

SUMMARY
The mTORC1 complex is central to the cellular response to changes in nutrient availability. The signaling adapter p62 contributes to mTORC1 activation in response to amino acids, and interacts with TRAF6, which is required for the translocation of mTORC1 to the lysosome and the subsequent K63-polyubiquitination and activation of mTOR. However, the signal initiating these p62-driven processes was previously unknown. Here we show that p62 is phosphorylated via a cascade that includes MEK3/6 and p38δ and is driven by the PB1-containing kinase MEKK3. This phosphorylation results in the recruitment of TRAF6 to p62, the ubiquitination and activation of mTOR, and the regulation of autophagy and cell proliferation. Genetic inactivation of MEKK3 or p38δ mimics that of p62 in that it leads to inhibited growth of PTEN-deficient prostate organoids. Analysis of human prostate cancer samples showed upregulation of these three components of the pathway, which correlated with enhanced mTORC1 activation.
Graphical abstract
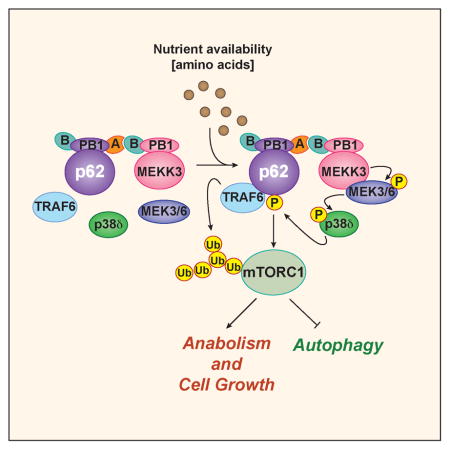
INTRODUCTION
Cell metabolism is responsive to the availability of environmental and intracellular nutrients. The mTORC1 kinase complex is a key nutrient sensor and an essential mediator of this response via its actions as a regulator of anabolism and autophagy (Hay and Sonenberg, 2004; Laplante and Sabatini, 2012). The aberrant activation of mTORC1 has important repercussions in several diseases, including cancer (Guertin and Sabatini, 2007; Laplante and Sabatini, 2012; Sabatini, 2006). An essential step in amino acid-induced activation of mTORC1 is its translocation to the lysosome, mediated by the Rag guanosine triphosphatases (GTPases), where it is activated by another GTPase termed Rheb (Duran and Hall, 2012; Sancak et al., 2010; Sancak et al., 2008; Yuan et al., 2013). A lysosomal pentameric complex termed ragulator, along with the vacuolar-ATPase, have been proposed to promote the exchange of GDP for GTP on RagA or RagB in amino acid-activated cells (Sancak et al., 2010). Additionally, the Rags have been shown to be regulated by other proteins including the GATOR1 complex (Bar-Peled et al., 2013), FLCN (Petit et al., 2013; Tsun et al., 2013), and sestrins (Budanov and Karin, 2008; Chantranupong et al., 2014). There is also evidence for Rag-independent mechanisms of mTORC1 activation. For example, it has recently been reported that Rab1A mediates mTORC1 activity in a Rag-independent manner through the formation of a Rheb-mTORC1 complex in the Golgi (Thomas et al., 2014). Moreover, RagA-null cells display a diffuse cytosolic localization of mTOR and RagC but can, nonetheless, maintain the activity of mTORC1 (Efeyan et al., 2014). Also, it has recently been shown that RheB-null cells retain significant levels of mTORC1 activity (Groenewoud et al., 2013). Moreover, very recent results suggest that the mechanism whereby glutamine activates mTORC1 differs from that of leucine as it is independent of the Rag-Ragulator cascade and is mediated by Arf-1 (Jewell et al., 2015). Therefore, it is clear that our comprehension of mTORC1 activation is still fragmentary and that more work is necessary to achieve a thorough understanding of the mechanisms that modulate its activity in response to nutrients.
The signaling adapter p62 (also known as SQSTM1) is central to cell survival and proliferation through the activation of mTORC1 (Duran et al., 2011; Duran et al., 2008; Linares et al., 2013; Moscat and Diaz-Meco, 2009, 2011; Valencia et al., 2014). This is achieved through the interaction of p62 with raptor, a distinctive component of the mTORC1 complex, and by facilitating mTORC1 translocation to the lysosome, a process that likely involves the interaction of p62 with the Rag proteins (Duran et al., 2011). Our recent data demonstrate that the E3-ubiquitin ligase TRAF6 is another important player in this process (Linares et al., 2013). That is, the interaction of TRAF6 with p62 facilitates the lysosomal recruitment of mTORC1, and catalyzes the K63-polyubiquitination of the mTOR subunit of the complex, which is required for its optimal activation by amino acids (Linares et al., 2013). Therefore, the p62/TRAF6 tandem must be considered an important modulator of nutrient sensing through mTORC1. Consistent with this notion, the loss of TRAF6, like that of p62, attenuated proliferation and the transforming properties of cancer cells, and led to enhanced autophagy, which could be rescued by the expression of a permanently active RagB mutant, indicating that p62 acts upstream of the Rag proteins (Linares et al., 2013). Thus, p62 acts as a scaffold bringing components involved in the control of mTORC1 signaling to the correct cellular location (Duran et al., 2011). How mTORC1 is linked to the upstream nutrient-sensing machinery and the potential role of p62 in that process are key unresolved issues.
Here we demonstrate that p62 phosphorylation at T269/S272 is a critical event in channeling the amino acid response to mTORC1 activation, likely due to its ability to orchestrate the binding of p62 with the different components of the mTORC1 signaling complex. That is, we found that p62 organizes a molecular platform with a kinase cascade that is initiated by MEKK3 and triggers the activation of p38δ, is driven by PB1-domain interactions between p62 and MEKK3, and is located on the lysosomal surface. These kinases, like p62, are selectively required for amino acid-induced mTORC1 activation while dispensable for insulin signaling, and are required for effective control of cell growth, autophagy, and transformation. These findings define a critical mechanism to transmit nutrient-sensing signals to the mTORC1 complex, which is mediated by phosphorylation of the scaffold protein p62.
