


Abstract
Mammalian nonsense-mediated mRNA decay (NMD) is a eukaryotic surveillance mechanism that degrades mRNAs containing premature translation termination codons. Phosphorylation of the essential NMD effector UPF1 by the phosphoinositide-3-kinase-like kinase (PIKK) SMG-1 is a key step in NMD and occurs when SMG-1, its two regulatory factors SMG-8 and SMG-9, and UPF1 form a complex at a terminating ribosome. Electron cryo-microscopy of the SMG-1–8–9-UPF1 complex shows the head and arm architecture characteristic of PIKKs and reveals different states of UPF1 docking. UPF1 is recruited to the SMG-1 kinase domain and C-terminal insertion domain, inducing an opening of the head domain that provides access to the active site. SMG-8 and SMG-9 interact with the SMG-1 C-insertion and promote high-affinity UPF1 binding to SMG-1–8–9, as well as decelerated SMG-1 kinase activity and enhanced stringency of phosphorylation site selection. The presence of UPF2 destabilizes the SMG-1–8–9-UPF1 complex leading to substrate release. Our results suggest an intricate molecular network of SMG-8, SMG-9 and the SMG-1 C-insertion domain that governs UPF1 substrate recruitment and phosphorylation by SMG-1 kinase, an event that is central to trigger mRNA decay.
INTRODUCTION
Nonsense-mediated mRNA decay (NMD) targets mRNAs with premature termination codons (PTC) to degradation and thus limits the expression of potentially harmful C-terminally truncated proteins (1–6). The NMD pathway involves a cascade of fine-tuned events. A current model suggests that a SURF complex comprising SMG-1, SMG-8, SMG-9, UPF1 and the eukaryotic Release Factors 1 and 3 (eRF1 and 3) is formed on a ribosome that encounters a PTC (7,8). Interaction of UPF1 with UPF2 and UPF3B (9) bound to the downstream EJC (7,10–12) triggers UPF1 phosphorylation by the SMG-1 kinase (7,8) and remodels the SURF complex to form the decay-inducing complex (7,13). UPF1 phosphorylation is considered to be a key event in NMD, as it leads to the release of eRF1 and eRF3 (14), the recruitment of SMG-5, 6 and 7 (14,15), the remodeling of the 3′ UTR mRNP by UPF1 and eventually the degradation of the PTC-containing mRNA (16–18).
The core NMD effector UPF1 is a 125 kDa protein consisting of a cysteine-histidine rich region (CH) and a 5′–3′ RNA helicase domain with nucleic acid-dependent adenosine triphosphatase (ATPase) activity (19) (Figure 1A). The C-terminal part comprises clusters of serine/threonine-glutamine (S/T-Q) motifs that are phosphorylated by phosphoinositide-3-kinase-like kinases (PIKKs). Two UPF1 phosphorylation sites are of prime importance in NMD: Thr28 in the unstructured N-terminal part and Ser1107 (isoform 1 numbering) in the C-terminal part of UPF1. Together they establish a binding platform for the NMD factors SMG-5, 6 and 7 (14). Mutation of these sites largely abrogates NMD (14). In crystal structures, UPF1 adopts different conformations depending on its binding partners, thus regulating its various functions (13,20,21). UPF1 bound to RNA and adenosine triphosphate (ATP) is characterized by a compact conformation, with the CH domain positioned close to the RNA (20). UPF1/UPF2 complex formation leads to an extended conformation of the UPF1 CH and helicase domains (13). This conformational change has been suggested to release UPF1's hold on the mRNA and to potentiate its ATPase and helicase activities, thus switching from an RNA-clamping to an unwinding mode (10,13,20). More recently, UPF1 phosphorylation by SMG-1 has been suggested to induce a conformation favoring higher helicase activity and mRNA decay (21).
Figure 1.
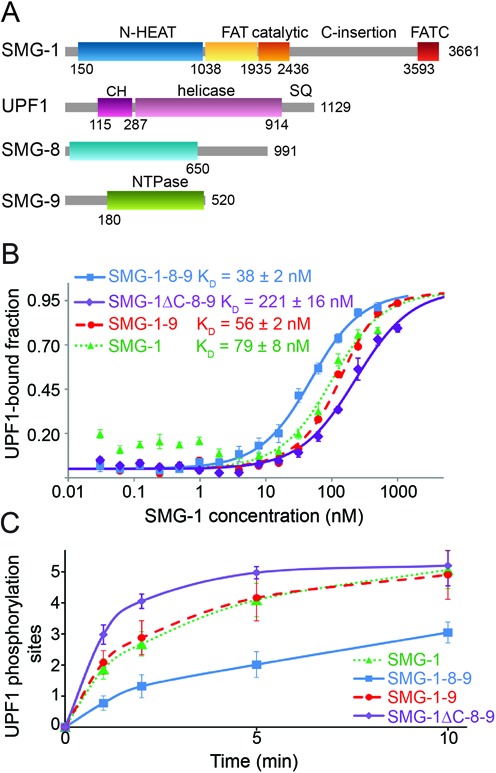
Binding and phosphorylation of UPF1 by SMG-1, SMG-1–9, SMG-1–8–9 and SMG-1ΔC-8–9. (A) Schematic representation of full-length SMG-1, UPF1, SMG-8 and SMG-9. Regions predicted to be structured are indicated with their domain boundaries. N-HEAT: N-terminal SMG-1 HEAT repeats, C-insertion: C-terminal insertion domain, CH: UPF1 cysteine-histidine-rich domain, SQ: UPF1 C-terminal unstructured region containing S/T-Q phosphorylation motifs. (B) Microscale thermophoresis experiments using fluorescently labeled UPF1 and increasing concentrations of SMG-1 (green), SMG-1–9 (red), SMG-1–8–9 (blue) or SMG-1ΔC-8–9 (violet). KD values from four independent experiments are indicated. (C) Kinetic in vitro UPF1 phosphorylation assays using SMG-1, SMG-1–9, SMG-1–8–9 and SMG-1ΔC-8–9. The number of phospho-sites in UPF1 was determined as a function of time in the presence of the same amount of SMG-1, SMG-1–9, SMG-1–8–9 or SMG-1ΔC-8–9, using ovalbumin as standard. Experiments were repeated at least three times.
The PIK-like kinase SMG-1 is a 410 kDa protein (22); its activity and stability is regulated by SMG-8 (110 kDa) and SMG-9 (60 kDa) (8). A kinase-stimulating activity was suggested for SMG-9 (23). SMG-8, which can only bind SMG-1 kinase in complex with SMG-9, seems to inhibit SMG-1 kinase activity in vitro (23). However, SMG-8 depletion in fibroblasts limited both NMD efficiency and UPF1 phosphorylation indicating that the situation in vivo is more complex (24). SMG-1 has a typical PIKK domain architecture with N-terminal HEAT repeats (22,25) (∼1000 residues) and a C-terminal kinase domain consisting of a FAT (FRAP-ATM-TRRAP) domain, a catalytic domain containing a FKBP12-rapamycin binding-like (FRB-like) domain and the FAT-C terminal domain (FATC) (Figure 1A). In the recent crystal structure of the mTOR (mammalian Target of Rapamycin) PIKK kinase domain, the catalytic domain displayed a constitutively active conformation (26), the activity of which was regulated by substrate access. Additionally, human SMG-1 contains a unique insertion of ∼1200 residues between the catalytic and the FATC domain (22) of to date unknown function (Figure 1A).
