


Abstract
Toxin-antitoxin (TA) systems play important roles in bacterial physiology, such as multidrug tolerance, biofilm formation, and arrest of cellular growth under stress conditions. To develop novel antimicrobial agents against tuberculosis, we focused on VapBC systems, which encompass more than half of TA systems in Mycobacterium tuberculosis. Here, we report that theMycobacterium tuberculosis VapC30 toxin regulates cellular growth through both magnesium and manganese ion-dependent ribonuclease activity and is inhibited by the cognate VapB30 antitoxin. We also determined the 2.7-Å resolution crystal structure of the M. tuberculosis VapBC30 complex, which revealed a novel process of inactivation of the VapC30 toxin via swapped blocking by the VapB30 antitoxin. Our study on M. tuberculosis VapBC30 leads us to design two kinds of VapB30 and VapC30-based novel peptides which successfully disrupt the toxin-antitoxin complex and thus activate the ribonuclease activity of the VapC30 toxin. Our discovery herein possibly paves the way to treat tuberculosis for next generation.
INTRODUCTION
Tuberculosis is a major global health problem. Globally, an estimated 9.0 million people developed tuberculosis in 2013, and ∼1.5 million people died from the disease (1). Furthermore, the emergence of multidrug-resistant (MDR) tuberculosis (2) and extensively drug-resistant (XDR) tuberculosis (3) is making tuberculosis more life-threatening and difficult to treat. Strains that are completely resistant to all anti-tuberculosis drugs have recently emerged (4–6), creating an urgent need for novel therapeutic targets and antimicrobial agents with novel mechanisms of action. As part of an effort to develop innovative antimicrobial agents to combat tuberculosis, our group focused on a toxin-antitoxin (TA) system chromosomally encoded by Mycobacterium tuberculosis, the causative agent of tuberculosis.
TA loci were first identified in a plasmid of Escherichia coli and confer stability of maintenance in host bacteria (7). Loss of TA loci-coding plasmids can result in growth inhibition and eventually lead to cell death because production of the labile antitoxin is not sufficient to neutralize the stable toxin under stress conditions. Since the initial discovery of TA loci in plasmids, TA loci in various bacterial chromosomes have been reported (8), and recent evidence suggests other several important cellular functions of TA systems such as multidrug tolerance (9–11), biofilm formation (12), and arrest of cellular growth under stress conditions (13).
TA systems can be grouped into three major categories (Types I, II and III) based on the antitoxin function (13). In Type I systems, an RNA antitoxin binds to the toxin-coding mRNA and inhibits translation by forming an RNA duplex. In Type II systems, a protein antitoxin binds to the protein toxin and neutralizes it by forming a stable TA protein complex. In Type III systems, an RNA antitoxin inhibits toxicity by binding to the protein toxin to form an RNA-protein complex. Type II TA systems have been studied most extensively. In a Type II system, the toxin and its cognate antitoxin molecules are encoded by two small genes and are organized in an operon. Under appropriate growth conditions, the antitoxin binds to its cognate toxin and forms a stable complex to inhibit the toxic effect (14,15). Under harsh extracellular conditions, such as elevated temperature or nutritional deprivation, stress-induced proteases degrade the antitoxin and allow its cognate toxin to be released, which eventually leads to cell death (16,17). The most prevalent Type II family is the VapBC family, which is defined by the presence of a PIN domain in the toxin component (VapC) (14,18). The PIN domain, which was named for its sequence similarity to the N-terminal domain of type IV pili protein, is a small protein domain (approximately 140 amino acids) containing four conserved acidic residues (19). Computational analysis first suggested that the conserved acidic residues in the PIN domain comprise an active site and are key functional residues in ribonuclease activity (20). Several detailed biochemical and biological studies have recently confirmed that VapC has ribo- or deoxyribonuclease activities in vitro (21–25).
TA systems have been suggested as attractive targets for the development of novel antibiotics (26–28) because artificially inducing prolonged action of the toxin by disrupting the interaction of the TA complex may lead cells to the point of no return and eventually to death (26,28,29). In M. tuberculosis, more than half of the TA systems belong to the VapBC family (47 of 88 putative TA systems) (30), and vapBC loci are tightly associated with virulence factors and pathogenicity factors (18). The VapBC family may therefore contribute to the extreme persistence and pathogenic success of M. tuberculosis (31). Although efforts to develop antimicrobial drugs based on TA systems are emerging, antimicrobial peptides based on a structural rationale remain limited to the toxin structure of Bacillus anthracis MoxT (32). Structural information on M. tuberculosis VapBC family members now includes the VapBC5, VapBC3 and VapBC15 complexes (33–35), although the detailed aspects of these structures and their functional mechanisms remain elusive.
In this work, we demonstrate that the M. tuberculosis VapC30 toxin regulates cellular growth via ribonuclease activity that requires magnesium or manganese ion as a cofactor and is inhibited by the cognate VapB30 antitoxin. We also determined the crystal structure of the M. tuberculosis VapBC30 complex at 2.7-Å resolution. Despite the structural similarity of M. tuberculosis VapBC family members, we identified distinct structural features of VapBC30 in the crystal structure, such as inactivation of the VapC30 toxin via swapped blocking by the VapB30 antitoxin. Based on our functional and structural study on M. tuberculosis VapBC30, we designed the novel antimicrobial peptides which successfully disrupt the toxin-antitoxin complex and thus activate the ribonuclease activity of the VapC30 toxin. Our two kinds of novel peptides (VapB30 and VapC30-based) may suggest a promising lead compound to develop antimicrobial peptides to treat tuberculosis.
MATERIALS AND METHODS
Cloning, expression, and protein purification
The gene encoding M. tuberculosis (strain H37Rv) VapB30 (rv0623) was amplified using primers VapB30-f/VapB30-r (Supplementary Table S1) and cloned into the expression vector pET28a (+) (Novagen) to enable the production of VapB30 with an N-terminal hexahistidine tag. The gene encoding M. tuberculosis VapC30 (rv0624) was amplified using primers VapC30-f/VapC30-r (Supplementary Table S1) and cloned into the expression vector pET21a (+) (Novagen), resulting in the production of VapC30 with an additional methionine residue at the N-terminus. Both recombinant plasmids were co-transformed into E. coli Rosetta2 (DE3). Cells were grown at 37°C in LB medium supplemented with ampicillin and kanamycin until the OD600 reached 0.5. Expression was induced by the addition of 0.5 mM isopropyl 1-thio-β-d-galactopyranoside, and the culture was grown at 37°C for 4 h. The cells were harvested by centrifugation at 5,600 g and lysed by sonication in buffer A (50 mM Tris–HCl at pH 8.0 and 500 mM NaCl) containing 10% (v/v) glycerol and protease inhibitor cocktail tablets (Roche), then centrifuged to remove cellular debris at 17,900 g. The supernatant was applied to an affinity chromatography column of nickel-nitrilotriacetic acid-agarose (Novagen) equilibrated in buffer A. The protein was eluted with buffer A containing 500 mM imidazole, and the eluted sample was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 prep-grade column (GE Healthcare). To cleave the hexahistidine tag, 100 units of thrombin from human plasma (Sigma-Aldrich) was added to 10 mg of purified protein and further incubated overnight at 20°C. The thrombin was then removed by passage over a HiLoad 16/60 Superdex 200 prep-grade column equilibrated in buffer A. The recombinant VapBC30 complex substituted with selenomethionine (SeMet) was expressed and purified as described above, except that cells were grown in M9 cell culture medium containing extra amino acids, including SeMet. For crystallization, the purified protein was concentrated to 15 mg/ml using an Amicon Ultra-15 centrifugal filter unit (Millipore). Attempts to obtain soluble M. tuberculosis VapC30 proteins using pET vectors for in vitro ribonuclease testing were unsuccessful. Therefore, the gene encoding VapC30 was amplified using primers VapC30-f/VapC30-r (Supplementary Table S1) and cloned into the pCOLD1 vector (Takara). Plasmids with the vapC30 insert were then transformed into E. coli BL21 cells carrying the plasmid pGro7 (Takara). After inoculating the cells into LB medium containing ampicillin and chloramphenicol, the expression of the GroES and GroEL chaperones was induced by the addition of 0.5 mg/ml L-arabinose. When the cells grown at 37°C reached mid-log phase, expression of the VapC30 protein was induced by the addition of 0.5 mM isopropyl 1-thio-β-D-galactopyranoside, and the cells were incubated for an additional 12 h at 15°C. The cells were then harvested by centrifugation at 5600 g, lysed by sonication in buffer A containing 10% (v/v) glycerol and protease inhibitor cocktail tablets (Roche), and centrifuged to remove cellular debris at 17,900 g. The supernatant was applied to an affinity chromatography column of nickel-nitrilotriacetic acid-agarose (Novagen) equilibrated in buffer A. The protein was eluted with buffer A containing 500 mM imidazole, and the eluted sample was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 prep-grade column (GE Healthcare), equilibrated with buffer A.
