

Abstract
Nitrosamines mediate their mutagenic effects by causing DNA damage, oxidative stress, lipid peroxidation, and pro-inflammatory cytokine activation, which lead to increased cellular degeneration and death. However, the very same pathophysiological processes comprise the “unbuilding” blocks of aging and insulin-resistance diseases including, neurodegeneration, diabetes mellitus (DM), and non-alcoholic steatohepatitis (NASH). Previous studies demonstrated that experimental exposure to streptozotocin, a nitrosamine-related compound, causes NASH, and diabetes mellitus Types 1, 2 and 3 (Alzheimer (AD)-type neurodegeneration). Herein, we review evidence that the upwardly spiraling trends in mortality rates due to DM, AD, and Parkinson's disease typify exposure rather than genetic-based disease models, and parallel the progressive increases in human exposure to nitrates, nitrites, and nitrosamines via processed/preserved foods. We propose that such chronic exposures have critical roles in the pathogenesis of our insulin resistance disease pandemic. Potential solutions include: 1) eliminating the use of nitrites in food; 2) reducing nitrate levels in fertilizer and water used to irrigate crops; and 3) employing safe and effective measures to detoxify food and water prior to human consumption. Future research efforts should focus on refining our ability to detect and monitor human exposures to nitrosamines and assess early evidence of nitrosamine-mediated tissue injury and insulin resistance.
Keywords: Alzheimer's disease, diabetes, environmental toxin, neurodegeneration, nitrosamine, obesity
INTRODUCTION
Nitrosamines and N-nitroso compounds are among the most potent and broad acting carcinogens that can also function as transplacental mutagens. Nitrosamine-mediated target organ damage and mutagenesis are heavily influenced by route of administration, dose, chemical nature of the compound, and frequency of exposure. Among the more than 300 N-nitroso or nitrosamine compounds tested, over 90% have been found to be carcinogenic in various organs including liver, gastrointestinal tract (from esophagus to rectum), lung, kidney, bladder, pancreas, prostate, and uterus. All mammals are susceptible. Fried bacon, cured meats, beer, cheese products, fish byproducts, nonfat dry milk, tobacco, gastric juices (dietary), and water are major regular sources of consumer exposure to nitrosamines. However, nitrosamine exposures also occur through manufacturing, processing, and utilization of rubber and latex products, fertilizers, pesticides, and cosmetics (for review, see [1]).
Nitrosamines (R1N(-R2)-N=O) are formed by a chemical reaction between nitrites and secondary amines or proteins. How does this occur, and what factors contribute to human dietary exposures? First, as a public health measure, sodium nitrite is deliberately added to meat and fish to prevent toxin production by Clostridium botulinum. Second, sodium nitrite is used to preserve, color, and flavor meats. Heating, acidification, or oxidation of nitrite leads to nitrous acid formation. The resulting nitrosonium cation (N=O+) is a nitrosating agent that reacts with dimethylamine to generate nitrosamines. Dimethylamine is commonly present in fish meal. Moreover, since ground beef, cured meats, and bacon in particular, contain abundant amines due to their high protein content, and they have significant levels of added nitrates and nitrites [2], nitrosamines are nearly always detectable in these food products. Finally, nitrosamines are easily generated under strong acid conditions, such as in the stomach, or at high temperatures associated with frying or flame broiling. Reducing sodium nitrite content definitely lowers nitrosamine formation in foods.
NITROSAMNE GENERATION: THE BASICS
Nitrate exposures pose human health problems because about 5% of ingested nitrates get chemically reduced, forming harmful nitrites by bacterial enzymes present in the oral cavity [3,4]. After entering the highly acidic gastric juice environment, nitrites can be converted to nitrous acid (HNO2), and nitrosating agents such as nitrous anhydride (N2O3) and nitric acid [5]. Nitrosating agents [6] and nitrites, either formed endogenously or ingested directly with food or water [7] react with amines to generate nitrosamines. Amine precursors of N-nitroso compounds are generated from dietary sources during the natural process of protein digestion. Alternatively, they can be consumed directly when present in overcooked or processed meats and over-ripened fruits and vegetables. High protein content foods yield high levels of amine [8]. Besides amines, choline, a nutrient found in most foods, including meats, vegetables, peanuts, and eggs, is essential for many biological functions [9], but can react with nitrites to form dimethylamine (DMA), the precursor to nitrosodimethylamine (NDMA) [4].
In essence, diets rich in amines, choline, and nitrates lead to increased nitrosamine production relative to diets that are low in nitrate, and include fish and seafood as the main sources of amines [3,10]. Human exposure to nitrates stems in part from the abundant use of nitrate-containing fertilizers for agriculture, which results in relatively high levels of nitrates in many crops, particularly root vegetables, such as potatoes and beets. Vegetables, especially potatoes, account for nearly 85% of our daily dietary intake of nitrates [3,6]. Nitrates can also leech from the soil and contaminate water supplies used for crop irrigation, food processing, and drinking. Since amines and choline are essential nutrients, and therefore cannot be eliminated from the diet, a more feasible approach for reducing exposures to nitrates, nitrites, and nitrosamines, would be to discontinue their deliberate addition and permitted contamination of our food and water sources.
