4 mm defect distal to the tibial tubercle. The allograft segment was then inserted into the defect and fixed in position using a 22 gauge needle inserted into the intramedullary canal from the proximal end of tibia, through the allograft, and into the distal end of tibia. Ketoprofen was administered subcutaneously at 3 mg/kg doses immediately post-surgery and every 24 h thereafter for up to 72 h. Rats were given free access to food and water and monitored for abnormalities and complications. At 2 weeks post-surgery, 3 rats randomly selected from each experimental group were euthanized by injection of 0.5 mL Nembutal into the heart. The left tibial bone was harvested for subsequent histological analysis.
2.5. X-ray and microCT analysis
Radiography was performed at week 0 and week 4 post-surgery. Rats were anesthetized by intraperitoneal (IP) injection with a cocktail of ketamine HCl (60 mg/kg) and xylazine (5 mg/kg) and then placed on top of an X-ray film inside the X-ray machine (90 s, 30 V exposure). Yohimbine was given afterward by IP injection to reverse anesthesia. At the end of the study period (8 weeks post-surgery), rats were euthanized and the tibial bones were harvested from each rat. The soft tissue surrounding the left tibia and the intermedullary fixation needle were removed prior to analysis using micro-computed tomography (microCT) with a VivaCT40 scanner (SCANCO Medical) using the following parameters: 38 μm voxel size, 55 kVp, 145 μA, medium resolution, 40.9 mm diameter field of view, and 200 ms integration time (73 mGy radiation per scan). After microCT scanning, the tibiae were stored at −20 °C before undergoing mechanical testing. For quantification of microCT images, a scoring system was developed to assess the extent of integration between host and allograft bone. For each two-dimensional microCT image, a bridging score between 0 and 4 was assigned. A blinded observer determined whether bridging had occurred in each of four pre-defined regions of interest. For each region, a binary score of ‘0’ or ‘1’ was assigned, with ‘1’ indicating the occurrence of bridging within that region. The four values (0 or 1) were tallied and an overall bridging score was assigned to each animal, with a maximum score of 4 possible. Subsequently, the minimum distance between the host and donor bone was quantified in each region of interest using ImageJ software, with all bridges being assigned a value of ‘0 mm’ to indicate that no space exists between host and donor bone.
2.6. Assessment of cell proliferation
MSCs were isolated from WT and chimeric SD rats and seeded in culture plates under growth media (α-MEM, 15% FBS, 1% Penicillin–Streptomycin (PS), 1% L-Glutamine) with 0, 0.15 nM or 15 nM FTY720. These concentrations were selected because low dose FTY720 (below 100 nM) was previously demonstrated to promote osteogenic differentiation of C2C12 myoblasts [17]. After 10 days of culture, cells were fixed with 10% formalin and subsequently stained with crystal violet. Subsequent imaging was performed using an upright microscope at 10× magnification and colony formation was measured by quantifying the number of pixels above a pre-determined threshold.
2.7. Alizarin red staining
MSCs harvested from WT and chimeric SD rats were seeded in culture plates and grown in growth media (DMEM F12, 10% FBS, 1% PS) for 3 days. The MSCs were then cultured under osteogenic differentiation media (DMEM-Low Glucose, 10% FBS, 1% PS, 100 nM Dexamethasone, 10 mM Beta-glycerol phosphate, 2 mM L-Glutamine) and 0.15 nM or 15 nM FTY720. After 10 days, the cells were fixed with 10% formalin and stained with Alizarin Red S Solution (2% w/v; pH 4.2). The plates were evaluated by light microscopy and mineralization was measured by quantifying the pixel intensity.
2.8. Tartrate-resistant acid phosphatase staining
The RAW264.7 murine macrophage cell line was used to assess the effect of FTY720 on osteoclastogenesis. Cells were seeded on a coverslip in a culture plate and cultured in growth media (α-MEM, 10% FBS, 1% PS) with 50 ng/ml murine sRANKL and 0.15 nM or 15 nM of FTY720. Following 5 days of culture, tartrate-resistant acid phosphatase (TRAP) staining was carried out according to the manufacturer’s instructions (Leukocyte TRAP Staining Kit 387A, Sigma Aldrich). Stained cells were counter-stained with Hematoxylin and imaged on an upright microscope.
2.9. Mechanical testing
At 8 weeks post-surgery, tibiae were extracted and wrapped in PBS-soaked gauze for storage at −20 °C until mechanical testing was performed. Immediately prior to testing, samples were thawed and any residual soft tissue was removed. The distal end of the tibia was trimmed to center the defect in the sample. The proximal and distal ends of the tibia were then embedded in end blocks using Wood’s metal (Alfa Aesar) and aligned using a custom fixture. The sample was loaded into holding brackets mounted on a Bose ElectroForce System (ELF 3200, Bose Endura TEC) fitted with a 2 Nm loading cell. Keeping the proximal end fixed, the distal end was rotated at 3° per second until fracture. Torque and rotation were recorded. Maximum torque was determined by locating the failure torque.
