

ABSTRACT
Vibrionaceae family members are interesting models for studying DNA replication initiation, as they contain two circular chromosomes. Chromosome II (chrII) replication is governed by two evolutionarily unique yet highly conserved elements, the origin DNA sequence oriCII and the initiator protein RctB. The minimum functional region of oriCII, oriCII-min, contains multiple elements that are bound by RctB in vitro, but little is known about the specific requirements for individual elements during oriCII initiation. We utilized undirected and site-specific mutagenesis to investigate the functionality of mutant forms of oriCII-min and assessed binding to various mutant forms by RctB. Our analyses showed that deletions, point mutations, and changes in RctB target site spacing or methylation all impaired oriCII-min-based replication. RctB displayed a reduced affinity for most of the low-efficacy origins tested, although its characteristic cooperative binding was generally maintained. Mutations that removed or altered the relative positions of origin components other than RctB binding sites (e.g., AT-rich sequence, DnaA target site) also abolished replicative capacity. Comprehensive mutagenesis and deep-sequencing-based screening (OriSeq) allowed the identification of a previously uncharacterized methylated domain in oriCII that is required for origin function. Together, our results reveal the remarkable evolutionary honing of oriCII and provide new insight into the complex interplay between RctB and oriCII.
IMPORTANCE
The genome of the enteric pathogen Vibrio cholerae consists of two chromosomes. While the chromosome I replication origin and its cognate replication initiator protein resemble those of Escherichia coli, the factors responsible for chromosome II replication initiation display no similarity to any other known initiation systems. Here, to enhance our understanding of how this DNA sequence, oriCII, and its initiator protein, RctB, function, we used both targeted mutagenesis and a new random-mutagenesis approach (OriSeq) to finely map the oriCII structural features and sequences required for RctB-mediated DNA replication. Collectively, our findings reveal the extraordinary evolutionary honing of the architecture and motifs that constitute oriCII and reveal a new role for methylation in oriCII-based replication. Finally, our findings suggest that the OriSeq approach is likely to be widely applicable for defining critical bases in cis-acting sequences.
INTRODUCTION
Most bacterial genomes consist of a single circular chromosome, the replication of which is governed by two evolutionarily conserved factors: DnaA, the initiator protein, and oriC, the replication origin (1). The bulk of our knowledge regarding how interactions between these two elements initiate chromosome replication stems from studies of the model bacterium Escherichia coli. Initiation is thought to be a multistep process. First, DnaA binds to a variety of binding sites within oriC, the most notable of which are 9-bp sequences known as DnaA boxes (2). Formation of a nucleoprotein complex between DnaA and the DnaA boxes results in the unwinding of an adjacent AT-rich region, which in turn leads to the recruitment and loading of the replicative helicase DnaB onto the open replication bubble (3). After DnaB widens the bubble, primase (DnaG) synthesizes RNA primers, allowing DNA polymerase III to load and begin chromosome replication.
Vibrio cholerae, the causative agent of the diarrheal disease cholera, was unexpectedly discovered to have a genome composed of two circular chromosomes in 1998, and all subsequently analyzed members of the Vibrionaceae family have been found to have the same genomic arrangement (4–6). In V. cholerae, the replication of chromosome I (chrI) is thought to initiate via processes similar to those of E. coli replication initiation; DnaAVc and oriCI bear sequence similarity to their E. coli counterparts and seem to function in a similar fashion (7–9). In contrast, neither the origin of chrII (oriCII) nor its cognate replication initiator protein RctB bears homology to functional analogs utilized by known chromosome or plasmid replication systems. However, both of these elements are conserved among all vibrio and photobacterial species and are likely to be essential for their replication (6, 8, 10). Studies of chrII replication have yielded knowledge of a novel mode of regulating DNA replication in bacteria and illuminated how bacteria can coordinate the replication of multipartite genomes (8, 9, 11–16).