NITROSAMINE SOURCES IN CONSUMABLES
The two main forms of nitrosamine that threaten human health are N-nitrosodimethylamine (DMN; NDMA) and N-nitrosodiethylamine (DEN; NDEA). NDMA and NDEA are highly toxic, semi-volatile, organic chemicals that contaminate food and water sources [11]. NDMA is a waste- or by-product of industrial processes such as organic nitrogen-containing wastewater chloramination, water chlorination, rocket fuel production, and anion exchange resin water treatment [12,13]. NDMA can also be found at low levels in tobacco smoke and foods such as cured meat, fish, and beer [2]. NDMA contamination of drinking water and food is problematic because, even at virtually undetectable levels, the compound can be harmful, particularly after long-term bioaccumulation. Experimentally, short-term, high-level exposure to NDMA is hepatotoxic and causes liver fibrosis [14–16], whereas chronic low-level or moderate exposure to NMDA causes liver tumors [14,17–19]. In humans, nitrosamine exposure from preserved meats and fish has been correlated with increased frequency of gastric cancer [20,21].
NDEA is a volatile water-soluble chemical that is used as an additive for gasoline and lubricants, and in the manufacturing of polymers, plastics, and pesticides. Human exposures often occur through ingestion, inhalation, or skin contact with these substances. Like NDMA, NDEA contaminates air,food, beverages, drinking water, tobacco smoke, herbicides, and pesticides, and is also an industrial pollutant. Appreciable levels of NDEA can be found in inhaled and side-stream tobacco smoke and cured meats. Although NDEA is widespread in the environment, sunlight and UV irradiation cause it to decompose, thereby reducing exposure from inhaled air and open bodies of water [22]. NDEA exposure causes cancer in liver, thyroid, gastrointestinal tract, and respiratory tract [22]. The main specific carcinogenic nitrosamines associated with exposure to tobacco products,including cigarettes,cigars, and chewing tobacco,include N-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone [23], the latter of which is produced by reaction of nitrite in saliva with tobacco alkaloids such as nicotine. Since nitrosonornicotine is also present in cigar and cigarette smoke, non-smokers can be exposed via second-hand smoke inhalation. Finally, nicotine replacement products can also serve as sources of nitrosamine exposure [23]. In essence, discontinued use of tobacco and tobacco-related products may be the single most important means of reducing nitrosamine exposure and related adverse effects on health status.
MECHANISMS OF CELLULAR INJURY
Nitrosamines exert their toxic and mutagenic effects by functioning as alkylating agents. In brief, metabolic processing converts nitrosamines into active methylating agents that are highly reactive with nucleic acids, altering gene expression and causing DNA damage. Activated nitrosamines, most commonly alkylate N-7 of guanine, lead to destabilization and increased breakage of DNA [17]. In addition, activated nitrosamines generate reactive oxygen species such as superoxide (O2-) and hydrogen peroxide (H2O2), causing increased oxidative stress, DNA damage, lipid peroxidation, and protein adduct formation [24]. Oxidative stress and DNA damage lead to activation of pro-inflammatory cytokines, insulin resistance, and aging, all of which are key elements in the pathogenesis of Type 2 diabetes mellitus (T2DM), neurodegeneration, and malignancy. The consequences of nitrosamine-mediated tissue injury may vary with the phenotypic properties of the target cells, i.e., proliferating cells may be more prone to undergo malignant transformation, whereas terminally differentiated cells may instead undergo senescence or degeneration.
NITRATES, NITRITES, NITROSAMINES, AND NITROSAMINE-RELATED COMPOUNDS IN DISEASE
Investigations about the role of nitrosamines in the pathogenesis of human disease have primarily focused on carcinogenesis, yet the basic cellular and molecular alterations produced by nitrosamine exposure are fundamentally similar to those that occur with aging, Alzheimer's disease (AD), non-alcoholic steatohepatitis (NASH), and T2DM. All of these non-neoplastic diseases are associated with increased insulin resistance, DNA damage, lipid peroxidation, oxidative stress, and pro-inflammatory cytokine activation [25–31]. The prevalence rates of AD, NASH, obesity, and T2DM have all increased exponentially over the past several decades, and thus far, show no hints of plateau [32–37]. Of particular note is that the rather short time interval associated with dramatic shifts in disease incidence and prevalence rates is more consistent with exposure-related, rather than genetic etiologies (discussed later).
An important clue regarding the probable connection between nitrosamine exposure and AD, NASH, and diabetes mellitus was provided by experimental data demonstrating that treatment with Streptozotocin [2-deoxy-2-(3-methyl-3-nitrosoureido-D-glucopyranose (C8H15N3O7)] (STZ), a glucosamine-nitrosourea compound and derivative of N-methyl-N-nitrosourea (MNU), causes AD-type neurodegeneration with cognitive impairment, T1DM, T2DM [38–44], or hepatic steatosis with chronic inflammation and scarring, i.e., NASH [45]. Of further interest is the finding that similar disease models can be produced by delivery of either a single large dose (40–60 mg/kg) [43], or repeated smaller doses [42] of STZ. STZ is taken up by cells via glucose transporters [46], and once metabolized, liberates N-nitrosoureido. STZ inhibits DNA synthesis and kills cells by lowering ATP and nicotine adenine dinucleotide (NAD+) content [43].
Structurally, STZ is quite similar to nitrosamines, and like other N-nitroso compounds, including NDEA and NDMA [47], STZ's MNU causes cellular injury and disease by functioning as: 1) an alkylating agent and potent mutagen resulting in cancer development in various organs [48]; 2) an inducer of DNA adducts, most significantly N7-methylguanine, which lead to increased apoptosis [49]; 3) a mediator of unscheduled DNA synthesis that triggers cell death [48]; 4) an inducer of single-strand DNA breaks; 5) a stimulus for nitric oxide (NO) formation following breakdown of its nitrosamine group [43]; and 6) an enhancer of the xanthine oxidase system leading to increased production of superoxide anion, H2O2, and OH− radicals [50]. In essence, STZ-induced cellular injury is mediated by the generation of reactive oxygen species with attendant increased levels of superoxide, nitric oxide, and lipid peroxidation, all of which cause DNA damage. Radical ion accumulation leads to inhibition of oxidative metabolism, mitochondrial dysfunction [43], decreased ATP production [51], activation of poly-ADP ribosylation, and finally cell death.
