


Abstract
Computational modeling is employed to provide a plausible structural explanation for the experimentally-observed differential global genome repair (GGR) propensity of the ALII-N2-dG and ALII-N6-dA DNA adducts of aristolochic acid II. Our modeling studies suggest that an intrinsic twist at the carcinogen–purine linkage of ALII-N2-dG induces lesion site structural perturbations and conformational heterogeneity of damaged DNA. These structural characteristics correlate with the relative repair propensities of AA-adducts, where GGR recognition occurs for ALII-N2-dG, but is evaded for intrinsically planar ALII-N6-dA that minimally distorts DNA and restricts the conformational flexibility of the damaged duplex. The present analysis on the ALII adduct model systems will inspire future experimental studies on these adducts, and thereby may extend the list of structural factors that directly correlate with the propensity for GGR recognition.
INTRODUCTION
Aristolochic acids (AAs, Figure 1a) are a group of structurally related phenanthrene carboxylic acids produced as secondary metabolites by the Aristolochiaceae family of angiosperms. Extracts from these plants mainly consist of AAI and AAII (Figure 1a), which have been implicated in the etiology of chronic renal diseases such as Chinese Herbal Nephropathy (1) and Balkan Endemic Nephropathy (2) (collectively known as Aristolochic Acid Nephropathy (3)), as well as upper urinary tract urothelial carcinoma (UUC) (4–7). Metabolic activation of AAs involves reduction of the nitro group to form N-hydroxylaristolactams (N-hydroxyl-ALs) (8–10). These intermediates are subsequently hydrolyzed to form reactive nitrenium ions that yield bulky AL-purine DNA adducts (addition products) at the exocyclic amino groups of the purines (11–13). The resulting adenine (AL-N6-dA, Figure 1b) and guanine (AL-N2-dG, Figure 1c) adducts misincorporate adenine during replication (14).
Figure 1.
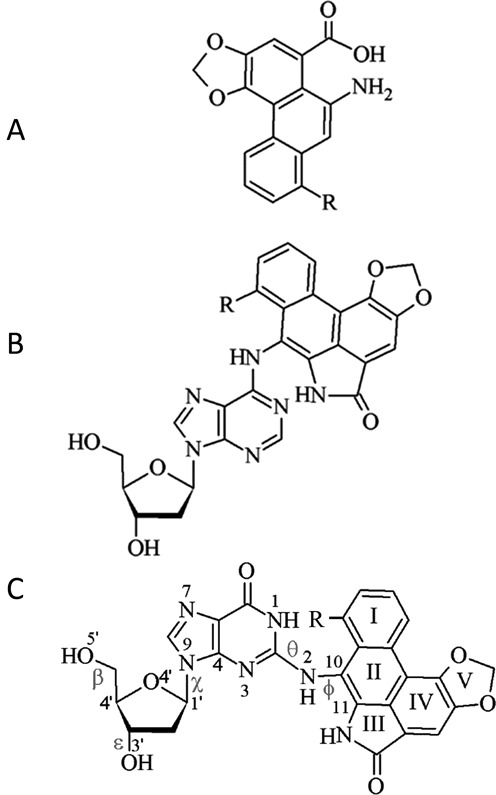
Structure of (A) aristolochic acids, as well as the corresponding (B) AL-N6-dA and (C) AL-N2-dG adducts (R = OCH3 for ALI and H for ALII). Definitions are provided for the θ (∠(N1C2N2C10)) and ϕ (∠(C2N2C10C11)) dihedral angles, which determine the orientation of the ALII moiety with respect to the base and χ (∠(O4′C1′N9C4)), which dictates the glycosidic bond orientation to be syn (90° > χ > −60°), high syn (120° > χ > 90°), anti (120° > χ > −90°) or high anti (−60° > χ > −90°). Dihedral angles β (∠(C4′C5′O5′H)) and ε (∠(C4′C3′O3′H)) govern the DNA sugar–phosphate backbone orientation.
Bulky DNA lesions, including the AL-DNA adducts, are commonly repaired through the nucleotide excision repair (NER) pathway, which removes a 24–32 nt long section of the lesion-containing strand (15). NER operates by two different mechanisms, viz. transcription-coupled repair (TCR) and global genome repair (GGR) (16–18), which primarily differ in two ways. First, TCR only repairs transcriptionally active regions, while GGR recognizes and repairs lesions spanning the entire genome. Second, damage recognition in TCR is associated with transcription inhibition (i.e., stalling of an RNA polymerase), while GGR involves specific damage recognition factors (i.e., XPC-RAD23B in eukaryotes). Nevertheless, the subsequent repair steps are similar in both mechanisms.
In a recent experimental study (19), Sidorenko et al. observed that cell lines with deficiencies in (CSB) genes associated with the TCR pathway lead to higher levels of both the ALII-N6-dA and ALII-N2-dG adducts compared to control cells, implying that both adducts are repaired by the TCR pathway. On the other hand, after exposure to 100 μM of AAII, a small, but significant, change was observed in the levels of the ALII-N2-dG adduct in cells deficient in (XPC) genes associated with GGR compared to control cells. This implies that the ALII-N2-dG adduct is repaired by both TCR and GGR, which correlates with the high rate of AL-N2-dG removal in target tissues (20). Further, investigations on the toxicity of ALII in cell lines with deficiencies in TCR or GGR or both pathways point towards the GGR resistance of ALII-N6-dA (19). Furthermore, a number of recent studies on patients exposed to AAs (5,21–23) have revealed that the signature A→T mutation of AL-N6-dA adduction is predominantly located in non-transcribed DNA strands (21,22). This conspicuous lack of A→T mutations in transcriptionally active regions of the genome strengthens the argument that the associated AL-N6-dA adducts are actively repaired by the TCR pathway (19). In addition, in vitro binding studies of the eukaryotic GGR damage sensing factor (XPC-RAD23B) to oligonucleotides containing ALII-N6-dA also reveal that these adducts lack GGR recognition (19). The proposed GGR resistance of the ALII-N6-dA adduct is in synchrony with its greater overall persistence (19–20,24) and mutagenicity (14,25) compared to ALII-N2-dG in cells. Despite these experimentally-observed differences in the GGR susceptibility of ALII-N6-dA and ALII-N2-dG, the structural basis for the differences in their GGR recognition and subsequent repair remains to be established.
GGR recognition in eukaryotes is believed to involve a dynamic two-step mechanism (26–28). The first step identifies distortions at the lesion site by XPC-RAD23B sensors, whereas the second step involves lesion verification and formation of a stable GGR recognition complex. In relation to the first step of GGR recognition, studies on a variety of bulky DNA adducts have revealed that GGR propensity correlates with changes in a number of local structural features upon DNA damage, including significant perturbations to helical parameters (such as minor groove dimension, rise and twist) (29–33), increases in helical (34–37) and backbone dynamics (29), and decreases in the stacking interactions at the lesion site (38,39), as well as global changes to the DNA helix, such as helix bending (40). Furthermore, a crystal structure of the yeast ortholog (Rad4) of XPC-RAD23B bound to DNA containing the cyclobutane-pyrimidine (CPD) dimer (41) indicates that GGR recognition is initiated by the insertion of a β-hairpin of the enzyme through the major groove side of damaged DNA. This insertion flips the bases opposing the lesion out of the helix and pushes the lesion into the DNA minor groove. Modeling studies on adducted DNA suggest lower free energy barriers for opposing base flipping in damaged duplexes compared to unmodified helices (42,43), indicating that ease of partner base flipping facilitates GGR (43). To complement this information, a recent crystal structure of Rad4 bound to undamaged DNA led to speculations that lesion recognition may depend on the time required to open a (damaged) DNA site (opening time) and the time the recognition factor spends at the damaged site (residence time) (44). In addition, the presence of distortions at the lesion site have been hypothesized to decrease the opening time and increase the residence time of the recognition factor (44). Most importantly, this literature suggests that the structural characteristics of damaged DNA are crucial for understanding the issue of the ‘repair versus persistence’ of bulky lesions.