Crystallization and X-ray data collection
Crystals of SeMet-labeled and native M. tuberculosis VapBC30 were grown by the sitting-drop vapor diffusion method at 20°C. Each sitting drop was prepared by mixing equal volumes (0.5 μl each) of the protein solution and the reservoir solution. The best crystals of SeMet-labeled M. tuberculosis VapBC30 were obtained using a reservoir solution of 0.2 M sodium acetate, 0.1 M sodium citrate pH 5.5, and 5% (w/v) PEG4000. The best crystals of native M. tuberculosis VapBC30 were obtained using a reservoir solution of 0.1 M sodium citrate tribasic dihydrate pH 5.6 and 1.0 M ammonium phosphate monobasic. The crystals of SeMet-labeled and native M. tuberculosis VapBC30 were transferred to a cryoprotectant solution containing 20% (v/v) glycerol in the reservoir solution. X-ray diffraction data for SeMet-labeled and native crystals were collected at 100 K using an ADSC Quantum 315r CCD detector system (Area Detector Systems Corporation, Poway, California) at the BL-5C experimental station of the Pohang Light Source, Korea. For each image, the crystal was rotated by 1°, and the raw data were processed and scaled using the program suite HKL2000 (36). The crystals of SeMet-labeled M. tuberculosis VapBC30 belong to space group P3121 with unit cell dimensions of a = 96.31 Å, b = 96.31 Å and c = 233.03 Å. The crystals of native M. tuberculosis VapBC30 complex also belong to space group P3121 with unit cell dimensions of a = 96.38 Å, b = 96.38 Å and c = 232.79 Å. Four VapBC30 heterodimers are present per asymmetric unit in both SeMet-labeled and native M. tuberculosis VapBC30, giving Matthews parameters of 3.33 and 3.34 Å3 Da−1 and solvent contents of 63.14% and 63.15% using the calculated masses of 9140 Da for VapB30 (residues 1–84) and 14 250 Da for VapC30 (residues 1–131), respectively. Supplementary Table S2 summarizes the data collection statistics.
Structure determination and refinement
Phase calculation and density modification were conducted using Autosol of PHENIX (37). Subsequent manual model building was conducted using the program Coot (38), and the model was refined using Refmac5 (39) and PHENIX (37), including the bulk solvent correction. Five percent of the data were randomly set aside as the test data for the calculation of Rfree (40). Water molecules were added using the program Coot and were manually inspected. All refined models were evaluated using MolProbity (41). A total of 92.49% and 93.99% of the residues in the models from SeMet-labeled and native crystals, respectively, lie in favored regions of the Ramachandran plot with no outliers. The overall geometry of the two final models ranked in the 98th and 99th percentiles, with MolProbity scores of 2.00 and 1.70, respectively. The 100th percentile is the best among structures of comparable resolution. Superpositions and calculations of structural deviations were performed using the secondary structure matching (SSM) function in Coot (38). Solvent-accessible surface areas were calculated using PISA (42). The refined model was visualized and drawn using PyMOL (43). Sequence alignments were performed using ClustalX 2.0 (44) and visualized using ESPript 3.0 (45). Electrostatic surface potentials were calculated using the CHARMM force field implemented in the server-based version of PDB2PQR (46). The resulting PQR file was used to calculate and display the electrostatic surface potential using the APBS method (47) implemented in PyMOL (43). The crystallographic and refinement statistics are summarized in Supplementary Table S2.
Size exclusion chromatography coupled with multi-angle light scattering
Size exclusion chromatography was performed on a BioSep SEC-s3000 size-exclusion column (Phenomenex) using a 1260 infinity HPLC system (Agilent Technologies) and multi-angle light scattering (MALS) was measured in line using a miniDAWN-TREOS instrument with an emission at 657.4 nm (Wyatt Technology). Scattering data were analyzed with the ASTRA 6.0.1.10 software (Wyatt Technology). VapBC30 samples (loading concentrations of 40 μM) were run in 50 mM Tris (pH 8.0), 500 mM NaCl, at room temperature.
Cell growth curve measurement
For growth curve measurement of the VapBC30 complex, the operon encoding both M. tuberculosis (strain H37Rv) VapB30 (rv0623) and VapC30 (rv0624) was amplified using primers VapBC30-f/VapBC30-r (Supplementary Table S1) and cloned into pCOLD1 vector (Takara). The resulting pCOLD1-VapBC30 plasmids were transformed into E.coli BL21 cells carrying plasmid Gro7 (Takara). For the growth curve measurement of VapC30, E. coli BL21 cells carrying plasmid pGro7 were transformed with pCOLD1-His6-VapC30 plasmids. E. coli cells inoculated from single colonies on freshly streaked plates were grown overnight and diluted 20-fold into pre-warmed M9 minimal medium containing 0.1% glucose. After dilution, expression of the chaperones GroES and GroEL were induced by the addition of 0.5 mg/ml l-arabinose and incubated at 37°C. When O.D600 reached 0.5 (hour 0), VapC30 or VapBC30 expression was induced by the addition of 0.5 mM isopropyl 1-thio-β-d-galactopyranoside. The cells were then incubated at 15°C, and OD600 was measured every 60 min up to 6 h.
In vitro ribonuclease assay
We have closely followed the method described by Sisido et al. (48) to synthesize M. tuberculosis H37Rv tRNAfMet. The DNA fragments containing the entire tRNAfMET gene were amplified by PCR using primers tRNAfMET_T7-f/ tRNAfMET_T7-r (Supplementary Table S1) to enable the incorporation of a T7 RNA polymerase promoter sequence upstream. The RNA sequence containing M. tuberculosis tRNAfMet was then transcribed from the template DNA containing the entire M. tuberculosis tRNAfMet gene using MEGAshotpscript T7 transcription kit (Ambion), following the manufacturer's protocol. The size and purity of the M. tuberculosis tRNAfMET transcript was analyzed by denaturing 15% acrylamide gel electrophoresis containing 8 M urea. To remove residual template DNA, 2 units of TURBO DNase (Ambion) were added and incubated at 37°C for 60 minutes. The M. tuberculosis tRNAfMET was then purified using the MEGAclear kit (Ambion), following the manufacturer's protocol. M. tuberculosis VapC30 protein was mixed with tRNAfMet and incubated at 37°C for 60 min. Digested RNA fragments were analyzed by denaturing 15% acrylamide gel electrophoresis containing 8 M urea. Ribonuclease activity was also measured by fluorescence quenching assay using an RNase Alert kit (IDT). In this assay system, a fluorophore is covalently linked to one end of a synthetic RNA strand and quenched by a quencher group at the other end. When ribonuclease is added to the synthetic RNA containing a fluorophore-quencher pair, digestion of the RNA results in the separation of fluorophore and quencher, causing an elevation of fluorescence at 520 nm upon excitation at 490 nm by a fluorometer. The resulting fluorescence (RFU) was monitored on a SPECTRAmax GEMINI XS spectrofluorometer.
RESULTS AND DISCUSSION
Structure determination of the M. tuberculosis VapBC30 complex
We initially attempted to determine the M. tuberculosis VapBC30 complex structure using molecular replacement with the TA complexes of M. tuberculosis VapBC3 and VapBC5 as search models, but the structure determination was not successful. Therefore, a set of SAD data from a crystal of the SeMet-substituted M. tuberculosis VapBC30 complex was used to solve the phase problem at 2.85-Å resolution and to refine the model of crystal form I (Supplementary Table S2). The structure of crystal form II was solved by the molecular replacement method employing the refined model of the crystal form I as a probe. The M. tuberculosis VapBC30 complex exhibits subtle but distinct structural differences from other VapBC complex structures. These differences as well as the low levels of sequence identity may explain why molecular replacement trials with monomer or dimer models of VapBC3 and VapBC5 as search models failed.
The native M. tuberculosis VapBC30 complex diffracted to 2.70-Å resolution and was refined to Rwork/Rfree values of 0.207/0.244. Structural dissimilarities were observed when the model from the SeMet-labeled crystal (crystal form I) and the model from the native crystal (crystal form II) were compared, as described in detail below (Supplementary Figure S1). Therefore, we built and refined both crystal models. The SeMet-labeled M. tuberculosis VapBC30 complex also diffracted to 2.70-Å resolution and was refined to Rwork/Rfree values of 0.245/0.281.