Structural studies revealed a common architecture for PIKKs with a large ‘head’ comprising the C-terminal part and a tubular ‘arm’ formed by the N-terminal HEAT repeats (27–30). In electron microscopy (EM) reconstructions of SMG-1 and SMG-1–9 and SMG-1–8–9 complexes, density corresponding to SMG-8 and SMG-9 was located in the arm next to the SMG-1 HEAT repeats (23,31). Recently, negative stain EM structures of the SMG-1–8–9-UPF1, SMG-1–8–9-UPF2 as well as SMG-1–8–9-UPF1-UPF2 complexes have been reported (31). Chemical cross-linking with glutaraldehyde was used to stabilize these rather unstable SMG-UPF complexes. The resulting negative stain EM structures revealed that UPF1 and UPF2 bind at two different sites to the head domain which comprises the SMG-1 kinase domain (31). The authors proposed that UPF1 binds to the kinase domain and UPF2 to the FRB-like domain of SMG-1 in the head and that UPF2 could be transferred to UPF1 within the SMG-1–8–9 complex (31).
Here, we determined the cryo-EM structure of the 720 kDa SMG-1–8–9-UPF1 complex and reveal the dynamic binding of UPF1 to SMG-1. UPF1 docking induces an opening of the SMG-1 head by displacing the C-insertion domain from the catalytic domain. In this complex, SMG-9 holds a central position where it likely stabilizes and properly arranges the complex components. SMG-8, SMG-9 and the SMG-1 C-insertion domain interact and jointly regulate UPF1 binding and phosphorylation of specific S/TQ sites by SMG-1. Deletion of the SMG-1 C-insertion domain in the SMG-1–8–9 complex leads to the functional deregulation of SMG-8's modulating effect on the kinase activity, indicating that SMG-8 exerts its function through interaction with the C-insertion domain of SMG1. The unique C-insertion domain thus serves as a scaffold to recruit substrates and regulate access to the kinase active site. Finally, we show that UPF2 is not required to stimulate SMG-1 kinase activity, but favors UPF1 release from the SMG-1–8–9 complex.
MATERIALS AND METHODS
Purification of UPF1 variants and UPF2 (121–1227)
UPF1 and its variants were cloned from the plasmid pCiNeo_UPF1 (11) into pFastBac-Htc (Life Technologies) using SalI and NotI and expressed in insect cells using the MultiBac baculovirus (32). UPF1(115–287) and UPF1(115–914) were expressed in Escherichia coli (13). Cells were lysed in 50 mM Hepes pH 8.0, 300 mM KCl, 5 mM β-mercaptoethanol and 10 mM imidazole supplemented with protease inhibitors (Roche). UPF1 proteins were purified using Ni-NTA affinity chromatography followed by ion exchange chromatography using a Q-Xl column (GE-Healthcare). UPF1 was eluted using a continuous salt gradient from 100 mM to 1 M KCl in 20 mM Hepes pH 8.0, 5 mM DTT. Finally, UPF1 was purified by SEC (Superdex-200, GE-Healthcare) in 20 mM Hepes pH 8.0, 300 mM KCl and 5 mM DTT. The monomer peak was concentrated, flash-frozen in liquid nitrogen and stored at −80°C. UPF2 (121–1227) was expressed and purified as described (33).
Purification of SMG-1, SMG-1–9, SMG-1–8–9 and SMG-1ΔC-8–9 complexes
The genes encoding full length SMG-1 or SMG-1 deleted from the residues 2442 to 3591 with an N-terminal streptavidin-binding peptide (SBP)-tag were introduced into the pLEXm plasmid (34). Genes encoding SMG-8 with an N-terminal hexahistidine-tag and SMG-9 were synthesized (GenScript) and subcloned into the pLEXm and the pcDNA5-frt plasmids, respectively. SMG-1 and the SMG-1–9, SMG-1–8–9 and SMG-1ΔC-8–9 complexes were expressed in HEK-293T cells following published protocols (34). A plasmid ratio of 2:1 and 2:1:1 in mass was used for the transfection of SMG-1–9 and SMG-1–8–9 (or SMG-1ΔC-8–9), respectively. After 48 h of transient transfection, cells were recovered by scrapping, pelleted by centrifugation at 1000 g (10 min), frozen in liquid nitrogen and stored at −80°C. The cells were lysed using cytobuster (Calbiochem) supplemented with protease and phosphatase inhibitors (Roche). After centrifugation (100 000 g, 30 min), the supernatant was mixed with streptavidin beads (Fisher Scientific) and incubated for 2 h at 4°C with mixing. Streptavidin beads were washed with 50 ml of SMG-1 buffer (20 mM Tris pH 9.0, 100 mM KCl, 25 mM glycine, 1 mM DTT and 5% sucrose). SMG-1, SMG-1–9, SMG-1–8–9 or SMG-1ΔC-8–9 were eluted by incubation for 30 min with SMG-1 buffer containing 2 mM biotin, followed by SEC (Superose-6, GE-Healthcare) using SMG-1 buffer. Monomeric SMG-1, SMG-1–9, SMG-1–8–9 and SMG-1ΔC-8–9 complexes were concentrated, flash-frozen in liquid nitrogen and stored at −80°C.
Negative-stain electron microscopy and random conical tilt (RCT) reconstructions
SMG-1–8–9 and SMG-1–8–9-UPF1 complexes were affinity purified using the SBP-tag and concentrated to 0.05 mg/ml. Complexes were adsorbed to carbon film for 30 s, followed by 1% uranyl acetate staining for 60 s. Hundred micrograph pairs were recorded at 45° and 0° tilt angles under low-dose conditions at room temperature on a Biotwin CM120 electron microscope (FEI) operating at 120 kV using a 4k × 4k CCD camera and a defocus of ∼−2 μm. The magnification used was 41 667× which corresponds to a pixel size of 3.6 Å. For both samples ∼12 000 tilt pairs were extracted manually by using tiltpicker (35). Untitled images were subjected to cycles of 2D multivariance statistical analysis and classification resulting in 400 (SMG-1–8–9) or 300 (SMG-1–8–9-UPF1) classes using IMAGIC-5 software (36) and XMIPP (37). For each 2D class, one random conical tilt (RCT) volume was calculated from its respective tilted particles. The RCT volumes were aligned and classified simultaneously into different classes in XMIPP MLtomo (37). For SMG-1–8–9, 390 RCT volumes resulted in six class volumes. For SMG-1–8–9-UPF1, 278 RCT volumes were used to generate three distinct volumes. The volumes for which projections correlated best with the reference-free 2D class averages were further refined using maximum likelihood 3D refinement in XMIPP (38) until convergence.
Electron cryo-microscopy and image processing
Quantifoil 300 mesh R 1.2/1.3 type grids (Quantifoil MicroTools GmbH) coated with a thin continuous carbon layer were glow discharged before usage. A total of 3.5 μl of freshly prepared SMG-1–8–9-UPF1 sample was applied to the carbon side of the grid and vitrified using a Vitrobot Mark IV (FEI, Eindhoven, Netherlands). Micrographs were recorded using the automatic data collection software EPU (FEI company) under low dose conditions on a Tecnai G2 Polara (FEI) operated at 100 kV with a Gatan 4k × 4k CCD camera and a nominal magnification of 59 000× in a defocus range between −1 and −4.5 μm. The final pixel size was 1.86 Å (object scale).