Owing to the remarkable nephrotoxic and carcinogenic potential of AAs, the structural properties of AL-DNA adducts are of great contemporary interest. To understand how the intrinsic structural features of the AL-DNA adducts influence the conformational preferences of damaged DNA and how the structural features of AL-adducted DNA influence damage recognition by GGR proteins, we use computational modeling to compare the structural properties of the repair-prone ALII-N2-dG adduct with the repair-resistant ALII-N6-dA adduct previously studied using nuclear magnetic resonance (NMR) (45) and computational methods (46). Specifically, within the same 11-mer oligonucleotide, both NMR and molecular dynamics (MD) simulations suggest a preference for an intercalated ALII-N6-dA adducted DNA conformation with the opposing thymine displaced into the major groove. However, no analogous data has been reported for the ALII-N2-dG adduct. Thus, models of the ALII-adducted nucleobases, nucleosides and nucleotides are initially considered in the present work using quantum mechanics (DFT) in order to determine intrinsic conformational differences between the damaged purines in terms of the orientation of the ALII and deoxyribose moieties with respect to the nucleobase. Subsequently, the conformational space of ALII-adducted DNA is explored using MD simulations on an 11-mer damaged DNA oligonucleotide. In the absence of experimental NMR data in the literature, our study utilizes a plausible computational model that mimics the structural properties of AA-damaged DNA. Our work suggests key differences in the structural features of ALII-N2-dG and ALII-N6-dA, as well as the associated adducted DNA helices, that likely play a critical role in dictating the observed differential GGR recognition of these adducts. Most importantly, our work points toward the effects of the intrinsic adduct conformation on GGR recognition and repair propensity. If verified by future experimental studies, our results will likely expand the list of previously identified factors that affect the structure and repair propensity of adducted DNA, such as substitution at the carcinogen–base linkage (47), stereochemistry (29,48–49), adduct ionization state (50), sequence context (35,47) and the identity of the partner base (51).
MATERIALS AND METHODS
The B3LYP/6–31G(d) potential energy surface (PES) was considered as a function of the θ and ϕ dihedral angles for the ALII-N2-G nucleobase adduct, and as a function of χ and θ for the nucleoside adduct by adding a 2′-deoxyribose to the lowest energy conformation obtained from the nucleobase model (dihedral angles are defined in Figure 1c). This build-up approach for the conformational scans has been validated in our previous study on phenolic DNA adducts (52), where Density Functional Theory (DFT) results correlate with experimental spectroscopic data. The minima identified on the PESs were subsequently optimized using dispersion-corrected B3LYP-D3/6–31G(d) and single-point energies were calculated using B3LYP-D3/6–311+G(2df,p), since the inclusion of the empirical dispersion correction using the DFT-D3 approach has been shown to perform reasonably well for systems where non-covalent interactions are important (53). Furthermore, since a previous reference study on non-covalent interactions indicates that adding the dispersion correction strongly diminishes the performance differences between DFT functionals (54), we chose B3LYP-D3 to allow comparisons with the previously reported B3LYP-D3 optimized structures of the AL-N6-dA adducts (46). The ALII-N2-dG nucleotide was built by adding a (Na+ ion neutralized) 5′–monophosphate unit to the lowest energy anti and syn nucleoside conformers. In this model, the phosphate oxygen bonded to the 5′-neighboring nucleotide in DNA was capped with a hydrogen atom and the Na+ ion was placed between the two non-capped phosphate oxygens. The structures were energy minimized in water using PCM-B3LYP-D3/6–31G(d), since our previous study has shown that the structural properties of modified nucleotides are best characterized in solvent (water) with a sodium ion to counter the phosphate charge (55). Single-point energy calculations were carried out using B3LYP-D3/6–311+G(2df,p) for the nucleobase and nucleoside minima and PCM-B3LYP-D3/6–311+G(2df,p) for the nucleotide minima. MD simulations were performed by incorporating the adduct into the 11-mer DNA sequence (5′-CGTACXCATGC, X = adduct) previously studied for ALII-N6-dA (45,46). Starting structures for the MD simulations were generated using a large number of trial simulations initiated using different possible locations of the ALII moiety in the helical environment, with GAFF parameters (56) describing the ALII moiety and parm99 parameters (57) describing the rest of DNA. Similar to the nucleotide model, the oligonucleotide model was neutralized by sodium ions described by parm94 parameters. All simulations were carried out in water containing 20 Na+ ions for 20 ns (excluding trial simulations) using the AMBER 11 (58) or 12 program (59). MM-PBSA free energy calculations were performed on simulation trajectories (60). Although free energy methods that are theoretically more rigorous than end-point free energy methods such as the MM-PBSA method are available in the literature (61,62), a previous study has shown that DNA conformational free energy differences obtained using the MM-PBSA approach are in close agreement with the potentials of mean force determined using the more rigorous umbrella sampling approach (63). Although potential inaccuracies in free energy calculations may arise due to the theoretical simplicity of the MM-PBSA approach (e.g., implicit treatment of solvent), as well as limitations in our conformational sampling, the MM-PBSA free energies are expected to provide at least qualitative estimates of the energetic differences between possible conformations of damaged DNA (64). Furthermore, the choice of this method for the present application stems from our desire to compare the conformational preferences of ALII-N2-dG adducted DNA with those of ALII-N6-dA adducted DNA determined using the same protocol (46), as well as the successful applications of this method in previous studies of damaged DNA (50,65–67). Full details of the computational protocol are provided in Supplementary Data (Section S1).
RESULTS AND DISCUSSION
ALII-N2-dG intrinsically prefers a twisted conformation, while ALII-N6-dA prefers a planar conformation, about the carcinogen–purine linkage
DFT energies plotted as a function of θ and ϕ for the ALII-N2-G nucleobase adduct suggest two low-energy conformations with a twisted orientation of ALII with respect to G (Figure 2a (right) and Supplementary Figure S1 and Section S2, Supplementary Data). This twist decreases steric repulsion between N1–H of damaged G and the lactam N–H group of ALII. In contrast, the lowest energy ALII-N6-A nucleobase minimum is planar (Figure 2a (left)) because of stabilizing hydrogen bonding between N1 of A and the lactam N–H of ALII (46). DFT nucleoside energies plotted as a function of χ and θ suggest that both ALII-N2-dG (Supplementary Figures S2 and S3 and Section S2, Supplementary Data) and ALII-N6-dA (46) intrinsically prefer a syn glycosidic orientation (by ∼14 and 25 kJ mol−1, respectively), mainly due to an interaction between the 5′-OH group and N3 (Figure 2b). Although such a conformational preference is relevant when the adduct is located at the 5′-terminal position (where the 5′-OH is not capped by a phosphate group), the presence of the 5′-phosphate group may alter the preferences at non-terminal DNA positions. Therefore, in order to neglect such spurious interactions, the 5′-OH was subsequently directed (and fixed) away from the nucleobase (Figure 2c, and Supplementary Figure S4a and b, and Section S2, Supplementary Data). This results in the anti conformation being slightly more stable than the syn for both ALII-N2-dG (by ∼7 kJ mol−1) and ALII-N6-dA (by ∼11 kJ mol−1 (46)). However, steric repulsion arising from the close proximity of the 5′-phosphate and ALII moieties in the syn orientation renders the anti ALII-N2-dG nucleotide 27 kJ mol−1 more stable than the syn conformer (Figure 2d, and Supplementary Figure S4c and d, and Section S2, Supplementary Data). In contrast, since the ALII moiety of the ALII-N6-dA nucleotide does not interact with the 5′-phosphate in either the syn or anti conformation, anti ALII-N6-dA is the most stable conformer by only ∼10 kJ mol−1 (46). Overall, regardless of the DFT model considered, ALII-N2-dG prefers a twisted conformation, while ALII-N6-dA adopts a planar conformation, at the carcinogen–purine linkage.