For both crystal forms I and II, four VapC30 toxins (chains A, C, E, and G) and four VapB30 antitoxins (chains B, D, F and H) were present in each crystallographic asymmetric unit (Supplementary Figure S2A). For the model of crystal form I, 623 amino acid residues in four VapC30 toxins (residues 1–131 for chains A, E and G; residues 1–129 for chain C), four VapB30 antitoxins (residues 47–79 for chain B; residues 48–79 for chain D; residues 50–73 for chain F; residues 51–74 for chain H) and 55 water molecules were included in the crystallographic asymmetric unit. For the model of crystal form II, 648 amino acid residues in four VapC30 toxins (residues 1–131 for chains A, B, C and D), four VapB30 antitoxins (residues 47–76 for chains B, D, F and H) and 103 water molecules were included in the crystallographic asymmetric unit. The electron density is clearly visible for nearly all residues of the four VapC30 toxins in crystal form II (residues 1–131 for chains A, E and G; residues 1–129 for chain C), whereas the electron density of residues Gly89 to Arg92 was not visible in crystal form I, suggesting that these residues are disordered. For VapB30, we were unable to build the residues Met1–Val46 and residues Tyr77–Ala84 because of ambiguous or missing electron density in crystal form II. In crystal form I, the electron density of the residues Met1–Val46 was also absent; however, additional residues (Tyr77–Glu79) in chains B and H were visible in the electron density map.
Two heterotetramers of the M. tuberculosis VapBC30 complex are present in each crystallographic asymmetric unit of crystal forms I and II (Supplementary Figure S2A). The heterotetramer is formed by two VapBC30 heterodimer complexes via a large VapC30-VapC30 interface related by pseudo-2-fold symmetry (Figure 1A). Although there was ambiguous or missing electron density in VapB30, we confirmed the presence of nearly full residues of VapB30 (theoretical molecular weight of 9551.9 Da) using matrix-associated laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (Supplementary Figure S2B). The apparent molecular weight of the complex determined by size exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) is 92.9 ± 0.6 kDa, which corresponds well to the theoretical molecular weight of a heterooctamer model of the VapBC30 complex (93.2 kDa) (Supplementary Figure S2C). Taken together, these observations suggest that the M. tuberculosis VapBC30 exists as a heterooctamer in solution.
Figure 1.
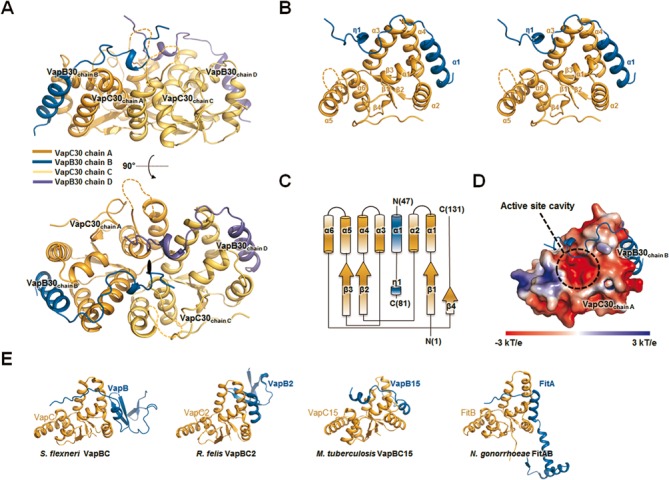
Overall structure of the Mycobacterium tuberculosis VapBC30 complex. (A) Ribbon representation of the VapBC30 heterotetramer of crystal form I, which is nearly identical to the VapBC30 heterotetramer of crystal form II. The models of the VapC30 toxin are colored in orange (chain A) and yellow (chain C), respectively. The models of the VapB30 antitoxin are colored in blue (chain B) and purple (chain D), respectively. The disordered regions (Gly89–Arg92) of the loop connecting α5 and α6 are depicted as dotted lines. The pseudo-2-fold axis is indicated as a black oval. (B) Stereo view of the M. tuberculosis VapBC30 heterodimer formed by chains A (orange) and B (blue) of crystal form I. The α–helices and β–strands of the VapC30 fold are labeled as α1 to α6 and β1 to β4, respectively. An α helix and a 310 helix of the VapB30 fold are labeled as α1 and η1, respectively. The disordered regions (Gly89–Arg92) of the loop connecting α5 and α6 are depicted as dotted lines. (C) A topology diagram of the VapBC30 complex. VapC30 and VapB30 are colored in orange and blue, respectively. The helices and strands are indicated by cylinders and arrows, respectively. (D) The electrostatic surface potential of VapC30 (chain A of crystal form I) is plotted at ±3 kT/e and shown with the ribbon representation of VapB30 (chain B of crystal form I, colored in blue). The active site cavity is indicated by black dotted circle. The electrostatic surface potential was calculated without Mg2+ ion (Please see Supplementary Figure S4A for the electrostatic surface potential calculated with Mg2+ ion). (E) Structures of homologs of M. tuberculosis VapBC30 [Shigella flexneri VapBC (PDB code 3TND); Rickettsia felis VapBC2 (PDB code 3ZVK); M. tuberculosis VapBC15; Neisseria gonorrhoeae FitAB (PDB code 2FE1)]. All structures are drawn in nearly the same orientation as in (B). The models of toxins and antitoxins are colored in orange and blue, respectively.
Overall architecture of the M. tuberculosis VapC30 toxin
M. tuberculosis VapC30 toxin features the PIN-domain motif; it contains a canonical α/β/α sandwich fold comprising four parallel β-strands in the order β4-β1-β2-β3, flanked on both sides by six α-helices (Figure 1B and C). The overall arrangement of the secondary structural elements of M. tuberculosis VapC30 is identical in crystal forms I and II. The eight M. tuberculosis VapC30 molecules in the asymmetric units of crystal forms I and II are highly similar to each other, with r.m.s. deviations of 0.4–0.5 Å for 118–127 Cα atoms. A structural similarity search of VapC30 against other protein structures using the DALI server (49) revealed homology with other PIN domain-containing proteins, despite a high level of variations in amino acid sequences (Figures 1E and 2). The closest structural homologues were (i) toxin VapC from Shigella flexneri (50) [PDB code 3TND (chains A, C, E and G), with root mean square (r.m.s.) deviations of 2.5–2.6 Å for 106 equivalent Cα positions, Z-scores of 15.6–15.9, and sequence identity of 13%]; (ii) toxin VapC from Rickettsia felis (51) [PDB code 3ZVK (chains A, B, C and D); r.m.s. deviations of 2.5–2.7 Å for 102–105 equivalent Cα positions, Z-scores of 15.2–15.6, and sequence identity of 11%]; (iii) toxin VapC15 from M. tuberculosis (35) [PDB code 4CHG (chains A, B, C, D, E and F); r.m.s. deviations of 2.1–2.4 Å for 100–105 equivalent Cα positions, Z-scores of 14.4–14.6, and sequence identity of 18%]; (iv) toxin VapC3 from Pyrobaculum aerophilum (52) [PDB code 2FE1 (chain A); r.m.s. deviations of 2.5 Å for 107 equivalent Cα positions, Z-scores of 13.7, and sequence identity of 20%] and (v) toxin FitB from Neisseria gonorrhoeae (53) [PDB codes 2H1C (chain A) and 2BSQ (chains A, B, C, and D); r.m.s. deviations of 2.8–3.0 Å for 105–108 equivalent Cα positions, Z-scores of 12.7–13.2, and sequence identities of 13–15%].
Figure 2.

Sequence alignment of PIN-domain family proteins S. flexneri VapC, R. felis VapC2, M. tuberculosis VapC15, Pyrobaculum aerophilum VapC3 and N. gonorrhoeae FitB. Strictly conserved residues and highly conserved residues are indicated by red and yellow colored boxes, respectively. The conserved acidic residues discussed in the text are marked with red asterisks. The residues involved in the binding of proximal and distal antitoxins are marked with green triangles and cyan circles, respectively.
While the overall architecture of M. tuberculosis VapC30 is similar to those of previously reported PIN domain-containing VapC family members, discrete structural dissimilarities between M. tuberculosis VapC30 and PIN domain-containing proteins were identified. First, the C-terminal residues (residues 115–131) of M. tuberculosis VapC30 do not form any secondary structure, whereas the corresponding residues of VapC family members typically form an α-helix followed by one β-strand, designated as α7 and β5, respectively (Supplementary Figure S3A). The C-terminal region of VapC30 covers one side of the active site, which is encompassed by α1, α3 and the loop (residues 87–96) between the α5 and α6 helices. Despite distinct structural differences in the C-terminal region of VapC30, one (Asp119) of the conserved key residues is located in the C-terminal region, which may play an important role in catalytic activity by constituting the active site and coordinating with a metal ion (14,18). In addition, the loop (residues 87–96) between the α5 and α6 helices of VapC30 differs significantly from previously reported VapC family members (Supplementary Figure S3B). Interestingly, positively charged residues (Lys88, Arg90, His91 and Arg92) are positioned in the loop between the α5 and α6 helices. The positively charged residues and Mg2+ ions comprise positively charged groove which is expected to interact with RNA molecules, similar to the shallow groove in the VapBC3 complex from M. tuberculosis (34) (Supplementary Figure S4A). The entire loop region is visible in crystal form II and has relatively high B-factors (Supplementary Figures S1A and S4B), while the positively charged regions (residues 89–92) are disordered in crystal form I. The loop is located in the vicinity of the entrance to the active site pocket and is involved in the hydrogen bonding network at the active site, as discussed below.