RESULTS
p62 Phosphorylation Is Required for the Activation of mTORC1 by Amino Acids
The mechanisms whereby mTORC1 respond to amino acids are not fully understood. Our previous data demonstrated that p62 is a scaffold in this pathway (Duran et al., 2011; Linares et al., 2013), but the process by which it senses amino acids remains to be elucidated. Recent studies have suggested phosphorylation as a regulatory mechanism for the control of p62 function (Ichimura et al., 2013; Linares et al., 2011; Matsumoto et al., 2011). Therefore, we hypothesized that nutrient-driven p62 phosphorylation might underlie p62-mediated activation of mTORC1 in response to amino acids. To address this possibility, we generated HEK293T cells stably expressing FLAG-tagged p62 or FLAG control. Cells were stimulated with amino acids, after which anti-FLAG immunoprecipitates were subjected to liquid chromatography coupled to mass spectrometry (LC-MS). We found that p62 exhibited low-abundance baseline phosphorylation at residues S28, T221, and S224 that was constitutive and not changed upon amino acid stimulation (Figure 1A, upper panel). In contrast, we found that phosphorylation at residues T269 and S272 was markedly induced by amino acids (Figure 1A, upper panel). These sites, and their surrounding sequences, were highly conserved across species (Figure 1A, lower panel). To establish the relevance of these phosphorylation events in mTORC1 signaling, we used a phosphospecific antibody generated against the human p62 peptide SRLT(P)PVS(P)PES(C), which allowed us to detect phospho-T269/S272. Interestingly, immunoblot analysis revealed the strong phosphorylation of p62 at T269/S272 upon re-addition of all amino acids to cells amino-acid starved (Figure 1B). Leucine and arginine, two key amino acids for mTORC1 stimulation, also promoted p62-T269/S272 phosphorylation, which correlated with the magnitude of mTORC1 activation, as measured by phosphorylation of S6K, (Figure 1C). Conversely, amino acid starvation resulted in a pronounced inhibition of p62-T269/S272 phosphorylation, concomitant with the decrease of mTORC1 activity (Figure 1D). Mutation of p62-T269/S272 sites to alanine (p62T269/S272AA) abolished amino acid-induced p62 phosphorylation, demonstrating that these are bona fide nutrient-sensitive p62 phosphorylation residues (Figure 1E). To explore whether phosphorylation of p62 has any impact on its ability to regulate mTORC1 activity, we used p62 KO MEFs reconstituted with either p62WT or p62T269/S272AA. Consistent with our previously published data, mTORC1 activation in response to amino acids was impaired in p62-deficient cells (Figure 1F) (Duran et al., 2011). Importantly, whereas re-expression of p62WT restored mTORC1 activity in the KO MEFs, that of p62T269/S272AA failed to do so, as demonstrated by phosphorylation of multiple mTORC1 downstream targets including S6K, S6, and 4EBP1 (Figure 1F). Furthermore, overexpression of an active RagB-GTP-bound mutant rescued the mTORC1 inhibition caused by p62T269/S272AA expression (Figure 1G). This is in agreement with p62 acting upstream of the Rag GTPases in the control of mTORC1 signaling in the amino acid pathway, and supports the notion that p62 phosphorylation also lies upstream of Rag activation (Duran et al., 2011). In keeping with a specific role for p62 in the amino acid, but not in the insulin pathway, insulin did not promote p62 phosphorylation, which, in turn, was not required for insulin-induced mTORC1 activity (Figure 1H). Notably, p62 phosphorylation was also critical for assembly of the amino acid-induced p62-mTORC1 complex, as demonstrated by the ability of p62T269/S272AA expression to inhibit the interaction of p62 with the different components of mTORC1, including mTOR, raptor, and TRAF6 (Figure 1I). Mutation of p62-T269/S272 sites to aspartic acid did not mimic p62 phosphorylation and did not restore mTORC1 activation or TRAF6 recruitment (Figures S1A and S1B). Together, these findings demonstrate that p62 phosphorylation is a key event in mTORC1 activation, selectively in response to amino acids.
Figure 1. p62 Phosphorylation at T269/S272 Was Required for mTORC1 Activation in Response to Amino Acids.

(A) p62 domain organization and phosphorylation sites identified by mass spec. Phosphorylation of T269 and S272 was induced by amino acids (upper panel). Alignment of the amino acid sequence of human p62 (264–276) with orthologs in other species is shown (lower panel).
(B) p62 phosphorylation at T269 and S272 is induced by amino acids. HEK293T cells were starved of amino acids for 50 min and restimulated with amino acids for the indicated durations. Cell lysates were analyzed by immunoblotting.
(C) p62 phosphorylation was determined in response to different amino acids. HEK293T cells were starved of amino acids and restimulated with the indicated amino acids for 30 min.
(D) Amino acid starvation inhibits p62 phosphorylation and mTORC1 activation. HEK293T cells were starved of amino acids for the indicated durations. Total cell lysates were analyzed by immunoblotting.
(E) Mutation of p62-T269/S272 sites to alanine (p62T269/S272AA) abolished p62 phosphorylation in response to amino acids. HEK293T cells stably expressing FLAG-p62WT or FLAG-p62T269/S272AA were treated and analyzed as in (B).
(F) The p62T269/S272AA mutant was not able to reconstitute mTOR activation in p62KO MEFs. WT and p62KO MEFs, reconstituted with p62WT or p62T269/S272AA, were treated as in (B). Cell lysates were analyzed by western blot.
(G) RagBGTP overexpression rescued the defects in mTOR activation by amino acids in cells stably expressing the p62T269/S272AA mutant. HEK293T cells stably expressing FLAG-p62WT, FLAG-p62T269/S272AA, or FLAG-p62T269/S272AA and FLAG-RagBGTP were treated as in (B), and immunoblotted for the specified proteins.
(H) p62 phosphorylation was not required for mTOR activation by insulin. HEK293T cells stably expressing FLAG-p62WT or FLAG-p62T269/S272AA were deprived of serum for 24 hr and stimulated with insulin for the indicated durations. Cell lysates were analyzed by western blot.
(I) The p62T269/S272AA mutant eliminated the interaction of p62 with different components of the mTORC1 complex. HEK293T cells stably expressing FLAG-p62WT or FLAG-p62T269/S272AA were treated as in (B). Cell lysates and FLAG-tagged immunoprecipitates were immunoblotted to detect the indicated proteins.