Functional analyses have revealed that oriCII can be divided into two parts (see Fig. 1) (8). One is the minimum sequence for oriCII-based replication (referred to below as oriCII-min), and the other is a regulatory region known as the incompatibility (Inc) region. oriCII-min and inc both contain a variety of RctB binding sites, including sites that consist of 12 bp (12-mer), 11 bp (11-mer), 39 bp (39-mer), and 29 bp (29-mer) (8, 14, 17, 18). The ~450-bp oriCII-min region consists of an array of six 12-mers, a DnaA box, and an AT-rich region that contains a 29-mer. The oriCII-min region is sufficient for the replication of oriCII-based plasmids in V. cholerae, as well as in E. coli, provided that RctB is supplied in trans (8, 10, 14, 19). The adjacent ~500-bp inc region negatively regulates oriCII replication, possibly by handcuffing, initiator titration, and/or initiator remodeling (8, 14, 18). It includes four 11-mers, one 12-mer, and a 39-mer. Both 11-mers and 12-mers (which have similar consensus sequences) contain GATC, a recognition sequence for the DNA adenine methyltransferase Dam (Fig. 1). Dam is essential both for V. cholerae viability and for oriCII replication, presumably because RctB binding to the 12-mer array in oriCII requires GATC methylation (8, 15, 17, 20). The importance of methylation for RctB binding to 11-mers has not been reported, nor has RctB’s relative affinity for 11-mers and 12-mers been described.
FIG 1 .

Schematic of V. cholerae oriCII. The size and spacing of the elements are to scale. Brown triangles are 11-mers, and purple triangles are 12-mers, the consensus sequences of which are shown at the bottom with differences in red. Blue squares are a 39-mer and a 29-mer. The orange triangle is a DnaA box. Green stars are GATC Dam methylation sites. The red region on the left is known as the incompatibility region, and it negatively regulates oriCII function. The blue-white box, including the DnaA box and six 12-mers, along with the AT-rich region (green box), is the minimal sequence required for oriCII-based replication, oriCII-min. Portions of oriCII present in EMSA fragments are indicated above the origin by black bars.
RctB mutants that are not subject to negative regulation by inc have been isolated, including several C-terminal truncations (e.g., RctB ΔC159) (14, 21, 22). The mutant’s lack of inhibition by inc appears to reflect the higher affinity of ΔC159 mutant RctB than wild-type (WT) RctB for a 12-mer and its lack of binding to the 39-mer motifs found in oriCII (14, 21). In addition, truncated forms of RctB have a lower capacity to dimerize than the full-length protein, and Chattoraj and colleagues have proposed that monomeric and dimeric forms of RctB promote and inhibit initiation at oriCII, respectively (23). The effects of the truncations are incompletely understood; nonetheless, their behavior has provided some insight into how RctB-mediated initiation at oriCII may be regulated (14, 21–23).
While some studies have explored potential mechanisms by which the inc region modulates oriCII-min activity, in depth investigation of the requirements for the various elements within oriCII-min for oriCII-based replication has not been carried out. Here, we engineered a large number of oriCII-min mutants to systematically investigate which elements of oriCII-min are required for replication and how the absence or presence of these elements affects the binding of RctB. We discovered that deleting, mutating, differentially spacing, or substituting 12-mers reduces or abolishes oriCII replicative function, while concomitantly affecting RctB’s binding kinetics. The presence and orientation of the DnaA box are also essential for initiation, as is the presence of a portion of the oriCII (and not oriCI) AT-rich region. Finally, we devised and applied a high-throughput deep-sequencing approach (OriSeq) to screen an extensive oriCII mutant library for oriCII features required for replication. This unbiased strategy confirmed findings from engineered origins, as well as hypotheses based on comparisons of diverse oriCII sequences. Furthermore, this approach led to the identification of a presumably methylated region in the oriCII AT-rich region that is essential for oriCII function. Together, our results reveal the remarkable evolutionary honing of oriCII—for the most part, its architecture and motifs do not tolerate mutations—and provide new insight into the complex interplay between RctB and oriCII.
RESULTS
RctB binds to individual 11-mer and 12-mer motifs.
The consensus 11-mer and 12-mer motifs show a high degree of sequence homology, differing by four bases located at their respective 3′ ends (Fig. 1). Despite their similarity, they are associated with opposing effects on chrII replication, as 11-mers reside solely within the inc region, whereas oriCII-min contains only 12-mers. To gain further insight into functional differences between 11-mers and 12-mers, we utilized electrophoretic mobility shift assays (EMSAs) to quantitatively assess how purified RctB interacts with these sequences. Binding was assessed for WT RctB (659 amino acids) and an N-terminal fragment consisting of amino acids 1 to 500 (RctB ΔC159). Previous work revealed that RctB ΔC159 binds to a 12-mer and a 39-mer with different affinities than WT (14, 21, 23), but it is not known if this RctB fragment binds to an 11-mer in a different manner than WT; this information might help explain why RctB ΔC159 is not subject to Inc-mediated negative regulation (14, 21, 23).