Figure 2.
DFT (B3LYP-D3/6–31G(d)) minimum energy conformations of ALII-N6-dA (left) (46) and ALII-N2-dG (right) according to (A) nucleobase, (B) nucleoside, (C) 5′-OH constrained nucleoside and (D) nucleotide models.
Twisted conformation about the carcinogen–purine linkage facilitates greater conformational heterogeneity of ALII-N2-dG adducted DNA compared to ALII-N6-dA adducted DNA
ALII-N2-dG adducted DNA is conformationally heterogeneous
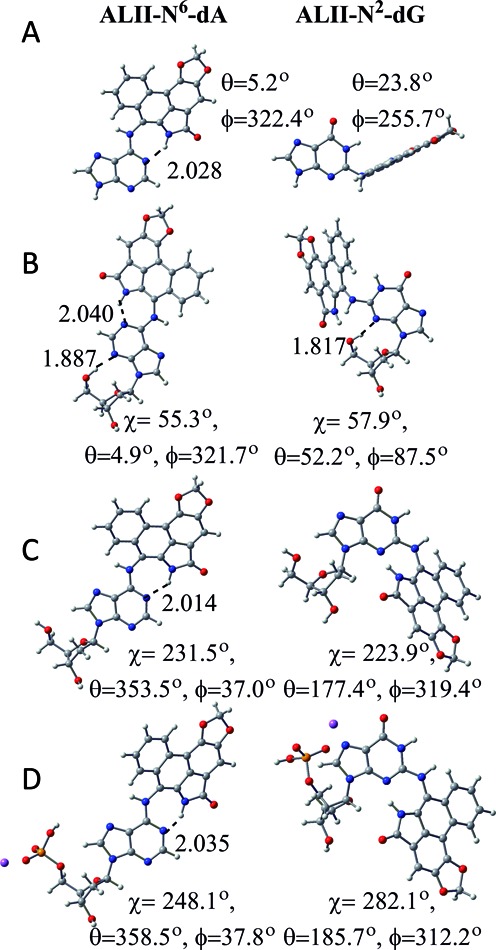
Using our detailed computational protocol, six distinct conformers were isolated from MD simulations on an 11-mer oligonucleotide model of ALII-N2-dG adducted DNA, which differ in the location of the ALII moiety and the (anti/syn) glycosidic orientation of the adducted nucleotide (Figure 3a). Two of these conformations (i.e., anti base−displaced intercalated and 3′−intercalated) correspond to the anti adduct glycosidic orientation, one (i.e., minor groove stacked) involves the high anti orientation and three (i.e., syn base−displaced intercalated, 5′−intercalated and 3′,5′−intercalated) correspond to the syn glycosidic orientation. Approximate MM-PBSA relative free energies indicate a close energetic separation between all six conformations (<21 kJ mol−1; Figure 3a). Salient features of each conformation are described below, with details provided in Supplementary Data (Section S2).
Figure 3.
Base-pair trimers containing the lesion site in representative MD structures obtained from 20 ns simulations carried out independently on each energetically-accessible conformation of (A) ALII-N2-dG and (B) ALII-N6-dA adducted DNA. The relative free energies (kJ mol−1) with respect to the lowest energy conformation for a given adduct are provided in parantheses.
Five of the accessible ALII-N2-dG adducted DNA conformations are analogous to those previously observed for other DNA adducts
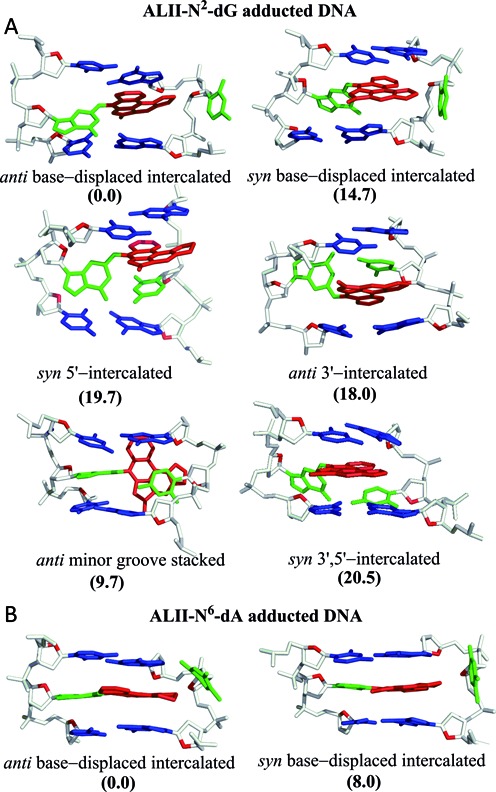
The anti base−displaced intercalated conformation of ALII-N2-dG adducted DNA is characterized by stacking of the ALII moiety between the 5′-base pair and 3′–flanking base in the opposing strand, the extrahelical (major groove) location of the opposing C and complete loss of Watson–Crick (WC) hydrogen bonding in the damaged base pair (Figure 3a). This structure is similar to the previously reported experimental structure of DNA containing the two stereoisomeric cis-anti-[BP]-N2-dG polycyclic aromatic hydrocarbon (PAH) adducts (68,69), which exhibit (major groove) displacement of the opposing cytosine and a twisted conformation at the carcinogen–dG linkage. Nevertheless, the origin of the twist differs between the PAH and ALII adducts. Specifically, the twist in ALII-N2-dG alleviates steric repulsion between N1–H of G and the lactam N–H group, while the twist in the PAH adducts arises due to the absence of extended conjugation between the damaged guanine and PAH moieties (i.e., sp3 hybridization of the linker C of the carcinogen). In contrast, DNA damaged by a heterocyclic aromatic amine carcinogenic adduct (i.e., IQ-N2-dG) adopts a base–displaced intercalated conformation with a nearly planar carcinogen–guanine linkage due to extended conjugation (i.e., sp2 hybridization of the linker C of the carcinogen) and the absence of intrinsic steric repulsion (70).
Although the damaged guanine retains stacking interactions with its 3′−flanking pair in the syn base−displaced intercalated conformation of ALII-N2-dG adducted DNA, only the ALII moiety stacks with the 5′−flanking pair (Figure 3a). To the best of our knowledge, no example exists in the literature of N2-dG adducted DNA in such a conformation. Nevertheless, this structure is analogous to the syn base−displaced intercalated conformation observed for damaged DNA corresponding to several C8-dG adducts (47,50,71–72) in terms of the (major groove) extrahelical opposing cytosine. However, damaged guanine is displaced towards the major groove in C8-dG adducted DNA versus the minor groove in ALII-N2-dG adducted DNA.