As described above, VapBC30 complexes exist as a heterooctamer both in solution and in crystals, which contain two tightly bound VapC30 homodimers. The interface areas of the VapC30 dimers of crystal forms I and II are 1100–1140 Å2 per monomer and formed by 29 residues on each monomer, representing strong interactions. The interactions in the VapC30 dimers of crystal forms I and II are nearly identical. The major contribution to the dimer formation is made by the residues on the α3, α4, α5 and α6 helices (Figure 3A and B). The dimer interfaces of chains A and C (or chains E and G) are formed by both hydrogen bonding networks and hydrophobic interactions. Among these interactions, Arg80 and Arg84 on α5 of chain A (or chain E) highly stabilize the VapC30 homodimer by forming multiple salt bridges with Glu46 (on α3’) and Asp58 (on α4’) of chain C (or chain G) within the dimeric interface (Figure 3B and C). The other hydrophilic interactions are contributed by Thr35, Ala36, Tyr38, Gly54, Val71, Asp77 and Tyr97 of the four chains (chains A, C, E and G) (Figure 3B and C). In the homodimeric interface, the hydrophobic interactions are mediated by thirteen amino acids from each monomer (Figure 3B and C): Ala36/Tyr38/Leu39/Leu43 on α3, Leu68 on β3, Ala70/Val71 on the loop connecting β3 and α3, Ala73/Ala76/Ala79/Tyr83 on α5 and Tyr97/Phe101 on α6. Interestingly, Tyr97 and Phe101 of chain A (or E) interact with additional residues on the neighboring chain C (or G) by fitting into a hydrophobic surface cavity that covers Ala36/Leu39/Leu43/Tyr97/Phe101 (Supplementary Figure S5). This observation indicates that Tyr97 and Phe101 may play a key role in stable homodimer formation.
Figure 3.
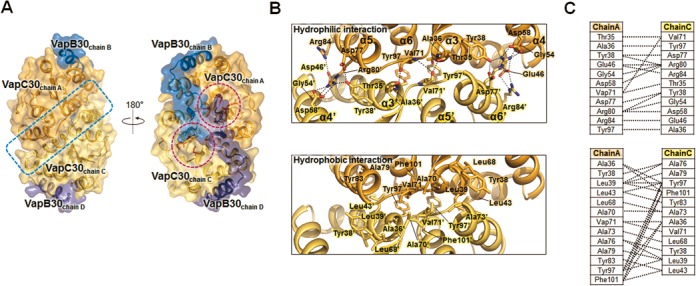
Detailed structures of the homodimeric interface of M. tuberculosis VapC30. (A) The homodimeric interface of VapC30 is indicated by a blue dotted rectangle. Each active site of VapC30 is indicated by pink dotted circles. The key residues in the active site are colored in red and also depicted in the figure. The detailed interface of the VapC30 homodimer is shown in Figure 3(B). (B) Details of the homodimeric interface of the VapC30 toxin between chains A and C. Upper: the residues involved in hydrophilic interactions at the homodimeric interface. Hydrogen bonds and salt bridges are shown as black dotted lines. Lower: the residues involved in hydrophobic interactions at the homodimeric interface. Residues participating in hydrophobic interactions are also shown in stick models and presented in the same orientation as in the upper panel. (C) Schematic diagrams of the hydrophilic interactions (upper) and hydrophobic interactions (lower) contributed by residues in the homodimeric interface of VapC30.
Distinct structural features of the M. tuberculosis VapB30 antitoxin
When we superimposed eight VapB30 monomers in the asymmetric units of the crystal forms I and II (Supplementary Figure S1B), characteristic structural features were identified. While the N-terminal regions (residues 47–51 to 70 in crystal form I; residues 47–70 in crystal form II) of the eight antitoxin models in crystal forms I and II have nearly identical structures comprising an α-helix and a loop, the C-terminal regions (residues 71 to 73–81 in crystal form I; residues 71–76 in crystal form II) of the antitoxin models revealed a variety of conformations, as described below. In the N-terminal region of both crystal forms I and II, all of the VapB30 monomers adopt α-helix conformations (residues 49–62) that exhibit minor structural differences with r.m.s. deviations of 0.2–0.3 Å for 14 Cα atoms (Supplementary Figure S1B). Notable differences were observed in the C-terminal regions of crystal forms I and II. In crystal form I, two helices (310 and α-helix) are only observed at residues Ala73–Leu75 in chain B and Ala71–Leu75 in chain D, respectively. The corresponding residues in the other six chains adopt random-coil conformations (chains F and H of crystal form I and chains B, D, F, and H of crystal form II) (Supplementary Figure S1B). While chains B and D of crystal form I are nearly identical, the other six chains exhibit different conformations, with r.m.s. deviations of 3.5–7.2 Å. In crystal form I, electron density corresponding to the C-terminal regions of the antitoxin is apparent, which allowed us to build additional residues (Tyr77–Glu79) in chains B and D. These residues are positioned in the active site of the distal VapC30 toxin, as discussed in detail below. The electron density of the corresponding residues in the other chains was not observed.
Although a structural similarity search of M. tuberculosis VapB30 with the DALI server (49) was unsuccessful, structural similarities among antitoxin molecules were observed among TA complexes when we superimposed the structure of M. tuberculosis VapB30 on those of four antitoxin molecules from other VapBC complexes. As shown in Figure 1E, the N-terminal α-helix of the M. tuberculosis VapB30 (residues 49–62) and corresponding regions of the other VapB family proteins are well matched to each other and also interact with their equivalent toxin molecules in identical positions. These molecules are (i) antitoxin VapB from S. flexneri [PDB code 3TND (chain D), r.m.s. deviation of 2.6 Å for 20 equivalent Cα positions] (50); (ii) antitoxin VapB from R. felis [PDB code 3ZVK (chains E, F, G and H), r.m.s. deviation of 0.5–1.2 Å for 13–14 equivalent Cα positions] (51); (iii) antitoxin VapB15 from M. tuberculosis [PDB code 4CHG (chains G, H, I and J), r.m.s. deviations of 0.6–1.9 Å for 8–13 equivalent Cα positions] (35); and (iv) antitoxin FitA from N. gonorrhoeae [PDB code 2H1C (chain B); r.m.s. deviations of 1.1–1.35 Å for 18–21 equivalent Cα positions] (53). With the exception of the N-terminal α-helix, the overall structure of M. tuberculosis VapB30 is distinct from those of other VapB family proteins (Figure 1E). The unique structural features of VapB30 and VapC30 and the interactions between them may be useful in designing effective antibiotic peptides or small molecules and may accelerate a rational approach to developing effective antibiotics specific for M. tuberculosis.
The active site structure of M. tuberculosis VapC30 toxin
TA systems containing PIN domains in the toxin components are classified as VapBC TA family members (14,18). PIN-domain proteins typically possess nuclease activity. The PIN-domain proteins were expected to be metallo-dependent RNases based on initial bioinformatics analysis (20) and were confirmed to have nuclease activities with various substrate specificities, as discussed below.
The active site of M. tuberculosis VapC30 is well matched to the active sites of VapC family members, despite low sequence identities (11–20%) among the VapC family members (Figure 2). The active sites of each VapC30 monomer are in close proximity and positioned in a concave surface of the VapC30 homodimeric interface. (Figure 3A and Supplementary Figure S4A). The active site of VapC30 is formed by the loop between α1 and β1, α3, α6 and the C-terminal loop (Figure 4A). Characteristic key residues essential for ribonuclease activity are present in the active site of VapC30, as observed for other PIN domain-containing proteins. The key residues are three strictly conserved acidic residues (Asp4, Glu40 and Asp99) and a fourth less well conserved acidic residue (Asp119) (Figure 2). They are positioned in a possible metal binding site that is nearly identical to equivalent residues in other VapC toxins (Figure 4B). To identify possible metal ions that might have been co-purified and co-crystallized with VapBC30 complex, dissolved crystals were analyzed by inductively coupled plasma mass spectrometry (ICP-MS) and found to contain ∼1.6 μM magnesium ion in ∼4.0 μM VapBC30 complex. Also, VapC30 showed ribonuclease activity independent of the magnesium ion concentration, when EDTA was not treated before the assay (Supplementary Figure S6). Taken together, we concluded that the major species bound in the active site of VapC30 are Mg2+ ions (Mg1). The key residues and Mg1 form extensive hydrogen bonding networks that may help to maintain structural rigidity or stabilize ionization states to modulate the turnover rate and control the local charge balance during the reactions. The hydrogen bonding network includes hydrophilic interaction clusters of Mg1/Asp4/Ser6/Ser102, Mg1/Glu40 and Mg1/Lys88 (only in crystal form II)/Asn96/Asp99/Asp119 (Figure 4A). The loop between α5 and α6 displays two different structures as shown in Supplementary Figure S1A, despite the high structural similarity of the active sites in crystal forms I and II. The entire region of the loop (residues 87–96) between α5 and α6 is visible in crystal form II, while the positively charged portion (residues 89–92) of the loop is disordered in crystal form I. This result indicates that the different orientations of Asn96 in crystal form I may arise from the flexibility of the loop and affect the active site conformation. Asn96 interacts with Asp99 and Asp119 in chains A and G, while the side chain of Asn96 in chains C and E exhibits a different orientation and does not interact with those residues (Supplementary Figure S7). In addition, the C-terminal loop (residues 115–131) does not form any secondary structure in the crystal and is located close to the active site, in contrast to the other VapC toxins described above.