The contrast transfer function (CTF) estimation and correction of micrographs was performed in bctf (Bsoft (39)). e2boxer from EMAN2 (40) was used to manually pick 131 456 particles. The SMG-1–8–9-UPF1 and the SMG-1–8–9 negative-stain 3D reconstructions were low-pass filtered to 60 Å to avoid model bias as recommended in the Xmipp tutorial (gold-standard refinement procedure) and used as starting volumes for heterogeneity sorting in XMIPP (41). Five independent volumes were initially reconstructed in parallel by projection matching and maximum likelihood 3D refinement until convergence. Three volumes showed additional density in the head region of SMG-1 and were therefore considered to be SMG-1–8–9 complexes with UPF1 bound. The corresponding pools of particles were subjected to subsequent rounds of 3D classification to clean up the data and thus improve the quality and the homogeneity of the particle pools (42). The three final maps resulted from subsets of the data which could not be sorted further into more homogenous particle pools as assessed by 3D variance analyses (Supplementary Figure S3I). Subsequently, SMG-1–8–9-UPF1 volume 1 was refined in XMIPP to an angular sampling of 5° reaching a resolution of 17 Å (FSC criterion 0.5) using 26 653 particles (Figure 2C and Supplementary Figure S3F). SMG-1–8–9-UPF1 volumes 2 and 3 were refined to an angular sampling of 10° resulting in a resolution of 19 Å using 10 451 particles and of 20 Å using 11 161 particles, respectively (Figure 2D and E and Supplementary Figure S3G and H). The final maps were filtered to their indicated resolution.
Figure 2.
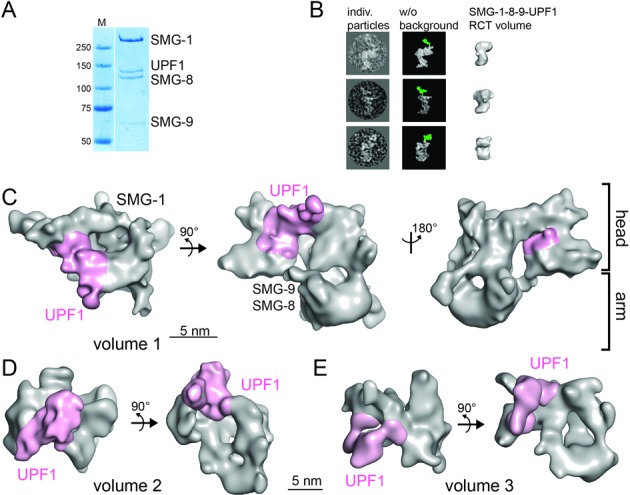
Cryo-EM of the SMG-1–8–9-UPF1 complex. (A) Coomassie-stained SDS-PAGE gel of purified SMG-1–8–9-UPF1. (B) Localization of UPF1 in the SMG-1–8–9-UPF1 complex using a polyclonal antibody against the UPF1 CH domain. Individual particles showing the anti-UPF1 antibody bound to the complex (left) are depicted next to the same particles after background removal (middle). Density corresponding to the antibody is colored in green. The RCT reconstruction of SMG-1–8–9-UPF1 is shown in a similar orientation (right). (C) Cryo-EM structure of SMG-1–8–9-UPF1 complex volume 1. SMG-8, SMG-9 and UPF1 are positioned based on results from antibody-labeling experiments ((23,31), this study). Density corresponding to SMG-1–8–9 is depicted in gray and UPF1 in magenta. (D and E) Cryo-EM structures of SMG-1–8–9-UPF1 volume 2 (D) and volume 3 (E) with UPF1 density colored in magenta.
Antibody labeling of the SMG-1–8–9 and SMG-1–8–9-UPF1 complexes
SMG-1–8–9 was mixed with a five-fold molar excess of an anti-SMG-1 antibody recognizing residues 2922–3032 in the C-insertion (Abcam 77304). After 1 h of incubation, negative stain grids were prepared. Hundred micrographs were recorded at a magnification of 40 000× using a JEOL 1200EX II. A total of 2816 particles were picked manually and subjected to 2D multivariate statistical analysis (MSA) and classification in IMAGIC-5 (36).
SMG-1–8–9-UPF1 was mixed with a five-fold molar excess of a hUPF1 antibody recognizing residues 250–300 of UPF1 (Abcam10510). After incubation (15 min), negative-stain grids were prepared. One-hundred and ten micrographs were recorded at a magnification of 40 000× using a JEOL 1200EX II. A total of 3284 particles were picked manually and subjected to 2D MSA and classification in IMAGIC-5 (36).
Chemical protein cross-linking coupled to mass spectrometry (CLMS) analysis is described in detail in Supplementary Material and Methods.
Generation of the hybrid quasi-atomic model of SMG-1–8–9-UPF1
The homology models of SMG-1 N-terminal HEAT repeats and kinase domain were generated using the HHpred server (43). In each case, best hits were used to generate the homology models. SMG-1 residues 150 to 782 were modeled based on their homology to importins (PDBs: 3W3W, 2BPT), TIP120 (PDB: 1U6G), Protein Phosphatase 2A (PDB: 4lAC) and one exportin (PDB: 3M1I). SMG-1 residues 1038 to 2436 and 3593 to 3661 were modeled using the mTOR kinase domain structure (PDB: 4JSN).
The antibody mapping results for SMG-8, SMG-9 and the SMG-1 FATC domain (23,31) were taken into account for fitting the SMG-1 N-terminal HEAT domain into the arm and the kinase domain into the back of the head. In both cases, Chimera (44) yielded unambiguous fittings with high correlation coefficients (cc = 0.85 and 0.83 respectively). The N-terminal HEAT repeat model was fitted into a continuously curved and tubular density with regular features stretching from the bottom of the arm toward the N-terminus of the kinase domain. The structure of UPF1 (13) was fitted interactively into the EM map with UCSF Chimera (44) (cc = 0.71 for helicase and cc = 0.76 for CH domain) taking into account the position determined by UPF1 antibody localization (Figure 2B and Supplementary Figure S2D) and the CLMS results for the helicase and CH domain arrangement (Figure 3B). The resulting SMG-1–8–9-UPF1 model was energy minimized in Crystallography & NMR System (CNS) Version 1.0 (45). The figures were generated with PyMOL (DeLano Scientific); the maps are displayed at ∼120% of their theoretical volume.
Figure 3.
