

Abstract
An established paradigm in current drug development is (i) to identify a single protein target whose inhibition is likely to result in the successful treatment of a disease of interest; (ii) to assay experimentally large libraries of small-molecule compounds in vitro and in vivo to identify promising inhibitors in model systems; and (iii) to determine whether the findings are extensible to humans. This complex process, which is largely based on trial and error, is risk-, time- and cost-intensive. Computational (virtual) screening of drug-like compounds simultaneously against the atomic structures of multiple protein targets, taking into account protein–inhibitor dynamics, might help to identify lead inhibitors more efficiently, particularly for complex drug-resistant diseases. Here we discuss the potential benefits of this approach, using HIV-1 and Plasmodium falciparum infections as examples. We propose a virtual drug discovery ‘pipeline’ that will not only identify lead inhibitors efficiently, but also help minimize side-effects and toxicity, thereby increasing the likelihood of successful therapies.
Promise for a new paradigm in drug discovery
Current therapeutic strategies for several diseases including human immunodeficiency virus type 1 (HIV-1) infection have evolved from an initial single-target treatment to a multitarget one [1]. Single antiretroviral drug regimens are no longer recommended for clinical use against HIV-1 owing to the rapid emergence of drug-resistant strains after initiation of therapy [2,3]. A combination of antiretroviral drugs (see Glossary) targeting different viral proteins is more effective at suppressing viral growth [4]. In many cases, however, these regimens are expensive and result in greater toxicity and in poor patient adherence [5–7]. New paradigms in multitarget drug discovery have emerged [8–11], particularly for the treatment of HIV-1 infection [12,13]. For example, the multitarget antiretroviral drug Cosalane has been developed to inhibit several HIV-1 proteins (gp120, integrase, protease and reverse transcriptase) simultaneously [14–19].
Computational screening of small-molecule compounds against protein targets implicated in a disease of interest has been widely used to discover potential inhibitors. This process typically involves identifying putative hits either by systematic chemical group perturbations to a compound already known to inhibit a target, as in quantitative structure–activity relationships (QSARs), or by ‘docking’ a molecule from a large database of compounds into the active site of the three dimensional (3D) structure of a protein target on the basis of the calculated binding affinity of the molecule to the target. As the number of high-resolution protein structures and computer processing capabilities have increased exponentially in recent years, so computational docking methods have been used to complement experimental high-throughput screening (HTS) methods to improve the efficiency and efficacy of discovering lead inhibitors. In addition, studies have shown that the success rates of HTS are increased several fold when compounds are pre-filtered by computational screening [20–22].
Here we describe a novel methodology with the capacity to catalyze drug discovery profoundly for all diseases. The opinion and evidence presented here are largely in the context of therapeutic targets in infectious disease; however, this computational multitarget approach can be readily extended to other complex human diseases such as cancer that require the inhibition of multiple proteins in a developmental pathway to be effective.
Summary and advantages of our computational paradigm
We have developed a new computational paradigm for the discovery of potential lead inhibitors that is based on a combination of three tenets (Figure 1) [23].
Figure 1.
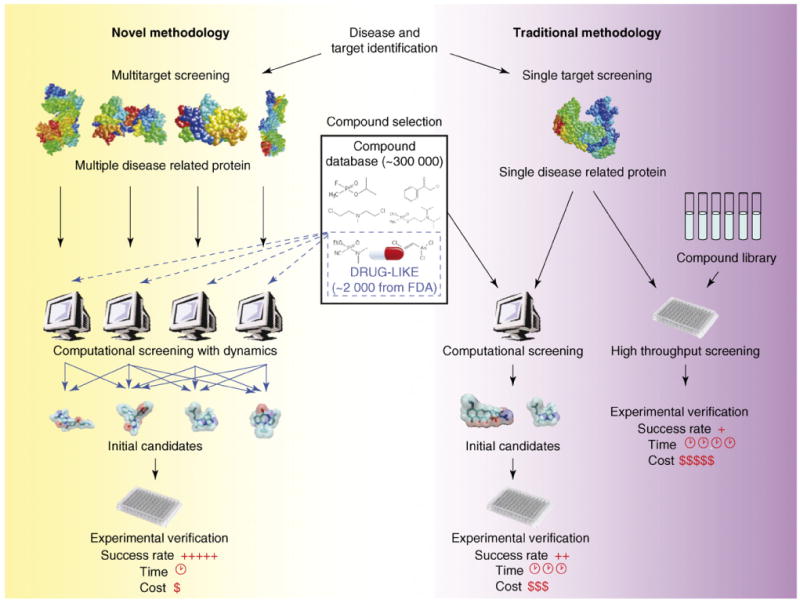
Comparison of our multitarget inhibitor discovery protocol with currently used traditional approaches. The advantages of using our novel broad-spectrum multitarget inhibitor discovery protocol (right) against key pathogens and diseases are contrasted with traditional approaches (left). The main differences in our protocol, corresponding to reasons why it is more effective, are as follows. (i) The use of a docking with molecular dynamics algorithm to take both protein and inhibitor flexibility completely into account (http://compbio.washington.edu/papers/therapeutics.html). This algorithm is effective because all molecules in biology undergo dynamic or thermal motion. Traditional rigid-docking approaches do not account for this motion, resulting in poor accuracy in predicting binding energies or inhibitory constants as compared with our approach. (ii) The use of compounds that bind to multiple targets simultaneously. The most effective drugs in humans (e.g. aspirin or Gleevec) inevitably interact with and bind to multiple proteins, a feature that traditional models based on single-target drugs fail to take into account. The multitarget approach is a necessary one because every drug has to be effective at its site of action (e.g. HIV-1 protease inhibitors have to bind and inhibit the protease molecule) and has to be readily metabolized by the body (e.g. the cytochrome P450 (CYP450) enzymes, which are responsible for metabolizing the majority of drugs) [54]. Computational screening for multitarget binding and inhibition is effective because it exploits the evolutionary fact that protein structure is conserved much more in nature than is function or sequence. (iii) The use of FDA-approved and experimental drug and drug-like compounds in the computational screening process. Screening drugs developed for other conditions against infectious diseases is likely to lead to fewer side-effects because the toxicity, absorption, distribution, metabolism and excretion pharmacokinetics is typically well established in human and in animal models. To our knowledge, this is the first time that these three elements have been combined to create an effective inhibitor and drug discovery protocol with predictions that have been experimentally verified to yield highly promising lead inhibitors for further drug development. The computational aspects of our protocol are fully automated, can be run completely in parallel and require only a fixed initial investment in the number of processors purchased (i.e. the greater the number of CPUs, the more targets and compounds that can be screened). Our novel protocol is extremely effective and increases success rates downstream in preclinical and clinical use with a considerable reduction in time, effort and cost expended.
Incorporation of protein side chains and main chain dynamics during the docking process to more accurately evaluate binding affinities.
Selection of single inhibitors that bind to multiple protein targets simultaneously.
Use of a screening library consisting of drug and drug-like compounds.
Each tenet increases the probability that a predicted compound will successfully inhibit the disease; furthermore, screening with drug-like compounds specifically increases pharmacological viability. Overall, this new paradigm produces hits that will more expediently and predictably become lead compounds that can potentially be developed further into viable drugs for all diseases (see Supplementary Table S1 online).