Results are representative of three experiments. See also Figure S1
MEKK3 Is a Critical Kinase for Amino Acid-Induced p62 Phosphorylation
An important question arising from these results is the identity of the amino acid-activated p62 kinase. In this regard, the structure of p62 includes a PB1 domain, which is a protein-protein interaction module that is present in kinases, such as the atypical PKCs (PKCζ and PKCλ/ι) and MEKK3, as well as in signaling adapters such as Par-6 and NBR1 (Moscat et al., 2006; Sanchez et al., 1998). Protein dimerization occurs by PB1-PB1 interactions through a β-grasp topology in a front-to-back orientation of the two PB1 domains. This involves electrostatic interactions of a cluster of basic residues in one of the PB1 domains that can bind clusters of acidic amino acids in the back of a second PB1 (Figure 2A) (Moscat et al., 2006; Sumimoto et al., 2007). The PB1 domains can be classified into three different types based on the presence of the acid cluster (Type I), the basic cluster (Type II), or both in the same domain (Type I/II) (Fig. 2A). The PB1 domain of p62 belongs to the Type I/II group and, therefore, can accommodate interactions through both faces (Moscat et al., 2006; Sumimoto et al., 2007). In this regard, we previously showed that proteins such as PKCζ, PKCλ/ι, or NBR1, which interact with p62 through its basic cluster, were not required for the activation of mTORC1 (Duran et al., 2011). In contrast, we found that disruption of the acidic cluster of p62, by mutation of D69/D73 to alanine, abolished p62 phosphorylation in response to amino acids (Figures 2B and 2C). This suggests that if a PB1-domain protein is involved in mTORC1 activation, it must interact with the acidic cluster of p62 using its basic cluster. Interestingly, the PB1 domain-containing kinase, MEKK3, harbors a type II PB1 domain and previous results have shown that the p62 D69A/D73A PB1-domain mutant is unable to interact with MEKK3 (Figure 2B) (Nakamura et al., 2010). Consistent with this, the overexpression of MEKK3 resulted in the phosphorylation of p62WT but not of p62T269/S272AA (Figure 2D). Furthermore, p62 phosphorylation in response to MEKK3 overexpression was eliminated in the p62 PB1-domain mutant (Figure 2E). MEKK3 overexpression, but not that of a kinase-dead mutant, was able to induce the phosphorylation of p62 at T269/S272 (Figure 2F), which correlated with mTORC1 activation (Fig. 2F), suggesting that MEKK3 could be a bona fide regulator of p62 phosphorylation and mTORC1 activity. Next, we determined whether MEKK3 is required for mTORC1 activation by using the clustered regularly interspaced short palindromic repeats (CRISPR/Cas9) system to generate MEKK3-deficient HEK-293T cells. Notably, the loss of MEKK3 severely reduced p62 phosphorylation and mTORC1 activation in response to amino acids, which were both rescued by the ectopic expression of MEKK3 (Figure 2G). Similar results were obtained with two independent sgMEKK3 clones, as well as by knocking down MEKK3 with an shRNA lentiviral vector in HEK293T, A549, and PC3 cells (Figure S2A–S2D). Of note, the effects of MEKK3 deficiency in mTORC1 activation were rescued by expression of active RagB (Figure 2H). The loss of MEKK3 did not affect insulin-activated mTORC1, consistent with the specificity of p62 in the amino acid pathway (Figure 2I). In keeping with the importance of MEKK3 in this process, we found that, upon cell stimulation by amino acids, endogenous MEKK3 was recruited to an endogenous complex containing p62 and mTOR (Figure 2J). In addition, Figure 2K demonstrates that the kinase activity of MEKK3 was stimulated in amino acid-treated cells. Collectively, these results demonstrate that p62 is phosphorylated in response to amino acids through a MEKK3-dependent mechanism that is critical for mTORC1 activation, and is mediated by the interaction of p62 and MEKK3 through their respective PB1 domains.
Figure 2. MEKK3 Was a Critical Kinase for p62 Phosphorylation and mTORC1 Activation in Response to Amino Acids.

(A) Schematic of the different types of PB1 domains based on the presence of an acid cluster (Type I), basic cluster (Type II), or both in the same domain (Type I/II).
(B) p62 and MEKK3 domain architecture and schematic of the interaction between the acidic cluster of the PB1 domain of p62 and the basic domain of MEKK3.
(C) Mutation of p62-D69/73 sites to alanine (p62D69/73AA) abolished p62 phosphorylation. HEK293T cells transfected with the indicated plasmids were starved of amino acids for 50 min and restimulated with amino acids for the indicated durations. Myc-tagged immunoprecipitates were analyzed by western blot.
(D) MEKK3 promoted p62 phosphorylation at T269/S272. HEK293T cells were transfected with the indicated plasmids and cell lysates were analyzed by western blot.
(E) MEKK3-induced p62 phosphorylation required the PB1 domain of p62. HEK293T cells were transfected with the indicated plasmids and cell lysates were immunoblotted to detect the specified proteins.
(F) Overexpression of MEKK3, but not that of MEKK3 kinase-dead mutant, induced p62 phosphorylation and mTORC1 activation by amino acids. HEK293T cells transfected with the indicated plasmids, were deprived of amino acids for 50 min and stimulated with amino acids for 15 min. Cells were analyzed by western blot.
(G) MEKK3 expression rescued p62 phosphorylation and mTOR activation in MEKK3-deficient cells. MEKK3-deficient HEK293T cells generated with the CRISPR/CAS9 system were reconstituted with MEKK3. Cells were deprived of amino acids for 50 min and then stimulated with amino acids for the indicated durations. Cell lysates were immunoblotted for the specified proteins.
(H) RagBGTP overexpression rescued mTOR activation by amino acids in MEKK3-deficient cells. Control and MEKK3-deficient HEK293T cells expressing FLAG-RagBGTP were treated as in (G) and immunoblotted to detect the specified proteins.
(I) MEKK3 was not required for mTOR activation by insulin. Control and MEKK3-deficient HEK293T cells were deprived of serum for 24 hr and stimulated with insulin for the indicated durations. Cell lysates were analyzed by western blot.
(J) MEKK3 is a component of the mTORC1 complex. mTOR immunoprecipitates and cell lysates from HEK293T cells, treated as in (F), were immunoblotted for the indicated proteins.