2.10. Immunohistochemical analysis
Animals from each treatment group were euthanized at 8 weeks, and the tibiae were extracted. Samples were decalcified and embedded in paraffin and 5 μm sections were prepared. Prior to staining, sections were deparaffinized and antigen retrieval was performed at 95 °C with 10 mM citrate buffer for 8 min. Sodium borohydride solution (100 mg/mL) was used to reduce background autofluorescence of the tissue. Samples were permeabilized with 0.2% Triton X-100 and then blocked using 10% donkey and goat serum to prevent non-specific antibody binding. Immunofluorescence staining was performed using anti-GFP (Abcam), anti-Osteocalcin (Abcam), anti-alpha Smooth Muscle Actin (Abcam), isolectin IB4 (Life Technologies), anti-CCR7 (Abcam), CD206 (Bioss), or CD90 (BioLegend). Samples were incubated with primary antibody overnight at 4 °C, and secondary antibody for one hour at room temperature. Slides were mounted in 100 mM glycine and images were taken on the Zeiss LSM 700 confocal microscope using Zen software (Zeiss). Quantification of immunohistochemistry was performed by counting the number of cells positive for the specified markers by identifying a threshold based on secondary only controls. For identification of newly formed bone, sections were stained with hematoxylin and eosin and then imaged for autofluorescence using an upright microscope.
3. Results
3.1. Development and characterization of eGFP rat chimeras
To gain insight into how circulating cell populations contribute to allograft incorporation in a critical sized rat tibial defect model, we generated chimeric rats expressing eGFP in the bone marrow compartment. eGFP−/− SD rats were lethally irradiated and transplanted with bone marrow derived from eGFP+/− litter mates, resulting in 100% survival of all transplant recipients (Fig. 1A). Flow cytometry analysis revealed that 75% of circulating white blood cells were GFP+ cells (Fig. 1B). Additionally, chimeric rats demonstrated reconstitution of CD11b+, CD29+, CD45+, and CD90+ lineages in blood at 4 weeks post-transplantation (Fig. 1C), signifying successful and stable transplantation with GFP+ bone marrow.
Fig. 1.
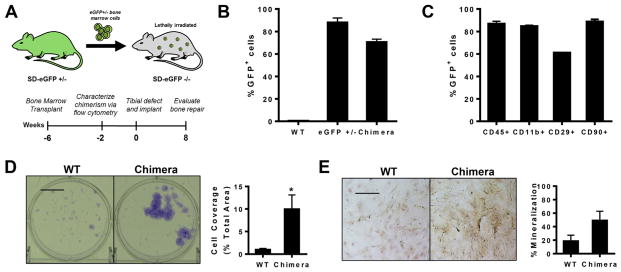
Rat bone marrow chimeras as a model for studying bone regeneration. (A) Sprague–Dawley rat bone marrow chimeras were generated by transplanting eGFP+ bone marrow into lethally irradiated eGFP−/− rats. Tibial defects were created 6 weeks post-transplantation. (B) Flow cytometry analysis of eGFP expression in blood 4 weeks post-bone marrow transplantation demonstrated stable reconstitution of the hematopoietic system with GFP+ cells, n = 3–4. (C) GFP+ cells display multi-lineage differentiation as assessed by flow cytometry analysis of blood taken 4 weeks post-transplantation, n = 3. (D) Bone marrow from chimeras after 7 days of culture as observed with crystal violet staining (left) and quantification of colony coverage (right), n = 6. (E) Bone marrow from eGFP chimeras after 7 days of culture in osteogenic media as observed with Alizarin red staining (left) and quantification of mineralized area (right), n = 3. Scale bar, 100 μm; *p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Bone marrow stromal cells from chimeric rats remain primarily uncharacterized in their proliferative and differentiation capacities. We compared colony formation and osteogenic differentiation of MSCs derived from WT and chimeric rats. Chimeric MSCs formed larger colonies that covered 5-fold more area and demonstrated a 5-fold increase in average size compared to WT MSCs (Fig. 1D), without impacting the overall colony number. MSCs from chimeric rats more highly stained with Alizarin Red, indicating that chimeric MSCs generate more mineralized matrix in vitro (Fig. 1E). This likely results either from an overall increase in the number of cells capable of mineralization or an increase in the average output per cell. These findings highlight an important difference in the behavior of MSCs derived from rats undergoing hematopoietic reconstitution with cells derived from eGFP+/− rats that should be considered when designing experiments that investigate the role of bone marrows cells in chimeric rats.
3.2. Bone growth and allograft incorporation in a long bone defect model
Critical sized segmental defects (4 mm) were generated in the tibiae of chimeric rats 6 weeks after lethal irradiation and bone marrow transplantation. Allograft incorporation was monitored at weeks 0 and 4 by X-ray and week 8 by ex vivo microCT scans. X-ray imaging indicates that animals treated with FTY720-coated allografts show better graft integration and callus formation as early as 4 weeks (Fig. 2A). MicroCT images taken at week 8 indicate that this leads to new mineralized bone formation and better healing in animals treated with grafts coated with FTY720 (Fig. 2B). In particular, the interface between donor and host bone is less distinct in animals receiving FTY720, indicating improved allograft incorporation. We scored the extent of bridging that occurred between allograft and host bone in two-dimensional microCT renderings and found that FTY720 significantly increases bridging relative to both uncoated and PLGA-coated grafts (Fig. 2C). Additionally, FTY720 reduces the overall distance between host and donor bone compared to uncoated controls (Fig. 2D). H&E staining of tibiae longitudinally sectioned through the defect site also show more bone formation and better graft integration in animals treated with FTY720-coated allografts compared to controls (Supplemental Fig. 1).