Figure 4.
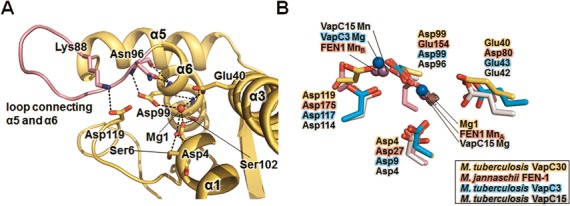
Detailed active site structures of M. tuberculosis VapC30 and homologs. (A) Close-up view of the M. tuberculosis VapC30 active site (chain A of crystal form II). The bound Mg2+ (Mg1) is shown as a red sphere, and the loop (residues 87–96) connecting α5 and α6 is colored in pink. The positively charged region of the loop (residues 89–92) is disordered in crystal form I. The key residues engaged in the hydrogen bonding network are also shown in the stick models. (B) Superposition of key conserved residues of M. tuberculosis VapC30 (yellow carbons) with M. jannaschii FEN-1 (pink), M. tuberculosis VapC3 (cyan), and M. tuberculosis VapC15 (grey). The Mn2+ ions (from M. jannaschii FEN-1 and M. tuberculosis VapC15) and Mg2+ ions (from M. tuberculosis VapC3 and VapC15) are represented as purple and blue spheres, respectively. Mg1 is represented as a red sphere with its difference electron density map (mFo-Fc; contoured at 1σ).
More importantly, a magnesium ion (Mg1) in the hydrogen bonding network interacts with the key residues Asp4, Glu40 and Asp99 and superimposes well with one manganese metal ion of Methanococcus jannaschii FEN-1 (Figure 4B). To understand the structural and functional implications of Mg1 ion, we superimposed M. tuberculosis VapC30 with Flap endonuclease-1(FEN-1) from M. jannaschii (PDB code 1A76), VapC3 from M. tuberculosis (PDB code 3H87), and VapC15 from M. tuberculosis (PDB code 4CHG), which feature two or one metal ions in each active site (34,35,54) (Figure 4B). The structures of FEN-1 from M. jannaschii, VapC3 from M. tuberculosis, and VapC15 from M. tuberculosis exhibit similarities with the structure of M. tuberculosis VapC30, including the overall architecture of the active site, with r.m.s. deviations of 3.3–3.4 Å for 76–77 equivalent Cα positions (for M. jannaschii FEN-1), 2.0 Å for 91 equivalent Cα positions (for M. tuberculosis VapC3), and 2.1–2.4 Å for 100–105 equivalent Cα positions (for M. tuberculosis VapC15). These structures include metal ions in the active sites that are important for their catalytic functions. A previous mutational study of the Mn2+ coordinating residues in M. jannaschii FEN-1 suggested that one metal ion (MnA; corresponding to Mg1 of M. tuberculosis VapC30) can accommodate phosphate hydrolysis, while the other ion (MnB) stabilizes the interactions between the protein and nucleic acids. Mg1 of M. tuberculosis VapC30 likely corresponds to MnA of M. jannaschii FEN-1 and may play a role in nucleophilic attack during the reaction. Consistent with this analysis, our ribonuclease experiments revealed that the ribonuclease activity was increased significantly upon the addition of both Mg2+ and Mn2+ ions, as described below. A possible metal ion corresponding to MnB was not observed in M. tuberculosis VapC30 due to the structural dissimilarity of the equivalent secondary structures. M. tuberculosis VapC30 may have a two-metal-ion mechanism similar to that previously suggested (54).
M. tuberculosis VapC30 toxin inhibits cellular growth through both Mg2+ and Mn2+-dependent ribonuclease activity
Most PIN domain-containing proteins, including VapC toxins, recognize specific substrates and exhibit ribonuclease activities. PAE2754 from P. aerophilum and VapC4 from M. tuberculosis both exhibit weak nuclease activities for single-stranded DNA and bacteriophage MS2 RNA, respectively, while FitB (the PIN domain toxin from the fast intracellular trafficking locus) from Neisseria gonorrhoeae has no nuclease activity (21,53,55). VapC from Haemophilus influenzae and the VapBC5 complex from M. tuberculosis exhibit ribonuclease activities for total RNA and double-stranded RNA as substrates, respectively (23,33). A more recent study of VapC toxins from S. flexneri and Salmonella typhimurium revealed specific cleavage of tRNAfMet at a single site between the stem and loop of its anticodon (25). In this work, we have identified M. tuberculosis VapC30 as a ribonuclease that regulates cellular growth and exhibits distinct features in its mode of action and metal dependency, despite common characteristics with the VapC toxins from S. flexneri and S. typhimurium.
To determine whether the expression of VapC30 causes growth arrest or cell death, we performed an expression test of VapC30 under the control of an isopropyl 1-thio-β-d-galactopyranoside-inducible promoter in E. coli. As shown in Figure 5A, E. coli cells expressing VapC30 did not grow when the media was supplemented with isopropyl 1-thio-β-d-galactopyranoside. When we co-expressed VapC30 with VapB30, cells expressing both VapC30 and VapB30 grew at the same rate as control cells, confirming that VapB30 neutralizes the inhibitory effect of VapC30 on E. coli cell growth.
Figure 5.
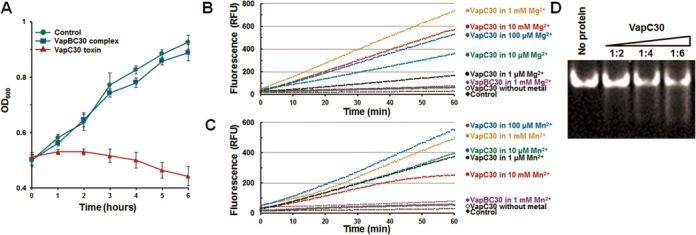
M. tuberculosis VapC30 regulates cellular growth through both Mg2+ and Mn2+-dependent ribonuclease activity. (A) Expression of the VapC30 or VapBC30 complex was induced at hour 0, and cell growth was monitored by measuring the optical density at 600 nm (OD600). Red curves indicate cells expressing the VapC30, and the blue curves indicate cells co-expressing the VapC30 and VapB30. The green curves indicate cells carrying empty pCOLD1 vector and pGro7 plasmids. The data (OD600) represent the average of experiments performed in triplicate, with standard deviations indicated by error bars. (B and C) Fluorescence quenching assay. In this assay system, fluorescence increases when the substrate RNA is cleaved. For the assay, 10 μM VapC30 (or VapBC30), 0.5 M NaCl, 20 mM Tris-HCl buffer at pH 8.0, 40 units of RiboLockTM RNase inhibitor (Thermo Scientific), and various concentrations of (B) MgCl2 or (C) MnCl2 were included in a 50 μl reaction volume and incubated at 37°C. For the assay, possible bound metal ions were removed before the assay (see main text for details). Control contained 10 mM MgCl2 (or 10 mM MnCl2), 0.5 M NaCl, 50 mM Tris-HCl buffer pH 8.0, and 40 units of RiboLockTM (Thermo scientific) RNase inhibitor. Each experiment was performed in triplicate. (D) M. tuberculosis tRNAfMET (1 μM) was used to analyze the ribonuclease activity of VapC30 (2, 4, and 6 μM, respectively). For the assay, 0.5 M NaCl, 20 mM Tris-HCl buffer at pH 8.0, and 40 units of RiboLockTM RNase inhibitor (Thermo Scientific) and 1 mM MgCl2 were included in a 10 μl reaction volume and incubated at 37°C for 60 minutes. Digested RNA fragments were analyzed by denaturing 15% acrylamide gel electrophoresis in the presence of 8 M urea.