(K) MEKK3 kinase activity was activated upon amino acid stimulation. HEK293T cells transfected with the indicated plasmids were treated as in (F). HA-tagged immunoprecipitates were used in an in vitro phosphorylation with ATPγS, with myelin basic protein (MyBP) as the substrate, followed by PNBM alkylation and immunoblotting to detect the indicated proteins.
Results are representative of three experiments. See also Figure S2.
MEK3/6 and p38δ Channel MEKK3-Induced Phosphorylation of p62 by Amino Acids
Based on these results, it is possible that p62 could be targeted directly by MEKK3. However, when bacterially expressed recombinant p62 was incubated with active recombinant MEKK3 in an in vitro kinase assay, we found that MEKK3 was not able to directly phosphorylate p62 (Figure S3A). These results strongly suggest the existence of other kinase that acts downstream of MEKK3 to phosphorylate p62 in response to amino acids. Our previously published data showed that CDK1 was able to phosphorylate p62 at residues T269/S272 during mitosis (Linares et al., 2011). However, a selective CDK1 inhibitor did not affect p62 phosphorylation in response to amino acids (Figure S3B). To identify that kinase, since MEKK3 is a MAP3K, we reasoned that a MAP2K/MAPK cascade could act downstream of MEKK3 to phosphorylate p62. To address this possibility, we individually knocked down all the members of the five distinct groups of MAPKs characterized in mammals (Figure 3A). Cells were stimulated with amino acids, as above, and the activation of mTORC1 was determined. Notably, only depletion of p38δ (MAPK13) severely impaired amino acid-induced activation of mTORC1, (Figures 3A and S3C). Importantly, knockout of p38δ by CRISPR/Cas9 severely abolished p62 phosphorylation and mTORC1 activation in cells stimulated with amino acids, which was rescued by the expression of active RagB (Figures 3B and 3C). Of note, p38δ-deficient cells displayed normal insulin-induced mTORC1 activation (Figure 3D). Similar results were obtained with two other independent CRISPR/Cas9-generated p38δ KO clones (Figure S3D). Furthermore, the pharmacological inhibition of p38δ severely abrogated mTORC1 activation and p62 phosphorylation by amino acids (Figure S3E). Interestingly, we also found that the overexpression of WT, but not of a kinase-inactive p38δ (T180A/Y182F) mutant, was able to induce the phosphorylation of p62WT but not of p62T269/S272AA (Figure 3E and 3F). Taken together, these results demonstrate that p38δ is responsible for p62 phosphorylation and mTORC1 activation by amino acids. To determine whether p62 is actually a direct substrate of p38δ, we incubated bacterially expressed recombinant p62 with active p38δ in an in vitro kinase assay, and found that p38δ directly phosphorylated p62 at T269/S272 (Figure 3G). To confirm that these residues account for p62 phosphorylation by p38δ, purified p62WT and p62T269/S272AA were phosphorylated in vitro with ATP-γ-S and recombinant active p38δ. Figure 3H demonstrates that p62 phosphorylation by p38δ was completely abolished in the p62T269/S272AA mutant, as compared to p62WT, indicating that p38δ is a bona fide direct p62 T269/S272 kinase that channels MEKK3 signals in amino acid-activated cells.
Figure 3. MEKK3/MEK3/6-p38δ Induced p62 Phosphorylation in Response to Amino Acids.

(A) p38δ was required for mTORC1 activation in response to amino acids. Results of siRNA screening of MAPKs in mTORC1 activation.
(B) p38δ was required for p62 phosphorylation at T269/S272 in response to amino acids. p38δ-deficient HEK293T cells generated with the CRISPR/CAS9 system. Cells were deprived of amino acids for 50 min and then stimulated with amino acids for 15 min. Cell lysates were immunoblotted for the indicated proteins.
(C) RagBGTP overexpression rescued amino acid-induced mTOR activation in p38δ-deficient cells. Control and p38δ-deficient HEK293T cells, expressing FLAG-RagBGTP, were treated as in (B). Cell lysates were immunoblotted for the indicated proteins.
(D) p38δ was not required for insulin-induced mTORC1 activation. Control and p38δ-deficient HEK293T cells were deprived of serum for 24 hr and then stimulated with insulin for the indicated durations. Cell lysates were immunoblotted to detect the indicated proteins.
(E) p38δ overexpression promotes p62 phosphorylation at T269/S272. HEK293T cells were transfected with the indicated plasmids and immunoblotted for the specified proteins.
(F) p38δ kinase activity was required for p62 phosphorylation. HEK293T cells were transfected with the indicated plasmids and immunoblotted for the specified proteins.
(G) p38δ directly phosphorylated p62 at T269/S272 in vitro. An in vitro phosphorylation assay using recombinant p62 and recombinant p38δ is shown.
(H) T269/S272 sites accounted for p62 phosphorylation by p38δ. FLAG-tagged immunoprecipitates from HEK293T cells were phosphorylated in vitro by recombinant p38δ with ATPγS, followed by PNBM alkylation and immunoblotting for the indicated proteins.
(I) p38δ kinase activity was activated by amino acids. HEK293T cells transfected with the indicated plasmids were treated as in (B). In vitro phosphorylation was carried out with the FLAG-tagged immunoprecipitates and MBP-p62 recombinant protein as a substrate.
(J) MEKK3 was required for p38δ-induced p62 phosphorylation by amino acids. shNT or shMEKK3 HEK293T cells transfected with the indicated plasmids were treated as in (B). In vitro phosphorylation was carried out with the FLAG-tagged immunoprecipitates and MBP-p62 recombinant protein was used as the substrate.
(K) MEK3/6 was required for p62 phosphorylation and mTORC1 activation in response to amino acids. HEK293T cells transfected with scramble siRNA or MEK3 and MEK6 siRNAs and FLAG-RagBGTP were treated as in (B). Cell lysates were immunoblotted for the indicated proteins.
(L) MEK3 is activated in response to amino acids. HEK293T cells transfected with the indicated plasmids were treated as in (B). FLAG-tagged MEK3 immunoprecipitates were used in an in vitro phosphorylation, using MyBP as substrate, with ATPγS followed by PNBM alkylation and immunoblotting for the indicated proteins.