Fig. 2.

FTY720-coated allografts promote bone formation and graft incorporation. (A) Representative X-rays of tibiae indicate the FTY720 improves integration between host and donor bone by week 4 post-injury. (B) Representative microCT reconstructed images taken at 8 weeks post-injury indicate that FTY720 increases bone deposition around implanted allgorafts (top row: 3D images, bottom row: 2D cross-sectional images). (C) FTY720 increases the bridge score assessed in 2D microCT images compared to uncoated and PLGA-coated allograft controls, n = 4–5. (D) FTY720 decreases the distance between host and donor bone compared to uncoated allografts, n = 4–5. (E) Representative torque plots from torsional mechanical testing of tibiae explanted 8 weeks post-injury indicate that FTY720 allografts better recapitulate the mechanical properties of uninjured contralateral control bone, n = 3–4. Scale bars, 500 mm; *p < 0.05.
In the case of load-bearing bone repair, newly-deposited bone must be able to support the mechanical load exerted by the rest of the body. We used torsional testing to measure the magnitude of forces sustained by newly-healed bone. While we experienced significant sample variability in mechanical testing, inspection of the force–displacement curves shows that FTY720-treated grafts exhibited mechanical properties more similar to uninjured contralateral bones (Fig. 2E). Across all tested samples, FTY720 best restores the strength of native bone, as the torque at failure is most similar to contralateral controls (Supplemental Fig. 2). No significant differences in stiffness between all groups were observed.
3.3. S1P signals regulate osteogenic processes
Given that polymer coatings releasing FTY720 promote allograft incorporation and mineralization, we assessed the presence of osteocalcin-positive (OC+) cells at the interface between allograft bone and host tissue. FTY720-coated allografts have a higher frequency of OC+ cells in the surrounding tissue than uncoated or PLGA-coated allografts as detected by immunohistochemistry (Fig. 3A, B). Additionally, in animals receiving FTY720, OC+ cells infiltrated void areas within the graft and closely associated with donor bone, enabling bone deposition at the graft surface. Fluorescence measurements of histological sections, in which new bone autofluoresces more brightly, indicate a greater amount of newly-formed bone in animals receiving FTY720 allografts (Supplemental Fig. 1). New bone formation is the cumulative result of a delicate balance between osteoblast deposition of mineralized matrix and osteoclast resorption of damaged bone. Consequently, we investigated the effect of S1P receptor signaling on both osteoblastic and osteoclastic behaviors in vitro. Low dose FTY720 appears to minimally impact colony formation of MSCs, as total colony coverage only slightly increased (Fig. 3C) and colony number was not impacted by the presence of 0.15 nM or 15 nM FTY720. The osteogenic capacity of MSCs appears to be enhanced by S1P signals, as the intensity of Alizarin Red staining increased in the presence of FTY720 and this was associated with a modest increase in the total percentage of mineralized regions (Fig. 3C and D). To study the effect of FTY720 on osteoclastogenesis, we treated RAW264.7 macrophages with RANKL and stained for TRAP. Low (0.15 nM) and high (15 nM) dose FTY720 increased osteoclastic differentiation, with the lower dose having the greatest effect (Fig. 3E). Taken together, these results indicate that FTY720 released from degradable allograft coatings likely alters the fate of osteoblastic and osteoclastic precursors in the peri-implant tissue.
Fig. 3.

FTY720 promotes osteogenesis and osteoclastogenesis. (A) Histological sections from tibiae explanted at week 8 post-injury and immunostained with anti-osteocalcin (OC) and DAPI demonstrate that FTY720 increases the frequency of OC+ cells and their proximity to the graft, n = 2–3. Scale bars, 100 μm; white arrows, OC+ cells; dotted line, graft edge. (B) Quantification of histological sections stained with OC, n = 2–3. (C) Bone marrow from wild type animals cultured for 7 days in the presence of low (0.15 nM) and high (15 nM) dose FTY720 and stained with crystal violet (top) or Alizarin red (bottom). (D) Quantification of colony cell coverage (left) or amount of mineralization (right) shows that FTY720 minimally impacts colony size, but increases mineralization of MSCs, n = 3–6. (E) FTY720 promotes osteoclastogenesis as assessed by TRAP staining of RAW264.7 cells cultured for 5 days in RANKL and low or high dose FTY720. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. FTY720 promotes vascular maturation in healing bone
We have previously demonstrated that local delivery of FTY720 increases the formation of vascular networks in remodeling bone after injury [19,22]. Here, the dynamics of FTY720’s effects on vascular ingrowth into transplanted allografts were assessed by visualizing lectin+ microvessels and mature SMA+ blood vessel formation after bone injury. At 2 weeks post-injury, SMA+ cells were primarily unassociated into vascular structures and were prominent in the tissue surrounding the graft (Fig. 4A). By week 8, the interfacial tissue appeared much more highly vascularized, with the appearance of lectin+ microvessels and large smooth muscle-coated SMA+ vessels having a diameter greater than 100 μm (Fig. 4B and C). Groups receiving FTY720 appear to have more lectin+ and SMA+ blood vessels surrounding the allograft and more SMA+ cells infiltrating donor bone at week 8 post-injury (Fig. 4B and C). Interestingly, these group differences were not observed in week 2 histological sections, indicating that FTY720 likely affects the maturation of nascent vasculature formed during early wound healing processes. FTY720 had a more robust effect on SMA+ blood vessels than lectin+ vessels, further supporting the hypothesis that FTY720 promotes vascular maturation. Moreover, FTY720’s effect on vascularization in and around implanted allografts likely contributes to improved allograft incorporation and bone regeneration.