Rationale for the use of dynamics
Biologically active proteins are in continuous motion, yet the majority of protein structure information is limited to the most stable form of a protein when crystallized in artificial conditions. The differing conformations of bound and non-bound crystallographic structures suggest that binding events and protein motions induce variance in the dimensions and electrostatics of the catalytic site. It is likely that, in physiological conditions, an inhibitor will bind to one of these variant conformations with an affinity higher than that observed for the artificially stabilized structure. Thus, dynamics simulations increase the possibility of surveying a physiologically relevant conformation beyond using the static crystal structure alone.
We perform docking with dynamics by (i) docking the ligand of interest, (ii) solvating in a water and salt shell, (iii) applying 100 steps of energy minimization, (iv) simulating protein movement through cycles of random structure perturbations and (v) selecting the most relaxed of these models with a knowledge-based scoring function. We have demonstrated that this dynamic docking method is successful, as compared with static docking methods, in targets of two important pathogens, HIV-1 [24–28] and P. falciparum [23].
Rationale for multitargeting
Functional promiscuity of a given compound, coupled with structural conservation of active sites and/or binding pockets, enables activity of that compound in multiple proteins, as indicated by large regulatory systems such as ATP-facilitated energy transmission [29,30]. This same principle creates a common susceptibility of proteins involved in functions essential to life, creating a niche for multitargeting drugs. From a computational perspective, a compound that is predicted to inhibit multiple targets in a disease has an additive probability of having pharmacological activity against that disease. Most importantly, inhibitor resistance is largely overcome by the exponentially decreased probability of resistant mutations simultaneously arising in genes encoding proteins corresponding to all targets [31].
Rationale for use of drug-like compounds
Living organisms have evolved in comparable chemical environments containing similar sets of organic molecules. This shared evolutionary chemical context sets the stage for various organisms to use the same compounds to control different processes, making one molecule relevant to diverse physiological activity.
This principle is supported by the evolutionary observation that structure is much more conserved than sequence or function. Similar structures with comparable active and binding sites but different chemistries are used to perform a host of diverse functions [32,33]. This observation that structural folds are largely conserved, even when sequence and function are not, provides logical evidence that one compound can be an excellent initial candidate for many different protein targets.
Example applications of our computational paradigm
We can compare the efficacy of lead compound identification by our multitarget computational screening approach to the traditional experimental HTS and single-target screening approaches (Figure 1), by using HIV-1 and its associated opportunistic pathogen infections and the malarial parasite P. falciparum infection as examples. As we first describe in this section, several compounds show simultaneous effectiveness against HIV-1 and associated opportunistic pathogen infections and thus are potential multitarget drugs (Table 1). We then show how our computational multitarget screening approach can be used to discover effective inhibitors against the malarial parasite P. falciparum. We argue that our multitarget approach is likely to result in higher success rates, in reduced costs and time and in the identification of new lead inhibitors. Lastly, we describe how our proposed approach might also be used to minimize side-effects and toxicity, thereby reducing risk in the drug discovery pipeline and increasing the likelihood of developing successful therapies against diseases of interest.
Table 1. Compounds with inhibitory activity against HIV-1 and other microorganisms or diseasesa.
| Drug | HIV-1 | Other microorganisms or diseases | Refs | ||
|---|---|---|---|---|---|
|
|
|||||
| Target | Inhibitory effect (μM) | Refs | |||
| Amphotericin B | Gp41 | IC50 >10 | [55] | Aspergillus fumigatus | [56] |
| Blastomyces dermatitidis | [57] | ||||
| Candida genus | [56] | ||||
| Cryptococcus neoformans | [56] | ||||
| Fusarium species | [58] | ||||
| Hepatitis B virus | [59] | ||||
| Histoplasma capsulatum | [60] | ||||
| Chloroquine | Integrase | IC50 = 5.14 | [61] | Cryptococcus neoformans | [62] |
| Reverse transcriptase | IC50 >300 | [63] | Mycobacterium tuberculosis | [64] | |
| Tat | IC50 <50 | [65] | Plasmodium berghei | [66] | |
| Plasmodium falciparum | [67] | ||||
| Anti-rheumatoid arthritis | [68] | ||||
| Curcumin | Integrase | IC50 = 30 | [69] | Anti-Alzheimer | [70] |
| Reverse transcriptase | NA | [71] | Anti-cancer | [72] | |
| Tat | IC50 <30 | [65] | Anti-inflammation | [73] | |
| Cyclosporin A | gag | IC50 <1 | [74] | Candida albicans | [75] |
| Cryptococcus neoformans | [76] | ||||
| Cryptosporidium parvum | [77] | ||||
| Hepatitis C virus | [78] | ||||
| Toxoplasma gondii | [79] | ||||
| Vaccinia virus | [80] | ||||
| Durhamycin A | Tat | IC50 = 0.0048 | [81] | Aspergillus fumigatus | [82] |
| Candida albicans | [82] | ||||
| Cryptococcus neoformans | [82] | ||||
| Histoplasma capsulatum | [82] | ||||
| Enviroxime | Unknown | EC50 >36.5 | [83] | Coxsackie virus | [84] |
| Human rhinovirus | [85] | ||||
| Polio virus | [86] | ||||
| Fumagillin | Vpr | EC50 = 0.015 | [87] | Encephalitozoon cuniculi | [88] |
| Unknown | EC50 >0.2 | [83] | Encephalitozoon intestinalis | [89] | |
| Enterocytozoon bieneusi | [90] | ||||
| Plasmodium falciparum | [91] | ||||
| Vittaforma corneae | [89] | ||||
| Hydroxychloroquine | Integrase | IC50 >100 | [61] | Plasmodium falciparum | [92] |
| Reverse transcriptase | NA | [93] | |||
| KNI-764 | Protease | IC50 >0.05 | [94] | Plasmodium malariae | [36] |
| Minocycline | Reverse transcriptase | IC50 = 1200 | [37] | Cryptosporidium parvum | [95] |
| Unknown | EC50 <22 | [38] | Enterococcus faecalis | [96] | |
| Enterococcus faecium | [96] | ||||
| Mycobacterium fortuitum | [97] | ||||
| Mycobacterium tuberculosis | [98] | ||||
| Mycoplasma pneumoniae | [99] | ||||
| Staphylococcus aureus | [96] | ||||
| Staphylococcus pyogenes | [96] | ||||
| Streptococcus pneumoniae | [96] | ||||
| Toxoplasma gondii | [100] | ||||
| Suramin | gp120 | ED50 = 7.7 | [101] | Cytomegalovirus | [102] |
| Integrase | IC50 = 2.4 | [103] | Herpes simplex virus | [104] | |
| Reverse transcriptase | IC50 = 1.4 | [105] | Influenza A virus | [106] | |
| Rhinovirus | [83] | ||||
| Sandfly fever virus | [107] | ||||
Abbreviations: EC50, effective concentration for half-maximum response; ED50, half-maximal effective dose; IC50, half-maximal inhibitory concentration.
The data show that one compound can target multiple diseases, including several AIDS-related opportunistic pathogens, simultaneously.
Diseases caused by multiple microorganisms: HIV-1 and AIDS-related opportunistic infections
Traditionally, the treatment of complex diseases such as acquired immune deficiency syndrome (AIDS) that involve several microorganisms, especially those with a high mutation rate, requires a therapeutic regimen based on several drugs, wherein each drug inhibits a single target in a particular microorganism. Multidrug regimens have been successfully used in several studies to treat complex diseases and to control the emergence of drug-resistant strains of infectious agents such as HIV-1 [1,4]. The use of several drugs in treatment regimens, however, typically causes serious adverse effects and is associated with low patient adherence owing to toxicity and high costs [5–7].