In the syn 5′−intercalated conformation, the ALII moiety stacks between the opposing (intrahelical) cytosine and the 5′–base pair with respect to the lesion (Figure 3a). Although this conformation has not been reported in the literature for other N2-dG adducts, some N6-linked PAH adducts acquire an analogous conformation (73–75). In contrast, the anti 3′–intercalated conformer (i.e., the ALII moiety stacks between the opposing (intrahelical) cytosine and the 3′–base pair with respect to the lesion; Figure 3a) has been observed for 14R(+)-trans-anti-DB[a,l]P-N2-dG PAH adducted DNA (76). However, the PAH adducted DNA conformation involves simultaneous stacking of the damaged guanine and PAH moieties with the flanking bases (due to flexibility imparted by the sp3 hybridization of the linker C of the PAH moiety), while only the ALII ring stacks with the flanking bases in ALII-N2-dG adducted DNA.
In the anti minor groove stacked conformation, cytosine opposing the lesion twists to form van der Waals (π–π stacking) interactions with the ALII moiety that is located in the minor groove (Figure 3a). A variant of this conformation was previously reported for 14S-cis-DB[a,l]N2-dG PAH adducted DNA (49). However, the minor groove stacked conformation associated with this PAH adduct ruptures WC pairing in both the damaged and flanking base pairs, while the ALII-N2-dG adducted DNA conformer retains WC pairing in the flanking pairs, and a N4–H•••O6 hydrogen bond in the lesion pair (Supplementary Table S1, Supplementary Data).
ALII-N2-dG adducted DNA adopts a unique (syn 3′,5′−intercalated) conformation
In addition to the conformations discussed above, ALII-N2-dG adducted DNA adopts a unique conformation that has not been reported in the literature for other DNA adducts to date. In this conformation, the modified guanine and opposing cytosine shift towards the minor groove, while the ALII moiety simultaneously stacks with the 5′ and 3′–flanking pairs, as well as the opposing cytosine (Figure 3a). The opposing cytosine maintains a N4–H•••O6 hydrogen bond with the damaged guanine and forms additional hydrogen bonds with the 3′–base pair with respect to the lesion (Supplementary Table S1, Supplementary Data).
Structural differences at the carcinogen–purine linkage of the ALII-N2-dG and ALII-N6-dA adducts alter the conformational heterogeneity of damaged DNA
Relative MM-PBSA free energies suggest that anti base–displaced intercalated conformer is the most stable orientation for both ALII-N2-dG and ALII-N6-dA adducted DNA (Figure 3). However, the total number of energetically-accessible conformations differs between the two types of adducted DNA. Specifically, although MM-PBSA suggests that five conformations lie within 21 kJ mol−1 of the most stable conformer for ALII-N2-dG adducted DNA (Figure 3a), only one other conformation, namely the syn base–displaced intercalated orientation, falls within this energetic separation for ALII-N6-dA adducted DNA (Figure 3b) (46). The reduced conformational heterogeneity of ALII-N6-dA arises since the planar adduct forms highly stabilizing van der Waals (stacking) interactions with the flanking base pairs when the opposing C is extrahelical (i.e., the base–displaced intercalated conformations). However, since the opposing C remains inside the helix in all other possible ALII-N6-dA adducted DNA conformations, the rise between the flanking pairs increases, stacking with the lesion decreases and the associated adducted DNA conformations become energetically unfavorable (greater approximately 35 kJ mol−1) (46). In contrast, since ALII-N2-dG is twisted, optimal stacking at the lesion site cannot be achieved in the corresponding base–displaced intercalated conformations, which prevents these conformers from becoming highly stabilized as for ALII-N6-dA adducted DNA. In fact, the conformational heterogeneity for ALII-N2-dG adducted DNA arises due to a complicated interplay between mutually compensating interactions arising from the twisted lesion, including van der Waals, steric and hydrogen-bonding interactions at the damaged site (Section 2, Supplementary Data). Although the approximate nature of the MM-PBSA method may result in inherent inaccuracies in the free energies, the relative free energy rankings of different conformations at least provide a qualitative picture for estimating the differences in the conformational preferences of ALII-N2-dG and ALII-N6-dA adducted DNA. Thus, our models suggest differences in the intrinsic (planar versus twisted) conformation at the carcinogen–purine linkage lead to differences in the conformational heterogeneity of ALII-N6-dA and ALII-N2-dG adducted DNA.
Increased lesion site distortions, diminished stacking and enhanced dynamics likely contribute to the greater propensity of GGR recognition for ALII-N2-dG compared to ALII-N6-dA
Twisted conformation of ALII-N2-dG leads to greater DNA distortions at the lesion site compared to ALII-N6-dA
Structural distortions at the lesion site are believed to be one of the important factors in GGR recognition (29–30,47). For example, the enhanced repair susceptibility of the (aromatic amine) AAF-C8-dG adduct has been partly attributed to its greater (structurally distorting) impact on DNA compared to the corresponding non-acetylated AF-C8-dG adduct (47). In the case of the ALII adducts, DFT calculations indicate that the ALII-N2-dG nucleotide intrinsically prefers a twisted conformation at the carcinogen–purine linkage. This twist distorts DNA at the lesion site according to the MD pseudostep parameters (i.e., step parameters calculated between the base step consisting of the 3′ and 5′−base pairs with respect to the lesion; see Section S1, Supplementary Data) and minor groove dimensions. Specifically, each accessible conformation of ALII-N2-dG adducted DNA significantly changes at least one of the lesion site pseudostep parameters compared to unmodified DNA (i.e., shift (up to 3Å), slide (up to 6Å), rise (up to 4Å), tilt (up to 10°), roll (up to 27°) or twist (up to 27°)) or the minor groove dimensions (up to 6Å). On the other hand, ALII-N6-dA prefers an intrinsically planar conformation of the ALII moiety with respect to the damaged A, and therefore ALII-N6-dA adducted DNA exhibits only minimal changes in the pseudostep parameters compared to unmodified DNA (i.e., shift (up to 1.4Å), slide (up to 1.5Å), rise (up to 0.2Å), tilt (up to 2°), roll (up to 10°) or twist (up to 10°)) and minor groove dimensions (up to 1Å; Figure 4) (46). This suggests that planar ALII-N6-dA is better accommodated in DNA than the (distorting) twisted ALII-N2-dG adduct.
Figure 4.
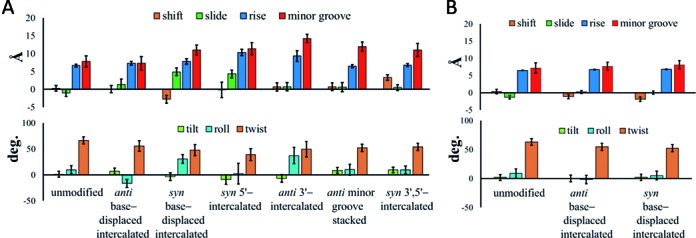
The pseudostep parameters and minor groove dimensions for different conformations of (A) ALII-N2-dG or (B) ALII-N6-dA (46) adducted DNA relative to the corresponding unmodified helix. The helical dynamics are indicated by error bars.