Fig. 4.

FTY720 increases the number of α-SMA+ blood vessels surrounding allografts. (A) Histological sections from tibiae explanted and immunostained with anti-α smooth muscle actin (SMA) and DAPI show that few mature blood vessels are present around allografts in all groups at 2 weeks post-injury. (B) FTY720 promotes formation of lectin+ microvessels by 8 weeks post-injury, n = 2–3. (C) FTY720 promotes formation of mature smooth muscle-coated blood vessels by 8 weeks post-injury, n = 2–3. Scale bars, 100 μm; dotted line, graft edge. *p < 0.05.
3.5. Fate of bone marrow-derived cells during bone regeneration
The precise role of circulating and resident cell populations, including inflammatory cells and mesenchymal progenitors, to bone healing remains unclear. While it is apparent that infiltrating inflammatory cells likely participate in repair processes, the relative contributions by blood and tissue compartments has not been characterized. To assess the frequency of bone marrow-derived cells that becomes M1-like or M2-like macrophages, histological sections stained with anti-GFP antibody were counterstained for CCR7 (for M1-like macrophages) or CD206 (for M2-like macrophages) expression (Fig. 5A). We quantified the ratio of GFP+CD206+ to GFP+CCR7+ cells to investigate whether FTY720 alters the fate of bone marrow-derived inflammatory cells. At 2 weeks post-injury, we measured a 30% increase in the ratio of GFP+CD206+ to GFP+CCR7+ cells, suggesting that FTY720 changes the phenotype of recruited macrophages (Fig. 5B). Additionally, in animals not receiving FTY720, CD206+ cells appear to be excluded from the tissue space that is immediately adjacent to implanted grafts, likely reducing their ability to guide repair processes and graft integration. GFP+ cells represent cells derived from transplanted bone marrow and thus correlate to circulation-derived cells or tissue-resident cells that were replenished by circulating cells during the 6-week pre-surgery period of hematopoietic reconstitution. Consequently, GFP-negative cells signify populations that were protected from irradiation, likely consisting of embryonically-derived tissue resident immune cells, stromal and parenchymal cells, and a small frequency (20–30%) of non-GFP+ circulating cells (Fig. 1B). GFP+ cells infiltrated the interfacial tissue space surrounding allografts, with a subset of cells also expressing CCR7 or CD206 (Fig. 5A, white arrows). Quantification across all animal groups indicates that CCR7+ M1-like macrophages encompass a larger frequency of GFP+ cells (30%) than CD206+ M2-like macrophages (20%), indicating that at 2 weeks post-injury, bone marrow-derived cells have a greater probability of adopting an inflammatory phenotype (Fig. 5C). Interestingly, the GFP+ population makes up an equal proportion of CCR7+ and CD206+ cells (Fig. 5D). These results indicate that both a circulating and a tissue resident, irradiation-resistant population contribute to the inflammatory cell profile during early stage bone regeneration and FTY720 may promote tissue repair by shifting the phenotype of recruited macrophages to a pro-regenerative M2-like profile.
Fig. 5.

The phenotype of bone marrow-derived macrophages is altered by FTY720. (A) Histological sections from tibiae explanted at week 2 post-injury and stained with anti-GFP, DAPI, and CCR7 (top row) or CD206 (bottom row). Scale bars, 50 μm; white arrows, GFP+CCR7+ or GFP+CD206+ cells; dotted line, graft edge. (B) FTY720 increases the ratio of CD206+GFP+ to CCR7+GFP+ cells at 2 weeks post-injury, n = 2–3. (C) Quantification of the percentage of CCR7+ and CD206+ cells within the total GFP+ cell population demonstrates that across all groups, GFP+ bone marrow-derived cells are more likely to become CCR7+ than CD206+ at 2 weeks post-injury, n = 7–8. (D) Quantification of the percentage of total CCR7+ and CD206+ cells that also express GFP demonstrates that across all groups, the bone marrow contributes equal proportions to both CCR7+ and CD206+ cells at 2 weeks post-injury, n = 7–8. *p < 0.05.