HIV-1, first discovered in 1981, is a pandemic human pathogen that has resulted in more than 25 million deaths caused by AIDS, in which the immune system ceases to function, leading to life-threatening opportunistic infections. Individuals infected with HIV-1 need a regimen consisting of drugs to treat both HIV-1 and opportunistic infections that arise because of immunosuppression. These individuals thus present a therapeutic challenge for which multitarget computational screening might provide an effective solution, because a regimen consisting of a single drug that could simultaneously inhibit protein targets from multiple microorganisms would be ideal for treatment and control of the complex combinations of infectious diseases present in these individuals.
HIV-1 infection is commonly co-morbid with opportunistic infection by bacterial, fungal, protozomal and viral pathogens. Pharmacological prophylaxis is available for several of these pathogens [34]. Cotrimoxazole is a broad-spectrum antibiotic that is effective at preventing many opportunistic infections. This drug is both cheap and widely available [35]; however, it does not inhibit HIV-1 replication. Because HIV-1 infection is a chronic disease that requires life-long antiretroviral treatment, a new generation of antiretroviral drugs that simultaneously control HIV-1 and opportunistic pathogens would benefit HIV-1-infected individuals, especially those with limited access to antiretroviral and prophylactic drugs.
Several drugs approved for the treatment of human diseases other than HIV-1 infection, in addition to other drug-like compounds, have been shown to inhibit HIV-1 protein targets either in vitro or in vivo (Table 1). These compounds include drugs developed primarily to treat Alzheimer's disease, cancer and infectious diseases caused by bacteria, fungi, protozoa and viruses. The multitargeting features of these compounds against HIV-1 and its opportunistic pathogens have been largely identified by HTS through serendipity. Computational multitarget screening using the X-ray diffraction structures of HIV-1 protein targets present in the Protein Data Bank (PDB; http://www.pdb.org), however, would have helped to facilitate the rational identification of these multitarget drugs.
The data in Table 1 provide evidence that one or more compounds can inhibit infection by multiple bacteria, fungi, protozoa and viruses, including HIV-1, simultaneously. A striking example is the inhibitor KNI-764 (also known as JE-2,164) (Table 1, row 36), which inhibits both HIV-1 protease and plasmepsin II enzyme from the malarial parasite Plasmodium malariae (Table 1) [36]. Complexes of both of these targets with KNI-764 have been solved by X-ray diffraction (PDB identifiers 1msm and 2anl, respectively); thus, this inhibitor provides strong evidence for the existence and utility of multitarget drugs, because the binding mode of a single inhibitor bound to targets from two very different and destructive pathogens has been elucidated to atomic detail. Another example is minocycline (Table 1, row 37), a broad-spectrum antibiotic that has been shown to possess inhibitory activity against HIV-1 in vitro (Table 1) [37,38]. Our docking simulations predict that it inhibits HIV-1 integrase [28], thereby illustrating how computational screening methods can be used to identify targets and binding modes of multitarget inhibitors discovered fortuitously.
Diseases caused by a single microorganism: the malarial parasite P. falciparum
We previously screened a library of 2344 drug and drug-like compounds against 14 proteins of the malarial parasite P. falciparum [23] (see Figure I in Box 1), by using a computational docking with dynamics protocol that predicts inhibitors of target protein structures by simultaneously considering protein–inhibitor flexibility and dynamics [24,26]. We have subsequently evaluated experimentally 16 of the top ranking multitarget predictions for P. falciparum growth inhibition, and five compounds predicted to have no inhibitory activity were used as negative predictions. Six (38%) of the sixteen top predictions had a half-maximal effective dose (ED50) of ≤1 μM against either the chloroquine-sensitive or the chloroquine-resistant strain of P. falciparum (see Figure Ia in Box 1). None of the five negative prediction compounds inhibited P. falciparum growth at the desired level, producing an overall prediction accuracy of 52% (11/21; see Table S1a online).
Two studies using structure-based single-target computational screening of different libraries of compounds against two P. falciparum cysteine proteases (falcipain-2 and falcipain-3) have shown low success rates [39,40]. Out of 355 000 compounds in the Available Chemical Directory (ACD) database, one study computationally predicted 100 putative inhibitors, of which 1 demonstrated an experimental antimalarial activity of ≤10 μM in vitro [39]. The second experiment carried out on the same targets with 241 000 compounds from the ChemBridge database predicted 84 putative inhibitors, of which 4 demonstrated an antimalarial activity of ≤10 μM [39]. The overall success of this screening method therefore yields a hit rate of 0.0008% (5/596 000), as compared with a hit rate of 0.3% (6/2344) at ≤1 μM activity by computational docking with dynamics screening of drug-like compounds [23]. Although 36 compounds in total showed an activity of ≤50 μM against the predicted target proteins, only 5 worked against P. falciparum, clearly demonstrating the benefit of starting with compounds with known biological activity: namely, these compounds are more likely to find their way to the site of biological interference.
Two recent experimental HTS studies demonstrate the increased success of our third tenet: that is, screening with drug-like compounds [41,42]. However, their experimental success rates of 0.7–1.7% for identifying inhibitors of P. falciparum growth points to the advantages of our other tenets: namely, multitargeting and docking with dynamics. In the first study, 2687 existing drugs were screened for P. falciparum growth inhibition and 19 novel antimalarial compounds with an activity of ≤1 μM were discovered, yielding a success rate of 0.7%, although five of these compounds were found to show an activity of ≤100 nM [41]. In the second study, 2160 compounds were screened and 36 novel antimalarial compounds with an activity of ≤1 μM were found, giving a success rate of 1.7% [42]. Thus, the experimental success rate (38%) of our multitarget screening approach represents a significant improvement over the previous success rates of single-target computational screening (2.7%) [39,40] and HTS (0.7–1.7%) [41,42].
Multitarget computational screening can also be applied to predict the potential targets of an inhibitor identified by HTS but with an unknown mechanism of action. This application is illustrated in Figure Ib in Box 1, which shows predicted targets for 12 experimental hits from the two HTS studies [28,41,42]. By our computational protocols, these hits are predicted to inhibit multiple P. falciparum targets and generally have low consensus weighted ranks. These predictions reveal a putative multitargeting function for 12 of the best inhibitors found by the HTS studies.
The high success rates of experiments guided by multitarget computational screening (38%; Table S1a) can be coupled with HTS screens to select the compounds to be followed up in the subsequent time-consuming and costly further characterization (54%; Table S1b). With our protocol, a smaller number of experiments will produce better hits at a fraction of the time, effort and cost that would have been required to follow protocols based on experimental HTS and subsequent characterization.
Toxicity and side-effect minimization
Although a multitarget inhibitor is expected to bind to multiple disease protein targets with high affinity, it might undesirably inhibit other human proteins, leading to toxicity (see above sections on rationales). Strategies to identify and to predict side-effects such as acute toxicity, mutagenicity and carcinogenicity have been extensively studied and reviewed [43–49].
In terms of computational screening, a library of either approved drug and drug-like compounds that have been evaluated in clinical trials or compounds with known toxicity profiles can be used to identify initial lead inhibitors, thereby reducing the likelihood of deleterious side-effects. Additional compounds could be selected from larger libraries containing synthetic and natural compounds if the whole library is filtered and categorized into groups according to their onset and severity of toxicity. The latter can be accomplished by using data in the TOXNET database (http://toxnet.nlm.nih.gov) [50] or by examining the absorption, distribution, metabolism and elimination profiles of the compounds [51]. Focusing on infectious disease targets that are not similar to essential proteins in humans also reduces the likelihood of a toxic reaction.