Twisted conformation of ALII-N2-dG diminishes lesion site stacking interactions
Previous experimental and computational studies indicate that DNA adducts prone to GGR recognition typically exhibit diminished stacking at the lesion site compared to GGR resistant adducts (29,47,49,77). In the ALII adducts, the intrinsic twist in ALII-N2-dG hinders simultaneous stacking of the damaged guanine and ALII moieties with the flanking bases in all accessible conformations of ALII-N2-dG adducted DNA. In contrast, the intrinsic planar conformation of ALII-N6-dA facilitates simultaneous stacking of both the ALII and modified adenine moieties between the flanking base pairs in DNA. As a result, the calculated van der Waals (stacking) interactions involving the lesion are consistently smaller (less negative) for the repair prone ALII-N2-dG than the repair resistant ALII-N6-dA in DNA (Figure 5). Although the trend in relative NER propensities of the two ALII-purine adducts is similar to that observed for the 14R-DB[a,l]P-N2-dG and 14R-DB[a,l]P-N6-dA adducts (i.e., the dG adduct is more repair prone than the dA adduct), the structural origin of the differential NER propensity differs between the two types of adducts. Specifically, the differential NER propensity of the PAH adducts has been interpreted in terms of structural distortions in the 14R-DB[a,l]P-N2-dG lesion originating from the loss of WC hydrogen bonding between the damaged pair and relatively weaker stacking interactions between the intercalated bulky moiety and the adjacent bases. On the other hand, the 14R-DB[a,l]P-N6-dA adduct retains WC hydrogen-bonding within the damaged pair and is associated with greater lesion site stacking interactions, which makes this adduct more repair resistant (49).
Figure 5.
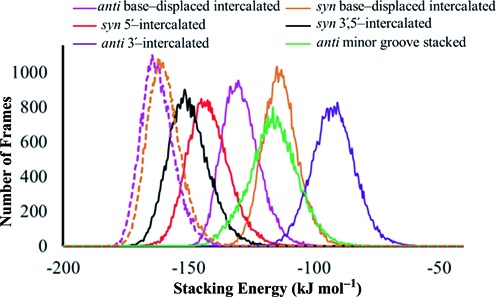
Comparison of lesion van der Waals (stacking) energies for anti or syn ALII-N2-dG (solid lines) and ALII-N6-dA (dashed lines) in different adducted DNA conformations over 20 ns MD simulations.
DNA containing ALII-N2-dG is structurally more dynamic than DNA containing ALII-N6-dA
Previous studies have shown that damage recognition by XPC-RAD23B is facilitated by increased lesion site dynamics (29,34–35). For example, the greater repair propensity of the (+)-trans-anti-[BP]N2-dG adduct in the TG*T over the CG*C sequence has been partly attributed to a more dynamic roll/bending and greater minor groove flexibility in the former sequence (35). Similarly, greater lesion site backbone dynamics of the 5′R compared to the 5′S enantiomer of the 5′,8-cyclo-2′-deoxypurine lesion correlates with the greater NER susceptibility of the 5′R enantiomer (29). For the ALII adducts, the standard deviations in the pseudostep parameters at the lesion site in ALII-N6-dA adducted DNA (maximum of 1.4Å for the translational parameters (i.e., shift, slide and rise), 1.2Å for the minor groove width and 6.3° for the rotational parameters (i.e., tilt, roll and twist; Figure 4) are similar to those for the corresponding unmodified DNA (maximum of 1.0Å for the translational parameters, 1.5Å for the minor grove width and 6.0° for the rotational parameters). However, all accessible conformations of ALII-N2-dG adducted DNA typically exhibit greater standard deviations in the lesion site parameters (maximum of 2.2Å for the translational parameters, 1.9Å for the minor grove width and 15° for the rotational parameters) than the corresponding unmodified DNA (maximum of 1.0Å for the translational parameters, 1.6Å for the minor grove width and 7.6° for the rotational parameters).
Differences in structural features of ALII-N2-dG and ALII-N6-dA adducted DNA may lead to better recognition of ALII-N2-dG in the cells
Our model suggests that ALII-N2-dG induces greater structural distortions to DNA and leads to smaller lesion site van der Waals stacking interactions compared to the ALII-N6-dA adduct. In addition, due to an intrinsically twisted carcinogen–purine linkage, the ALII-N2-dG adduct induces significantly greater helical flexibility at the lesion site compared to the ALII-N6-dA adduct, which may contribute to its enhanced recognition by the GGR pathway. This provides a correlation between a twisted carcinogen–purine linkage and GGR propensity. If verified using experimental techniques in the future, our models will likely expand the current list of factors that affect the GGR of DNA adducts (e.g., substitution at the carcinogen–base linkage (47), stereochemistry (29,48–49), adduct ionization state (50), sequence context (35,47) and the identity of the partner base (51)).
The differences in the structural features of ALII-N2-dG and ALII-N6-dA adducted DNA identified from our modeling studies also provide information about XPC-RAD23 binding to adducted DNA. Specifically, the significant structural distortions and diminished van der Waals (stacking) interactions at the lesion site in all accessible conformations of ALII-N2-dG adducted DNA will likely decrease the opening time and increase the residence time of the recognition factor at the damage site, which will in turn facilitate lesion recognition. In contrast, the smaller structural distortions and enhanced stacking interactions at the lesion site in ALII-N6-dA adducted DNA compared to ALII-N2-dG adducted DNA, due to the planar ALII–dA linkage (45,46), will likely increase the opening time and decrease the residence time of the recognition factor and thereby render this adduct resistant to GGR. Furthermore, the major groove intercalation of the bulky moiety may block β-hairpin insertion through the major groove side of DNA in the most stable (anti base–displaced intercalated) ALII-N6-dA adducted DNA conformation, while the minor groove intercalation of the bulky moiety in the most stable ALII-N2-dG adducted DNA conformer may facilitate repair. Nevertheless, the conformational heterogeneity of ALII-N2-dG found in our models allows the bulky moiety to adopt a number of helical positions and therefore underscores that differences in key structural features at the lesion site are likely the primary explanation for the enhanced GGR of ALII-N2-dG over ALII-N6-dA.
CONCLUSION
The present computational study provides comprehensive structural details of the conformational preferences of ALII-N2-dG adducted DNA with the help of MD simulations. The calculations suggest an intrinsic twist at the linkage between the damaged G and ALII moieties, which leads to conformational heterogeneity of ALII-N2-dG adducted DNA. Within the constraints of our model, comparison of the structural features of ALII-N2-dG and ALII-N6-dA adducted DNA provides a structural explanation for the observed differential GGR recognition propensities of these purine adducts. Specifically, the intrinsic twisted conformation of ALII-N2-dG induces destabilizing distortions to DNA at the lesion site, reduces van der Waals (stacking) interactions with the neighboring base pairs and enhances the helical dynamics at the damaged site, which may collectively facilitate GGR recognition. In contrast, smaller distortions, greater stacking stabilization and decreased dynamics at the lesion site likely render ALII-N6-dA adducted DNA resistant to GGR. This GGR resistance is likely the primary factor responsible for the greater abundance and nephrotoxic potential of ALII-N6-dA compared to ALII-N2-dG in cells affected by aristolochic acids. This effect of the carcinogen–nucleobase linkage on the repair propensity may not have been previously reported in the literature since adduct conformation strongly depends on the chemical composition of the lesion and experimental structural studies have yet to be completed on the ALII-N2-dG adduct. In this context, although the structural properties of ALII-N6-dA adducted DNA have been previously validated using NMR, the predicted structural differences between the ALII-N6-dA and ALII-N2-dG adducts that lead to their differential GGR propensity in DNA must be validated through future NMR studies on the ALII-N2-dG adduct within the same sequence. Nevertheless, in the absence of experimental structural data on ALII-N2-dG adducted DNA, the present study suggests the critical role of the conformation at the carcinogen–DNA linkage in determining GGR recognition and excision propensities.