NITROSAMINE COMPOUNDS AND INSULIN RESISTANCE-MEDIATED DISEASES
STZ treatment causes T1DM [44,52], T2DM [53–55], steatohepatitis [45], and neurodegeneration [38–41]. The spectrum of disease produced is dictated by the dose and route of administration of STZ. Intraperitoneal delivery of STZ causes T1DM, T2DM, or NASH, with high doses usually resulting in T1DM, and lower repeated doses causing T2DM or steato-hepatitis. Intracerebral (ic) STZ administration causes neurodegeneration in the absence of T1DM, T2DM, or NASH, indicating that the brain can be a selective target of MNU/nitrosamine-mediated injury or neurotoxicity. Importantly, these disease entities share a common theme, but vary with respect to nature and degree of target-organ injury, insulin deficiency and/or insulin resistance, increased oxidative stress and DNA damage, increased levels of nitric oxide and free radicals,pro-inflammatory cytokine activation, and mitochondrial dysfunction with deficiencies in ATP production. Specific effects of ic-STZ-induced neurodegeneration include: increased amyloid-β protein precursor-amyloid-β (AβPP-Aβ) deposition, AβPP gene expression, and tau phosphorylation, accompanied by cognitive impairment [38,41,56], deficits in acetylcholine homeostasis [38,41] and neurotrophin gene expression [57], as typically occur in AD [58]. Since the cognitive deficits and neurodegeneration caused by ic-STZ can be abrogated by concurrent delivery of insulin [59], or early intervention with peroxisome-proliferator activated receptor (PPAR) agonists, which function as both insulin-sensitizer and anti-inflammatory/antioxidant agents [38], this AD model mechanistically overlaps with both T1DM and T2DM, prompting us to coin the term, “Type 3 Diabetes” [38,41].
In essence, at the core of AD pathology, DM, and NASH is insulin resistance with associated deficits in glucose utilization and energy metabolism, and increased levels of chronic inflammation and oxidative stress [60–65]. Although for years, disturbances in metabolic function have been recognized features of both DM and AD [60], the mediators of these abnormalities were not understood. However, the more recent and growing interest in the roles of insulin resistance and insulin deficiency syndromes, including AD, has opened doors to more diversified investigational approaches. Correspondingly, epidemiological trend analysis demonstrated significant associations between AD and T2DM, revealing the nearly two-fold increased risk of developing AD in subjects with T2DM [25,65–71]. In addition, other studies showed associations between obesity or peripheral insulin resistance and cognitive impairment [72,73]. Since NASH and AD are associated with T2DM, T2DM and NASH are associated with obesity and metabolic syndrome, while AD and T1DM are associated with insulin deficiency, there appears to be a 3-way Venn-diagram type overlap among AD, T2DM, and NASH. But, is there a common cause?
EVIDENCE THAT DIABETES MELLITUS AND ALZHEIMER's ARE EXPOSURE-RELATED DISEASES
We were prompted to consider the potential role of nitrosamines as environmental toxins mediating insulin-resistance diseases, including NASH, T2DM, and AD because STZ has a methylnitrosourea group and its effects on DNA and cellular functions are similar to those of nitrosamine compounds, including NDEA and NDMA (Fig. 1). However, since in low, submutagenic doses, STZ causes insulin resistance, we considered the concept that low-dose chronic exposure to NDEA, NDMA, and other nitrosamines may cause insulin-resistance mediated diseases rather than malignancy. To assess the validity of an exposure hypothesis in relation to the pathogenesis of T2DM and AD, it was first necessary to demonstrate epidemio-logical trends that mirrored effects of exposure-related diseases such as infectious diseases, rather than genetic diseases which remain relatively stable over a period of decades. Therefore, we graphed and analyzed time-dependent, age-stratified mortality rates from all causes, AD, Parkinson's disease (PD), diabetes mellitus, cerebrovasculardisease, chronic liver disease, lymphomas, leukemias, and HIV-AIDS from 1968 to 2005. HIV-AIDS served as a positive control for disease that is caused by exposure to an infectious agent. The data source was the US Department of Health and Human Services, Centers for Disease Control and Prevention National Center for Health Statistics database (www.cdc.gov/nchs/hus.htm). To first determine the degree to which death rates from these diseases correlated with increasing age, we computed areas under the curves corresponding to death rates over time versus age group, i.e., 45–54, 55–64, 65–74, 75–84, and 85+ years (Fig. 2). The graphs demonstrate that the highest rates of death from nearly all of these disease occurred in the older age groups, indicating that they are aging-associated. Exceptions included chronic liver disease and HIV-AIDS, which had higher death rates in the earlier age groups.
Fig. 1.
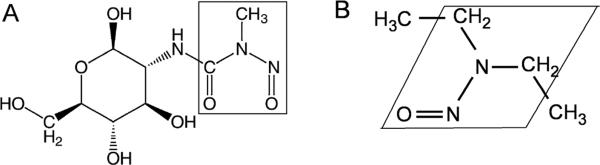
Structural similarity between (A) Streptozotocin and (B) N-nitrosodiethylamine. Boxed regions delineate regions of homology. These compounds are alkylating agents and potent mutagens that cause cancer in various organs, induce DNA adducts leading to increased apoptosis, promote single-strand DNA breaks, and enhance production of superoxide anion, H2O2, and OH− radicals
Fig. 2.