Monocyte-derived macrophages represent a population of blood- and bone marrow-derived progenitor cells that play critical roles in dictating the capacity of an injury to heal. In the context of bone repair, these cells participate in crosstalk with osteoblasts and mesenchymal progenitors, as well as vascular endothelial cells [8]. Consequently, we assessed whether progenitor cells surrounding allograft implants express GFP. In all treatment groups, CD90+ cells that co-stained for GFP were observed in sections taken at week 2 post-injury, indicating that a subset of these progenitor cells originates from bone marrow (Fig. 6D). Moreover, we were interested in whether more mature osteoblastic cells were derived from the bone marrow compartment. Interestingly, we observed cells that stained positive for both OC and GFP, indicating that a fraction of OC+ osteoblastic cells are derived from the bone marrow compartment 8 weeks after injury (Fig. 6A). FTY720 does not appear to affect the absolute number of GFP+OC+ bone marrow-derived osteoblatic cells in the tissue surrounding allograft (Fig. 6B); however, the proximity of these cells to the graft and intensity of osteocalcin staining is enhanced in FTY720 animals (Fig. 6A). Quantification of histological sections from all groups indicated that as many as 25% of OC+ cells were derived from bone marrow (Fig. 6C). While this is much lower than the contribution of WT cells to the OC+ population in healing bone, this data indicates that there is likely a significant role for osteogenic progenitor cells derived from bone marrow in healing bone.
Fig. 6.

Bone marrow contributes to mature bone-forming cells and osteogenic progenitor cells. (A) Histological sections from tibiae explanted at week 8 post-injury and stained with anti-GFP, OC, and DAPI. White arrows, GFP+OC+ cells; dotted line, graft edge. (B) FTY720 does not impact the number of GFP+OC+ cells in histological sections imaged around the graft, n = 2–3. (C) Across all groups, GFP+ bone marrow-derived cells contribute significantly less (about 25%) than unlabeled wild type (WT) cells (about 75%) to OC+ populations detected 8 weeks post-injury, n = 7. (D) Histological sections from tibiae explanted at week 2 post-injury and stained with anti-GFP, anti-CD90, and DAPI. White arrows, GFP+CD90+ cells. Scale bars, 50 μm; *p < 0.05.
4. Discussion
The precise role of bone marrow- and circulation-derived progenitor cells in bone healing remains poorly understood, and thus the design of strategies that exploit the relative contributions of these cells has been limited. Peripheral blood mononuclear cells encompass a heterogeneous cell population containing many different cell types that may participate in bone healing, including monocytes/macrophages, neutrophils, endothelial progenitor cells, and osteogenic progenitor cells. Elucidation of the respective roles for various tissue compartments in bone repair is critical for designing novel therapeutics that harness these cell populations to promote healing. The current work systematically examines the contributions of GFP+ cells that are derived from reconstituted bone marrow during healing of critical-sized tibial defects in chimeric rats and examines whether lipid-based therapies can improve post-trauma functional recovery of damaged bone by directing progenitor cell fate.
Macrophages represent a multi-faced population of cells that differ in recruitment kinetics and function in a wide range of injury contexts. These cells are required for efficient bone repair, as systemic depletion of macrophages with clodronate liposomes significantly impairs tibial defect healing [28]. Macrophages exhibit a wide range of diverse phenotypes that have been primarily categorized into “classically activated” M1 macrophages, which possess pro-inflammatory, phagocytic, and proteolytic functions essential for removal of damaged tissues, and “alternatively activated” M2 macrophages, which are anti-inflammatory and participate in tissue regeneration, angiogenesis, and matrix synthesis [29]. In vivo, injury-responsive macrophages are extremely heterogeneous, existing along a spectrum of phenotypes, with the M1 and M2 phenotypes representing opposite extremes. An additional level of complexity exists due to the fact that macrophages present at inflammatory sites may be derived from either circulation or tissue resident macrophage pools that are embryonically-derived and self-renewing during homeostasis [30].
Our data indicates that bone marrow-derived GFP+ cells are more likely to become CCR7+ than CD206+ at 2 weeks post-injury (Fig. 5C). The kinetics of specific macrophage phenotypes is not well-characterized, particularly in the context of bone repair. However, the current work indicates that specification down an M1-like lineage is more prominent during early bone repair, suggesting that the characteristic pro-inflammatory phase may be extended beyond a couple of days to several weeks during bone healing. Interestingly, GFP+ cells contribute equal proportions to both CCR7+ and CD206+ populations (Fig. 5D), likely indicating that the injury microenvironment is the primary determinant of macrophage phenotype, as there is no difference in the fate of either recruited or tissue resident populations. We have previously reported that local delivery of FTY720 skews the inflammatory profile to generate a pro-regenerative injury niche, both in soft tissue and craniofacial bone [18,19]. In particular, FTY720 increases the frequency of anti-inflammatory M2 macrophages both at early (3 days) and later (3 weeks) time points, which facilitates neo-vascularization and tissue repair. In the current work, FTY720 increased the ratio of GFP+ M2:M1 macrophages at 2 weeks post-injury, which likely leads to improvements in vascularization and allograft bridging observed by 8 weeks (Fig. 5B). FTY720 more robustly induced the formation of SMA+ mature blood vessels than lectin+ vessels around implanted allografts (Fig. 4B and C), indicating that FTY720 likely promotes vascular maturation, which could be initiated by pro-arteriogenic macrophages. Additionally, myeloid cells are a key player in coupling osteogenesis to angiogenesis [8] and the precise ratio of M2:M1 macrophages likely dictates the ability of injured tissue to regenerate [31,32].