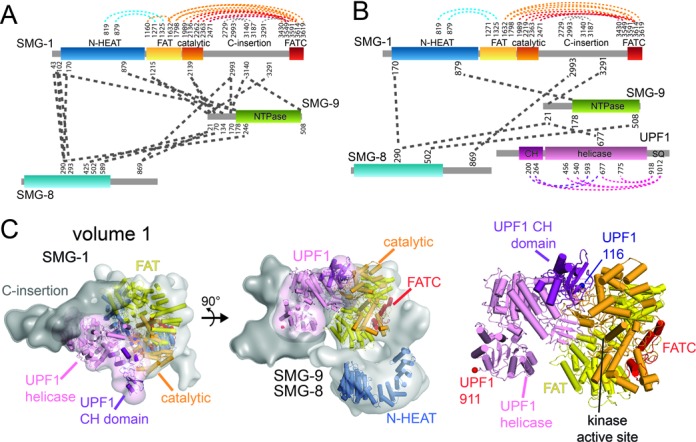
Crosslinking-mass spectrometry and model of the SMG-1–8–9-UPF1 complex volume 1. (A) Schematic representation of intra- and inter-protein cross-links identified in the SMG-1–8–9 complex. Residues involved in cross-links are indicated. (B) Schematic representation of intra- and inter-protein cross-links identified in the SMG-1–8–9-UPF1 complex. (C) Quasi-atomic model of the SMG-1–8–9-UPF1 in a top (left) and a side view (middle). The EM density of volume 1 is colored in gray for SMG-1, dark gray for the C-insertion domain and pink for UPF1. SMG-1 N-terminal HEAT repeats are colored blue, the FAT domain yellow, the catalytic domain orange, the FATC domain red, the UPF1 CH domain purple and the helicase light pink. Right: closeup view of the SMG-1-UPF1 model in the head domain. The N-terminus of the UPF1 CH domain and the C-terminus of the UPF1 helicase are indicated. See text for details.
SMG-1 kinase assays
A total of 0.05 μg of SMG-1, 0.055 μg of SMG-1–9, 0.07 μg of SMG-1–8–9 or 0.055 μg SMG-1ΔC-8–9 were mixed with 0.5 μg of UPF1 variants in SMG1 buffer. The reaction was started by the addition of 5 mM MnCl2 and 5 mM ATP (final concentration). The reaction was stopped by the addition of protein gel loading buffer and subsequent boiling of the samples for 5 min. For the UPF1 phosphorylation experiments in the presence of UPF2, 0.5 μg of UPF2 (121–1227) was included in the reaction. Proteins were separated on 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels, stained first with Pro-Q Diamond staining (Life Technologies) and then with Coomassie staining. The Pro-Q stained gel was imaged with a Typhoon scanner using 532 nm and 580 nm as excitation and emission wavelength, respectively. The bands were quantified with the ImageQuant software. The number of UPF1 phosphorylation sites was determined using the ovalbumin signal as a standard and knowing that ovalbumin contains two phosphorylated sites per molecule.
SMG-1–8–9 pull-down experiments
Six microgram of SMG-1–8–9 were mixed in SMG-1 buffer with approximately five-fold molar excess of the UPF1 variants or of the UPF1-UPF2 complex and incubated for 2 h at room temperature. In the case of the sequential addition of UPF1 and UPF2, SMG-1–8–9 was incubated for 1.5 h at room temperature with approximately five-fold molar excess UPF1, which was followed by the addition of ∼10-fold molar excess of UPF2 and 1.5 h incubation at room temperature. After centrifugation (10 min, 13 000 g at 4°C), 30 μl of streptavidin beads were added to the supernatant and incubated for 2 h at 4°C. The beads were washed three times with SMG-1 buffer and the complexes were eluted in 30 μl buffer supplemented with 2 mM biotin. Input and elution samples were analyzed on 8% SDS-PAGE gels stained with Coomassie.
Surface plasmon resonance experiments
Surface plasmon resonance (SPR) experiments were performed on a BIAcore3000 using streptavidin-coated sensor chips (GE-Healthcare). The first flow-cell was not functionalized and used as a control; the second flow-cell was functionalized with purified SMG-1–8–9 to a density of about 2000 RU. The assay buffer contained 20 mM Tris pH 9.0, 100 mM KCl, 25 mM glycine, 1 mM DTT, 0.01% Tween-20. UPF1 variants were injected at 30 μl/min during 3 min followed by a 10 min dissociation phase. To determine the binding sites, all UPF1 variants were injected at 1 nM on both flow-cells. Between each UPF1 injection, the surface was regenerated by 30 s injection of assay buffer containing 0.5 M KCl. Data were analyzed by subtracting both the control flow cell of any UPF1 injection and the buffer injection curve.
Microscale thermophoresis measurements
To obtain comparable affinity constants, full length UPF1 was fluorescently labeled and microscale thermophoresis experiments were performed. UPF1 was labeled using the nanotemper protein labeling kit reacting on the amines. A solution containing 10 μM of UPF1 and 100 μM NT-547 dye in 20 mM Hepes pH 8.0, 300 mM KCl, 2 mM DTT was incubated for 30 min at room temperature in the dark. The mixture was then loaded on a NAP-5 column (GE-Healthcare) to remove the excess free dye. The protein was recovered in 20 mM Tris pH 9.0, 100 mM KCl, 25 mM glycine, 1 mM DTT and 0.02% Tween 20. The protein contained on average one fluorescent moiety per UPF1 molecule. A constant concentration of 20 nM labeled UPF1 was mixed with increasing concentrations of SMG-1, SMG-1–9, SMG-1–8–9 or SMG-1ΔC-8–9 ranging from 0.03 to 500 nM for SMG-1 and SMG-1–8–9 and to 1 μM for SMG-1–9 and SMG-1ΔC-8–9. About 3–5 μl of each sample were loaded into hydrophilic-treated glass capillaries (Nanotemper) and the thermophoresis analysis was performed using a Nanotemper Monolith NT115 instrument (20% LED power and 80% IR-laser power). The MST curves were fitted using the Nanotemper analysis software to obtain KD values. In order to be able to superimpose the three curves the data were represented in terms of UPF1-bound fractions.
SEC-MALLS experiments are described in the Supplementary Material and Methods part.
Limited proteolysis
Purified SMG-1 C-insertion at 0.1 μg.μl−1 was digested by trypsin at a weight ratio of 1 to 100 in 20 mM Hepes pH 8, 150 mM NaCl and 2 mM β-mercaptoethanol for 2 h at room temperature. Aliquots corresponding to 1 μg of SMG-1 C-insertion were taken at different time points and analyzed on 12% SDS-PAGE gels stained with Coomassie.
RESULTS
UPF1 binding and phosphorylation is regulated by SMG-8, SMG-9 and the SMG-1 C-insertion
To investigate the impact of SMG-8 and SMG-9 on substrate binding, we performed microscale thermophoresis experiments using highly purified SMG-1, SMG-1–9 and SMG-1–8–9 complexes (Supplementary Figure S1A and B). Dissociation constants (KD) of 75, 56 and 38 nM for the affinity of SMG-1, SMG-1–9 and SMG-1–8–9, respectively, to full-length UPF1 were determined (Figure 1B and Supplementary Figure S1C, D and E). Thus, the affinity of UPF1 for SMG-1 increases by a factor of two in the presence of SMG-8 and SMG-9.
Previous experiments suggested that SMG-8 restricts the catalytic activity of SMG-1 (23). We performed kinetic phosphorylation experiments using SMG-1, SMG-1–9 or SMG-1–8–9 and full-length UPF1 as substrate. All kinase complexes were able to efficiently phosphorylate UPF1 (Figure 1C). SMG-1 and SMG-1–9 were about two-fold faster during the first five minutes of the reaction leading to four phosphorylated sites in UPF1, whereas the SMG-1–8–9 complex yielded only two phosphorylated sites under the same conditions (Figure 1C and Supplementary Figure S1F). This indicates that SMG-8 regulates SMG-1 kinase activity by restriction of UPF1 phosphorylation sites.