The sequences of the DNA probes used for these initial assays were derived from the inc region. Probe DNA was isolated from plasmids grown in a Dam+ strain and contained an individual 11-mer or 12-mer, as well as adjacent sequences (Fig. 1 and 2A). At most of the protein concentrations tested, WT RctB bound to the Inc 11 and Inc 12 probes as a single species (Fig. 2B and C, green arrow). However, at very high concentrations, a second, faster-migrating band appeared before the probes became completely saturated and shifted to the well (Fig. 2B and C, orange and blue arrows, respectively). This pattern was observed previously with WT RctB bound to a 12-mer probe, and binding by dimeric and monomeric forms of RctB was proposed to generate the upper and lower bands, respectively (21). In contrast, a single band shift was observed when using RctB ΔC159 and either Inc 11 or Inc 12 (Fig. 2D and E, green arrows).
FIG 2 .
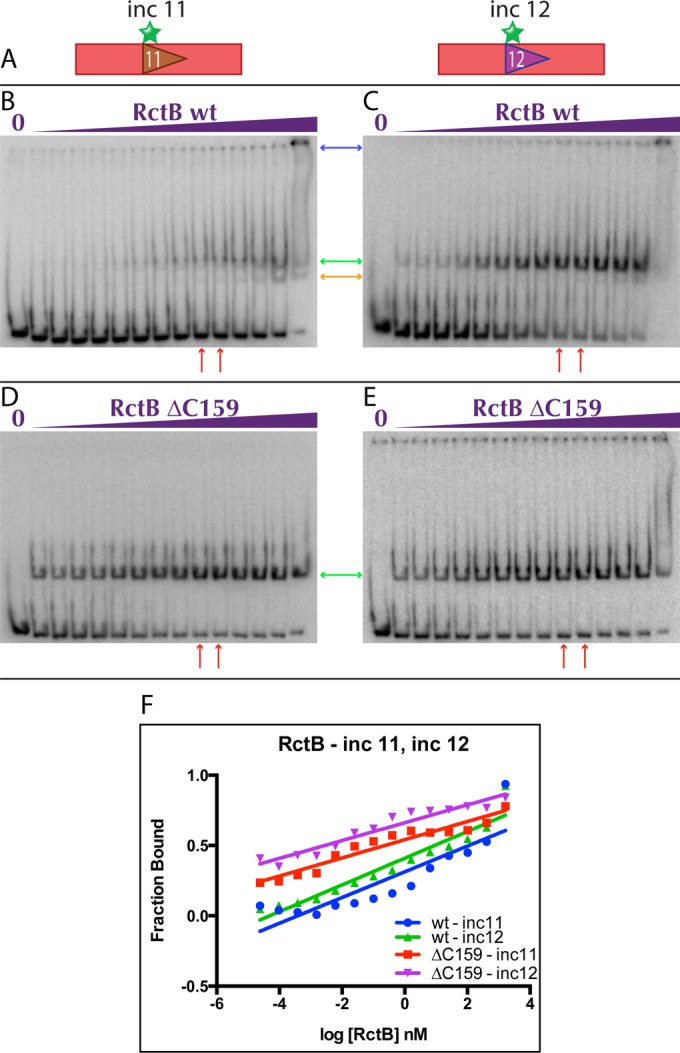
RctB binding to probes containing a single 11-mer or 12-mer from the inc region. (A) Schematics of the probes used in the EMSAs. (B to E) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 (D and E) with methylated Inc 11 (B and D) and Inc 12 (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. (F) Representative binding curves of EMSA quantifications. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively.