While the occurrence of myeloid cell infiltration from the blood during bone repair is well-documented, the existence and role of circulating osteogenic progenitor cells is less understood. Peripheral blood cells that express osteogenic markers such as osteocalcin or alkaline phosphatase have been detected both in murine and human samples [33,34]. In studies using parabiotic mice in which a WT and a chimeric mouse containing GFP-labeled bone marrow were conjoined, BMP-2-laden pellets were subcutaneously implanted. OC+ GFP+ cells were found at surrounding ectopic bone in the WT mouse [35] and in similar parabiotic studies using a fracture model, indicating the existence of circulation-derived GFP+ bone-forming cells. Additionally, administration of compounds that increase the frequency of osteogenic progenitor cells in circulation such as the CXCR4 inhibitor AMD3100, increases ectopic bone formation [36] and improves fracture healing [37]. These results indicate that bone marrowand circulation likely contribute to bone repair, but the precise contribution may be context-dependent, where both the size and location of injury are important. In the present work, we observed that the bone marrow compartment significantly contributes to the pool of tissue OC+ cells, with as many as 25% being GFP+ at week 8 post-injury (Fig. 6C). The source of these cells is not clear and could derive from circulating osteogenic cells or contiguous marrow that migrates in response to injury-induced chemotactic cues [6]. It is likely that at least a population of these cells were derived from a pool of GFP+CD90+ progenitor cells that appeared at the injury site at week 2 post-surgery (Fig. 6D). The reported mechanisms of OC+ cell infiltration appear to be conserved across multiple types of bone injury, as fracture healing also induces bone marrow-derived OC+ cell infiltration [38]. FTY720 localizes OC+ cells to the graft surface (Fig. 3A), which may be through enhanced recruitment of progenitor cells, as we have previously reported that local FTY720 increases tissue levels of chemokines such as SDF-1 and RANTES [18].
Healing damaged bone is a function of both the amount of osteogenic cells active within the defect area, as well as the osteogenic capacity of each individual cell. While allografts provide a scaffold for cell and tissue ingrowth into the defect area, non-union remains a major concern, in part due to insufficient infiltration of osteogenic cells. Consequently, small molecule-based approaches for increasing the recruitment and differentiation of bone-forming cells represents a viable and translational approach for improving allograft functionality [39]. In this study, we show that in addition to an increase in the frequency of OC+ cells at the allograft integration site, the osteogenic capacity of MSCs is also elevated after FTY720 treatment (Fig. 3B). The osteogenic effects of FTY720 enabled newly formed bone to sustain higher torsional forces and exhibited a deformation pattern similar to that sustained by native, unbroken bone (Fig. 2E).
The current work advances our understanding of the role that bone marrow-derived progenitor cells play in bone regeneration and allograft incorporation. While cell infiltration from blood and bone marrow are likely context- and time-dependent, a significant contribution to both osteogenic and inflammatory cell populations from irradiation-sensitive hematopoietic tissues was observed. The development of novel strategies to improve clinical bone reconstruction and regeneration will likely depend on an understanding of endogenous mechanisms of cell recruitment and their in situ roles in repair. Therapies derived from engineering progenitor cell migration within the injury microenvironment represent a promising and powerful approach for bone repair.
5. Conclusions
Development of tissues that fully interface with damaged host tissue remains a significant challenge for the field of tissue engineering. Allografts, which recapitulate some of the structure and function of native bone, would likely have a better clinical outcome if coupled to strategies that specifically guide endogenous cell recruitment. The current work demonstrates that bone marrow-derived progenitor cells infiltrate rat critical sized bone defects and significantly contribute to inflammatory and bone-forming cell populations. Bioactive allograft coatings containing FTY720 improve graft vascularization, bone formation, and integration with host tissue, likely through local immune modulation that improves angiogenic–osteogenic coupling. Future strategies for engineering bone regeneration will likely rely on an understanding of endogenous mechanisms of repair to identify therapeutic targets and maximizing cellular contributions.
Supplementary Material
Acknowledgments
We thank the core facilities staff of the Parker H. Petit Institute for Bioscience and Bioengineering for technical expertise and assistance. Sources of support for this study include the Department of Defense grant W81XWH-10-1-0928 to Dr. Botchwey; National Institutes of Health grants R01AR056445-01A2 and R01DE019935-01 to Dr. Botchwey; and the National Science Foundation grant NSF GRFP DGE-1148903 to Claire Segar.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2015.06.019.