To investigate the role of the SMG-1 C-insertion (Figure 1A), we generated a mutant lacking this domain (SMG-1ΔC-8–9). In thermophoresis experiments, SMG-1ΔC-8–9 shows a five-fold lower affinity for UPF1 (KD = 221 nM) than SMG-1–8–9 (Figure 1B). Intriguingly, despite its lower substrate affinity the kinase activity is increased leading to complete phosphorylation of five sites in UPF1 within five minutes (Figure 1C and Supplementary Figure S1F). Apparently, SMG-8 cannot exert its regulatory effect in SMG-1ΔC-8–9. We conclude that the SMG-1 C-insertion domain together with SMG-8 controls UPF1 recruitment and phosphorylation.
Cryo-EM of the SMG-1–8–9-UPF1 complex
To understand the molecular interplay within the SMG-1–8–9-UPF1 complex, we studied its architecture by cryo-EM. The complex was prepared by mixing excess UPF1 with the SMG-1–8–9 complex, followed by streptavidin affinity purification (Figure 2A). Initial models were obtained by RCT reconstruction of SMG-1–8–9 and SMG-1–8–9-UPF1 (Supplementary Figure S2A and B). The two negative stain maps reveal an S-shape composed of the head and arm domains characteristic for PIKKs. Superimposition of the reconstructions reveals a larger, less compact head domain in the presence of UPF1 (Supplementary Figure S2C). UPF1 binding to the head was confirmed by using an antibody directed against the UPF1 CH domain. In negative-stain EM, additional density corresponding to the antibody was detected above the head domain in side and front views of raw particles (Figure 2B, green density) indicating that UPF1 is located in the front half of the head domain. These results were corroborated by 2D MSA analysis (Supplementary Figure S2D) and are in agreement with the recent SMG-1–8–9-UPF1 negative stain EM structure (31). In summary, UPF1 can be localized in the head domain, but the RCT volume does not show the complete UPF1 density due to conformational variability (see below).
For cryo-EM, 131 456 particles (Supplementary Figure S3A, B and C) were aligned against the SMG-1–8–9-UPF1 and SMG-1–8–9 RCT reconstructions as initial reference models. Subsequent heterogeneity sorting using XMIPP (37) led to three volumes possessing the typical PIKK head and arm features that contain additional density in the head domain (red arrows in Supplementary Figure S3D; and Figure 2C, D and E). The volumes were validated by comparing reference-free 2D class averages of the sub-pool of particles with projections from the respective volumes (Supplementary Figure S3E). Volume 1 was refined to 17 Å resolution (Fourier Shell Correlation criterion 0.5 (Supplementary Figure S3F)). It revealed a tubular density in the arm domain which comprises the SMG-1 HEAT repeats (Figure 2C, right view). The volumes 2 and 3 were refined to 19 Å and 20 Å resolution (Figure 2D, E and Supplementary Figure S3G and H), respectively. Moreover, we observe a second connection between the head and arm domains in each volume (Figure 2C middle view, 2D and 2E right view).
Molecular architecture of SMG-1–8–9-UPF1
Lysine-specific chemical CLMS analysis revealed inter- and intra-molecular interactions in the complex (Figure 3A and B). SMG-8 and SMG-9 cross-link to each other, in agreement with the formation of a stable binary complex. Consistent with their localization in the arm domain, the N-terminal part of SMG-1 was cross-linked to SMG-8 and SMG-9 (23,31). Importantly, SMG-8 and SMG-9 both cross-link to the C-insertion of SMG-1 (Figure 3A and B) providing additional evidence for their co-regulation of SMG-1 kinase activity.
SMG-9 has a central position in the complex which agrees with its observed stabilizing effect and activation of SMG-1 kinase by SMG-9. SMG-9 is cross-linked to SMG-1 kinase domain in absence of UPF1 and to the UPF1 helicase domain in its presence (Figure 3A and B). We therefore propose a different placement for SMG-8 and SMG-9 compared to the recently published SMG-1–8–9 model (31) where SMG-9 is closer to the head domain (Figure 2C). Direct cross-links between UPF1 and SMG-1 were not identified, likely due to the low number of lysines present in the UPF1 N- and C-terminal parts (see below).
To generate a quasi-atomic model, we placed the SMG-1 N-terminal HEAT repeats (residues 150 to 782) into the tubular density in the arm domain and the FAT-kinase-FATC homology model (residues 1038–2436 and 3593–3661) into the head domain of volume 1 (Figure 3C) as previously suggested (23,31). After fitting the FAT-kinase-FATC domain into the 3D reconstruction of the SMG-1–8–9 complex, a large part of the head domain remained unfilled (Supplementary Figure S4A). We localized the SMG-1 C-insertion region by negative stain EM using an antibody directed against a peptide in the middle of the C-insertion domain. Additional density for the antibody is detected next to the head domain, pointing to the front and side of the head (Supplementary Figure S4B). Therefore, we attribute the density corresponding to the front half of the head to the SMG-1 C-insertion. Small angle X-ray scattering and limited proteolysis indicate that the C-insertion contains several structured domains connected by flexible linkers (Supplementary Figure S5A), confirming the plasticity of this domain (Figure 3C and below).
The hydrodynamic radii, determined by SEC-MALLS (size exclusion chromatography-multi-angle laser light scattering), of the SMG-8–9 complex and of the C-terminal insertion confirmed their elongated shape (Supplementary Table S1). This is consistent with their localization in the elongated EM density next to the HEAT repeats reaching toward the C-insertion domain (Figure 3C).
Intra-UPF1 crosslinks indicate that the CH domain relative to the helicase domain adopts an intermediate conformation compared to the extended UPF1-UPF2 crystal structure (13) and the compact UPF1 crystal structure with RNA and ATP (20) (Supplementary Figure S4C). We placed UPF1 in the head domain of volume 1 (Figure 3C) such that the CH domain contacts the SMG-1 catalytic domain and the helicase domain the SMG-1 C-insertion. This placement agrees with our crosslinks and explains the observed enlargement of the head in the SMG-1–8–9-UPF1 complex (Supplementary Figure S2C).
UPF1 binding induces conformational changes in the head domain
In cryo-EM volume 2, the UPF1 helicase domain binds to the SMG-1 C-insertion and to the kinase domain (Figure 2D and Supplementary Figure S4D), whereas the CH domain virtually does not contact SMG-1. UPF1 docking occurs from the top and the head domain is more compact compared to volume 1. An intermediate conformation is observed in volume 3 (Figure 2E) with an enlarged head domain and two contact sites between SMG-1 and UPF1. UPF1 is not well defined in volume 3, but it is similarly positioned as in the recent glutaraldehyde-stabilized SMG-1–8–9-UPF1 negative stain EM structure (31).
UPF1 binding results in conformational changes in the SMG-1 C-insertion, leading to a less compact head domain. We suggest that UPF1 binds SMG-1 first on top of the head domain (volume 2) (Figure 2D and Supplementary Figure S4D). Subsequently, a rotational movement of UPF1 and dislocation of the C-insertion from the catalytic domain (volume 3) give rise to a stable complex (volume 1, Figure 3C) where UPF1 is tightly bound to the head of the SMG-1–8–9 complex.