Comparison of trends in death rates from (A) all causes, (B) Alzheimer's disease (AD), (C) Parkinson's disease (PD), (D) diabetes mellitus, (E) cerebrovascular disease, (F) chronic liver disease, (G) Human immunodeficiency virus-AIDS (HIV), (H) lymphomas, and (I) leukemias (all types) with respect to age from 1968–2005. Graphs were generated by calculating the area under curve (AUC) corresponding age-adjusted mortality rates over time. Results demonstrate that the majority of deaths from these diseases occur in the older age groups. Exceptions include chronic liver disease and HIV-AIDS.
To determine the degree to which the mortality rates had shifted upward or downward over time, we graphed age-stratified death rates for each disease. This approach enables direct comparison of death rates from any given disease in, for example, 65–74 year olds in 1970 versus the same age group in 2005. Moreover, by superimposing or aligning graphs for all age groups within a disease category, it was possible to determine the rank order of death rates with respect to age group (Fig. 3). However, to clearly visualize specific disease trends, the death rates corresponding to individual age brackets were re-plotted to scale on separate graphs (Fig. 4). Among individuals 45–54, 55–64, 65–74, 75– 84, and 85+ years old, the death rates from all causes progressively declined over time (Fig. 3A). Similarly, death rates from cerebrovascular disease (Figs 3E, 4A–4E) and chronic liver disease (Fig. 3F) also declined within each age group over time. Nonetheless, for each of these diseases, the death rates were highest among the 85+ group followed by the 75–84 year olds, while the other three groups were lower but tightly clustered. In contrast, the trends for lymphomas and leukemias remained relatively stable (Fig. 3G, 3H), except for the time-dependent upward drift in death rate from lymphoma among 85+ year olds (Fig. 3G).
Fig. 3.

Comparison of trends in death rates from (A) all causes, (B) Alzheimer's disease (AD), (C) Parkinson's disease (PD), (D) diabetes mellitus, (E) cerebrovascular disease, (F) chronic liver disease, (G) Human immunodeficiency virus-AIDS, (H) lymphomas, and (I) leukemias (all types) over time. Note time-dependent declines in death rates within each age group for (A) all causes, (E) cerebrovascular disease and (F) chronic liver disease, and relatively stable trends with respect to (H) lymphomas and (I) leukemias, and sharp increases followed by a rapid fall in death rate due to widespread treatment with anti-retroviral agents. Only (B) AD, (C) PD, and (D) diabetes mellitus exhibit prominent increases in death rate for most age groups over time.
Fig. 4.

Detailed comparison of trends in death rates for (A-E) cerebrovascular disease, (F-J) HIV, (K-O) diabetes mellitus, (P-T) Alzheimer's disease, and (U-Y) Parkinson's disease. Note the similar shapes of curves for nearly all age groups within each disease category, indicating that overall trends were not due to aging of the population.
Death rates from HIV-AIDS exhibited classical exposure-associated increases from 1985 to 1995, followed by precipitous declines due to increased availability of effective anti-retroviral drugs (Figs 3F, 4F–4J). Trends mapping death rates from DM were U-shaped for all age groups in that the rates sharply declined after 1968, reaching a nadir in ~1980, but subsequently increased to levels that were similar to, or higher than in 1968, with modest hints of plateau over the last 3–4 years (Figs 3D, 4K–4O). Death rates from AD (Figs 3B, 4P–4T) and PD (Figs 3C, 4U–4Y) exhibited opposite trends relative to cerebrovascular disease and all causes of death due to increased rather than decreased rates within each age group over time. Of note is that between 1968 and 2005, the death rates from AD increased by 4-fold in the 55–64, 20-fold in the 64–74, 150-fold in the 75–84, and 800-fold in the 85+ year old groups. With respect to PD, the death rates increased about 2-fold in the 65–74,3-fold in the 75–84,and nearly 6-fold in the 85+ age groups from 1980–2005 (PD death rates were not tracked prior to 1980). Therefore, besides bucking the trends with respect to other major aging-associated diseases, the relatively short intervals associated with dramatic increases in death rates from DM, AD, and PD are more consistent with exposure-related rather than genetic etiologies. Moreover, the strikingly higher and climbing mortality rates in older age brackets suggest that aging and/or longer durations of exposure have greater impacts on progression and severity of these diseases. The U-shaped curve for DM is particularly disconcerting because presumably the recognition and treatments are better today than they were in 1980, yet mortality is higher. Perhaps factors contributing to the pathogenesis of DM prior to 1970 differ from those in modern times,which seem to render DM a more “malignant” and uncontrollable disease.
Since human exposures to nitrates, nitrites, and nitrosamines are likely to occur due to fertilizer use, consumption of processed and preserved foods, and agricultural products that heavily rely in fertilizer use (e.g., grains), we examined US population growth (Fig. 5A), annual use/consumption of nitrate-containing fertilizers (Fig. 5B), annual sales for a popular fast food franchise (Fig. 5C), sales for a major meat processing company (Fig. 5D), consumption of grain (Fig. 5E), and consumption of watermelon and cantaloupe (Fig. 5F). Watermelon and cantaloupe served as controls since they are not typically associated with nitrate or nitrite exposure, DM, or AD. Business sales data were obtained from the companies’ websites or stock exchange databases, and agricultural sales/consumption data were obtained from USDA websites. The results demonstrated that the US population nearly doubled between 1955 and 2005. Although nitrogen-containing fertilizer consumption increased by 230% over the same interval, its usage doubled between 1960 and 1980, just preceding the insulin-resistance epidemics. Sales from a popular fast food franchise and a major meat processing company increased more than 8-fold from 1970 to 2005, and grain consumption increased 5-fold. In contrast, watermelon consumption remained relatively flat, while cantaloupe consumption nearly doubled, paralleling population growth, although overall per capita consumption remains relatively low compared with processed foods.