Toxicity filtering can also be done by structural similarity comparison or by a SMILES strings similarity search [52] between successful lead candidates and compounds with known toxicity profiles. The purpose of categorizing compounds is to prioritize the experimental verification of the computational screening results for a given set of targets or diseases. Compounds with moderate toxicity could be included in our screening library for diseases that require short courses of treatment. By contrast, the same compounds might be eliminated from our library for chronic diseases.
Potential side-effects can also be predicted by using computational multitarget screening to screen lead inhibitors against essential human proteins with known structure. Lead inhibitors can also be screened against proteins involved in human drug metabolism (such as the cytochrome P450 family of enzymes) to ensure their proper metabolism and to minimize the risk of producing toxic metabolites.
Comparison of our computational paradigm with traditional pharmaceutical methods
Although some screens using the latest technologies can screen up to 50 000 compounds per day, the blind pharmaceutical approach has severe disadvantages compared to computational ones. Screening compounds efficiently against either the target or the organism necessitates the design of a specific assay, whereas our computational protocol is completely general and applicable to any target with an experimentally determined structure or with a close homolog (>50% sequence identity) of known structure. Distant homologs can also be used, but docking accuracy decreases as the sequence similarity decreases. Furthermore, the pharmaceutical screens do not identify the targets or binding mode, so any optimization is typically done in a blind manner. Lastly, large-scale HTS screens create monumental environmental waste, a problem that is mostly avoidable in computational multitarget screening.
QSARs correlate chemical structure similarity with biological activity. This type of approach finds compounds related to those of known function through pattern recognition of structure and functional groups. Variation in the catalytic site can limit functional analogy for even tiny chemical changes made in such pharmacophore-based screening. Docking simulations such as INVDOCK [53] resolve these high-resolution challenges by comparing the electrochemical topography of ligand and target, thereby avoiding the necessity of starting with a known active compound and raising the possibility of finding completely new families of drugs for a disease. Moreover, our computational docking with dynamics approach assesses this relationship in physiologically relevant movements, leading to higher experimental success. The current limitations of this method are thought to be the scoring functions and protein dynamics movements; however, our scoring functions and dynamics simulations do increase success rates. Overall, our method can be used either to discover completely new hits, or in conjunction with pharmacophore methods such as QSAR to modify a compound computationally in accordance with medicinal chemistry rules and to assess immediately the affinity of the newly designed compound, leading to improved multitarget selectivity.
In terms of optimization of processing speed, the first protocol that we developed is rather naive, whereas our second-generation protocol is capable of screening hundreds of thousands of existing compounds, or designing new ones computationally on the basis of medicinal chemistry rules, on a single central processing unit (CPU) in a single day. Nonetheless, our current computer cluster can be arbitrarily scaled to include a larger number of CPUs and to screen within six hours up to a million compounds with the first-generation version of our protocol because it can be run completely in parallel. The cost of such an installation would be only a few hundred thousand dollars initially, and it has the advantages of being completely general (i.e. applicable to any protein target of interest) and easily maintained (our ‘farm’ is maintained with <25% of the time of a single staff member, minimal parts replacements and no cost for screened compounds or reagents).
Conclusion and future directions
Multitarget computational screening using a docking with dynamics protocol and a drug-like compound library has the promise to enhance significantly the identification of lead inhibitors for drug development. This protocol can identify inhibitors that simultaneously and selectively bind to multiple targets with high affinity, in contrast to most drug development strategies that focus on only single-target inhibition. The efficacy and efficiency of multitarget computational screening have the potential to reduce time, effort and cost considerably to obtain promising lead candidates for drug development.
We have provided evidence that multitarget inhibitors exist for complex diseases, including AIDS that involve several microorganisms such as HIV-1 and associated opportunistic pathogen infections, and that these lead compounds are excellent starting points for further chemical modification to improve potency and specificity against targets of interest. We have also demonstrated that computationally predicted multitarget antimalarial inhibitors show high potency at inhibiting P. falciparum growth in vitro with a success rate higher than that of single-target computational screening and experimental HTS. The onset of drug resistance, a considerable problem in both HIV-1 and P. falciparum infection, might be significantly delayed by inhibiting multiple targets simultaneously.
Box 1. Multitargeting predictions.
We computationally evaluated the ability of 2344 compounds from a library of known physiologically active compounds to inhibit a multitargeting combination of 14 P. falciparum proteins, using a computational docking with dynamics protocol simultaneously considering protein–inhibitor flexibility and dynamics [23]. Thirteen of the potential drug targets were selected because of their known identity as enzymes necessary for the P. falciparum life cycle and because high-quality structures were available for these proteins. Another available P. falciparum protein structure, erythrocyte-binding antigen, was chosen for its known role in pathogenesis.
The screened compounds were ranked according to the consensus weighted rank (the average of the ranks of the compound observed in all simulations divided by the number of proteins predicted to be inhibited by that compound; the lower the rank, the better the predicted efficacy), which is a measure of the multitargeting capability of a compound.
We experimentally evaluated the 16 top ranking compounds from these predictions, along with five compounds predicted to have no inhibitory activity as negative predictions. Experimental verification was performed against the chloroquine-sensitive strain 3D7 and the chloroquine-resistant strain K1 of P. falciparum cultures. Six of the sixteen top predictions had an ED50 of ≤1 μM activity against either the 3D7 or K1 strain (Figure Ia).
By experimentally screening only 16 predictions from a computational library of 2344 compounds [23], six lead candidates with submicromolar antimalarial activity were obtained at a fraction of the time, effort and cost that would have been required to perform experimental HTS. Overall, the experimental success rate of ∼38% for the multitarget computational screening is significantly higher than the rates of 0.7–1.7% produced by the two experimental HTS studies for identifying antimalarial inhibitors [41,42].
In addition, we compared the multitarget computational screening predictions to the two experimental HTS studies to discover potential targets of P. falciparum growth inhibitors [41,42]. Twelve of the compounds for which experimental inhibition values were provided and were considered by the HTS screening studies to be valid antimalarial hits are predicted to inhibit multiple proteins (Figure Ib). This application of multitarget computational screening is therefore useful in prioritizing targets for further study, for compounds with unknown inhibitory mechanisms.
Figure I.