In the quasi-atomic model corresponding to volume 1 (Figure 3C) residue 116 at the N-terminal end of the CH domain is located within a distance of ∼40 Å from the SMG-1 active site. Thus, the N-terminal phosphorylation site (Thr28) (14) could reach into the active site. Residue 911 at the C-terminus of the helicase domain is located ∼85 Å away from the SMG-1 active site in our model. In fact, the helicase domain is followed by an unstructured region, which is sufficiently long to enable the phosphorylation sites in the C-terminal part (including Ser1107) to reach into the active site (Figure 3C). We actually detected intra-UPF1 cross-links between the C-terminal part and the helicase and the CH domains (Figure 3B) supporting that the UPF1 C-terminal part traverses the helicase and the CH domains and reaches toward the SMG-1 active site.
SMG-1–8–9 binding regions in UPF1 and phosphorylation sites
Next, we biochemically defined the UPF1 regions essential for SMG-1–8–9 interaction. We generated truncated versions of UPF1 lacking N- or C-terminal sequences or both, an isoform lacking the CH domain (UPF1 ΔCH) and a version that comprises only the CH-domain (UPF1 CH) (Figure 4A). We performed pull-down experiments using SBP-tagged SMG-1 as well as SPR experiments with the different UPF1 variants (Figure 4B and C). The CH and helicase domains of UPF1 apparently do not contribute to SMG-1–8–9 binding (Figure 4B, C and Supplementary Figure S5B). Thus, the weaker binding of the UPF1 ΔCH mutant may be explained by mis-positioning of the N-terminal part (Figure 4C) and possibly by a missing, weak contact site of the CH domain to the SMG-1–8–9 complex (Figure 3C). A reduced binding was also observed for variants lacking part of the C-terminal SQ rich region (UPF1 stop1084) or 40 residues of the N-terminus (UPF1 ΔN). Furthermore, SPR clearly showed that UPF1 stop 985 bound very weakly to SMG-1–8–9 (Figure 4C). In summary, we found that UPF1 binding to SMG-1–8–9 critically depends on the presence of the C-terminal residues 985–1084.
Figure 4.

Binding and phosphorylation of UPF1 variants to SMG-1–8–9. (A) Schematic representation of UPF1 constructs used for binding and phosphorylation experiments. Deletions are indicated by amino acid residue numbers. (B) Pull-down experiments of UPF1 variants using SBP-tagged SMG-1–8–9 and streptavidin beads. Input samples (top) and eluate fractions (bottom) are shown on the Coomassie-stained SDS-PAGE. Red star: UPF1 variants. (C) SPR sensorgrams of full-length (light purple), ΔN (light blue), stop1084 (orange), ΔCH (purple), stop985 (green) CH-helicase (red) and CH (blue) variants of UPF1 injected at a concentration of 1 nM. SMG-1–8–9 was immobilized on a streptavidin-coated surface. (D) UPF1 in vitro phosphorylation assay. The signal was quantified relative to the signal of full-length UPF1 and of ovalbumin. The graph summarizes the average phosphorylation signal and the standard deviation from the quantification of four independent experiments. UPF1 4S-A denotes a quadruple mutant UPF1(SSSS-1084/1089/1107/1127-AAAA). See also Supplementary Figure S5C. (E) Streptavidin pull-down of UPF1, UPF2, UPF1-UPF2 complex or the sequential addition of UPF1 and UPF2 mixed with SMG-1–8–9. Input samples (left) and eluate fractions (right) are shown on the Coomassie-stained polyacrylamide gel. UPF1 and UPF2 proteins in the elutions are marked (red box). (F) In vitro UPF1 phosphorylation assays using SMG-1–8–9 in the presence of UPF2. The signal was quantified using ovalbumin as standard and represented as the number of phospho-sites in UPF1 after 0, 1 and 5 min. The experiment was repeated three times. See also Supplementary Figure S5D.
In phosphorylation experiments, a single 15 min time point was chosen, which corresponds to four phosphorylation sites for UPF1 full-length. The UPF1 ΔCH construct reached a level corresponding to three phosphorylation sites in 15 min (Figure 4D and Supplementary Figure S5C). It is conceivable that due to the steric constraints caused by deletion of the CH domain, the UPF1 N-terminal part (Thr28) cannot be phosphorylated. The other variants showed less than two sites phosphorylated in UPF1 (Figure 4D and Supplementary Figure S5C). UPF1 ΔN reached about two sites phosphorylated in UPF1 C-terminus, while mutation or deletion of the C-terminal phosphorylation sites (UPF1 4S-A, stop1084, stop985) led to UPF1 phosphorylation in one site only, probably Thr28. In conclusion, the UPF1 N-terminal part weakly contributes to SMG-1–8–9 binding, but it is reproducibly phosphorylated by SMG-1–8–9. Consistent with the finding that UPF1 CH-helicase does not bind SMG-1–8–9, this UPF1 isoform was not detectably phosphorylated (Figure 4D).
UPF2 releases UPF1 from the SMG-1–8–9 complex
In order to understand the role of UPF2 in the context of UPF1 phosphorylation by SMG-1–8–9, we analyzed complex formation by pull-down experiments. While UPF1 efficiently bound SMG-1–8–9 and can be pull-downed (Figure 4E), UPF2 gives rise to a weak, sub-stoichiometric band in the eluate samples. Apparently, UPF2 is mostly eluted from the complex during the three washing steps from the SMG-1–8–9 complex. A preformed UPF1-UPF2 complex purified by gel filtration is also not able to form a stable SMG-1–8–9-UPF complex. Notably, the addition of UPF2 to a preformed SMG-1–8–9-UPF1 complex disassembles UPF1 from SMG-1–8–9 (Figure 4E).
In order to understand the influence of UPF2 on UPF1 phosphorylation by SMG-1–8–9, we performed kinase assays in the presence of UPF2. We observed similar phosphorylation levels for UPF1 after 1 and 5 min in the presence and absence of UPF2 (Figure 4F and Supplementary Figure S5D), indicating that UPF2 does not activate SMG-1–8–9 kinase activity. We conclude that upon UPF2 binding UPF1 adopts a conformation incompatible with SMG-1–8–9 binding. We thus propose that UPF2 favors the release of phosphorylated UPF1 trapped in a tight complex within SMG-1–8–9. Our results are consistent with the recently published data showing a SMG-1–8–9-UPF1-UPF2 complex containing sub-stoichiometric amounts of UPF1 in the complex (31). For structural studies, the authors trapped this SMG-1–8–9-UPF1-UPF2 complex, which is rather unstable, using chemical cross-linking.
DISCUSSION
UPF1 phosphorylation by the PIK-like kinase SMG-1 is a central step in NMD as phosphorylated UPF1 provides the platform for recruitment of the mRNA decay factors. Our understanding of the underlying processes is restricted by the transient nature and the difficulty to produce these complexes. Here, we describe the dynamic interaction of SMG-1 with its regulatory factors SMG-8 and SMG-9 and the substrate UPF1. We identify a novel role for the C-terminal insertion domain of SMG-1 in substrate recruitment and regulation of kinase activity.