To quantify binding, we assessed the fraction of unbound probe and calculated the dissociation constant, Kd, as the concentration of protein where 50% of the probe was shifted. Full-length RctB bound to both Inc 11 and Inc 12 with significantly lower affinity (~1,000×) than that of truncated RctB (Fig. 2), but all of the probe-protein combinations displayed linear interactions with similar increases in binding for a given change in protein concentration. Interestingly, both proteins bound to Inc 12 with approximately 100-fold greater affinity than to Inc 11 (Table 1). Overall, these binding analyses are consistent with Chattoraj’s proposals that full-length RctB, but not C-terminally truncated RctB ΔC159, is competent at dimerization and that dimerization impairs binding to DNA, so that dimers bind only at high protein concentrations and the effective affinity of the WT protein is lower. The basis for differential binding to Inc 11 and Inc 12 cannot be determined from these data, because it could be attributable to sequence differences between either the core 11- or 12-mers or the associated flanking sequences in each probe.
TABLE 1 .
Dissociation constants of WT RctB and RctB ΔC159 for various DNA fragments
| DNA probe | Avg Kd (nM) ± SDa of: |
|
|---|---|---|
| WT RctB | RctB ΔC159 | |
| 12×6 Dam+ | 0.0227 ± 0.0198 | 0.0094 ± 0.004 |
| 12×6 Dam− | NDb | 13.84 ± 3.86 |
| Inc 11 | 197.6 ± 252.3 | 0.061 ± 0.11 |
| Inc 12 | 2.98 ± 0.70 | 0.00016 ± 0.00025 |
| 11×6 | 8.50 ± 6.92 | 5.45 ± 2.87 |
| 11+1×6 | 7.85 ± 8.13 | 5.35 ± 6.25 |
| Rev12×6 | 0.22 ± 0.16 | 0.29 ± 0.20 |
| 12×2 | 10.16 ± 7.14 | 0.26 ± 0.029 |
| 12×4 | 0.023 ± 0.0071 | 0.036 ± 0.029 |
| 12-1 | 0.036 ± 0.026 | 0.0081 ± 0.0025 |
| 12-2 | 0.092 ± 0.11 | 0.012 ± 0.06 |
| 12-3 | 0.06 ± 0.015 | 0.032 ± 0.0025 |
| 12-4 | 0.11 ± 0.021 | 0.04 ± 0.029 |
| 12-5 | 0.087 ± 0.03 | 0.038 ± 0.025 |
| 12-6 | 0.093 ± 0.013 | 0.17 ± 0.033 |
| 12-135 | 1.07 ± 0.23 | 0.35 ± 0.59 |
| 12-246 | 2.13 ± 0.40 | 1.20 ± 0.96 |
| 12-12456 | 42.62 ± 22.23 | 5.66 ± 2.97 |
Average values and standard deviations were obtained by quantitation of at least three independent binding experiments.
ND, not determined.
To confirm the importance of GATC methylation in the binding of RctB to target sites, we also isolated Inc 11 and Inc 12 probe DNA from plasmids propagated in dam mutant hosts. As previously reported (17), we found that Dam methylation of Inc 12 was required for both WT RctB and RctB ΔC159 to bind to the probe (see Fig. S1B and D in the supplemental material). Similarly, Dam methylation of Inc 11 was required for RctB binding (see Fig. S1C and E).
WT RctB, but not RctB ΔC159, binds to the 12-mer array in a cooperative manner.
EMSAs were also used to explore the binding of WT RctB and RctB ΔC159 to a probe containing the six-12-mer array and the DnaA box from the min part of oriCII (12×6). The two proteins exhibited dramatically different patterns of binding to this probe, which also differed from the binding observed with the Inc 12 probe. WT RctB displayed a largely all-or-nothing pattern of binding to the multisite probe (Fig. 3B), in contrast to the gradual increase in bound probe seen with the single 12-mer. Furthermore, most of the probe was either unbound or completely shifted to the well (Fig. 3B, blue arrow), which likely reflects coordinate occupancy of all six potential 12-mer binding sites by RctB. In contrast, the truncated protein did not appear to occupy all of the target sites simultaneously; instead, multiple, distinct protein-DNA complexes were visible, with increasing shifts accompanying increased protein concentrations (Fig. 3C, green arrows). Maximal binding of the 12×6 probe was observed at similar concentrations of both WT RctB and RctB ΔC159, approximately 6.4 to 25.6 nM (Fig. 3B and C, red arrows); however, low-level binding could be observed when using far lower concentrations of RctB ΔC159 (0.00006125 nM) than WT RctB (0.03125 to 0.0625 nM). Both proteins had markedly reduced affinity for the 12-mer array when this probe was unmethylated (Fig. 3E and F). Overall, these data suggest that WT RctB cannot initiate binding to the 12-mer array as readily as RctB ΔC159, a trend that was also observed when using the individual 12-mers (Fig. 2C and E); however, once initiated, binding of the WT protein appears to be extremely cooperative, i.e., occupancy of the first site facilitates the occupancy of adjacent sites.