Supplementary Material
Acknowledgments
Computational resources provided by the New Up-scale Cluster for Lethbridge to Enable Innovative Chemistry (NUCLEIC), as well as Westgrid and Compute/Calcul Canada, are greatly appreciated.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada [249598-07]; Canada Research Chain Program [950-228175]; Canadian Foundation of Innovation [22770]; University of Lethbridge Research Fund [13368]. Funding for open access charge: Natural Sciences and Engineering Research Council of Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1.Debelle F.D., Vanherweghem J.-L., Nortier J.L. Aristolochic acid nephropathy: a worldwide problem. Kidney Int. 2008;74:158–169. doi: 10.1038/ki.2008.129. [DOI] [PubMed] [Google Scholar]
- 2.Grollman A.P., Shibutani S., Moriya M., Miller F., Wu L., Moll U., Suzuki N., Fernandes A., Rosenquist T., Medverec Z., et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12129–12134. doi: 10.1073/pnas.0701248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Broe M.E. Chinese herbs nephropathy and Balkan endemic nephropathy: toward a single entity, aristolochic acid nephropathy. Kidney Int. 2012;81:513–515. doi: 10.1038/ki.2011.428. [DOI] [PubMed] [Google Scholar]
- 4.Arlt V.M., Stiborová M., vom Brocke J., Simões M.L., Lord G.M., Nortier J.L., Hollstein M., Phillips D.H., Schmeiser H.H. Aristolochic acid mutagenesis: molecular clues to the aetiology of balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis. 2007;28:2253–2261. doi: 10.1093/carcin/bgm082. [DOI] [PubMed] [Google Scholar]
- 5.Chen C.-H., Dickman K.G., Moriya M., Zavadil J., Sidorenko V.S., Edwards K.L., Gnatenko D.V., Wu L., Turesky R.J., Wu X.-R., et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8241–8246. doi: 10.1073/pnas.1119920109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cosyns J.-P., Jadoul M., Squifflet J.-P., Wese F.-X., van Ypersele de Strihou C. Urothelial lesions in chinese-herb nephropathy. Am. J. Kidney Dis. 1999;33:1011–1017. doi: 10.1016/S0272-6386(99)70136-8. [DOI] [PubMed] [Google Scholar]
- 7.Wu F., Wang T. Risk assessment of upper tract urothelial carcinoma related to aristolochic acid. Cancer Epidemiol. Biomarkers Prev. 2013;22:812–820. doi: 10.1158/1055-9965.EPI-12-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sidorenko V.S., Attaluri S., Zaitseva I., Iden C.R., Dickman K.G., Johnson F., Grollman A.P. Bioactivation of the human carcinogen aristolochic acid. Carcinogenesis. 2014;35:1814–1822. doi: 10.1093/carcin/bgu095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinek V., Kubickova B., Arlt V.M., Frei E., Schmeiser H.H., Hudecek J., Stiborova M. Comparison of activation of aristolochic acid I and II with NADPH: quinone oxidoreductase, sulphotransferases and N-acetyltranferases. Neuro Endocrinol. Lett. 2010;32:57–70. [PubMed] [Google Scholar]
- 10.Stiborová M., Mareîs J., Frei E., Arlt V.M., Martínek V., Schmeiser H.H. The human carcinogen aristolochic acid I is activated to form DNA adducts by human NAD(P)H: quinone oxidoreductase without the contribution of acetyltransferases or sulfotransferases. Environ. Mol. Mutagen. 2011;52:448–459. doi: 10.1002/em.20642. [DOI] [PubMed] [Google Scholar]
- 11.Schmeiser H.H., Bieler C.A., Wiessler M., van Ypersele de Strihou C., Cosyns J.-P. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with chinese herbs nephropathy. Cancer Res. 1996;56:2025–2028. [PubMed] [Google Scholar]
- 12.Pfau W., Schmeiser H.H., Wiessler M. 32P-postlabelling analysis of the DNA adducts formed by aristolochic acid I and II. Carcinogenesis. 1990;11:1627–1633. doi: 10.1093/carcin/11.9.1627. [DOI] [PubMed] [Google Scholar]
- 13.Pfau W., Schmeiser H.H., Wiessler M. Aristolochic acid binds covalently to the exocyclic amino group of purine nucleotides in DNA. Carcinogenesis. 1990;11:313–319. doi: 10.1093/carcin/11.2.313. [DOI] [PubMed] [Google Scholar]
- 14.Attaluri S., Bonala R.R., Yang I.-Y., Lukin M.A., Wen Y., Grollman A.P., Moriya M., Iden C.R., Johnson F. DNA adducts of aristolochic acid II: total synthesis and site-specific mutagenesis studies in mammalian cells. Nucleic Acids Res. 2010;38:339–352. doi: 10.1093/nar/gkp815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubbi C.P., Milner J. Analysis of nucleotide excision repair by detection of single-stranded DNA transients. Carcinogenesis. 2001;22:1789–1796. doi: 10.1093/carcin/22.11.1789. [DOI] [PubMed] [Google Scholar]
- 16.de Laat W.L., Jaspers N.G.J., Hoeijmakers J.H.J. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 17.Gillet L.C.J., Schärer O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2005;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 18.Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 19.Sidorenko V.S., Yeo J.E., Bonala R.R., Johnson F., Schärer O.D., Grollman A.P. Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of lactam-DNA adducts. Nucleic Acids Res. 2012;40:2494–2505. doi: 10.1093/nar/gkr1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernando R., Schmeiser H., Scherf H., Wiessler M. Formation and persistence of specific purine DNA adducts by 32P-postlabelling in target and non-target organs of rats treated with aristolochic acid I. IARC Sci. Publ. 1992:167–171. [PubMed] [Google Scholar]
- 21.Poon S.L., Pang S.T., McPherson J.R., Yu W., Huang K.K., Guan P., Weng W.-H., Siew E.Y., Liu Y., Heng H.L., et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci. Transl. Med. 2013;5:197ra101. doi: 10.1126/scitranslmed.3006086. [DOI] [PubMed] [Google Scholar]
- 22.Hoang M.L., Chen C.H., Sidorenko V.S., He J., Dickman K.G., Yun B.H., Moriya M., Niknafs N., Douville C., Karchin R., et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. 2013;5:197ra102. doi: 10.1126/scitranslmed.3006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moriya M., Slade N., Brdar B., Medverec Z., Tomic K., Jelaković B., Wu L., Truong S., Fernandes A., Grollman A.P. TP53 mutational signature for aristolochic acid: an environmental carcinogen. Int. J. Cancer. 2011;129:1532–1536. doi: 10.1002/ijc.26077. [DOI] [PubMed] [Google Scholar]
- 24.Schmeiser H.H., Nortier J.L., Singh R., Gamboa da Costa G., Sennesael J., Cassuto-Viguier E., Ambrosetti D., Rorive S., Pozdzik A., Phillips D.H., et al. Exceptionally long-term persistence of dna adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int. J. Cancer. 2014;135:502–507. doi: 10.1002/ijc.28681. [DOI] [PubMed] [Google Scholar]
- 25.Broschard T.H., Wiessler M., von der Lieth C.-W., Schmeiser H.H. Translesional synthesis on DNA templates containing site-specifically placed deoxyadenosine and deoxyguanosine adducts formed by the plant carcinogen aristolochic acid. Carcinogenesis. 1994;15:2331–2340. doi: 10.1093/carcin/15.10.2331. [DOI] [PubMed] [Google Scholar]