Recently, a negative stain EM structure of the SMG-1–8–9-UPF1 complex was described (31). The flexibility and instability of the complex was addressed using glutaraldehyde crosslinking which is known to reduce compositional and conformational heterogeneity and to stabilize compact conformations of the sample. Melero et al. report a complex where UPF1 was bound to the side of the SMG-1 head domain (31). Here, we study the native SMG-1–8–9-UPF1 complex by cryo-EM. Using state-of-the-art computational sorting we were able to address the heterogeneity in this complex and to determine the structure of the native complex embedded in vitreous ice. We identify three distinct conformational states of UPF1 docking to SMG-1–8–9, providing valuable insights into the dynamism of the kinase complex such as the opening of the head domain. One of the cryo-EM structures (volume 3) shows UPF1 bound on the side of the SMG-1 head domain, in a similar position as observed in the negative stain EM SMG-1–8–9-UPF1 structure (31).
Based on our biophysical and structural characterizations we propose the following model for UPF1 phosphorylation by SMG-1–8–9: UPF1 binds to the initially rather compact head of the SMG-1–8–9 complex (Figure 5, Supplementary Figures S2A and S4A). The UPF1 C-terminal part (residues 985–1129) which contains the (S/T-Q)-phosphorylation motifs is crucial for high-affinity binding to the SMG-1–8–9 complex (Figure 4). Our biochemical experiments show that the C-terminal insertion is essential for high-affinity substrate binding by SMG-1 (Figure 1B). It may also serve as an initial binding site for substrates. UPF1 docking induces a dislocation of the C-terminal insertion from the catalytic domain resulting in an overall opening of the head (Figure 5). This conformational change in SMG-1 may be a prerequisite for UPF1 to gain access to the kinase active site, which is restricted by the C-insertion domain. Accordingly, deletion of the C-insertion leads to hyper-activation of the kinase (Figure 1C). The C-terminal insertion appears to exert its function together with SMG-8–9 which similarly influence substrate recruitment and kinase activity (Figure 1B and C). Moreover, SMG-8 and SMG-9 interact directly with the C-insertion domain (Figure 3A and B). Full docking of UPF1 results in binding between the SMG-1 C-insertion and the kinase domain and leads to efficient phosphorylation (Figure 5). Data from CLMS indicate a novel arrangement of the UPF1 CH and helicase domains (Figures 3B and 5 and Supplementary Figure S4C), which is compatible with mRNA binding by the UPF1 helicase domain (20) as its RNA binding cleft is accessible.
Figure 5.
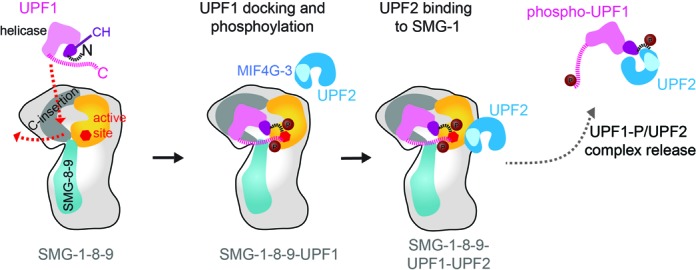
Model of substrate recruitment, phosphorylation and release by SMG-1–8–9. UPF1 is recruited to SMG-1 via the kinase domain and a binding site in the C-insertion domain. UPF1 binding induces a dislocation of the C-insertion from the kinase catalytic domain (red arrow). This rearrangement allows stable UPF1 binding and phosphorylation of N- and the C-terminal parts in the SMG-1 active site. UPF2 contacts SMG-1–8–9 next to the kinase catalytic domain via its MIF4G-3 domain (33) and facilitates release of phosphorylated UPF1.
The NMD factor UPF2 was recently shown to bind to the SMG-1 kinase domain (31,33) and to influence UPF1 binding (31). In agreement with the work by Melero et al. (31), we find that UPF2 destabilizes the SMG-1–8–9-UPF1 complex and interferes with UPF1 binding to SMG-1 in pull-down experiments (Figure 4E). In contrast to previous work (7,31), we do not observe strong UPF2 binding to SMG-1–8–9 in pull-down assays and our data suggest that UPF2 enhances the release of UPF1 from the SMG-1–8–9 complex (Figure 5) rather than activating SMG-1 kinase activity. In fact, we do not observe an additional stimulation of UPF1 phosphorylation by SMG-1 kinase in the presence of UPF2 indicating that SMG-1 is fully active without UPF2 (Figure 4F). We conclude that upon binding of UPF2 to the SMG-1–8–9-UPF1 complex, UPF1 is released together with UPF2 from SMG-1–8–9, switches to an RNA-unwinding conformation and thus triggers the re-modeling of the 3′ mRNP and the recruitment of SMG-5,-6,-7 to degrade the PTC-containing mRNA.
Our model of SMG-1–8–9 function (Figure 5) shares striking similarities with mTOR kinase function as suggested based on the crystal structure of its kinase domain (26). mTOR is the founding member of the PIK-like kinase family. Interestingly, mTOR kinase is in an intrinsically active conformation. Regulation of mTOR activity is suggested to occur through restricting substrate access (26). In mTOR complex 1 (mTORC1), the raptor subunit is suggested to provide a secondary binding site for substrate recruitment and to direct substrates to the kinase. In addition, the FRB domain of mTOR may act as a gatekeeper restricting access of substrates to the kinase active site (26). Similarly, SMG-1 kinase seems to be in an intrinsically active state. Here, we show that SMG-1 substrates are recruited by the C-terminal insertion domain. The C-insertion domain and SMG-8–9 together control kinase activity, likely by restricting access to the active site.
Notably, the 125 kDa SMG-1 C-insertion between the catalytic domain and the FATC domain is unique to mammalian SMG-1 and not found in other PIKKs. In lower eukaryotes lacking a SMG-1 C-insertion, SMG-8 has no function in NMD (46). This further supports our view of a regulatory network of SMG-8–9 and C-insertion to control substrate recruitment, to fine-tune SMG-1 activity and to thus provide an additional level of regulation in mammalian NMD.
ACCESSION NUMBERS
The cryo-EM maps have been deposited in the 3D-EM database under the accession codes EMD-3065 and EMD-3066.
Supplementary Material
Acknowledgments
We are grateful to Guy Schoehn, Marc Jamin, Wim Hagen and Maria Bacia for technical assistance and advice, and Imre Berger for discussions. We thank Stephen Cusack for plasmids pPROEXHtb-UPF1(115–287) and pPROEXHtb-UPF1(115–914). This work used the platforms of the Grenoble InstructCenter (ISBG : UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).The EM facility is supported by the Rhône-Alpes Region, the Fondation Recherche Medicale (FRM), the fonds FEDER, the Centre National de la Recherche Scientifique (CNRS), the CEA, the University of Grenoble, EMBL, and the GIS-Infrastructures en Biologie Sante et Agronomie (IBISA). We thank Radu Aricescu and Yuguang Zhao for technical support to implement mammalian cell expression.
Footnotes
Present addresses:
Aurélien Deniaud, CEA, iRTSV, Laboratoire de Chimie et Biologie des Métaux, 38000 Grenoble, France.
Simonas Masiulis, Henry Wellcome Centre for Gene Function, Department of Physiology, Anatomy and Genetics, University of Oxford, Parks Road, Oxford OX1 3PT, UK.
Kathrin Kerschgens, Thermo Fisher Scientific Biosciences GmbH, Opelstraße 9, 68789 St Leon-Rot, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
We thank the EC FP7 I3 project P-CUBE for financial support. A.D.was supported by an EC-EMBL CoFUNDS EIPOD fellowship. C.S. is supported by the European Research Council Starting grant (ComplexNMD, 281331); and by a Sinergia grant from the Swiss National Science Foundation (CRSII3_136254 /1).