FIG 3 .
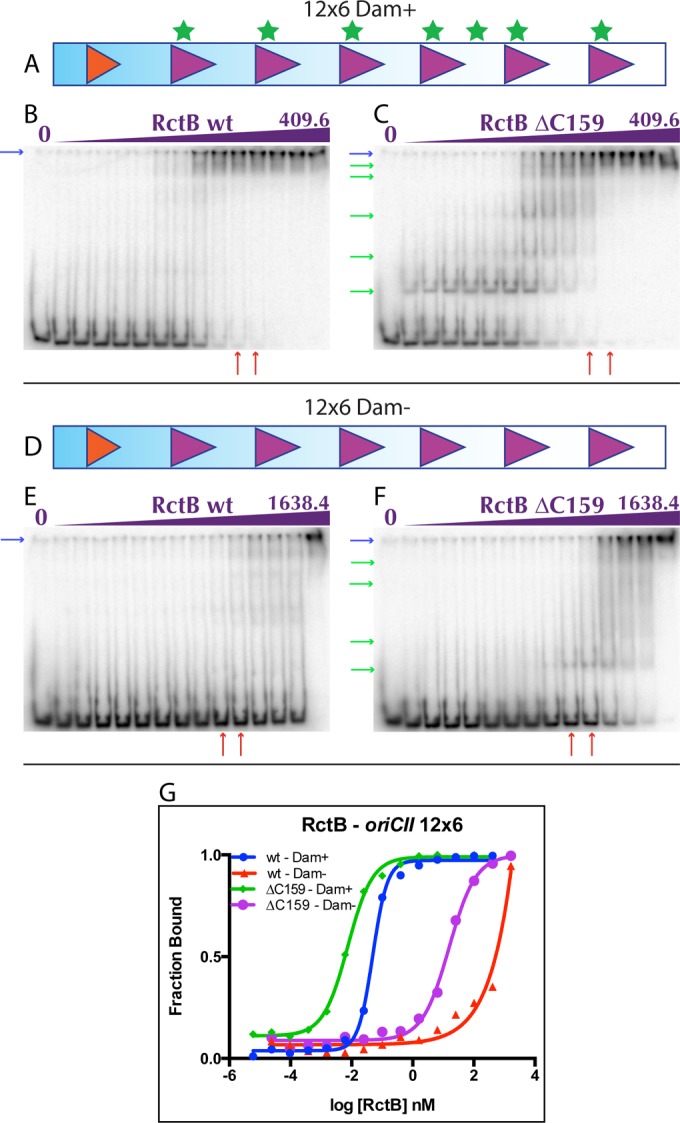
RctB binding to the 12×6 array of oriCII. Methylated (A) and unmethylated (D) probes used in the EMSAs shown in panels B and C and panels E and F, respectively. (B, C, E, and F) Representative EMSAs of WT RctB (B and E) and RctB ΔC159 (C and F) with methylated (B and C) and unmethylated (E and F) 12×6 probes. (G) Representative binding curves of EMSA quantifications. The concentrations of RctB ranged from 0.000006125 to 409.6 nm (B and C) and from 0.000025 to 1,638.4 nM (E and F) in 4-fold dilution series. Red arrows below the gels indicate RctB concentrations where near-maximal binding with the methylated 12×6 probe is shown in panels B and C.
Ideally, this somewhat qualitative characterization of RctB binding could be augmented by quantitative analyses of the individual protein-DNA interactions. However, given the multiplicity of binding sites within the 12×6 probe, coupled with WT RctB’s (but not RctB ΔC159’s) apparent capacity for cooperative binding, comparative analyses of binding are challenging. For WT RctB, only the extent of unbound or maximally shifted probe can be assessed, and any decrease in the amount of unbound probe is largely mirrored by an increase in the maximally shifted probe. Thus, any reaction constant will reflect the net interconversion of these two species, although the absence of incompletely shifted probe suggests that binding of the first protein molecule is the limiting step in allowing cooperative occupation of the remaining binding sites. In contrast, decreases in unbound probe when using RctB ΔC159 are not mirrored by the appearance of maximally shifted probe but instead are reflected in the appearance of partially saturated probe; consequently, the extent to which unbound probe disappears measures a rather different phenomenon.