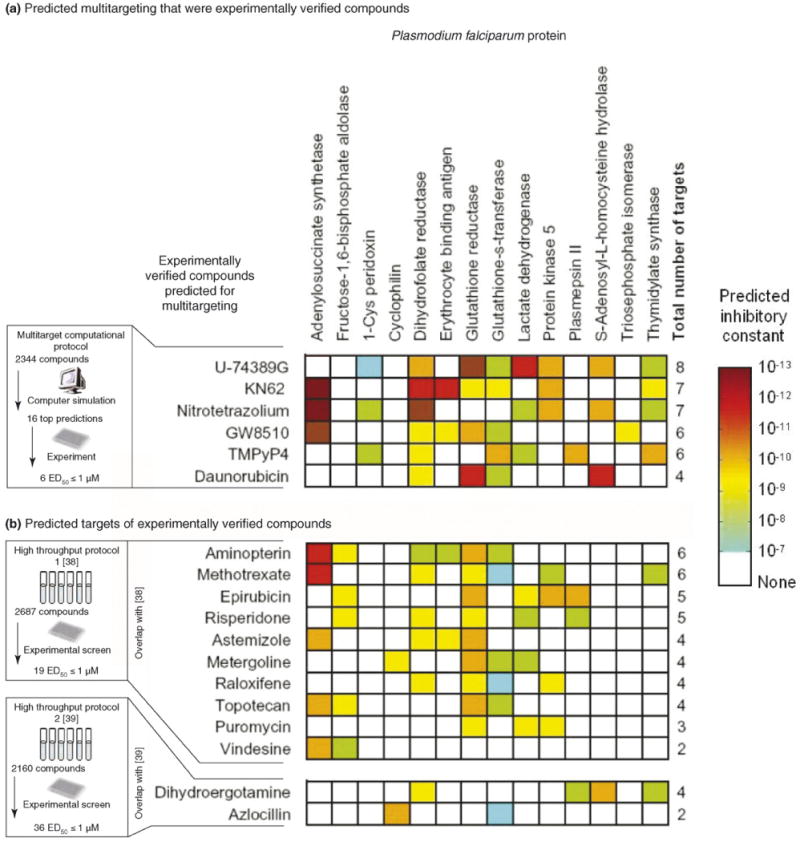
Multitargeting predictions for 18 antimalarial compounds. (a) Six compounds predicted for multitargeting by our computational screening study [23] that were subsequently verified for experimental antimalarial activity (see supplementary material). (b) Twelve compounds selected for antimalarial activity by two HTS studies were subsequently predicted computationally as multitargeting [41,42]. Shown for each compound are their predicted inhibitory constants against each of 14 P. falciparum proteins (shaded boxes; dark brown indicates highest inhibition) and the total number of proteins predicted to be inhibited. Some proteins have inhibitors in the mid-picomolar range (e.g. Glutathione reductase) but others have predicted inhibitors in the micromolar range (e.g. 1-Cys peridoxin). Our predictions indicate that a compound such as U-74389G is more likely to inhibit Glutathione reductase and Lactate dehydrogenase (all picomolar inhibitory constants) than 1-Cys peridoxin, Dihydrofolate reductase, Glutathione-s-transferase, Protein kinase-5, S-Adenosyl-L- homocysteine hydrolase or Thymidylate synthase (micromolar to nanomolar inhibitory constants). Predicted inhibitory constants from docking with dynamics simulations reflect hypothetical local concentrations, such that a given compound will inhibit its target at the predicted concentration in a simple in vitro experiment, but the whole organism might require different concentrations for toxicity.
An important application of multitarget computational screening is in identification of the potential targets of a drug with an unknown inhibitory mechanism. Starting with drug and drug-like compounds that are well characterized in terms of their pharmacological properties will increase the probability that an identified lead will be successful as a drug further down the development pipeline. Modification of lead compounds using medicinal chemistry rules can be performed computationally. Screening for side-effects against essential human proteins (a chief focus in structure determination; many such structures are available) can also be performed computationally to refine potential candidates, and screening against important human enzymes involved in eliminating drugs from the body can help to ensure proper metabolism without a build-up of toxic metabolites.
Developing a comprehensive computational pipeline that integrates the concepts described here not only will lead to the discovery of new inhibitors but also has the potential to facilitate significant advances in the efficacy and efficiency of the whole process of drug discovery and development, from in vitro and in vivo preclinial studies to clinical trials.
Supplementary Material
Acknowledgments
This work was partly support by NIH grants GM068152 and F30DE017522, NSF grant DBI-0217241, a NSF CAREER award, a Searle Scholar Award, and the Puget Sound Partners in Global Health. The authors would like to thank Brady Bernard, Roger Bumgarner, Michael Lagunoff, Renee Ireton and members of the Samudrala group for valuable discussions and comments.
Glossary
- Drug
a compound with Food and Drug Administration (FDA) approval for human use.
- Drug-like compound
a compound (including research and experimental drugs) that has been shown to have physiological activity in at least non-human in vivo systems.
- Hit
a compound that inhibits (or has high binding affinity for) one or more targets. In our case, the hits are initially virtual – that is, computationally derived.
- Lead
a hit that has been well-characterized experimentally. For example, one that has been shown to have a high dissociation constant (Kd) for the target of interest such that the functional activities of the target are decreased on binding and/or has demonstrated effectiveness of treatment against disease in an animal model.
- Potential lead
a computationally predicted hit that has been shown to work experimentally against in vitro (cell culture) disease models of the organism.
Footnotes
Disclosure: A U.S. patent invention comprising the protocol described in Figure 1 has been filed with the U.S. Patent and Trademark Office. The University of Washington, being the legal owner of this invention, will own all rights to this invention depending on the claims granted. The patent application has been filed but not approved and no decision has been made at the time of publication. All research was performed independently of the University of Washington's patent application, without any related bias on the part of the authors or inventors involved in this article.
Supplementary data: Supplementary data associated with this article can be found, in the online version, at 10.1016/j.tips.2007.11.007.
References
- 1.Hammer SM, et al. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society-U. S. A. panel. J Am Med Assoc. 2006;296:827–843. [PubMed] [Google Scholar]
- 2.Volberding PA, et al. A comparison of immediate with deferred zidovudine therapy for asymptomatic HIV-infected adults with CD4 cell counts of 500 or more per cubic millimeter. AIDS Clinical Trials Group. N Engl J Med. 1995;333:401–407. doi: 10.1056/NEJM199508173330701. [DOI] [PubMed] [Google Scholar]
- 3.Rusconi S, et al. Viral load, viral phenotype modification, zidovudine susceptibility and reverse transcriptase mutations during the first 6 months of zidovudine monotherapy in HIV-1-infected people. Antivir Ther. 1996;1:211–219. [PubMed] [Google Scholar]
- 4.Metzner KJ, et al. Rapid selection of drug-resistant HIV-1 during the first months of suppressive ART in treatment-naive patients. AIDS. 2007;21:703–711. doi: 10.1097/QAD.0b013e3280121ac6. [DOI] [PubMed] [Google Scholar]
- 5.Jones R, Gazzard B. The cost of antiretroviral drugs and influence on prescribing policies. Int J STD AIDS. 2006;17:499–506. doi: 10.1258/095646206778145587. [DOI] [PubMed] [Google Scholar]
- 6.Bisson GP, et al. Out-of-pocket costs of HAART limit HIV treatment responses in Botswana's private sector. AIDS. 2006;20:1333–1336. doi: 10.1097/01.aids.0000232245.36039.2b. [DOI] [PubMed] [Google Scholar]
- 7.Moya J, et al. Limitations of a simplification antiretroviral strategy for HIV-infected patients with decreasing adherence to a protease inhibitor regimen. HIV Clin Trials. 2006;7:210–214. doi: 10.1310/hct0704-210. [DOI] [PubMed] [Google Scholar]
- 8.Faivre S, et al. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 9.Millan MJ. The role of monoamines in the actions of established and “novel” antidepressant agents: a critical review. Eur J Pharmacol. 2004;500:371–384. doi: 10.1016/j.ejphar.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 10.Wong ML, et al. Identification, characterization, and gene expression profiling of endotoxin-induced myocarditis. Proc Natl Acad Sci U S A. 2003;100:14241–14246. doi: 10.1073/pnas.2336220100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Celotti F, Laufer S. Anti-inflammatory drugs: new multitarget compounds to face an old problem. The dual inhibition concept. Pharmacol Res. 2001;43:429–436. doi: 10.1006/phrs.2000.0784. [DOI] [PubMed] [Google Scholar]