(M) Schematic showing that MEK3/6-p38δ channels MEKK3-induced phosphorylation of p62 and mTORC1 activation by amino acids.
Results are representative of three experiments. See also Figure S3.
If this model is correct, then p38δ should be activated by amino acids in a MEKK3-dependent manner. To determine whether this was the case, HEK293T cells were transfected with FLAG-tagged p38δ, after which cells were treated with amino acids at different times as described above. Transfected p38δ was immunoprecipitated with an anti-FLAG antibody, and its ability to phosphorylate recombinant p62 was determined in an in vitro kinase assay. Interestingly, p38δ from shNT cells that were stimulated with amino acids displayed higher enzymatic activity towards recombinant p62 than p38δ from unstimulated shNT cells (Figure 3I). The finding that amino acid stimulation did not increase the activity of p38δ in shMEKK3 cells (Figure 3J) clearly established that p38δ is a critical downstream target of MEKK3 in the nutrient-sensing cascade that activates mTORC1 through p62 phosphorylation. To identify the kinase that links MEKK3 to p38δ we tested whether MEK3 and MEK6 might be the MAP2Ks upstream of p38δ. Notably, the simultaneous depletion of MEK3 and MEK6 severely impaired amino acid-induced mTORC1 activation and p62 phosphorylation, which was rescued by the expression of active RagB (Figure 3K). Furthermore, the kinase activity of MEK3 was stimulated in amino acid-treated cells (Figure 3L). Collectively, these results demonstrate that MEKK3 is the apical kinase in an amino acid-sensing cascade that includes MEK3/MEK6 and p38δ, and that leads to p62 phosphorylation, which is a critical step for mTORC1 activation in response to amino acids (Figure 3M).
MEKK3 and p38δ Control Lysosomal Translocation of mTOR
To be activated by amino acid stimulation, mTORC1 must undergo translocation from the cytoplasm to the lysosome (Sancak et al., 2008). Since MEKK3 and p38δ are necessary for amino acid-induced mTORC1 activity, we next investigated the subcellular localization of MEKK3 and p38δ by confocal immunofluorescence microscopy in both starved and amino acid-stimulated cells. Double staining of endogenous MEKK3 or p38δ, and lysosomal-associated membrane protein 2 (LAMP2), revealed the localization of both kinases at the lysosome, which was independent of nutrient availability (Figures 4A and 4B). The antibodies used in this experiment were validated for immunofluorescence in MEKK3 or p38δ knocked-down cells (Figure S4). Cell fractionation confirmed the constitutive localization of MEKK3, p38δ, and p62, along with Lamp2, in the heavy membrane lysosomal fraction (Figure 4C). Of great functional relevance, the knockdown of either MEKK3 or p38δ impaired the colocalization of mTORC1 with LAMP2 in response to amino acids (Figure 4D), demonstrating that p38δ and MEKK3, like p62, are required for mTORC1 translocation to the lysosome.
Figure 4. MEKK3/p38δ Cascade Is Required for the Lysosomal Translocation of mTOR.

(A–B) MEKK3 and p38δ localize at the lysosome in an amino acid-independent manner. Images of HeLA cells co-immunostained for MEKK3 and LAMP2 (A) or p38δ and LAMP2 (B). Cells were starved for 50 min and then stimulated with amino acids for 10 min before processing. In all images, graphs show the areas of staining overlap (merge). Scale bars=10μm. The quantification of colocalization was carried out on at least 15 cells per condition from 2 independent experiments. Results are shown as means ± SEM.
(C) MEKK3, p38δ and p62 were present in the lysosomal fraction. HEK293T cells were treated as in (A) and lysates were separated into heavy membrane and light/cytosolic fractions.
(D) MEKK3 and p38δ deficiency prevented amino acid-induced translocation of mTOR to lysosomes. Images of Control, MEKK3-deficient, or p38δ-deficient HEK293T cells treated and analyzed as in (A) that were co-immunostained to detect mTOR and LAMP2. Scale bars= 10μm. Results are shown as means ± SEM. ***p<0.001.
Images are representative of two independent experiments. See also Figure S4.
Role of the MEKK3/p38δ Cascade in the Polyubiquitination of mTOR
TRAF6 is recruited to the p62-mTORC1 complex upon amino acid stimulation, and that this promoted the K63-polyubiquitination of mTOR, a key event in amino acid-dependent activation of mTORC1 (Linares et al., 2013). Our current results link this process to the MEKK3/p38δ cascade, since depletion of either kinase severely impaired the interaction of TRAF6 with p62 (Figures 5A and 5B). This suggests a critical role for the phosphorylation of p62 by the MEKK3/p38δ cascade in the binding of TRAF6 to p62, consistent with the fact that p62T269/S272AA did not interact with TRAF6 in amino acid-stimulated cells (Figure 1I). As predicted by this model, endogenous polyubiquitination of mTOR in response to amino acids was severely inhibited by the deficiency of either MEKK3 or p38δ (Figures 5C and 5D). Furthermore, the endogenous polyubiquitination of mTOR in response to amino acids was inhibited in cells expressing the p62T269/S272AA mutant compared with those expressing p62WT (Figure 5E). Collectively, these data demonstrate that p62 phosphorylation by p38δ is a key event in the recruitment of mTORC1 to the lysosome, and in its subsequent activation by TRAF6-mediated polyubiquitination.
Figure 5. The MEKK3/p38δ Cascade Was Required for TRAF6-Catalyzed K63-Polyubiquitination of mTOR in Response to Amino Acids.

(A–B) Knockdown of MEKK3 or p38δ impaired the interaction of TRAF6 with p62 in response to amino acids. shNT, shMEKK3, or shp38δ HEK293T cells were deprived of amino acids for 50 min and then restimulated with amino acids for 30 min. Cell lysates and p62 immunoprecipitates were analyzed by western blot for the indicated proteins.
(C–E) MEKK3, p38δ, and p62 phosphorylation were required for polyubiquitination of mTOR in response to amino acids. shNT, shMEKK3, and shp38δ HEK293T cells or cells stably expressing FLAG-p62WT or FLAG-p62T269/S272AA were treated as in (A). Cell lysates and mTOR immunoprecipitates were immunoblotted for the indicated proteins.
Results are representative of three experiments.