To identify the ribonuclease activity and metal dependency of VapC30, we performed a fluorescence quenching assay in vitro. PIN domain-containing VapC family members use divalent metal ions such as Mg2+ and Mn2+ for enzymatic catalysis. For M. tuberculosis VapC30, the cleavage of RNA molecules increased significantly upon the addition of Mg2+ or Mn2+ ions (Figure 5B and C). To determine the optimal concentrations of Mg2+ and Mn2+ ions, we have added varying amounts of Mg2+ or Mn2+ ions (1 μM, 10 μM, 100 μM, 1 mM and 10 mM) to VapC30 proteins (10 μM) and performed fluorescence quenching assay (Figure 5B and C). Possible bound metal ions were removed before the assay by adding 10 mM EDTA to protein solutions, followed by dialysis against 20 mM Tris-HCl buffer containing 500 mM NaCl, pH8. The cleavage of RNA molecules significantly increased upon the addition of Mg2+ or Mn2+ ions. Also, it was observed that addition of 1 mM Mg2+ offered the highest fluorescence compared to addition of 1 μM, 10 μM, 100 μM and 10 mM of Mg2+ (Figure 5B). The optimal concentration of Mn2+ was determined to be 100 μM (Figure 5C). This result indicates an important role of Mg2+ and Mn2+ ions in enzymatic catalysis. The Mg2+ and Mn2+ dependency of M. tuberculosis VapC30 is consistent with crystal structures of VapC15 from M. tuberculosis (35), which revealed the presence of both metal ions in their active sites. The ribonuclease activity of the M. tuberculosis VapBC30 complex was significantly lower than the VapC30 toxin. However, we did not observe complete inhibition of the ribonuclease activity of the VapBC30 complex. This partial inhibition is likely due to the presence of detergent in the assay buffer, which may sequester the TA complex, as reported previously (33).
M. tuberculosis VapC30 did not exhibit a nuclease activity against DNA molecules, including the vapC30 gene region. However, M. tuberculosis VapC30 cleaved M. tuberculosis tRNAfMet, as reported for S. flexneri and S. typhimurium (25) (Figure 5D). However, M. tuberculosis VapC30 exhibits a slightly different mode for the cleavage of tRNAfMet substrate. The M. tuberculosis tRNAfMet molecules were totally degraded by increasing the amount of M. tuberculosis VapC30, and small fragmented tRNAfMet was not observed. For S. flexneri and S. typhimurium, VapC degraded E. coli tRNAfMet and produced small fragmented tRNAfMet. The experimental result also indicates that M. tuberculosis VapC30 is an endoribonuclease similar to S. flexneri VapC and S. typhimurium VapC.
Structural insights into the novel inactivation of the VapC30 toxin via swapped blocking by the VapB30 antitoxin
VapB antitoxin blocks the enzymatic function of the cognate VapC toxin by forming the VapBC toxin-antitoxin complex. The inactivation of VapC proteins occurs via the unique structural mechanism of complex formation between the toxin and antitoxin. For example, a single Rickettsia felis VapB2 binds to a VapC2 homodimer and interrupts the access of substrate molecules to each active site of the VapC2 homodimer (51). In M. tuberculosis VapBC3 and VapBC5, a single antitoxin binds tightly to its single cognate toxin and directly blocks the active sites of both VapC3 and VapC5 (33,34). In contrast, our structure of M. tuberculosis VapBC30 reveals a novel inactivation of VapC30 via swapped blocking by VapB30, as described below. In addition, the VapB30 structures in crystal forms I and II reveal multiple conformations of the C-terminal regions, suggesting a possible mechanism for the inactivation of VapC30 by VapB30.
The interface areas per monomer between VapB30 and VapC30 are calculated to be 800–1100 Å2 for crystal form I (or 930–1070 Å2 for crystal form II), indicating that VapBC30 is a stable complex. The interactions between VapB30 and VapC30 are formed entirely through the complex and can be classified into two regions according to the interacting regions in VapB30: (i) N-terminal regions (residues 49–69) comprising an α-helix (residues 49–62) and a loop and (ii) C-terminal regions (residues 71–81 in chains B and D, residues 71–73 and 71–74 in chains F and H, respectively, of crystal form I; residues 71–76 in chains B, D, F and H of crystal form II). In the N-terminal region, the interactions between VapB30 and VapC30 are highly conserved in the eight monomers in crystal forms I and II. The residues contributing to hydrophobic interactions in the N-terminal region of VapB30 are Leu53/Ile56/Leu63/Val65/Val66/Ile74/Leu75, which interact with Leu12/Val25/Leu43/Val44/Ala47/Phe49/Gly50/Pro52/Leu59/Trp60 in VapC30. The residues involved in hydrophilic interactions in the N-terminal region of VapB30 are Leu49/Arg50/Glu52/Arg57/Arg59/Val66/Asp67 and interact with Asp14/Glu22/Arg48/Glu56/Arg63 in VapC30 (Figure 6A and B). Interactions mediated by Leu49 or Arg50 of VapB30 are observed only in crystal form II, and the interaction contributed by Glu52 is observed only in chain B of crystal form I. Among the residues contributing to interactions between VapB30 and VapC30, Arg57 in VapB30 exhibits strong interactions with both Asp14 and Glu22 in VapC30.
Figure 6.
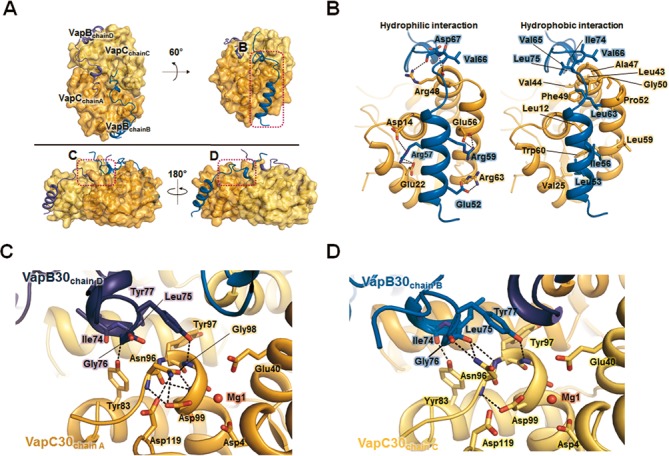
Interactions between M. tuberculosis VapC30 and VapB30. (A) Overview of the heterotetrameric complex shown in surface representations. The pink dotted rectangles indicate the views shown in (B), (C) and (D). (B) The interface between VapC30 and its proximal VapB30 is shown for hydrophilic interactions (left) and hydrophobic interactions (right). Residues participating in hydrophilic interactions (left) are shown in the stick models, and hydrogen bonds and salt bridges are shown as black dotted lines. Residues participating in hydrophobic interactions (right) are shown in the stick models. VapC30 and VapB30 are colored as in Figure 1. (C and D) Recognition and inhibition of VapC30 chain A by its distal VapB30 chain D (C) and VapC30 chain C by its distal VapB30 chain B (D). Residues participating in hydrophilic interactions are shown in the stick models, and hydrogen bonds are shown as black dotted lines.
While the N-terminal region of VapB30 is structurally conserved in other VapB family members, the structure of the C-terminal region of VapB30 is clearly distinct from those of other VapB family members, as discussed above. The C-terminal residues exhibit large deviations ranging from 3.2 to 9.4 Å between equivalent Cα atoms when VapB30 is compared with other homologs. The residue with the greatest deviation is Gly76, with an r.m.s. deviation of 9.4 Å between equivalent Cα atoms. Surprisingly, our VapBC30 complex structures revealed that the C-terminal region of the VapB30 antitoxin interacts with residues of the distal VapC30 toxin. The C-terminal regions of VapB30 may interrupt enzymatic catalysis of the distal toxin via a swapped inactivation process (Figure 6A, C and D). The interactions between the C-terminal regions of antitoxin VapB30 (chains B and D) and its distal toxin VapC30 are shown in the Figure 6C and D. When the interactions of VapB30 (chain D) and VapC30 (chain A) (Figure 6C) with VapB30 (chain B) and VapC30 (chain C) (Figure 6D) were compared, some conformational differences were revealed. In chain A of VapC30, Asn96 engages in intramolecular hydrogen bonding with active site residues Asp99 and Asp119 (Figure 6C). However, in chain C of VapC30, Asn96 is hydrogen bonded to the main chain carbonyl oxygens of both Leu75 and Gly76 of VapB30 (chain B). These interactions reorient the side chain of Asn96 of VapC30 by a distance of 2.8 Å, pushing Asn96 away from the key active site residues of Asp99 and Asp119 (Figure 6D). This conformational change may orient the key active site residues in a catalytically unfavorable position. In addition, a few but distinct hydrophobic interactions were observed between two VapB30 antitoxins (chains B and D). The interactions are contributed by both Leu75 and Tyr77 in chains B and D of two VapB30 monomers and may strengthen the inactivation of VapC30 toxins through the cooperative actions of two VapB30 antitoxins. The C-terminal regions of VapB30 antitoxins in the other chains have fewer interactions with their distal VapC30 toxins (Supplementary Figure S8). However, interactions between VapB30 and its distal VapC30, mediated by VapC30 Tyr83 and VapB30 Ala73 (or Ile74), are observed in seven of the eight chains of crystal forms I and II, indicating an important role of VapC30 Tyr83.