References
- 1.Giannoudis PV, Dinopoulos H, Tsiridis E. Bone substitutes: an update. Injury. 2005;36(Suppl 3):S20–S27. doi: 10.1016/j.injury.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 2.Toolan BC. Current concepts review: orthobiologics. Foot Ankle Int. 2006;27:561–566. doi: 10.1177/107110070602700715. [DOI] [PubMed] [Google Scholar]
- 3.Bhatt RA, Rozental TD. Bone graft substitutes. Hand Clin. 2012;28:457–468. doi: 10.1016/j.hcl.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 4.De Long WG, Jr, Einhorn TA, Koval K, McKee M, Smith W, Sanders R, Watson T. Bone grafts and bone graft substitutes in orthopaedic trauma surgery. A critical analysis. J Bone Jt Surg Am. 2007;89:649–658. doi: 10.2106/JBJS.F.00465. [DOI] [PubMed] [Google Scholar]
- 5.Grayson WL, Bunnell BA, Martin E, Frazier T, Hung BP, Gimble JM. Stromal cells and stem cells in clinical bone regeneration. Nat Rev Endocrinol. 2015;11(13):140–150. doi: 10.1038/nrendo.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pignolo RJ, Kassem M. Circulating osteogenic cells: implications for injury, repair, and regeneration. J Bone Mineral Res: Off J Am Soc Bone Mineral Res. 2011;26:1685–1693. doi: 10.1002/jbmr.370. [DOI] [PubMed] [Google Scholar]
- 7.Wu AC, Raggatt LJ, Alexander KA, Pettit AR. Unraveling macrophage contributions to bone repair. BoneKEy Reports. 2013;2:373. doi: 10.1038/bonekey.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, Li C, Xie L, Crane J, Wan M, Zhen G, Bian Q, Yu B, Chang W, Qiu T, Pickarski M, Duong le T, Windle JJ, Luo X, Liao E, Cao X. Pdgf-bb secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med. 2014;20:1270–1278. doi: 10.1038/nm.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Yu YY, Lieu S, Yang F, Lang J, Lu C, Werb Z, Hu D, Miclau T, Marcucio R, Colnot C. Mmp9 regulates the cellular response to inflammation after skeletal injury. Bone. 2013;52:111–119. doi: 10.1016/j.bone.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, Maylin ER, Ripoll VM, Hume DA, Pettit AR. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181:1232–1244. doi: 10.4049/jimmunol.181.2.1232. [DOI] [PubMed] [Google Scholar]
- 11.Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germain RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458:524–528. doi: 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogle ME, Sefcik LS, Awojoodu AO, Chiappa NF, Lynch K, Peirce-Cottler S, Botchwey EA. Engineering in vivo gradients of sphingosine-1-phosphate receptor ligands for localized microvascular remodeling and inflammatory cell positioning. Acta Biomater. 2014;10(11):4704–4714. doi: 10.1016/j.actbio.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor s1p1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL. Edg-1, the g protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryu J, Kim HJ, Chang EJ, Huang H, Banno Y, Kim HH. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006;25:5840–5851. doi: 10.1038/sj.emboj.7601430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immuno-modulator fty720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278:47281–47290. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- 17.Sato C, Iwasaki T, Kitano S, Tsunemi S, Sano H. Sphingosine 1-phosphate receptor activation enhances bmp-2-induced osteoblast differentiation. Biochem Biophys Res Commun. 2012;423:200–205. doi: 10.1016/j.bbrc.2012.05.130. [DOI] [PubMed] [Google Scholar]
- 18.Awojoodu AO, Ogle ME, Sefcik LS, Bowers DT, Martin K, Brayman KL, Lynch KR, Peirce-Cottler SM, Botchwey E. Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to micro-vessels during implant arteriogenesis. Proc Natl Acad Sci U S A. 2013;110:13785–13790. doi: 10.1073/pnas.1221309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das A, Segar CE, Hughley BB, Bowers DT, Botchwey EA. The promotion of mandibular defect healing by the targeting of s1p receptors and the recruitment of alternatively activated macrophages. Biomaterials. 2013;34:9853–9862. doi: 10.1016/j.biomaterials.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrie Aronin CE, Sefcik LS, Tholpady SS, Tholpady A, Sadik KW, Macdonald TL, Peirce SM, Wamhoff BR, Lynch KR, Ogle RC, Botchwey EA. Fty720 promotes local microvascular network formation and regeneration of cranial bone defects. Tissue Eng Part A. 2010;16:1801–1809. doi: 10.1089/ten.tea.2009.0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sefcik LS, Aronin CE, Awojoodu AO, Shin SJ, Mac Gabhann F, MacDonald TL, Wamhoff BR, Lynch KR, Peirce SM, Botchwey EA. Selective activation of sphingosine 1-phosphate receptors 1 and 3 promotes local microvascular network growth. Tissue Eng Part A. 2011;17:617–629. doi: 10.1089/ten.tea.2010.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Das A, Barker D, Tholpady S, Wang T, Cui Q, Ogle R, Botchwey E. Local delivery of fty720 accelerates cranial allograft incorporation and bone formation. Cell Tissue Res. 2012;347:553–566. doi: 10.1007/s00441-011-1217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajikawa Y, Morihara T, Watanabe N, Sakamoto H, Matsuda K, Kobayashi M, Oshima Y, Yoshida A, Kawata M, Kubo T. Gfp chimeric models exhibited a biphasic pattern of mesenchymal cell invasion in tendon healing. J Cell Physiol. 2007;210:684–691. doi: 10.1002/jcp.20876. [DOI] [PubMed] [Google Scholar]