Fig. 5.

Exposure trends in the US from 1960 or 1970 to 2005. (A) US population increased nearly two-fold between 1955 and 2005. (B) Fertilizer consumption more than doubled over nearly the same interval. (C) Sales from one popular fast food franchise and (D) one major meat processing company increased more than 8-fold from 1970 to 2005. (E) In contrast, watermelon consumption remained relatively flat, and (F) cantaloupe consumption nearly doubled in parallel with population growth, but sales were minor compared with fertilizer, fast foods, and processed meats.
HYPOTHESIS
Epidemiological data revealed staggering growths in prevalence and/or mortality rates for T2DM, obesity, NASH, and AD, and significant overlap between AD or NASH and T2DM. Basic, translational, and clinical research studies have demonstrated that AD neurodegeneration, like T2DM and NASH, is associated with insulin resistance as well as increased oxidative stress, DNA damage, NO-mediated injury, and ATP deficits caused by mitochondrial dysfunction and impaired insulin signaling, but with prominent or selective involvement of the brain. Experimental animal models provided evidence that STZ administration causes Type 1, Type 2, or Type 3 diabetes, depending on dose and route of administration. In addition, STZ treatment causes hepatic steatosis associated with insulin resistance, pro-inflammatory cytokine activation, increased oxidative stress, DNA damage, lipid peroxidation, and cell death culminating in fibrogenesis, i.e., a model of NASH [48,74].
Returning to the epidemiological data, the time course of increasing prevalence rates of T2DM, NASH, and AD cannot be explained on the basis of gene mutations, and instead, mirror the classical trends of exposure-relateddiseases. Since STZ is structurally related to nitrosamines and produces similar biochemical and molecular lesions in cells and tissues, it is conceivable that chronic exposure to relatively low levels of nitrites and nitrosamines through processed foods, water, and fertilizers, is responsible for our current epidemics and probably also the pandemics of T2DM, NASH, and AD. The link to obesity is through excessive consumption of nutritionally irresponsible processed foods. If this hypothesis is correct, potential solutions would include: 1) eliminating the use of nitrites and nitrates in food processing, food preservation, and agriculture; 2) taking steps to prevent formation of nitrosamines; and 3) employing safe and effective measures to detoxify food and water prior to human consumption. In addition, further development of highly sensitive measures to detect the footprints, e.g., measures of amine nitrosation, and assess levels of nitrosamine exposure in body fluids such as urine or cerebrospinal fluid, and in tissues such as liver, brain, adipose tissue, and muscle, could help identify individuals at risk for developing T2DM, NASH, and/or AD.
CONCLUSIONS
Nitrosamines are potent mutagens that cause DNA damage, oxidative stress, cell death, and cancer. However, the structural relatedness of nitrosamines to STZ demands attention because STZ is a proven mutagen, but in low doses causes Types 1 and 2 diabetes mellitus, hepatic steatohepatitis, or AD-type neurodegeneration. We hypothesize that widespread exposures to nitrites and nitrosamines in our environment have critical roles in the pathogenesis of these insulin-resistance and insulin-deficiency related diseases. Epidemiological trends support exposure rather than genetic causes of these diseases, and exposures to nitrites and ni trosamines through food, water, agriculture have increased just prior to and within the same interval due to proliferation of food processing, increased requirement for food preservation to enable “safe” storage and long distance shipping, and the use of fertilizers to enhance crop growth and meet growing demand for produce. Tobacco use contributes to the problem, but public health measures have already been taken to reduce public exposure. Sincere efforts should be made to significantly curtail or eliminate human exposure to nitrates and nitrites, and refine extant biotechnology to monitor exposure, metabolite formation, and associated cellular and tissue injury linked to nitrosamine-mediated insulin resistance-related diseases, including T2DM, NASH, and AD.
ACKNOWLEDGMENTS
Supported by AA-02666, AA-02169, AA-11431, AA-12908, and K24-AA-16126 from the National Institutes of Health.
REFERENCES
- 1.Kearney PC. Nitrosamines and pesticides: A special report on the occurrence of nitrosamines as terminal residues resulting from agricultural use of certain pesticides. Pure Appl Chem. 1980;52:499–526. [Google Scholar]
- 2.Lijinsky W. N-Nitroso compounds in the diet. Mutat Res. 1999;443:129–138. doi: 10.1016/s1383-5742(99)00015-0. [DOI] [PubMed] [Google Scholar]