- 12.Bugatti A, et al. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of HIV-1 Tat and gp120 proteins. Antimicrob Agents Chemother. 2007;51:2337–2345. doi: 10.1128/AAC.01362-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Litovchick A, et al. Neomycin B-arginine conjugate, a novel HIV-1 Tat antagonist: synthesis and anti-HIV activities. Biochemistry. 2001;40:15612–15623. doi: 10.1021/bi0108655. [DOI] [PubMed] [Google Scholar]
- 14.Santhosh KC, et al. Correlation of anti-HIV activity with anion spacing in a series of cosalane analogues with extended polycarboxylate pharmacophores. J Med Chem. 2001;44:703–714. doi: 10.1021/jm000290u. [DOI] [PubMed] [Google Scholar]
- 15.Kuchimanchi KR, et al. Binding of cosalane–a novel highly lipophilic anti-HIV agent–to albumin and glycoprotein. J Pharm Sci. 2001;90:659–666. doi: 10.1002/1520-6017(200105)90:5<659::aid-jps1022>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 16.De Clercq E. New developments in anti-HIV chemotherapy. Curr Med Chem. 2001;8:1543–1572. doi: 10.2174/0929867013371842. [DOI] [PubMed] [Google Scholar]
- 17.Casimiro-Garcia A, et al. Synthesis and anti-HIV activity of cosalane analogues incorporating nitrogen in the linker chain. Bioorg Med Chem. 2000;8:191–200. doi: 10.1016/s0968-0896(99)00269-2. [DOI] [PubMed] [Google Scholar]
- 18.Cushman M, et al. Cosalane analogues with enhanced potencies as inhibitors of HIV-1 protease and integrase. J Med Chem. 1995;38:443–452. doi: 10.1021/jm00003a007. [DOI] [PubMed] [Google Scholar]
- 19.Cushman M, et al. Design, synthesis, and biological evaluation of cosalane, a novel anti-HIV agent which inhibits multiple features of virus reproduction. J Med Chem. 1994;37:3040–3050. doi: 10.1021/jm00045a008. [DOI] [PubMed] [Google Scholar]
- 20.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins JL, et al. Virtual screening to enrich hit lists from high-throughput screening: a case study on small-molecule inhibitors of angiogenin. Proteins. 2003;50:81–93. doi: 10.1002/prot.10270. [DOI] [PubMed] [Google Scholar]
- 22.Mestres J. Virtual screening: a real screening complement to high-throughput screening. Biochem Soc Trans. 2002;30:797–799. doi: 10.1042/bst0300797. [DOI] [PubMed] [Google Scholar]
- 23.Jenwitheesuk E, Samudrala R. Identification of potential multitarget antimalarial drugs. J Am Med Assoc. 2005;294:1490–1491. doi: 10.1001/jama.294.12.1490. [DOI] [PubMed] [Google Scholar]
- 24.Jenwitheesuk E, Samudrala R. Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations. BMC Struct Biol. 2003;3:2. doi: 10.1186/1472-6807-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenwitheesuk E, et al. Improved accuracy of HIV-1 genotypic susceptibility interpretation using a consensus approach. AIDS. 2004;18:1858–1859. doi: 10.1097/00002030-200409030-00020. [DOI] [PubMed] [Google Scholar]
- 26.Jenwitheesuk E, Samudrala R. Prediction of HIV-1 protease inhibitor resistance using a protein-inhibitor flexible docking approach. Antivir Ther. 2005;10:157–166. [PubMed] [Google Scholar]
- 27.Jenwitheesuk E, et al. PIRSpred: a web server for reliable HIV-1 protein-inhibitor resistance/susceptibility prediction. Trends Microbiol. 2005;13:150–151. doi: 10.1016/j.tim.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Jenwitheesuk E, Samudrala R. Identification of potential HIV-1 targets of minocycline. Bioinformatics. 2007;23:2797–2799. doi: 10.1093/bioinformatics/btm424. [DOI] [PubMed] [Google Scholar]
- 29.Rao S, Rossmann M. Comparison of super-secondary structures in proteins. J Mol Biol. 1973;76:241–256. doi: 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- 30.Scheeff E, Bourne P. Structural evolution of the protein kinase-like superfamily. PLoS Comput Biol. 2005;1:e49. doi: 10.1371/journal.pcbi.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hopkins AL, et al. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16:127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–198. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 33.Hubbard TJ, et al. SCOP: a structural classification of proteins database. Nucleic Acids Res. 1997;25:236–239. doi: 10.1093/nar/25.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovacs JA, Masur H. Prophylaxis against opportunistic infections in patients with human immunodeficiency virus infection. N Engl J Med. 2000;342:1416–1429. doi: 10.1056/NEJM200005113421907. [DOI] [PubMed] [Google Scholar]
- 35.Pitter C, et al. Cost-effectiveness of cotrimoxazole prophylaxis among persons with HIV in Uganda. J Acquir Immune Defic Syndr. 2007;44:336–343. doi: 10.1097/QAI.0b013e31802f12b5. [DOI] [PubMed] [Google Scholar]
- 36.Clemente JC, et al. Structure of the aspartic protease plasmepsin 4 from the malarial parasite Plasmodium malariae bound to an allophenylnorstatine-based inhibitor. Acta Crystallogr D Biol Crystallogr. 2006;62:246–252. doi: 10.1107/S0907444905041260. [DOI] [PubMed] [Google Scholar]
- 37.Wondrak EM, et al. Inhibition of HIV-1 RNA-dependent DNA polymerase and cellular DNA polymerases alpha, beta and gamma by phosphonoformic acid and other drugs. J Antimicrob Chemother. 1988;21:151–161. doi: 10.1093/jac/21.2.151. [DOI] [PubMed] [Google Scholar]
- 38.Lemaitre M, et al. Protective activity of tetracycline analogs against the cytopathic effect of the human immunodeficiency viruses in CEM cells. Res Virol. 1990;141:5–16. doi: 10.1016/0923-2516(90)90052-k. [DOI] [PubMed] [Google Scholar]
- 39.Desai PV, et al. Identification of novel parasitic cysteine protease inhibitors by use of virtual screening. 2. The available chemical directory. J Med Chem. 2006;49:1576–1584. doi: 10.1021/jm0505765. [DOI] [PubMed] [Google Scholar]
- 40.Desai PV, et al. Identification of novel parasitic cysteine protease inhibitors using virtual screening. 1. The ChemBridge database. J Med Chem. 2004;47:6609–6615. doi: 10.1021/jm0493717. [DOI] [PubMed] [Google Scholar]
- 41.Chong CR, et al. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2:415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- 42.Weisman JL, et al. Searching for new antimalarial therapeutics amongst known drugs. Chem Biol Drug Des. 2006;67:409–416. doi: 10.1111/j.1747-0285.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barratt MD. Prediction of toxicity from chemical structure. Cell Biol Toxicol. 2000;16:1–13. doi: 10.1023/a:1007676602908. [DOI] [PubMed] [Google Scholar]
- 44.Benigni R, Zito R. Designing safer drugs: (Q)SAR-based identification of mutagens and carcinogens. Curr Top Med Chem. 2003;3:1289–1300. doi: 10.2174/1568026033452023. [DOI] [PubMed] [Google Scholar]