- 26.Clement F.C., Camenisch U., Fei J., Kaczmarek N., Mathieu N., Naegeli H. Dynamic two-stage mechanism of versatile DNA damage recognition by xeroderma pigmentosum group C protein. Mutat. Res-Fund. Mol. M. 2010;685:21–28. doi: 10.1016/j.mrfmmm.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Hess M.T., Schwitter U., Petretta M., Giese B., Naegeli H. Bipartite substrate discrimination by human nucleotide excision repair. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6664–6669. doi: 10.1073/pnas.94.13.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maillard O., Camenisch U., Blagoev K.B., Naegeli H. Versatile protection from mutagenic DNA lesions conferred by bipartite recognition in nucleotide excision repair. Mutat. Res-Rev. Mutat. 2008;658:271–286. doi: 10.1016/j.mrrev.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Kropachev K., Ding S., Terzidis M.A., Masi A., Liu Z., Cai Y., Kolbanovskiy M., Chatgilialoglu C., Broyde S., Geacintov N.E., et al. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014;42:5020–5032. doi: 10.1093/nar/gku162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geacintov N.E., Broyde S., Buterin T., Naegeli H., Wu M., Yan S., Patel D.J. Thermodynamic and structural factors in the removal of bulky DNA adducts by the nucleotide excision repair machinery. Biopolymers. 2002;65:202–210. doi: 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- 31.Rechkoblit O., Kolbanovskiy A., Malinina L., Geacintov N.E., Broyde S., Patel D.J. Mechanism of error-free and semi targeted mutagenic bypass of an aromatic amine lesion by Y-family polymerase Dpo4. Nat. Struct. Mol. Biol. 2010;17:379–388. doi: 10.1038/nsmb.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schärer O.D. Achieving broad substrate specificity in damage recognition by binding accessible nondamaged DNA. Mol. Cell. 2007;28:184–186. doi: 10.1016/j.molcel.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Sugasawa K., Okamoto T., Shimizu Y., Masutani C., Iwai S., Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maillard O., Camenisch U., Clement F.C., Blagoev K.B., Naegeli H. DNA repair triggered by sensors of helical dynamics. Trends Biochem. Sci. 2007;32:494–499. doi: 10.1016/j.tibs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Cai Y., Patel D.J., Geacintov N.E., Broyde S. Dynamics of a Benzo[a]pyrene-derived guanine DNA lesion in TGT and CGC sequence contexts: enhanced mobility in TGT explains conformational heterogeneity, flexible bending, and greater susceptibility to nucleotide excision repair. J. Mol. Biol. 2007;374:292–305. doi: 10.1016/j.jmb.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blagoev K.B., Alexandrov B.S., Goodwin E.H., Bishop A.R. Ultra-violet light induced changes in DNA dynamics may enhance TT-dimer recognition. DNA Repair. 2006;5:863–867. doi: 10.1016/j.dnarep.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Isaacs R.J., Spielmann H.P. A Model for Initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair. 2004;3:455–464. doi: 10.1016/j.dnarep.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Reeves D.A., Mu H., Kropachev K., Cai Y., Ding S., Kolbanovskiy A., Kolbanovskiy M., Chen Y., Krzeminski J., Amin S. Resistance of bulky DNA lesions to nucleotide excision repair can result from extensive aromatic lesion–base stacking interactions. Nucleic Acids Res. 2011;39:8752–8764. doi: 10.1093/nar/gkr537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair. 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Jain V., Hilton B., Lin B., Patnaik S., Liang F., Darian E., Zou Y., MacKerell A.D., Cho B.P. Unusual sequence effects on nucleotide excision repair of arylamine lesions: DNA bending/distortion as a primary recognition factor. Nucleic Acids Res. 2013;41:869–880. doi: 10.1093/nar/gks1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Min J.-H., Pavletich N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 42.Zheng H., Cai Y., Ding S., Tang Y., Kropachev K., Zhou Y., Wang L., Wang S., Geacintov N.E., Zhang Y., et al. Base flipping free energy profiles for damaged and undamaged DNA. Chem. Res. Toxicol. 2010;23:1868–1870. doi: 10.1021/tx1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai Y., Zheng H., Ding S., Kropachev K., Schwaid A.G., Tang Y., Mu H., Wang S., Geacintov N.E., Zhang Y., et al. Free energy profiles of base flipping in intercalative polycyclic aromatic hydrocarbon-damaged DNA duplexes: energetic and structural relationships to nucleotide excision repair susceptibility. Chem. Res. Toxicol. 2013;26:1115–1125. doi: 10.1021/tx400156a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen X., Velmurugu Y., Zheng G., Park B., Shim Y., Kim Y., Liu L., Van Houten B., He C., Ansari A., et al. Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nat. Commun. 2015;6 doi: 10.1038/ncomms6849. doi:10.1038/ncomms6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lukin M., Zaliznyak T., Johnson F., de los Santos C. Structure and stability of DNA containing an aristolactam II-dA lesion: implications for the NER recognition of bulky adducts. Nucleic Acids Res. 2012;40:2759–2770. doi: 10.1093/nar/gkr1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kathuria P., Sharma P., Abendong M., Wetmore S.D. Conformational preferences of DNA following damage by aristolochic acids: structural and energetic insights into the different mutagenic potential of the ALI and ALII-N6-dA adducts. Biochemistry. 2015;15:2414–2428. doi: 10.1021/bi501484m. [DOI] [PubMed] [Google Scholar]
- 47.Mu H., Kropachev K., Wang L., Zhang L., Kolbanovskiy A., Kolbanovskiy M., Geacintov N.E., Broyde S. Nucleotide excision repair of 2-acetylaminofluorene- and 2-aminofluorene-(C8)-guanine adducts: molecular dynamics simulations elucidate how lesion structure and base sequence context impact repair efficiencies. Nucleic Acids Res. 2012;40:9675–9690. doi: 10.1093/nar/gks788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai Y., Geacintov N.E., Broyde S. Nucleotide excision repair efficiencies of bulky carcinogen-DNA adducts are governed by a balance between stabilizing and destabilizing interactions. Biochemistry. 2012;51:1486–1499. doi: 10.1021/bi201794x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kropachev K., Kolbanovskiy M., Liu Z., Cai Y., Zhang L., Schwaid A.G., Kolbanovskiy A., Ding S., Amin S., Broyde S., et al. Adenine–DNA adducts derived from the highly tumorigenic dibenzo[a,l]pyrene are resistant to nucleotide excision repair while guanine adducts are not. Chem. Res. Toxicol. 2013;26:783–793. doi: 10.1021/tx400080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma P., Manderville R.A., Wetmore S.D. Structural and energetic characterization of the major DNA adduct formed from the food mutagen ochratoxin A in the NarI hotspot sequence: influence of adduct ionization on the conformational preferences and implications for the NER propensity. Nucleic Acids Res. 2014;42:11831–11845. doi: 10.1093/nar/gku821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mu H., Kropachev K., Chen Y., Zhang H., Cai Y., Geacintov N.E., Broyde S. Role of structural and energetic factors in regulating repair of a bulky DNA lesion with different opposite partner bases. Biochemistry. 2013;52:5517–5521. doi: 10.1021/bi4009177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Millen A.L., McLaughlin C.K., Sun K.M., Manderville R.A., Wetmore S.D. Computational and experimental evidence for the structural preference of phenolic C-8 purine adducts. J. Phys. Chem. A. 2008;112:3742–3753. doi: 10.1021/jp712058a. [DOI] [PubMed] [Google Scholar]