Still, a comparison of the disappearance of probe when using each protein should at least reveal the likelihood of the first binding reaction. Our analyses indicate that half-maximal binding by WT RctB occurs with 0.0227 nM protein, while half-maximal binding of RctB ΔC159 (to its first target site) requires 0.0094 nM protein, consistent with the idea that RctB ΔC159 has a higher affinity than the WT protein for the first site to be occupied within the 12-mer array (Table 1).
Most oriCII elements and their specific architecture are essential for replication.
To begin to ascertain which molecular features of oriCII-min are required for it to serve as a substrate for initiation of replication, we generated a large series of oriCII mutants (Fig. 4) and assessed their capacity to support replication by using a previously developed transformation assay (14). For this assay, the mutant oriCII sequences were ligated to a kanamycin resistance cassette and then the ligation products were transformed into E. coli expressing either WT RctB or RctB ΔC159. The number of colonies of each mutant was normalized to both a control plasmid and the WT oriCII-min fragment. The different oriCII constructs were engineered to investigate whether the nature (11-mer versus 12-mer), orientation, number, and spacing of the various elements in the oriCII sequence are necessary for its origin function. The results of multiple independent experiments are shown in Fig. 4.
FIG 4 .

Few structural changes in oriCII-min maintain its replicative capacity. (A) CFU count for each oriCII pseudo-origin normalized to both a kanamycin control plasmid and a WT oriCII fragment. Mean ratios and standard errors of the means shown were obtained from three independent transformation experiments. (B) Schematics of the pseudo-oriCIIs. The symbols are the same as in Fig. 1; in addition, the red rectangles represent 5-bp insertions, green rectangles represent 10-bp insertions, yellow rectangles represent 21-bp insertions, and red stars indicate Dam methylation sites that were mutated to GTAC.
Since the sequences of the 11-mers and 12-mers are similar and we found that RctB bound to these single motifs in a similar fashion, albeit with different affinities (Fig. 2), we initially tested if 11-mers could substitute for 12-mers in oriCII-min and enable replication. Two different substitutions of 11-mers for 12-mers were created. In one, each of the six 12-mers in the array was replaced with an 11-mer (designated 11×6 in Fig. 4); in the other (designated 11+1×6), the last base of the 12-mer consensus sequence was added to the 11-mer sequence to maintain the spacing and helical phasing of the binding sites. Surprisingly, neither of the 11-mer substitutions yielded transformants, demonstrating that an 11-mer cannot replace the function of a 12-mer for replication, despite the fact that RctB is able to bind to either individual motif, albeit with a lower affinity for the 11-mer.
In oriCII-min, the 12×6 array is located between the DnaA box and the AT-rich region. The 12-mers, which are not palindromic, all have the same orientation, raising the possibility that the set of RctB monomers bound to the 12-mer array form a directional oligomer. To test whether the orientation of the RctB oligomer (with respect to the DnaA box and the AT-rich region) is important for oriCII function, we created Rev12×6, a mutant form of oriCII-min in which the orientation of the 12-mer array is reversed, so that the 5′ end of the 12-mer array, which is ordinarily closest to the DnaA box, was flipped to become the 3′ end of the array closest to the AT-rich region (Fig. 4). We also created constructs in which the DnaA box was deleted or reversed (Delta-DnaA, Rev-DnaA). No transformants were detected with any of these constructs. These results are consistent with the previous finding that DnaA is required for replication (8) and also suggest that there may be critical interactions between proteins bound to the array and the DnaA box that necessitate specific relative orientations of these elements. In several plasmid replication systems, interactions between initiator proteins and DnaA are required for recruitment and/or loading of the replicative helicase DnaB (24, 25).