- 45.Dearden JC. In silico prediction of drug toxicity. J Comput Aided Mol Des. 2003;17:119–127. doi: 10.1023/a:1025361621494. [DOI] [PubMed] [Google Scholar]
- 46.Greene N. Computer systems for the prediction of toxicity: an update. Adv Drug Deliv Rev. 2002;54:417–431. doi: 10.1016/s0169-409x(02)00012-1. [DOI] [PubMed] [Google Scholar]
- 47.Roche O, Guba W. Computational chemistry as an integral component of lead generation. Mini Rev Med Chem. 2005;5:677–683. doi: 10.2174/1389557054368826. [DOI] [PubMed] [Google Scholar]
- 48.Swamidass SJ, et al. Kernels for small molecules and the prediction of mutagenicity, toxicity and anti-cancer activity. Bioinformatics. 2005;21(Suppl 1):i359–i368. doi: 10.1093/bioinformatics/bti1055. [DOI] [PubMed] [Google Scholar]
- 49.Zmuidinavicius D, et al. Progress in toxinformatics: the challenge of predicting acute toxicity. Curr Top Med Chem. 2003;3:1301–1314. doi: 10.2174/1568026033451989. [DOI] [PubMed] [Google Scholar]
- 50.Wexler P. TOXNET: the National Library of Medicine's toxicology database. Am Fam Physician. 1995;52:1677–1678. [PubMed] [Google Scholar]
- 51.Li AP. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today. 2001;6:357–366. doi: 10.1016/s1359-6446(01)01712-3. [DOI] [PubMed] [Google Scholar]
- 52.Vidal D, et al. A novel search engine for virtual screening of very large databases. J Chem Inf Model. 2006;46:836–843. doi: 10.1021/ci050458q. [DOI] [PubMed] [Google Scholar]
- 53.Chen YZ, Ung CY. Computer automated prediction of potential therapeutic and toxicity protein targets of bioactive compounds from Chinese medicinal plants. Am J Chin Med. 2002;30:139–154. doi: 10.1142/S0192415X02000156. [DOI] [PubMed] [Google Scholar]
- 54.Al Omari A, Murry DJ. Pharmacogenetics of the cytochrome P450 enzyme system: review of current knowledge and clinical significance. J Pharm Pract. 2007;20:206–218. [Google Scholar]
- 55.Pereira FB, et al. Membrane fusion induced by the HIV type 1 fusion peptide: modulation by factors affecting glycoprotein 41 activity and potential anti-HIV compounds. AIDS Res Hum Retroviruses. 1997;13:1203–1211. doi: 10.1089/aid.1997.13.1203. [DOI] [PubMed] [Google Scholar]
- 56.Oki T, et al. Pradimicins A, B and C: new antifungal antibiotics. II. In vitro and in vivo biological activities. J Antibiot (Tokyo) 1990;43:763–770. doi: 10.7164/antibiotics.43.763. [DOI] [PubMed] [Google Scholar]
- 57.Utz JP. Systemic fungal infections amenable to chemotherapy. Dis Mon. 1963;35:1–52. doi: 10.1016/s0011-5029(63)80008-5. [DOI] [PubMed] [Google Scholar]
- 58.Tzatzarakis MN, et al. Comparison of in vitro activities of amphotericin, clotrimazole, econazole, miconazole, and nystatin against Fusarium oxysporum. J Environ Sci Health B. 2001;36:331–340. doi: 10.1081/PFC-100103573. [DOI] [PubMed] [Google Scholar]
- 59.Kessler HA, et al. Effects of amphotericin B on hepatitis B virus. Antimicrob Agents Chemother. 1981;20:826–833. doi: 10.1128/aac.20.6.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kohler S, et al. Comparison of the echinocandin caspofungin with amphotericin B for treatment of histoplasmosis following pulmonary challenge in a murine model. Antimicrob Agents Chemother. 2000;44:1850–1854. doi: 10.1128/aac.44.7.1850-1854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fesen MR, et al. Inhibitors of human immunodeficiency virus integrase. Proc Natl Acad Sci U S A. 1993;90:2399–2403. doi: 10.1073/pnas.90.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levitz SM, et al. Chloroquine induces human mononuclear phagocytes to inhibit and kill Cryptococcus neoformans by a mechanism independent of iron deprivation. J Clin Invest. 1997;100:1640–1646. doi: 10.1172/JCI119688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai WP, et al. Inhibition of human immunodeficiency virus infectivity by chloroquine. AIDS Res Hum Retroviruses. 1990;6:481–489. doi: 10.1089/aid.1990.6.481. [DOI] [PubMed] [Google Scholar]
- 64.Crowle AJ, May MH. Inhibition of tubercle bacilli in cultured human macrophages by chloroquine used alone and in combination with streptomycin, isoniazid, pyrazinamide, and two metabolites of vitamin D3. Antimicrob Agents Chemother. 1990;34:2217–2222. doi: 10.1128/aac.34.11.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang MC, et al. Inhibition of HIV-1 Tat-mediated transactivation by quinacrine and chloroquine. Biochem Biophys Res Commun. 1996;226:1–7. doi: 10.1006/bbrc.1996.1302. [DOI] [PubMed] [Google Scholar]
- 66.Ang KK, et al. In vivo antimalarial activity of the beta-carboline alkaloid manzamine A. Antimicrob Agents Chemother. 2000;44:1645–1649. doi: 10.1128/aac.44.6.1645-1649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fivelman QL, et al. The effect of artesunate combined with standard antimalarials against chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum in vitro. Trans R Soc Trop Med Hyg. 1999;93:429–432. doi: 10.1016/s0035-9203(99)90147-5. [DOI] [PubMed] [Google Scholar]
- 68.Mackenzie AH. An appraisal of chloroquine. Arthritis Rheum. 1970;13:280–291. doi: 10.1002/art.1780130310. [DOI] [PubMed] [Google Scholar]
- 69.Artico M, et al. Geometrically and conformationally restrained cinnamoyl compounds as inhibitors of HIV-1 integrase: synthesis, biological evaluation, and molecular modeling. J Med Chem. 1998;41:3948–3960. doi: 10.1021/jm9707232. [DOI] [PubMed] [Google Scholar]
- 70.Yang F, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 71.Hooker CW, et al. Inhibitors of human immunodeficiency virus type 1 reverse transcriptase target distinct phases of early reverse transcription. J Virol. 2001;75:3095–3104. doi: 10.1128/JVI.75.7.3095-3104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li M, et al. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007;67:1988–1996. doi: 10.1158/0008-5472.CAN-06-3066. [DOI] [PubMed] [Google Scholar]
- 73.Srivastava KC, et al. Curcumin, a major component of food spice turmeric (Curcuma longa) inhibits aggregation and alters eicosanoid metabolism in human blood platelets. Prostaglandins Leukot Essent Fatty Acids. 1995;52:223–227. doi: 10.1016/0952-3278(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 74.Braaten D, et al. Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J Virol. 1996;70:5170–5176. doi: 10.1128/jvi.70.8.5170-5176.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Onyewu C, et al. Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob Agents Chemother. 2003;47:956–964. doi: 10.1128/AAC.47.3.956-964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Odom A, et al. The immunosuppressant FK506 and its nonimmunosuppressive analog L-685,818 are toxic to Cryptococcus neoformans by inhibition of a common target protein. Antimicrob Agents Chemother. 1997;41:156–161. doi: 10.1128/aac.41.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perkins ME, et al. Cyclosporin analogs inhibit in vitro growth of Cryptosporidium parvum. Antimicrob Agents Chemother. 1998;42:843–848. doi: 10.1128/aac.42.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakagawa M, et al. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem Biophys Res Commun. 2004;313:42–47. doi: 10.1016/j.bbrc.2003.11.080. [DOI] [PubMed] [Google Scholar]