The MEKK3/p38δ Cascade Contributes to Cell Proliferation and Autophagy
A well-established function of mTORC1 is to control cell size (Fingar et al., 2002). In keeping with a critical role for MEKK3 and p38δ in the activation of mTORC1, cells deficient in MEKK3, p38δ, or p62 were significantly smaller than WT controls (Figure 6A). On the other hand, it is known that mTORC1 activation promotes cell proliferation and transformation while inhibiting autophagy (Kim et al., 2011; Yu et al., 2010). Consistent with this, the knockdown of MEKK3 or p38δ in PC3 prostate cancer (PCa) cells significantly reduced cell proliferation under normal growing conditions (Figures 6B and 6C), and this effect was rescued by the expression of a constitutively active mutant of RagB (Figure 6D and 6E). Since nutrient starvation induces autophagy through inhibition of mTORC1 (Kim et al., 2011; Yu et al., 2010), we knocked down MEKK3 or p38δ and determined the effect on autophagy. Interestingly, reduction in the levels of either of these kinases resulted in enhanced LC3 processing, which was even more apparent when cells were incubated with Bafilomycin A1, an inhibitor of autophagosomal and lysosomal fusion (Figures 6F–6I). We also analyzed autophagic flux using the reporter GFP-mCherry-LC3 (Kimura et al., 2007), which allows the identification of autolysosomes (mCherry positive/GFP negative; red dots) and autophagosomes (mCherry-positive/GFP-positive; yellow dots). Our data showed that the total numbers of autophagosomes and autolysosomes under basal and amino acid-starvation conditions were higher in the MEKK3- and p38δ-deficient cells (Figures 6J and 6K). Taken together, these results demonstrate that the MEKK3/p38δ cascade modulates autophagy in response to nutrient starvation, consistent with its role in the regulation of mTORC1 activation.
Figure 6. The MEKK3/p38δ Cascade Controlled Cell Proliferation and Autophagy through mTORC1 Activation.

(A) MEKK3, p62, or p38δ deficiencies reduced cell size. Results are shown as means ± SEM (n=3) *p< 0.05, **p<0.01.
(B–C) Knock-down of MEKK3 or p38δ reduced cell proliferation under normal growing conditions. shNT, shMEKK3, or shp38δ PC3 cells were cultured under normal growing conditions, and cell viability was determined by trypan blue exclusion assay. Results are shown as means ± SEM (n=3). *p< 0.05, **p<0.01.
(D–E) RagBGTP overexpression rescued the defects in cell proliferation in MEKK3- or p38δ-knockdown cells. PC3 cells stably expressing FLAG-RagBGTP were infected with shNT, shMEKK3, or shp38δ lentiviral vectors. Cell lysates were analyzed by western blot, and cell viability was determined as in (C). Results are shown as means ± SEM (n=3). *p< 0.05, **p<0.01.
(F–I) Knockdown of MEKK3 or p38δ promoted autophagy in response to nutrient deprivation. shNT, shMEKK3, or shp38δ PC3 cells were deprived of amino acids and serum for 4 hr in the absence or presence of bafilomycin A1. Cell lysates were immunoblotted for the indicated proteins. Graphs represents LC3-II/actin ratio as measured by densitometry.
(J–K) Knockdown of MEKK3 or p38δ promoted increased autophagic flux. Images of shNT, shMEKK3, or shp38δ cells stably expressing GFP-mCherry-LC3 and treated as in (G). Scale bars=10μm. Quantification of the number of autophagosomes and autolysosomes per cell is shown. Results are shown as means ± SEM (n=20). *p< 0.05 **p< 0.01, ***p<0.001.
Results are representative of three experiments.
Relevance of the MEKK3/p38δ/p62/mTOR Axis in Prostate Cancer
To investigate the relevance of the p62/MEKK3/p38δ cascade in the activation of mTOR in PCa, we profited from a recently developed technology for creating 3D prostate organoid cultures (Gao et al., 2014; Karthaus et al., 2014). Murine prostate organoids faithfully recapitulate the in vivo phenotypes of genetic PCa mouse models, and can be easily manipulated (Gao et al., 2014; Karthaus et al., 2014). Thus, we isolated prostate epithelial cells from PTEN-deficient mice, and subjected them to lentiviral infection to selectively knock down MEKK3, p38δ, or p62, and then cultured them in 3D organoid conditions. Interestingly, we found that the inactivation of MEKK3, p38δ, or p62 decreased the efficiency of organoid formation and size of organoids, and reverted the hyperplastic phenotype of the PTEN−/− organoids (Figures 7A and 7B). This strongly suggests an important role for the MEKK3/p38δ cascade in PCa. Notably, the deficiency in p62, MEKK3, or p38δ in these organoids resulted in severe impairment of S6K and 4EBP1 phosphorylation in this model (Figure 7C). Consistent with these observations, immunohistochemical analysis of prostates from PTEN+/− mice showed increased expression of MEKK3, p38δ, and p62, as well as the activation of S6 phosphorylation, which was used as a surrogate marker of mTORC1 activity, in PIN areas of the prostate, as compared to normal glands (Figure 7D). Furthermore, we used double immunofluorescence to analyze the colocalization of p38δ either with p62 or with pS6 in sections of human PCa and normal prostate tissue. Of great interest, we found that p62 and p38δ levels were increased, and co-localized with enhanced pS6 staining, in tumor tissues as compared to normal controls (Figure 7E). We next analyzed the expression of MEKK3, p38δ, p62, and phospho-S6 in PCa tissue microarrays by immunohistochemistry. Interestingly, our data showed much stronger expression of all these proteins in aggressive tumors with high Gleason score (GS 7-10) than in those with low Gleason score (GS 2-6) (Figures 7F). Importantly, MEKK3 and p38δ expression significantly correlated with p62 and phospho-S6 in these human PCa samples (Figure 7G). Taken together, these results established that the PB1-driven MEKK3/p38δ/p62/mTOR pathway is relevant to PCa.
Figure 7. MEKK3/p38δ/p62/mTOR Is Relevant to Prostate Cancer.