Several constructs were created to test whether all six of the 12-mers in oriCII-min are essential for replication. These included constructs in which multiple 12-mers were deleted (12×2, 12×4), as well as constructs with point mutations (GATC to GTAC) that prevented their methylation in either single (12-1, 12-2, 12-3, 12-4, 12-5, 12-6) or multiple (12-135, 12-246, 12-12456) 12-mers (Fig. 4, red stars). Both WT RctB and RctB Δ159 bound to an individual 12-mer point mutant (Inc 12-GTAC) probe in the same manner as an unmethylated WT Inc 12 probe, supporting their use for disruption of RctB binding to specific 12-mers in the 12×6 array (see Fig. S1C and E and S2C and E in the supplemental material). These experiments suggest that at least five 12-mers must be present and intact for oriCII function; no transformants were observed with the 12×2, 12×4, 12-135, 12-246, and 12-12456 constructs. Transformants were obtained from each construct with a single 12-mer mutated, although fewer than with oriCII-min, and the mutations did not have identical effects. Mutation of the two 12-mers closest to the DnaA box and the AT-rich region caused a greater reduction in transformants than mutations to the middle two 12-mers. This may be a reflection of the cooperative nature of RctB binding to the 12-mer array, where weaker binding to the internal 12-mers could be stabilized by stronger associations on either side. Unexpectedly, the results suggest that the first, second, and fifth 12-mers are more important for replication mediated by RctB ΔC159 than by WT RctB. These three 12-mers differ from the consensus sequence, notably starting with T instead of A, raising the possibility that RctB ΔC159 may bind to these sequences differently than to the consensus 12-mers (see Fig. S3 in the supplemental material).
Spacers of 5, 10, or 21 bases were also inserted at various positions within the 12×6 array and between the array and the AT-rich region to investigate whether the spacing and helical phasing between these elements are important for replication. Addition of five bases after individual 12-mers in the 12×6 array, which is predicted to disrupt the helical phasing between the neighboring elements by half a turn, was not tolerated at any of the positions we tested (1+5, 3+5, 6+5), suggesting that the RctB binding sites must be on the same face of the DNA helix to support replication. Consistent with this idea, insertion of 10 bases between 12-mers, which preserves helical phasing, did not abolish replication mediated by WT RctB, though there were reduced numbers of transformants compared to oriCII-min (1+10 and 3+10 in Fig. 4). However, insertion of 10 bases after the sixth 12-mer in the array (6+10), i.e., between the array and the AT-rich region, abolished replication, suggesting that RctB may be unable to unwind this region when it is farther away, even though the phasing is unchanged. Both proper helical phasing and proper spacing are required between initiator binding sites and the AT-rich region for DnaA and oriC as well (26, 27). Insertions between 12-mers of larger (21-bp) spacers that preserved phasing (i.e., 1+21, 3+21) also abolished oriCII-min replication; thus, there appears to be a limit to the extent that tandem RctB binding sites can be separated and still mediate replication initiation. RctB ΔC159-mediated replication was more sensitive to changes in the spacing between 12-mers than replication dependent upon the WT protein; fewer transformants were observed for the 3+10 construct when using the truncated protein. This result could reflect the lower extent of dimerization and cooperativity of RctB ΔC159, which might lessen its ability to form stable, replication-competent complexes across more dispersed binding sites.
We also created oriCII-min constructs to investigate whether the distance and helical phasing between the DnaA box and the 12×6 array are important for replication. As seen for other regions of oriCII-min, the helical phasing between the DnaA box and the 12×6 array appears to be critical for replication; the DnaA+5 construct, which contains five additional bases between the DnaA box and the array, yielded no transformants. In contrast, the DnaA+10 and DnaA+21 constructs, which alter the spacing but not the phasing between these sequences, were able to support replication. It is unclear why the addition of 21 bases at this location yielded a greater number of transformants than the addition of 10 bases, but it is possible that two helical turns allow DnaA more freedom to contact other elements within oriCII required for replication.
The final set of oriCII constructs facilitated exploration of the importance of the AT-rich region, the presumed site of helix opening. The Delta-AT construct, which contains no AT-rich segment, yielded no transformants, presumably because there is no site for the DNA helix to unwind when RctB is bound. Somewhat unexpectedly, we found that the specific sequence of the AT-rich region, rather than simply the presence of a sequence with a relatively low melting temperature, is required for oriCII-based replication. No transformants were obtained when using oriCI-AT, a construct that contains the AT-rich region from oriCI in place of the oriCII AT-rich sequence. This result suggests that either RctB is unable to melt this stretch of DNA or the oriCII AT-rich region plays an additional role(s) during initiation. The latter possibility is supported by the observation that replication is also reduced by deletion of the 29-mer RctB binding site, which lies downstream of the −35 and −10 boxes of the rctB promoter, from this region (Delta-29-mer in Fig. 4). Together, these observations reveal that very few changes can be made within oriCII without impairing replication; nearly all of its component parts and their arrangement are of critical importance for oriCII to serve as a functional origin.