- 79.Silverman JA, et al. Characterization of anti-Toxoplasma activity of SDZ 215-918, a cyclosporin derivative lacking immunosuppressive and peptidyl-prolyl-isomerase-inhibiting activity: possible role of a P glycoprotein in Toxoplasma physiology. Antimicrob Agents Chemother. 1997;41:1859–1866. doi: 10.1128/aac.41.9.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Damaso CR, Moussatche N. Inhibition of vaccinia virus replication by cyclosporin A analogues correlates with their affinity for cellular cyclophilins. J Gen Virol. 1998;79:339–346. doi: 10.1099/0022-1317-79-2-339. [DOI] [PubMed] [Google Scholar]
- 81.Jayasuriya H, et al. Durhamycin A, a potent inhibitor of HIV Tat transactivation. J Nat Prod. 2002;65:1091–1095. doi: 10.1021/np010642f. [DOI] [PubMed] [Google Scholar]
- 82.Gordon MA, Lapa EW. Durhamycin, a Pentaene Antifungal Antibiotic from Streptomyces durhamensis sp. n. Appl Microbiol. 1966;14:754–760. doi: 10.1128/am.14.5.754-760.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.http://dtp.nci.nih.gov (2007)
- 84.Nikolaeva L, Galabov AS. Antiviral effect of the combination of enviroxime and disoxaril on coxsackievirus B1 infection. Acta Virol. 2000;44:73–78. [PubMed] [Google Scholar]
- 85.Ninomiya Y, et al. Comparative studies on the antirhinovirus activity and the mode of action of the rhinovirus capsid binding agents, chalcone amides. Antiviral Res. 1990;13:61–74. doi: 10.1016/0166-3542(90)90022-y. [DOI] [PubMed] [Google Scholar]
- 86.Nikolaeva L, Galabov AS. Synergistic inhibitory effect of enviroxime and disoxaril on poliovirus type 1 replication. Acta Virol. 1995;39:235–241. [PubMed] [Google Scholar]
- 87.Watanabe N, et al. Fumagillin suppresses HIV-1 infection of macrophages through the inhibition of Vpr activity. FEBS Lett. 2006;580:2598–2602. doi: 10.1016/j.febslet.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 88.Franssen FF, et al. Susceptibility of Encephalitozoon cuniculi to several drugs in vitro. Antimicrob Agents Chemother. 1995;39:1265–1268. doi: 10.1128/aac.39.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Didier ES. Effects of albendazole, fumagillin, and TNP-470 on microsporidial replication in vitro. Antimicrob Agents Chemother. 1997;41:1541–1546. doi: 10.1128/aac.41.7.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Molina JM, et al. Potential efficacy of fumagillin in intestinal microsporidiosis due to Enterocytozoon bieneusi in patients with HIV infection: results of a drug screening study. The French Microsporidiosis Study Group. AIDS. 1997;11:1603–1610. doi: 10.1097/00002030-199713000-00009. [DOI] [PubMed] [Google Scholar]
- 91.Zhang P, et al. Angiogenesis inhibitors specific for methionine aminopeptidase 2 as drugs for malaria and leishmaniasis. J Biomed Sci. 2002;9:34–40. doi: 10.1007/BF02256576. [DOI] [PubMed] [Google Scholar]
- 92.Warhurst DC, et al. Hydroxychloroquine is much less active than chloroquine against chloroquine-resistant Plasmodium falciparum, in agreement with its physicochemical properties. J Antimicrob Chemother. 2003;52:188–193. doi: 10.1093/jac/dkg319. [DOI] [PubMed] [Google Scholar]
- 93.Sperber K, et al. Inhibition of human immunodeficiency virus type 1 replication by hydroxychloroquine in T cells and monocytes. AIDS Res Hum Retroviruses. 1993;9:91–98. doi: 10.1089/aid.1993.9.91. [DOI] [PubMed] [Google Scholar]
- 94.Mimoto T, et al. Structure-activity relationship of small-sized HIV protease inhibitors containing allophenylnorstatine. J Med Chem. 1999;42:1789–1802. doi: 10.1021/jm980637h. [DOI] [PubMed] [Google Scholar]
- 95.Giacometti A, et al. In-vitro activity of macrolides alone and in combination with artemisin, atovaquone, dapsone, minocycline or pyrimethamine against Cryptosporidium parvum. J Antimicrob Chemother. 1996;38:399–408. doi: 10.1093/jac/38.3.399. [DOI] [PubMed] [Google Scholar]
- 96.Boucher HW, et al. In vitro activities of the glycylcycline GAR-936 against gram-positive bacteria. Antimicrob Agents Chemother. 2000;44:2225–2229. doi: 10.1128/aac.44.8.2225-2229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wallace RJ, Jr, et al. Comparison of the in vitro activity of the glycylcycline tigecycline (formerly GAR-936) with those of tetracycline, minocycline, and doxycycline against isolates of nontuberculous mycobacteria. Antimicrob Agents Chemother. 2002;46:3164–3167. doi: 10.1128/AAC.46.10.3164-3167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arai S, et al. Antimycoplasmal activities of new quinolones, tetracyclines, and macrolides against Mycoplasma pneumoniae. Antimicrob Agents Chemother. 1992;36:1322–1324. doi: 10.1128/aac.36.6.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chang HR, et al. Activity of minocycline against Toxoplasma gondii infection in mice. J Antimicrob Chemother. 1991;27:639–645. doi: 10.1093/jac/27.5.639. [DOI] [PubMed] [Google Scholar]
- 101.Yahi N, et al. Suramin inhibits binding of the V3 region of HIV-1 envelope glycoprotein gp120 to galactosylceramide, the receptor for HIV-1 gp120 on human colon epithelial cells. J Biol Chem. 1994;269:24349–24353. [PubMed] [Google Scholar]
- 102.Baba M, et al. Selective inhibition of human cytomegalovirus replication by naphthalenedisulfonic acid derivatives. Antiviral Res. 1993;20:223–233. doi: 10.1016/0166-3542(93)90022-b. [DOI] [PubMed] [Google Scholar]
- 103.Tewtrakul S, et al. HIV-1 integrase inhibitory substances from Coleus parvifolius. Phytother Res. 2003;17:232–239. doi: 10.1002/ptr.1111. [DOI] [PubMed] [Google Scholar]
- 104.Aguilar JS, et al. The polysulfonated compound suramin blocks adsorption and lateral difusion of herpes simplex virus type-1 in vero cells. Virology. 1999;258:141–151. doi: 10.1006/viro.1999.9723. [DOI] [PubMed] [Google Scholar]
- 105.Debyser Z, et al. An antiviral target on reverse transcriptase of human immunodeficiency virus type 1 revealed by tetrahydroimidazo-[4,5,1-jk] [1,4]benzodiazepin-2 (1H)-one and -thione derivatives. Proc Natl Acad Sci U S A. 1991;88:1451–1455. doi: 10.1073/pnas.88.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hui EK, Nayak DP. Role of G protein and protein kinase signalling in influenza virus budding in MDCK cells. J Gen Virol. 2002;83:3055–3066. doi: 10.1099/0022-1317-83-12-3055. [DOI] [PubMed] [Google Scholar]
- 107.Crance JM, et al. Inhibition of sandfly fever Sicilian virus (Phlebovirus) replication in vitro by antiviral compounds. Res Virol. 1997;148:353–365. doi: 10.1016/s0923-2516(97)89132-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.