- 53.Grimme S., Ehrlich S., Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 54.Goerigk L., Kruse H., Grimme S. Benchmarking density functional methods against the S66 and S66×8 datasets for non-covalent interactions. ChemPhysChem. 2011;12:3421–3433. doi: 10.1002/cphc.201100826. [DOI] [PubMed] [Google Scholar]
- 55.Millen A.L., Manderville R.A., Wetmore S.D. Conformational flexibility of C8-Phenoxyl-2′-deoxyguanosine nucleotide adducts. J. Phys. Chem. B. 2010;114:4373–4382. doi: 10.1021/jp911993f. [DOI] [PubMed] [Google Scholar]
- 56.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 57.Cheatham T.E., Cieplak P., Kollman P.A. A modified version of the cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 58.Case D.A., Darden T.A., Cheatham T.E. III, Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M., et al. AMBER 11. San Francisco, CA: University of California; 2010. [Google Scholar]
- 59.Case D.A., Darden T.A., Cheatham T.E. III, Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M., et al. AMBER 12. San Francisco, CA: University of California; 2012. [Google Scholar]
- 60.Miller B.R., McGee T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 61.Wojtas-Niziurski W., Meng Y., Roux B., Bernèche S. Self-learning adaptive umbrella sampling method for the determination of free energy landscapes in multiple dimensions. J. Chem. Theory Comput. 2013;9:1885–1895. doi: 10.1021/ct300978b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beveridge D.L., DiCapua F. Free energy via molecular simulation: applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 1989;18:431–492. doi: 10.1146/annurev.bb.18.060189.002243. [DOI] [PubMed] [Google Scholar]
- 63.Brice A.R., Dominy B.N. Analyzing the robustness of the MM/PBSA free energy calculation method: application to DNA conformational transitions. J. Comput. Chem. 2011;32:1431–1440. doi: 10.1002/jcc.21727. [DOI] [PubMed] [Google Scholar]
- 64.Yildirim A., Sharma M., Varner B.M., Fang L., Feig M. Conformational preferences of DNA in reduced dielectric environments. J. Phys. Chem. B. 2014;118:10874–10881. doi: 10.1021/jp505727w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan S., Wu M., Buterin T., Naegeli H., Geacintov N.E., Broyde S. Role of base sequence context in conformational equilibria and nucleotide excision repair of benzo[a]pyrene diol epoxide−adenine adducts†. Biochemistry. 2003;42:2339–2354. doi: 10.1021/bi0270081. [DOI] [PubMed] [Google Scholar]
- 66.Cai Y., Patel D.J., Geacintov N.E., Broyde S. Dynamics of a benzo[a]pyrene-derived guanine DNA lesion in TGT and CGC sequence contexts: enhanced mobility in TGT explains conformational heterogeneity, flexible bending, and greater susceptibility to nucleotide excision repair. J. Mol. Biol. 2007;374:292–305. doi: 10.1016/j.jmb.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sproviero M., Verwey A.M.R., Rankin K.M., Witham A.A., Soldatov D.V., Manderville R.A., Fekry M.I., Sturla S.J., Sharma P., Wetmore S.D. Structural and biochemical impact of C8-aryl-guanine adducts within the NarI recognition DNA sequence: influence of aryl ring size on targeted and semi-targeted mutagenicity. Nucleic Acids Res. 2014;42:13405–13421. doi: 10.1093/nar/gku1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cosman M., Hingerty B.E., Luneva N., Amin S., Geacintov N.E., Broyde S., Patel D.J. Solution conformation of the (−)-cis-anti-Benzo[a]pyrenyl-dG adduct opposite dC in a DNA duplex: intercalation of the covalently attached BP Ring into the helix with base displacement of the modified deoxyguanosine into the major groove. Biochemistry. 1996;35:9850–9863. doi: 10.1021/bi9605346. [DOI] [PubMed] [Google Scholar]
- 69.Cosman M., Fiala R., Hingerty B.E., Laryea A., Lee H., Harvey R.G., Amin S., Geacintov N.E., Broyde S., Patel D. Solution conformation of the (+)-trans-anti-[BPh] dA adduct opposite dT in a DNA duplex: intercalation of the covalently attached benzo[c]phenanthrene to the 5′-side of the adduct site without disruption of the modified base pair. Biochemistry. 1993;32:12488–12497. doi: 10.1021/bi00097a029. [DOI] [PubMed] [Google Scholar]
- 70.Stavros K.M., Hawkins E.K., Rizzo C.J., Stone M.P. Base-displaced intercalation of the 2-amino-3-methylimidazo 4,5-f quinolone N-2-dG adduct in the NarI DNA recognition sequence. Nucleic Acids Res. 2014;42:3450–3463. doi: 10.1093/nar/gkt1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang F., DeMuro N.E., Elmquist C.E., Stover J.S., Rizzo C.J., Stone M.P. Base-displaced intercalated structure of the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in the recognition sequence of the NarI restriction enzyme, a hotspot for −2 bp deletions. J. Am. Chem. Soc. 2006;128:10085–10095. doi: 10.1021/ja062004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mao B., Hingerty B.E., Broyde S., Patel D.J. Solution structure of the aminofluorene [AF]-intercalated conformer of the syn-[AF]-C8-dG adduct opposite dC in a DNA duplex. Biochemistry. 1998;37:81–94. doi: 10.1021/bi972257o. [DOI] [PubMed] [Google Scholar]
- 73.Cai Y., Ding S., Geacintov N.E., Broyde S. Intercalative conformations of the 14R (+)- and 14S (−)-trans-anti-DB [a,l] P-N6-dA adducts: molecular modeling and MD simulations. Chem. Res. Toxicol. 2011;24:522–531. doi: 10.1021/tx1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Z., Mao H., Kim H.-Y., Tamura P.J., Harris C.M., Harris T.M., Stone M.P. Intercalation of the (−)-(1R,2S,3R,4S)-N6-[1-Benz[a]anthracenyl]-2'-deoxyadenosyl Adduct in an oligodeoxynucleotide containing the human N-ras codon 61 sequence. Biochemistry. 1999;38:2969–2981. doi: 10.1021/bi982072x. [DOI] [PubMed] [Google Scholar]
- 75.Li Z., Tamura P.J., Wilkinson A.S., Harris C.M., Harris T.M., Stone M.P. Intercalation of the (1R,2S,3R,4S)-N6-[1-(1,2,3,4- Tetrahydro-2,3,4-trihydroxybenz[a]anthracenyl)]-2'-deoxyadenosyl adduct in the N-ras codon 61 sequence: DNA sequence effects. Biochemistry. 2001;40:6743–6755. doi: 10.1021/bi002785r. [DOI] [PubMed] [Google Scholar]
- 76.Tang Y., Liu Z., Ding S., Lin C.H., Cai Y., Rodriguez F.A., Sayer J.M., Jerina D.M., Amin S., Broyde S., et al. Nuclear magnetic resonance solution structure of an N2-guanine DNA adduct derived from the potent tumorigen dibenzo[a,l]pyrene: intercalation from the minor groove with ruptured Watson–Crick base pairing. Biochemistry. 2012;51:9751–9762. doi: 10.1021/bi3013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan S., Shapiro R., Geacintov N.E., Broyde S. Stereochemical, structural, and thermodynamic origins of stability differences between stereoisomeric benzo [a] pyrene diol epoxide deoxyadenosine adducts in a DNA mutational hot spot sequence. J. Am. Chem. Soc. 2001;123:7054–7066. doi: 10.1021/ja0043035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.