- 3.Vermeer IT, Pachen DM, Dallinga JW, Kleinjans JC, van Maanen JM. Volatile N-nitrosamine formation after intake of nitrate at the ADI level in combination with an amine-rich diet. Environ Health Perspect. 1998;106:459–463. doi: 10.1289/ehp.106-1533225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeisel SH, DaCosta KA, Fox JG. Endogenous formation of dimethylamine. Biochem J. 1985;232:403–408. doi: 10.1042/bj2320403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scanlan RA. Formation and occurrence of nitrosamines in food. Cancer Res. 1983;43:2435s–2440s. [PubMed] [Google Scholar]
- 6.Winter JW, Paterson S, Scobie G, Wirz A, Preston T, McColl KE. N-nitrosamine generation from ingested nitrate via nitric oxide in subjects with and without gastroesophageal reflux. Gastroenterology. 2007;133:164–174. doi: 10.1053/j.gastro.2007.04.047. [DOI] [PubMed] [Google Scholar]
- 7.Sander J, Burkle G. [Induction of malignant tumors in rats by simultaneous feeding of nitrite and secondary amines]. Z Krebsforsch. 1969;73:54–66. [PubMed] [Google Scholar]
- 8.Felician O, Sandson TA. The neurobiology and pharmacotherapy of Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 1999;11:19–31. doi: 10.1176/jnp.11.1.19. [DOI] [PubMed] [Google Scholar]
- 9.Blusztajn JK. Choline, a vital amine. Science. 1998;281:794–795. doi: 10.1126/science.281.5378.794. [DOI] [PubMed] [Google Scholar]
- 10.van Maanen JM, Pachen DM, Dallinga JW, Kleinjans JC. Formation of nitrosamines during consumption of nitrate- and amine-rich foods, and the influence of the use of mouthwashes. Cancer Detect Prev. 1998;22:204–212. doi: 10.1046/j.1525-1500.1998.0oa26.x. [DOI] [PubMed] [Google Scholar]
- 11.Hotchkiss JH. Sources of N-nitrosamine contamination in foods. Adv Exp Med Biol. 1984;177:287–298. doi: 10.1007/978-1-4684-4790-3_14. [DOI] [PubMed] [Google Scholar]
- 12.Levallois P, Ayotte P, Van Maanen JM, Desrosiers T, Gingras S, Dallinga JW, Vermeer IT, Zee J, Poirier G. Excretion of volatile nitrosamines in a rural population in relation to food and drinking water consumption. Food Chem Toxicol. 2000;38:1013–1019. doi: 10.1016/s0278-6915(00)00089-2. [DOI] [PubMed] [Google Scholar]
- 13.Najm I, Trussell RR. NDMA formation in water and wastewater. AWWA. 2001;93:92–99. [Google Scholar]
- 14.Lim IK. Spectrum of molecular changes during hepatocarcinogenesis induced by DEN and other chemicals in Fisher 344 male rats. Mech Ageing Dev. 2003;124:697–708. doi: 10.1016/s0047-6374(03)00010-1. [Mech Ageing Dev 123 (2002) 1665-1680] [DOI] [PubMed] [Google Scholar]
- 15.Jezequel AM, Mancini R, Rinaldesi ML, Macarri G, Venturini C, Orlandi F. A morphological study of the early stages of hepatic fibrosis induced by low doses of dimethylnitrosamine in the rat. J Hepatol. 1987;5:174–181. doi: 10.1016/s0168-8278(87)80570-6. [DOI] [PubMed] [Google Scholar]
- 16.Guo L, Enzan H, Hayashi Y, Miyazaki E, Jin Y, Toi M, Kuroda N, Hiroi M. Increased iron deposition in rat liver fibrosis induced by a high-dose injection of dimethylnitrosamine. Exp Mol Pathol. 2006;81:255–261. doi: 10.1016/j.yexmp.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Swann PF, Magee PN. Nitrosamine-induced carcino-genesis. The alklylation of nucleic acids of the rat by N-methyl-N-nitrosourea, dimethylnitrosamine, dimethyl sulphate and methyl methanesulphonate. Biochem J. 1968;110:39–47. doi: 10.1042/bj1100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai DY, Arcos JC. Minireview: dialkylnitrosamine bioactivation and carcinogenesis. Life Sci. 1980;27:2149–2165. doi: 10.1016/0024-3205(80)90379-3. [DOI] [PubMed] [Google Scholar]
- 19.Lijinsky W, Reuber MD, Riggs CW. Dose response studies of carcinogenesis in rats by nitrosodiethylamine. Cancer Res. 1981;41:4997–5003. [PubMed] [Google Scholar]
- 20.Sailer D. [Carcinogens in the food]. Fortschr Med. 1978;96:446–452. [PubMed] [Google Scholar]
- 21.Jakszyn P, Bingham S, Pera G, Agudo A, Luben R, Welch A, Boeing H, Del Giudice G, Palli D, Saieva C, Krogh V, Sacer-dote C, Tumino R, Panico S, Berglund G, Siman H, Hallmans G, Sanchez MJ, Larranaga N, Barricarte A, Chirlaque MD, Quiros JR, Key TJ, Allen N, Lund E, Carneiro F, Linseisen J, Nagel G, Overvad K, Tjonneland A, Olsen A, Bueno-de-Mesquita HB, Ocke MO, Peeters PH, Numans ME, Clavel-Chapelon F, Trichopoulou A, Fenger C, Stenling R, Ferrari P, Jenab M, Norat T, Riboli E, Gonzalez CA. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogen-esis. 2006;27:1497–1501. doi: 10.1093/carcin/bgl019. [DOI] [PubMed] [Google Scholar]
- 22.Scanlan RA. Nitrosamine and Cancer. Publication Review. 2003;14:34–35. [Google Scholar]
- 23.Stepanov I, Jensen J, Hatsukami D, Hecht SS. Tobacco-specific nitrosamines in new tobacco products. Nicotine Tob Res. 2006;8:309–313. doi: 10.1080/14622200500490151. [DOI] [PubMed] [Google Scholar]
- 24.Espey MG, Miranda KM, Thomas DD, Xavier S, Citrin D, Vitek MP, Wink DA. A chemical perspective on the interplay between NO, reactive oxygen species, and reactive nitrogen oxide species. Ann N Y Acad Sci. 2002;962:195–206. doi: 10.1111/j.1749-6632.2002.tb04068.x. [DOI] [PubMed] [Google Scholar]
- 25.Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes mellitus and dementia. Diabetes Metab. 2006;32:403–414. doi: 10.1016/s1262-3636(07)70298-7. [DOI] [PubMed] [Google Scholar]