- 24.Kanematsu A, Yamamoto S, Iwai-Kanai E, Kanatani I, Imamura M, Adam RM, Tabata Y, Ogawa O. Induction of smooth muscle cell-like phenotype in marrow-derived cells among regenerating urinary bladder smooth muscle cells. Am J Pathol. 2005;166:565–573. doi: 10.1016/S0002-9440(10)62278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomiyama K, Murase N, Stolz DB, Toyokawa H, O’Donnell DR, Smith DM, Dudas JR, Rubin JP, Marra KG. Characterization of transplanted green fluorescent protein+ bone marrow cells into adipose tissue. Stem Cells. 2008;26:330–338. doi: 10.1634/stemcells.2007-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePaula CA, Truncale KG, Gertzman AA, Sunwoo MH, Dunn MG. Effects of hydrogen peroxide cleaning procedures on bone graft osteoinductivity and mechanical properties. Cell Tissue Bank. 2005;6:287–298. doi: 10.1007/s10561-005-3148-2. [DOI] [PubMed] [Google Scholar]
- 27.Petrie Aronin CE, Shin SJ, Naden KB, Rios PD, Jr, Sefcik LS, Zawodny SR, Bagayoko ND, Cui Q, Khan Y, Botchwey EA. The enhancement of bone allograft incorporation by the local delivery of the sphingosine 1-phosphate receptor targeted drug fty720. Biomaterials. 2010;31:6417–6424. doi: 10.1016/j.biomaterials.2010.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander KA, Chang MK, Maylin ER, Kohler T, Muller R, Wu AC, Van Rooijen N, Sweet MJ, Hume DA, Raggatt LJ, Pettit AR. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Mineral Res: Off J Am Soc Bone Mineral Res. 2011;26:1517–1532. doi: 10.1002/jbmr.354. [DOI] [PubMed] [Google Scholar]
- 29.Mokarram N, Bellamkonda RV. A perspective on immunomodulation and tissue repair. Ann Biomed Eng. 2014;42:338–351. doi: 10.1007/s10439-013-0941-0. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mokarram N, Merchant A, Mukhatyar V, Patel G, Bellamkonda RV. Effect of modulating macrophage phenotype on peripheral nerve repair. Biomaterials. 2012;33:8793–8801. doi: 10.1016/j.biomaterials.2012.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown BN, Londono R, Tottey S, Zhang L, Kukla KA, Wolf MT, Daly KA, Reing JE, Badylak SF. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012;8:978–987. doi: 10.1016/j.actbio.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eghbali-Fatourechi GZ, Lamsam J, Fraser D, Nagel D, Riggs BL, Khosla S. Circulating osteoblast-lineage cells in humans. N Engl J Med. 2005;352:1959–1966. doi: 10.1056/NEJMoa044264. [DOI] [PubMed] [Google Scholar]
- 34.Kumagai K, Vasanji A, Drazba JA, Butler RS, Muschler GF. Circulating cells with osteogenic potential are physiologically mobilized into the fracture healing site in the parabiotic mice model. J Orthop Res: Off Publ Orthop Res Soc. 2008;26:165–175. doi: 10.1002/jor.20477. [DOI] [PubMed] [Google Scholar]
- 35.Otsuru S, Tamai K, Yamazaki T, Yoshikawa H, Kaneda Y. Circulating bone marrow-derived osteoblast progenitor cells are recruited to the bone-forming site by the cxcr4/stromal cell-derived factor-1 pathway. Stem Cells. 2008;26:223–234. doi: 10.1634/stemcells.2007-0515. [DOI] [PubMed] [Google Scholar]
- 36.Wise JK, Sumner DR, Virdi AS. Modulation of stromal cell-derived factor-1/cxc chemokine receptor 4 axis enhances rhbmp-2-induced ectopic bone formation. Tissue Eng Part A. 2012;18:860–869. doi: 10.1089/ten.tea.2011.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toupadakis CA, Granick JL, Sagy M, Wong A, Ghassemi E, Chung DJ, Borjesson DL, Yellowley CE. Mobilization of endogenous stem cell populations enhances fracture healing in a murine femoral fracture model. Cytotherapy. 2013;15:1136–1147. doi: 10.1016/j.jcyt.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taguchi K, Ogawa R, Migita M, Hanawa H, Ito H, Orimo H. The role of bone marrow-derived cells in bone fracture repair in a green fluorescent protein chimeric mouse model. Biochem Biophys Res Commun. 2005;331:31–36. doi: 10.1016/j.bbrc.2005.03.119. [DOI] [PubMed] [Google Scholar]
- 39.Segar CE, Ogle ME, Botchwey EA. Regulation of angiogenesis and bone regeneration with natural and synthetic small molecules. Curr Pharm Des. 2013;19:3403–3419. doi: 10.2174/1381612811319190007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.