As we have described above, a role of the Mg2+ ion in the enzymatic catalysis of VapC30 is also suggested by the crystal structure of VapC30. The locations of Mg1 molecules in our crystal structure and interacting residues are in good agreement with those in other reported structures (34,35,54). Interestingly, no Mg1 molecule is observed in chains E and G of VapC30 (crystal form I) or in chain G of VapC30 (crystal form II). Among these three chains, the entire region of the loop between α5 and α6 is only visible in chain G of VapC30 (crystal form II). The clear electron density map of the loop can be explained by a loss of flexibility, which may be affected by the closely positioned VapB30. By contrast, the four loops between α5 and α6 are disordered in crystal form II. This flexibility of the loop may not allow interaction with Asp119 on the C-terminal loop. Moreover, only chain G of VapC30 (crystal form II) exhibits an intermolecular interaction between Tyr83 and Lys88. The conformation of the active site in chain G of VapC30 (crystal form II) may explain the absence of Mg1 and resulting inactivation of VapC30. Our two crystal forms (crystal forms I and II) may explain how VapB30 stabilizes the loop between α5 and α6 and affects the catalytic activity of VapC30 by disrupting metal binding.
Disruption of VapB30-VapC30 interaction via designed peptides
The crystal structure of VapBC30 shows that the heterodimeric interface of the VapBC30 complex is mainly composed of three structural components; α1-helix of VapB30 (residues 49–62), α2-helix of VapC30 (residues 17–27), and α4-helix of VapC30 (residues 52–65) (Figure 6B). Any potential disruptor of the toxin-antitoxin interactions should be designed to block the interface between toxin protein and antitoxin protein, without affecting the toxicity of toxin protein. We therefore designed peptides either to mimic the α1-helix of VapB30, competing with VapB30 for interaction with VapC30, or to mimic the α2 and α4-helices of VapC30, competing with VapC30 for interaction with VapB30. Three designed peptides are 8-residue peptide I (Glu-Leu-Ala-Ala-Ile-Arg-His-Arg), 17-residue peptide II (Asp-Glu-Pro-Asp-Ala-Glu-Arg-Phe-Glu-Ala-Ala-Val-Glu-Ala-Asp-His-Ile), and 9-residue peptide III (Arg-Phe-Gly-Glu-Pro-Gly-Gly-Arg-Glu). Peptide I mimics the α1-helical region of VapB30 (residues 52–59), which contains five (Glu52, Leu53, Arg57, Arg59 and Ile56) of eight residues engaged in interactions with VapC30. Peptide II mimics the α2-helical region of VapC30 (residues 14–30), which contains four (Asp14, Glu22, Val25 and Glu26) of 17 residues engaged in interactions with VapB30. Peptide III mimics the α4-helical region of VapC30 (residues 48–56), which contains five (Arg48, Glu56, Phe49, Gly50 and Pro52) of nine residues engaged in interactions with VapB30.
To investigate the ability to disrupt the VapB30-VapC30 interaction, in vitro, the three peptides were mixed with VapBC30 complex, and the subsequent ribonuclease activities were measured (Figure 7 and Supplementary Figure S9). Compared with the VapBC complex, all three peptides were proven its capability of disrupting the toxin-antitoxin interaction to some extent in a concentration dependent manner (10–100 μM). Interestingly, peptides II and III, which mimic the VapC30, were shown to disrupt the VapBC30 complex more effectively than peptide I, which mimics the VapB30. Although the previously reported peptides modulated the toxin-antitoxin interaction by mimicking the structure of antitoxin (32,56), our results indicate that mimicking the toxin structure could be a more effective strategy to design an antimicrobial peptide targeting the toxin-antitoxin interactions. Of the two peptides which mimic the VapC30, peptide III (9-residue peptide containing five residues engaged in interactions with VapB30) was shown to disrupt the VapBC30 complex more effectively than peptide II (17-residue peptide containing four residues engaged in interactions with VapB30). The peptide III may allow increased binding as the shorter peptides need a smaller configurational entropic cost to overcome (57). In addition, the hydrophobic interactions (Phe49/Gly50/Pro52 in VapC30 interact with Leu63/Val65/Val66 in VapB30) observed between the α4 of VapC30 and the C-terminal region of VapB30 may contribute to the specific inter-molecular interactions between peptide III and VapB30.
Figure 7.
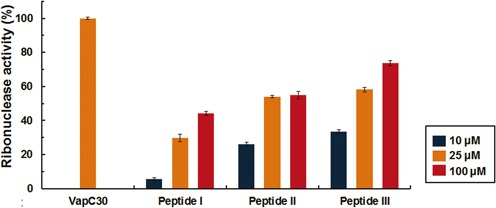
Fluorescence quenching assays with 10 μM VapC30 (or VapBC30 complex) and various concentrations of the peptides [10 μM (blue), 25 μM (orange), and 100 μM (red)]. VapBC30 complex and the peptides were incubated at 37°C for 60 min before measuring the fluorescence. Fluorescence (RFU) obtained with the 10 μM VapC30 was taken as 100% and fluorescence (RFU) obtained with the 10 μM VapBC30 complex was taken as 0%. For the assay, 0.5 M NaCl, 20 mM Tris–HCl buffer at pH 8.0, 1 mM MgCl2, and 40 units of RiboLock™ RNase inhibitor (Thermo Scientific) were included in a 50 μl reaction volume. Error bars represent the standard deviation of three replicate reactions.
CONCLUSION
M. tuberculosis is the causative agent of tuberculosis and contains an unusually high number of VapBC systems (30). The VapBC system, which is implicated in dormant state formation (31), virulence (58), and stress response (59), is an interesting target for the rational design of an antimicrobial peptide against tuberculosis (26). Via biochemical analysis, we confirmed that M. tuberculosis VapC30 is an Mg2+ and Mn2+ dependent ribonuclease that inhibits cellular growth. Although the crystal structures of VapC from P. aerophilum (52) and VapBC complex from M. tuberculosis (35) revealed the presence of manganese ions in their active sites, previous in vitro studies of VapC from various organisms have indicated a dependence of VapC ribonuclease activity on Mg2+ ions (25,33,35,60,61). We also determined the crystal structure of the M. tuberculosis VapBC30 complex at 2.7-Å resolution. The crystal structure reveals a novel inactivation of VapC30 via swapped blocking by VapB30. Finally, based on structural and functional studies of the M. tuberculosis VapBC30 complex, we designed the peptides to disrupt the toxin-antitoxin interactions. Our designed peptides mimic the helical regions of the VapB30 or VapC30 in the heterodimer interface of the VapBC30 complex, and they were capable of disrupting the interactions between VapBC30 complex in vitro. Our approach described here may form a foundation for the design of antimicrobial peptides targeting toxin-antitoxin systems, and the designed peptides may prove to be viable candidates in the development of anti-tuberculosis drugs.
ACCESSION NUMBERS
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under the accession codes 4XGR for the model from SeMet-labeled crystal (Crystal form I) and 4XGQ for the model from native crystal (Crystal form II).
Supplementary Material
Acknowledgments
We thank the beamline staffs at Pohang Light Source, Korea (BL-5C and BL-7A) and Photon Factory, Japan (BL-5A and BL-17A) for assistance during X-ray diffraction experiments.
Footnotes
Present address: Ji-Hun Kim, Research Institute for Biomedical & Health Science, College of Biomedical and Health Science, Konkuk University, Chungju, Chungbuk 380-701, Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea [NRF-2007–0056817, 2012R1A2A1A01003569, 2015R1A2A1A05001894]; Korean Government, Korea Healthcare Technology R&D project, Ministry for Health, Welfare and Family Affairs, Republic of Korea [A092006]; 2014 BK21 plus Project for Medicine, Dentistry and Pharmacy.
Conflict of interest statement. None declared.