Conflict of interest statement. None declared.
REFERENCES
- 1.Bidou L., Allamand V., Rousset J.P., Namy O. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol. Med. 2012;18:679–688. doi: 10.1016/j.molmed.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 3.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 4.Popp M.W., Maquat L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rebbapragada I., Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate. Curr. Opin. Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Stalder L., Mühlemann O. The meaning of nonsense. Trends Cell Biol. 2008;18:315–321. doi: 10.1016/j.tcb.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Kashima I., Yamashita A., Izumi N., Kataoka N., Morishita R., Hoshino S., Ohno M., Dreyfuss G., Ohno S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–367. doi: 10.1101/gad.1389006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamashita A., Izumi N., Kashima I., Ohnishi T., Saari B., Katsuhata Y., Muramatsu R., Morita T., Iwamatsu A., Hachiya T., et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009;23:1091–1105. doi: 10.1101/gad.1767209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lykke-Andersen J., Shu M.D., Steitz J.A. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell. 2000;103:1121–1131. doi: 10.1016/s0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 10.Chamieh H., Ballut L., Bonneau F., Le Hir H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008;15:85–93. doi: 10.1038/nsmb1330. [DOI] [PubMed] [Google Scholar]
- 11.Ivanov P.V., Gehring N.H., Kunz J.B., Hentze M.W., Kulozik A.E. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008;27:736–747. doi: 10.1038/emboj.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melero R., Buchwald G., Castano R., Raabe M., Gil D., Lazaro M., Urlaub H., Conti E., Llorca O. The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3 ‘ end. Nat. Struct. Mol. Biol. 2012;19:498–505. doi: 10.1038/nsmb.2287. [DOI] [PubMed] [Google Scholar]
- 13.Clerici M., Mourao A., Gutsche I., Gehring N.H., Hentze M.W., Kulozik A., Kadlec J., Sattler M., Cusack S. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28:2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada-Katsuhata Y., Yamashita A., Kutsuzawa K., Izumi N., Hirahara F., Ohno S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012;40:1251–1266. doi: 10.1093/nar/gkr791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohnishi T., Yamashita A., Kashima I., Schell T., Anders K.R., Grimson A., Hachiya T., Hentze M.W., Anderson P., Ohno S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell. 2003;12:1187–1200. doi: 10.1016/s1097-2765(03)00443-x. [DOI] [PubMed] [Google Scholar]
- 16.Kervestin S., Jacobson A. NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schweingruber C., Rufener S.C., Zund D., Yamashita A., Mühlemann O. Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim. Biophys. Acta. 2013;1829:612–623. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Yamashita A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes Cells. 2013;18:161–175. doi: 10.1111/gtc.12033. [DOI] [PubMed] [Google Scholar]
- 19.Czaplinski K., Weng Y., Hagan K.W., Peltz S.W. Purification and characterization of the Upf1 protein—a factor involved in translation and messenger-RNA degradation. RNA. 1995;1:610–623. [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarti S., Jayachandran U., Bonneau F., Fiorini F., Basquin C., Domcke S., Le Hir H., Conti E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell. 2011;41:693–703. doi: 10.1016/j.molcel.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 21.Fiorini F., Boudvillain M., Le Hir H. Tight intramolecular regulation of the human Upf1 helicase by its N- and C-terminal domains. Nucleic Acids Res. 2013;41:2404–2415. doi: 10.1093/nar/gks1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lempiainen H., Halazonetis T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–3073. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arias-Palomo E., Yamashita A., Fernandez I.S., Nunez-Ramirez R., Bamba Y., Izumi N., Ohno S., Llorca O. The nonsense-mediated mRNA decay SMG-1 kinase is regulated by large-scale conformational changes controlled by SMG-8. Genes Dev. 2011;25:153–164. doi: 10.1101/gad.606911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Usuki F., Yamashita A., Shiraishi T., Shiga A., Onodera O., Higuchi I., Ohno S. Inhibition of SMG-8, a subunit of SMG-1 kinase, ameliorates nonsense-mediated mRNA decay-exacerbated mutant phenotypes without cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 2013;110:15037–15042. doi: 10.1073/pnas.1300654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry J., Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112:151–155. doi: 10.1016/s0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 26.Yang H.J., Rudge D.G., Koos J.D., Vaidialingam B., Yang H.J., Pavletich N.P. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adami A., Garcia-Alvarez B., Arias-Palomo E., Barford D., Llorca O. Structure of TOR and its complex with KOG1. Mol. Cell. 2007;27:509–516. doi: 10.1016/j.molcel.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 28.Rivera-Calzada A., Maman J.P., Spagno L., Pearl L.H., Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) Structure. 2005;13:243–255. doi: 10.1016/j.str.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Sibanda B.L., Chirgadze D.Y., Blundell T.L. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams D.R., Lee K.J., Shi J., Chen D.J., Stewart P.L. Cryo-EM structure of the DNA-dependent protein kinase catalytic subunit at subnanometer resolution reveals alpha helices and insight into DNA binding. Structure. 2008;16:468–477. doi: 10.1016/j.str.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melero R., Uchiyama A., Castano R., Kataoka N., Kurosawa H., Ohno S., Yamashita A., Llorca O. Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure. 2014;22:1105–1119. doi: 10.1016/j.str.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 32.Fitzgerald D.J., Schaffitzel C., Berger P., Wellinger R., Bieniossek C., Richmond T.J., Berger I. Multiprotein expression strategy for structural biology of eukaryotic complexes. Structure. 2007;15:275–279. doi: 10.1016/j.str.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Clerici M., Deniaud A., Boehm V., Gehring N.H., Schaffitzel C., Cusack S. Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res. 2014;42:2673–2686. doi: 10.1093/nar/gkt1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aricescu A.R., Lu W.X., Jones E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D. 2006;62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- 35.Voss N.R., Yoshioka C.K., Radermacher M., Potter C.S., Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.vanHeel M., Harauz G., Orlova E.V., Schmidt R., Schatz M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 37.Scheres S.H.W., Nunez-Ramirez R., Sorzano C.O.S., Carazo J.M., Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheres S.H.W., Nunez-Ramirez R., Gomez-Llorente Y., Martin C.S., Eggermont P.P.B., Carazo J.M. Modeling experimental image formation for likelihood-based classification of electron microscopy. Structure. 2007;15:1167–1177. doi: 10.1016/j.str.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heymann J.B., Belnap D.M. Bsoft: Image processing and molecular modeling for electron microscopy. J. Struct. Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 41.Scheres S.H.W., Gao H.X., Valle M., Herman G.T., Eggermont P.P.B., Frank J., Carazo J.M. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods. 2007;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- 42.Shen B., Cheng B., Liao H., Frank J. Quantitative analysis in iterative classification schemes for Cryo-EM application. In: Herman GT, Frank J, editors. Computational Methods for Three Dimensional Microscopy Reconstruction. NY: Springer; 2014. pp. 67–96. [Google Scholar]
- 43.Soding J., Biegert A., Lupas A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF chimera— a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 46.Rosains J., Mango S.E. Genetic characterization of smg-8 mutants reveals No Role in C. elegans nonsense mediated decay. PLoS One. 2012;7:e49490. doi: 10.1371/journal.pone.0049490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.