- 26.Nicolls MR. The clinical and biological relationship between Type II diabetes mellitus and Alzheimer's disease. Curr Alzheimer Res. 2004;1:47–54. doi: 10.2174/1567205043480555. [DOI] [PubMed] [Google Scholar]
- 27.Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;128:837–847. doi: 10.1309/RTPM1PY6YGBL2G2R. [DOI] [PubMed] [Google Scholar]
- 28.Marchesini G, Marzocchi R. Metabolic syndrome and NASH. Clin Liver Dis. 2007;11:105–117. ix. doi: 10.1016/j.cld.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Papandreou D, Rousso I, Mavromichalis I. Update on non-alcoholic fatty liver disease in children. Clin Nutr. 2007;26:409–415. doi: 10.1016/j.clnu.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J Gastroenterol Hepatol 22 Suppl. 2007;1:S20–27. doi: 10.1111/j.1440-1746.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- 31.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rector RS, Thyfault JP, Wei Y, Ibdah JA. Non-alcoholic fatty liver disease and the metabolic syndrome: an update. World J Gastroenterol. 2008;14:185–192. doi: 10.3748/wjg.14.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pradhan A. Obesity, metabolic syndrome, and type 2 diabetes: inflammatory basis of glucose metabolic disorders. Nutr Rev. 2007;65:S152–156. doi: 10.1111/j.1753-4887.2007.tb00354.x. [DOI] [PubMed] [Google Scholar]
- 34.Launer LJ. Next steps in Alzheimer's disease research: interaction between epidemiology and basic science. Curr Alzheimer Res. 2007;4:141–143. doi: 10.2174/156720507780362155. [DOI] [PubMed] [Google Scholar]
- 35.Wang XP, Ding HL. Alzheimer's disease: epidemiology, genetics, and beyond. Neurosci Bull. 2008;24:105–109. doi: 10.1007/s12264-008-0105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delgado JS. Evolving trends in nonalcoholic fatty liver disease. Eur J Intern Med. 2008;19:75–82. doi: 10.1016/j.ejim.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 37.Nugent C, Younossi ZM. Evaluation and management of obesity-related nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4:432–441. doi: 10.1038/ncpgasthep0879. [DOI] [PubMed] [Google Scholar]
- 38.de la Monte SM, Tong M, Lester-Coll N, Plater M, Jr., Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer's disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 39.Hoyer S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv Exp Med Biol. 2004;541:135–152. doi: 10.1007/978-1-4419-8969-7_8. [DOI] [PubMed] [Google Scholar]
- 40.Hoyer S, Lannert H, Noldner M, Chatterjee SS. Damaged neuronal energy metabolism and behavior are improved by Ginkgo biloba extract (EGb 761). J Neural Transm. 1999;106:1171–1188. doi: 10.1007/s007020050232. [DOI] [PubMed] [Google Scholar]
- 41.Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- 42.Rossini AA, Like AA, Chick WL, Appel MC, Cahill GF., Jr. Studies of streptozotocin-induced insulitis and diabetes. Proc Natl Acad Sci U S A. 1977;74:2485–2489. doi: 10.1073/pnas.74.6.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50:537–546. [PubMed] [Google Scholar]
- 44.Doi K. [Studies on the mechanism of the diabetogenic activity of streptozotocin and on the ability of compounds to block the diabetogenic activity of streptozotocin (author's transl)]. Nippon Naibunpi Gakkai Zasshi. 1975;51:129–147. doi: 10.1507/endocrine1927.51.3_129. [DOI] [PubMed] [Google Scholar]
- 45.Iwai S, Murai T, Makino S, Min W, Morimura K, Mori S, Hagihara A, Seki S, Fukushima S. High sensitivity of fatty liver Shionogi (FLS) mice to diethylnitrosamine hepatocarcinogenesis: comparison to C3H and C57 mice. Cancer Lett. 2007;246:115–121. doi: 10.1016/j.canlet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Elsner M, Guldbakke B, Tiedge M, Munday R, Lenzen S. Relative importance of transport and alkylation for pancreatic beta-cell toxicity of streptozotocin. Diabetologia. 2000;43:1528–1533. doi: 10.1007/s001250051564. [DOI] [PubMed] [Google Scholar]
- 47.Robbiano L, Mereto E, Corbu C, Brambilla G. DNA damage induced by seven N-nitroso compounds in primary cultures of human and rat kidney cells. Mutat Res. 1996;368:41–47. doi: 10.1016/s0165-1218(96)90038-5. [DOI] [PubMed] [Google Scholar]
- 48.Bolzan AD, Bianchi MS. Genotoxicity of streptozotocin. Mutat Res. 2002;512:121–134. doi: 10.1016/s1383-5742(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 49.Murata M, Takahashi A, Saito I, Kawanishi S. Site-specific DNA methylation and apoptosis: induction by diabetogenic streptozotocin. Biochem Pharmacol. 1999;57:881–887. doi: 10.1016/s0006-2952(98)00370-0. [DOI] [PubMed] [Google Scholar]
- 50.Nukatsuka M, Sakurai H, Yoshimura Y, Nishida M, Kawada J. Enhancement by streptozotocin of O2-radical generation by the xanthine oxidase system of pancreatic beta-cells. FEBS Lett. 1988;239:295–298. doi: 10.1016/0014-5793(88)80938-4. [DOI] [PubMed] [Google Scholar]