(A) Knockdown of MEKK3, p38δ, or p62 led to a reduction in the efficiency of organoid formation, size, and hyperplastic phenotype of PTEN-null prostate organoids. Representative images of organoids and H&E staining are shown. Prostate organoids were prepared from PTENfl/fl-PBcre mice and infected with lentiviral vectors for shNT, shp62, shMEKK3, and shp38δ. Organoids were analyzed after 7 days in culture. Scale bars = 100 μm.
(B) Quantification of the efficiency of organoid formation and size in the experiment shown in (A). Scale bars =100 μm. Results are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
(C) Knockdown of MEKK3, p38δ, or p62 in PTEN-null prostate organoids led to decreased mTORC1 activation. Cell lysates from (A) were immunoblotted for the indicated proteins.
(D) Increased expression of MEKK3, p38δ, p62, and S6 phosphorylation in PIN areas (red arrows) of PTEN+/− prostates, compared with normal prostate glands (asterisks). Representative images of MEKK3, p38δ, p62, and phospho-S6 staining of primary PCa samples from PTEN+/− mice are shown. Scale bars = 25 μm.
(E) Human prostate shows increased expression of p38δ, p62, and S6 phosphorylation in tumor tissue, as compared with normal tissue. Images of human prostate co-immunostained for p38δ and p62, or and p38δ and p-S6 are shown. Scale bars=100 μm.
(F) Increased expression of MEKK3, p38δ, p62, and S6 phosphorylation in human prostate tissue microarray (TMA). Box plot graphs showing a statistical analysis of MEKK3 (p=0.006), p38δ (p=0.004), p62 (p=0.003), or phospho-S6 (p=0.049) expression in prostate tumors with GS 7-10 compared to prostate tissue with GS 2-6. Results are presented as mean ± SEM. *p < 0.05, **p < 0.01.
(G) MEKK3 and p38δ expression in human prostate tumors was correlated with p62 and phospho-S6. Correlation plots showing the relationship between MEKK3/p62, MEKK3/phosho-S6, p38δ/p62, and p38δ/phospho-S6 (arbitrary units). The coefficient of correlation (r) and the p value (p) are indicated.
DISCUSSION
The mechanisms whereby cells couple nutrient availability to anabolism and cell growth are being progressively unveiled, and the role of mTORC1 in these processes is well established (Jewell and Guan, 2013; Laplante and Sabatini, 2012; Shimobayashi and Hall, 2014). A major breakthrough in the field was the identification of the lysosome as a critical organelle where mTORC1 is recruited via the Rag proteins, and then activated by as-yet-undefined mechanisms. In addition, how mTORC1 senses the availability of nutrients, and specifically of amino acids, is a fundamental problem in the field that needs to be resolved. Here we show that the signaling adapter p62, which we previously demonstrated to contribute to mTORC1 activation by amino acids (Duran et al., 2011), is phosphorylated at two specific residues by p38δ through a MEKK3/MEK3/6 driven cascade that enables the recruitment of TRAF6 to the lysosome. This creates a signaling scaffold with the core mTORC1 complex that results in the K63-polyubiquitination of mTOR and its activation in response to amino acids, but independent of insulin. Therefore, p62 emerges as a platform that facilitates the recruitment and efficient activation of mTORC1. Interestingly, the specificity of this process is provided by the selective interaction of MEKK3 with p62 through their respective PB1 domains. In this regard, although different MAPKs have previously been implicated in the negative or positive control of mTORC1, primarily in response to stress stimuli (Cully et al., 2010; Li et al., 2003; Wu et al., 2011; Zheng et al., 2011), our data reveal a specific role for p38δ in mTOR activation in the nutrient cascade as part of a PB1-directed complex. Our studies contribute to a better understanding of the activation of mTORC1 by amino acids, but also they provide context for previously reported p62 phosphorylation events. That is, recent data demonstrate that p62 is phosphorylated at S351, which serves to increase its binding affinity for Keap1 and competitively inhibits the Keap1-Nrf2 interaction (Ichimura et al., 2013). This results in the stabilization of Nrf2 and the subsequent expression of genes encoding antioxidant proteins and anti-inflammatory enzymes (Ichimura et al., 2013). Interestingly, at least one of the kinases that can target p62’s S351 is mTOR itself. Therefore, it is tempting to speculate that the MEKK3-directed phosphorylation of p62 at T269/S272 serves to activate mTORC1, which then phosphorylates p62’s S351 to activate Nrf2 to protect cells from oxidative stress. In cancer, this could be highly relevant because tumor cells need to remove excess ROS while maintaining high levels of proliferation. Therefore, our model predicts that p62 is a crucial regulator of cancer cell proliferation by influencing cell growth through mTORC1, and cell survival through an mTORC1-p62-driven anti-oxidative mechanism.
Interestingly, we have recently reported that MEKK3 is part of another PB1 complex that activates a canonical MEK4/JNK cascade to regulate inflammation in macrophages in response to lipids, another type of nutrients that can trigger an inflammatory response when present in excess (Hernandez et al., 2014). This distinct MEKK3 pathway is orchestrated by the interaction of MEKK3 with NBR1 through their respective PB1 domains (Hernandez et al., 2014). Therefore, two PB1 scaffolds, p62 in mTORC1, and NBR1 in inflammation, use MEKK3 to deliver their respective signals in response to different nutrients. How the interaction of MEKK3 with either p62 or NBR1 orchestrates the p38δ or the JNK pathways, respectively, is not clear and will likely need more detailed structural studies to be fully understood.
In summary, the work presented here describes a nutrient-sensing pathway that is selectively activated in response to amino acids and is also operative in cancer cells. This kinase cascade is organized by a platform that depends on p62 PB1-domain interactions and is highly upregulated during cancer progression. Different components of the cascade are overexpressed in PCa in a manner that is correlated with tumor stage, which suggests that the cascade is essential for tumor development. Since kinases are eminently druggable targets, our findings have the potential to open new avenues for designing novel treatments for cancer.
EXPERIMENTAL PROCEDURES
Mice
PTEN+/− and PTENfl/fl-PBcre mice were described previously (Fernandez-Marcos et al., 2009). Both mouse strains were generated in a C57BL/6 background. All mice were born and maintained under pathogen-free conditions. Animal handling and experimental procedures conformed to institutional guidelines (Sanford-Burnham Medical Research Institute Institutional Animal Care and Use Committee).