REFERENCES
- 1.World Health Organization (WHO) Global Tuberculosis Report 2014. World Health Organization; 2014. [Google Scholar]
- 2.Centers for Disease Control (CDC) Nosocomial transmission of multidrug-resistant tuberculosis among HIV-infected persons–Florida and New York, 1988–1991. MMWR. Morbidity Mortality Weekly Rep. 1991;40:585. [PubMed] [Google Scholar]
- 3.Gandhi N.R., Moll A., Sturm A.W., Pawinski R., Govender T., Lalloo U., Zeller K., Andrews J., Friedland G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368:1575–1580. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]
- 4.Migliori G., De Iaco G., Besozzi G., Centis R., Cirillo D. First tuberculosis cases in Italy resistant to all tested drugs. Eur. Surveill. 2007;12:E070517. doi: 10.2807/esw.12.20.03194-en. [DOI] [PubMed] [Google Scholar]
- 5.Udwadia Z.F., Amale R.A., Ajbani K.K., Rodrigues C. Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 6.Velayati A.A., Masjedi M.R., Farnia P., Tabarsi P., Ghanavi J., ZiaZarifi A.H., Hoffner S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest. 2009;136:420–425. doi: 10.1378/chest.08-2427. [DOI] [PubMed] [Google Scholar]
- 7.Ogura T., Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. U.S.A. 1983;80:4784–4788. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerdes K., Rasmussen P.B., Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc. Natl. Acad. Sci. U.S.A. 1986;83:3116–3120. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vázquez-Laslop N., Lee H., Neyfakh A.A. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 2006;188:3494–3497. doi: 10.1128/JB.188.10.3494-3497.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falla T.J., Chopra I. Joint tolerance to β-lactam and fluoroquinolone antibiotics in Escherichia coli results from overexpression of hipA. Antimicrob. Agents Chemother. 1998;42:3282–3284. doi: 10.1128/aac.42.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korch S.B., Hill T.M. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J. Bacteriol. 2006;188:3826–3836. doi: 10.1128/JB.01740-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X., Wood T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011;77:5577–5583. doi: 10.1128/AEM.05068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi Y., Inouye M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 2011;9:779–790. doi: 10.1038/nrmicro2651. [DOI] [PubMed] [Google Scholar]
- 14.Sevin E.W., Barloy-Hubler F. RASTA-Bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol. 2007;8:R155. doi: 10.1186/gb-2007-8-8-r155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi Y., Inouye M. mRNA Interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009;85:467–500. doi: 10.1016/S0079-6603(08)00812-X. [DOI] [PubMed] [Google Scholar]
- 16.Christensen S.K., Maenhaut-Michel G., Mine N., Gottesman S., Gerdes K., Van Melderen L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 2004;51:1705–1717. doi: 10.1046/j.1365-2958.2003.03941.x. [DOI] [PubMed] [Google Scholar]
- 17.Lehnherr H., Yarmolinsky M.B. Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3274–3277. doi: 10.1073/pnas.92.8.3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arcus V.L., Rainey P.B., Turner S.J. The PIN-domain toxin–antitoxin array in mycobacteria. Trends Microbiol. 2005;13:360–365. doi: 10.1016/j.tim.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Arcus V.L., McKenzie J.L., Robson J., Cook G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin–antitoxin array. Protein Eng., Des. Sel. 2011;24:33–40. doi: 10.1093/protein/gzq081. [DOI] [PubMed] [Google Scholar]
- 20.Clissold P.M., Ponting C.P. PIN domains in nonsense-mediated mRNA decay and RNAi. Curr. Biol. 2000;10:R888–R890. doi: 10.1016/s0960-9822(00)00858-7. [DOI] [PubMed] [Google Scholar]
- 21.Arcus V.L., Bäckbro K., Roos A., Daniel E.L., Baker E.N. Distant structural homology leads to the functional characterization of an archaeal PIN domain as an exonuclease. J. Biol. Chem. 2004;279:16471–16478. doi: 10.1074/jbc.M313833200. [DOI] [PubMed] [Google Scholar]
- 22.Winther K.S., Gerdes K. Ectopic production of VapCs from Enterobacteria inhibits translation and trans-activates YoeB mRNA interferase. Mol. Microbiol. 2009;72:918–930. doi: 10.1111/j.1365-2958.2009.06694.x. [DOI] [PubMed] [Google Scholar]
- 23.Daines D.A., Wu M.H., Yuan S.Y. VapC-1 of nontypeable Haemophilus influenzae is a ribonuclease. J. Bacteriol. 2007;189:5041–5048. doi: 10.1128/JB.00290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robson J., McKenzie J.L., Cursons R., Cook G.M., Arcus V.L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 2009;390:353–367. doi: 10.1016/j.jmb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Winther K.S., Gerdes K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7403–7407. doi: 10.1073/pnas.1019587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams J.J., Hergenrother P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012;20:291–298. doi: 10.1016/j.tim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park S.J., Son W.S., Lee B.-J. Structural overview of toxin-antitoxin systems in infectious bacteria: A target for developing antimicrobial agents. Biochim. Biophys. Acta, Proteins Proteomics. 2013;1834:1155–1167. doi: 10.1016/j.bbapap.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Engelberg-Kulka H., Sat B., Reches M., Amitai S., Hazan R. Bacterial programmed cell death systems as targets for antibiotics. Trends Microbiol. 2004;12:66–71. doi: 10.1016/j.tim.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Lioy V.S., Rey O., Balsa D., Pellicer T., Alonso J.C. A toxin-antitoxin module as a target for antimicrobial development. Plasmid. 2010;63:31–39. doi: 10.1016/j.plasmid.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Ramage H.R., Connolly L.E., Cox J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demidenok O.I., Kaprelyants A.S., Goncharenko A.V. Toxin-antitoxin vapBC locus participates in formation of the dormant state in Mycobacterium smegmatis. FEMS Microbiol. Lett. 2014;352:69–77. doi: 10.1111/1574-6968.12380. [DOI] [PubMed] [Google Scholar]
- 32.Verma S., Kumar S., Gupta V.P., Gourinath S., Bhatnagar S., Bhatnagar R. Structural basis of Bacillus anthracis MoxXT disruption and the modulation of MoxT ribonuclease activity by rationally designed peptides. J. Biomol. Struct. Dyn. 2014;33:606–624. doi: 10.1080/07391102.2014.899924. [DOI] [PubMed] [Google Scholar]
- 33.Miallau L., Faller M., Chiang J., Arbing M., Guo F., Cascio D., Eisenberg D. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 2009;284:276–283. doi: 10.1074/jbc.M805061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min A.B., Miallau L., Sawaya M.R., Habel J., Cascio D., Eisenberg D. The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg2+ ion in the active site and a putative RNA-binding site. Protein Sci. 2012;21:1754–1767. doi: 10.1002/pro.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das U., Pogenberg V., Subhramanyam U.K.T., Wilmanns M., Gourinath S., Srinivasan A. VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J. Struct. Biol. 2014;188:249–258. doi: 10.1016/j.jsb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z., Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunger A.T. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 41.Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J.Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 43.Delano W.L. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 44.Larkin M.A., Blackshields G., Brown N., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 45.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: an automated pipeline for the setup, execution, and analysis of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sisido M., Ninomiya K., Ohtsuki T., Hohsaka T. Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids. Methods. 2005;36:270–278. doi: 10.1016/j.ymeth.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Holm L., Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dienemann C., Bøggild A., Winther K.S., Gerdes K., Brodersen D.E. Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J. Mol. Biol. 2011;414:713–722. doi: 10.1016/j.jmb.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maté M.J., Vincentelli R., Foos N., Raoult D., Cambillau C., Ortiz-Lombardía M. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res. 2012;40:3245–3258. doi: 10.1093/nar/gkr1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bunker R.D., McKenzie J.L., Baker E.N., Arcus V.L. Crystal structure of PAE0151 from Pyrobaculum aerophilum, a PIN-domain (VapC) protein from a toxin-antitoxin operon. Proteins: Struct. Funct. Bioinf. 2008;72:510–518. doi: 10.1002/prot.22048. [DOI] [PubMed] [Google Scholar]
- 53.Mattison K., Wilbur J.S., So M., Brennan R.G. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing PIN domains and ribbon-helix-helix motifs. J. Biol. Chem. 2006;281:37942–37951. doi: 10.1074/jbc.M605198200. [DOI] [PubMed] [Google Scholar]
- 54.Hwang K.Y., Baek K., Kim H.-Y., Cho Y. The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat. Struct. Mol. Biol. 1998;5:707–713. doi: 10.1038/1406. [DOI] [PubMed] [Google Scholar]
- 55.Sharp J.D., Cruz J.W., Raman S., Inouye M., Husson R.N., Woychik N.A. Growth and translation inhibition through sequence-specific RNA binding by Mycobacterium tuberculosis VapC toxin. J. Biol. Chem. 2012;287:12835–12847. doi: 10.1074/jbc.M112.340109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agarwal S., Mishra N.K., Bhatnagar S., Bhatnagar R. PemK toxin of Bacillus anthracis is a ribonuclease an insight into its active site, structure, and function. J. Biol. Chem. 2010;285:7254–7270. doi: 10.1074/jbc.M109.073387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.London N., Movshovitz-Attias D., Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18:188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 58.Ren D., Walker A.N., Daines D.A. Toxin-antitoxin loci vapBC-1 and vapXD contribute to survival and virulence in nontypeable Haemophilus influenzae. BMC Microbiol. 2012;12:263. doi: 10.1186/1471-2180-12-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahidjo B.A., Kuhnert D., McKenzie J.L., Machowski E.E., Gordhan B.G., Arcus V., Abrahams G.L., Mizrahi V. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One. 2011;6:e21738. doi: 10.1371/journal.pone.0021738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maezato Y., Daugherty A., Dana K., Soo E., Cooper C., Tachdjian S., Kelly R.M., Blum P. VapC6, a ribonucleolytic toxin regulates thermophilicity in the crenarchaeote Sulfolobus solfataricus. RNA. 2011;17:1381–1392. doi: 10.1261/rna.2679911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenzie J.L., Robson J., Berney M., Smith T.C., Ruthe A., Gardner P.P., Arcus V.L., Cook G.M. A VapBC toxin-antitoxin module is a posttranscriptional regulator of metabolic flux in mycobacteria. J. Bacteriol. 2012;194:2189–2204. doi: 10.1128/JB.06790-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.