- 51.West IC. Radicals and oxidative stress in diabetes. Diabet Med. 2000;17:171–180. doi: 10.1046/j.1464-5491.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- 52.Weiland D, Mondon CE, Reaven GM. Evidence for multiple causality in the development of diabetic hypertriglyceridaemia. Diabetologia. 1980;18:335–340. doi: 10.1007/BF00251016. [DOI] [PubMed] [Google Scholar]
- 53.Okamoto T, Kanemoto N, Ohbuchi Y, Okano M, Fukui H, Sudo T. Characterization of STZ-induced Type 2 diabetes in Zucker fatty fats. Exp Anim. 2008;57:335–345. doi: 10.1538/expanim.57.335. [DOI] [PubMed] [Google Scholar]
- 54.Takada J, Machado MA, Peres SB, Brito LC, Borges-Silva CN, Costa CE, Fonseca-Alaniz MH, Andreotti S, Lima FB. Neonatal streptozotocin-induced diabetes mellitus: a model of insulin resistance associated with loss of adipose mass. Metabolism. 2007;56:977–984. doi: 10.1016/j.metabol.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 55.Comte C, Bellenger S, Bellenger J, Tessier C, Poisson JP, Narce M. Effects of streptozotocin and dietary fructose on delta-6 desaturation in spontaneously hypertensive rat liver. Biochimie. 2004;86:799–806. doi: 10.1016/j.biochi.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci. 1998;112:1199–1208. doi: 10.1037//0735-7044.112.5.1199. [DOI] [PubMed] [Google Scholar]
- 57.Grunblatt E, Koutsilieri E, Hoyer S, Riederer P. Gene expression alterations in brain areas of intracerebroventricular streptozotocin treated rat. J Alzheimers Dis. 2006;9:261–271. doi: 10.3233/jad-2006-9305. [DOI] [PubMed] [Google Scholar]
- 58.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 59.Magarinos AM, Jain K, Blount ED, Reagan L, Smith BH, McEwen BS. Peritoneal implantation of macroencapsulated porcine pancreatic islets in diabetic rats ameliorates severe hyperglycemia and prevents retraction and simplification of hippocampal dendrites. Brain Res. 2001;902:282–287. doi: 10.1016/s0006-8993(01)02400-3. [DOI] [PubMed] [Google Scholar]
- 60.de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- 61.Garcia-Galiano D, Sanchez-Garrido MA, Espejo I, Montero JL, Costan G, Marchal T, Membrives A, Gallardo-Valverde JM, Munoz-Castaneda JR, Arevalo E, De la Mata M, Muntane J. IL-6 and IGF-1 are independent prognostic factors of liver steatosis and non-alcoholic steatohepatitis in morbidly obese patients. Obes Surg. 2007;17:493–503. doi: 10.1007/s11695-007-9087-1. [DOI] [PubMed] [Google Scholar]
- 62.Gholam PM, Flancbaum L, Machan JT, Charney DA, Kotler DP. Nonalcoholic fatty liver disease in severely obese subjects. Am J Gastroenterol. 2007;102:399–408. doi: 10.1111/j.1572-0241.2006.01041.x. [DOI] [PubMed] [Google Scholar]
- 63.Liew PL, Lee WJ, Lee YC, Wang HH, Wang W, Lin YC. Hepatic histopathology of morbid obesity: concurrence of other forms of chronic liver disease. Obes Surg. 2006;16:1584–1593. doi: 10.1381/096089206779319392. [DOI] [PubMed] [Google Scholar]
- 64.Nobili V, Manco M. Measurement of advanced glycation end products may change NASH management. J Gastroenterol Hepatol. 2007;22:1354–1355. doi: 10.1111/j.1440-1746.2007.05019.x. [DOI] [PubMed] [Google Scholar]
- 65.Whitmer RA. Type 2 diabetes and risk of cognitive impairment and dementia. Curr Neurol Neurosci Rep. 2007;7:373–380. doi: 10.1007/s11910-007-0058-7. [DOI] [PubMed] [Google Scholar]
- 66.Haan MN, Wallace R. Can dementia be prevented? Brain aging in a population-based context. Annu Rev Public Health. 2004;25:1–24. doi: 10.1146/annurev.publhealth.25.101802.122951. [DOI] [PubMed] [Google Scholar]
- 67.Launer LJ. Diabetes and brain aging: epidemiologic evidence. Curr Diab Rep. 2005;5:59–63. doi: 10.1007/s11892-005-0069-1. [DOI] [PubMed] [Google Scholar]
- 68.Luchsinger JA, Reitz C, Patel B, Tang MX, Manly JJ, Mayeux R. Relation of diabetes to mild cognitive impairment. Arch Neurol. 2007;64:570–575. doi: 10.1001/archneur.64.4.570. [DOI] [PubMed] [Google Scholar]
- 69.Luchsinger JA, Tang MX, Mayeux R. Glycemic load and risk of Alzheimer's disease. J Nutr Health Aging. 2007;11:238–241. [PubMed] [Google Scholar]
- 70.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–1192. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 71.Whitmer RA, Gunderson EP, Quesenberry CP, Jr., Zhou J, Yaffe K. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr Alzheimer Res. 2007;4:103–109. doi: 10.2174/156720507780362047. [DOI] [PubMed] [Google Scholar]
- 72.Jeong SK, Nam HS, Son MH, Son EJ, Cho KH. Interactive effect of obesity indexes on cognition. Dement Geriatr Cogn Disord. 2005;19:91–96. doi: 10.1159/000082659. [DOI] [PubMed] [Google Scholar]
- 73.Geroldi C, Frisoni GB, Paolisso G, Bandinelli S, Lamponi M, Abbatecola AM, Zanetti O, Guralnik JM, Ferrucci L. Insulin resistance in cognitive impairment: the InCHIANTI study. Arch Neurol. 2005;62:1067–1072. doi: 10.1001/archneur.62.7.1067. [DOI] [PubMed] [Google Scholar]
- 74.Koulmanda M, Qipo A, Chebrolu S, O'Neil J, Auchincloss H, Smith RN. The effect of low versus high dose of streptozotocin in cynomolgus monkeys (Macaca fascilularis). Am J Transplant. 2003;3:267–272. doi: 10.1034/j.1600-6143.2003.00040.x. [DOI] [PubMed] [Google Scholar]