Generation of Knockout Cell Lines
To knock out genes in cell lines, guide RNAs targeting MEKK3, p38δ and p62 were designed using the CRISPR design tool at http://crispr.mit.edu/ and cloned into a bicistronic expression vector (PX458) containing human codon-optimized Cas9 fused to EGFP through T2A sequence and the RNA components. (Addgene). Additional detailed procedures are described in the Supplemental Experimental Procedures.
Statistical Analysis
All the statistical tests are justified for every Figure. Data are presented as the mean ± SEM. Significant differences between groups were determined using a Student’s t-test (two-tailed unpaired) when the data met the normal distribution tested by D’Agostino test. If the data did not meet this test, a Mann-Whitney test was used. The significance level for statistical testing was set at p < 0.05. All experiments were performed at least two or three times.
Supplementary Material
Acknowledgments
NIH Grants R01CA132847 (J.M.), R01CA172025 (J.M.), R01CA134530 (M.T.D.-M.), R01CA192642 (M.T.D.-M.) and 5P30CA030199 (M.T.D-M. and J.M.) funded this work. Additional support was provided by DoD Grants W81XWH-13-1-0353 (M.T.D-M.) and W81XWH-13-1-0354 (J.M.). We thank Maryellen Daston for editing this manuscript, Diantha LaVine for the artwork, and Wei Liu and the personnel of the Cell Imaging, Animal Facility, Histology, Functional Genomics, Proteomics and Viral Vectors Shared Resources at SBMRI for technical assistance.
Footnotes
AUTHOR CONTRIBUTIONS
J.F.L performed most of experiments of this study. A.D. contributed the prostate organoid experiments and IHC staining; M.R-C. generated CRISPR/CAS9 clones. P.A contributed to the siRNA screening. A.C. performed the proteomic analysis. M.T.D-M. and J.M conceived and supervised the project; M.T.D-M., J.F.L. and J.M. wrote the manuscript with assistance from all the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins Interact with GATOR2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of mTORC1. Cell Rep. 2014;9:1–8. doi: 10.1016/j.celrep.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M, Genevet A, Warne P, Treins C, Liu T, Bastien J, Baum B, Tapon N, Leevers SJ, Downward J. A role for p38 stress-activated protein kinase in regulation of cell growth via TORC1. Mol Cell Biol. 2010;30:481–495. doi: 10.1128/MCB.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Duran RV, Hall MN. Regulation of TOR by small GTPases. EMBO Rep. 2012;13:121–128. doi: 10.1038/embor.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Schweitzer LD, Bilate AM, Chang S, Kirak O, Lamming DW, Sabatini DM. RagA, but not RagB, is essential for embryonic development and adult mice. Dev Cell. 2014;29:321–329. doi: 10.1016/j.devcel.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Abu-Baker S, Joshi J, Galvez A, Castilla EA, Canamero M, Collado M, Saez C, Moreno-Bueno G, Palacios J, et al. Simultaneous inactivation of Par-4 and PTEN in vivo leads to synergistic NF-kappaB activation and invasive prostate carcinoma. Proc Natl Acad Sci U S A. 2009;106:12962–12967. doi: 10.1073/pnas.0813055106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewoud MJ, Goorden SM, Kassies J, Pellis-van Berkel W, Lamb RF, Elgersma Y, Zwartkruis FJ. Mammalian target of rapamycin complex I (mTORC1) activity in ras homologue enriched in brain (Rheb)-deficient mouse embryonic fibroblasts. PLoS One. 2013;8:e81649. doi: 10.1371/journal.pone.0081649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hernandez ED, Lee SJ, Kim JY, Duran A, Linares JF, Yajima T, Muller TD, Tschop MH, Smith SR, Diaz-Meco MT, et al. A macrophage NBR1-MEKK3 complex triggers JNK-mediated adipose tissue inflammation in obesity. Cell Metab. 2014;20:499–511. doi: 10.1016/j.cmet.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci. 2013;38:233–242. doi: 10.1016/j.tibs.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347:194–198. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159:163–175. doi: 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Inoki K, Vacratsis P, Guan KL. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J Biol Chem. 2003;278:13663–13671. doi: 10.1074/jbc.M300862200. [DOI] [PubMed] [Google Scholar]
- Linares JF, Amanchy R, Greis K, Diaz-Meco MT, Moscat J. Phosphorylation of p62 by cdk1 controls the timely transit of cells through mitosis and tumor cell proliferation. Mol Cell Biol. 2011;31:105–117. doi: 10.1128/MCB.00620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–296. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 2011;147:724–727. doi: 10.1016/j.cell.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Albert A, Campuzano S. Cell signaling and function organized by PB1 domain interactions. Mol Cell. 2006;23:631–640. doi: 10.1016/j.molcel.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kimple AJ, Siderovski DP, Johnson GL. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF-kappaB activation. J Biol Chem. 2010;285:2077–2089. doi: 10.1074/jbc.M109.065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol. 2013;202:1107–1122. doi: 10.1083/jcb.201307084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol. 1998;18:3069–3080. doi: 10.1128/mcb.18.5.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Sumimoto H, Kamakura S, Ito T. Structure and function of the PB1 domain, a protein interaction module conserved in animals, fungi, amoebas, and plants. Sci STKE. 2007;2007:re6. doi: 10.1126/stke.4012007re6. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Zhang YJ, Wei YH, Cho JH, Morris LE, Wang HY, Zheng XF. Rab1A Is an mTORC1 Activator and a Colorectal Oncogene. Cancer cell. 2014;26:754–769. doi: 10.1016/j.ccell.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52:495–505. doi: 10.1016/j.molcel.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA, Metallo CM, Diaz-Meco MT, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer cell. 2014;26:121–135. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XN, Wang XK, Wu SQ, Lu J, Zheng M, Wang YH, Zhou H, Zhang H, Han J. Phosphorylation of Raptor by p38beta participates in arsenite-induced mammalian target of rapamycin complex 1 (mTORC1) activation. J Biol Chem. 2011;286:31501–31511. doi: 10.1074/jbc.M111.233122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013;49:379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Wang YH, Wu XN, Wu SQ, Lu BJ, Dong MQ, Zhang H, Sun P, Lin SC, Guan KL, et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol. 2011;13:263–272. doi: 10.1038/ncb2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.