Deletion of 12-mers from the oriCII array reduces RctB binding affinity and/or cooperativity.
We assessed the ability of RctB to bind to many of the oriCII constructs to gain more mechanistic insight into their various capacities to support replication. Reversal of the 12×6 array caused a roughly 10-fold reduction in the affinity of WT RctB and RctB ΔC159 for origin sequences (Table 1). In contrast, both WT RctB and RctB ΔC159 had markedly lower affinity (>300×) for the two 11-mer constructs (11×6 and 11+1×6) than for the WT probe (Fig. 5; see Fig. S4 in the supplemental material). The latter result likely accounts for the failure of these constructs to support replication. Interestingly, although WT RctB displays markedly reduced affinity for the 11-mer constructs, it still appears to interact with these probes in a cooperative fashion once the initial binding step occurs (Fig. 5B and C).
FIG 5 .
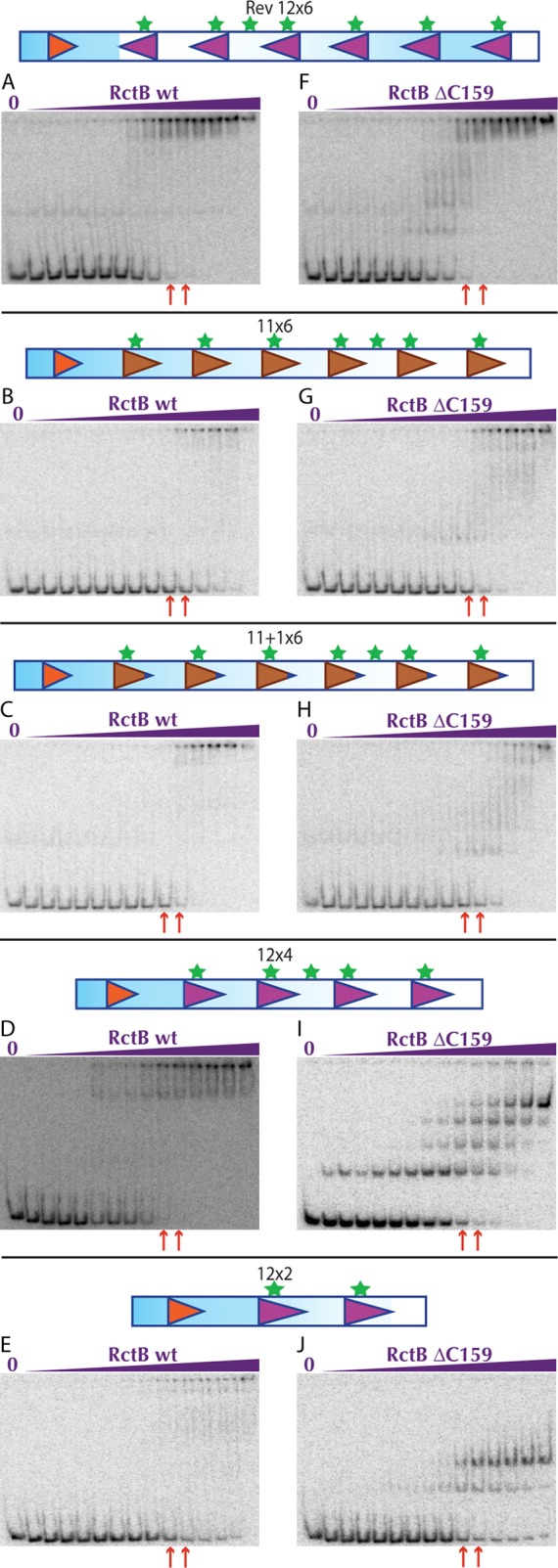
Twelve-mer substitutions and deletions reduce RctB binding affinity and/or cooperativity. Representative EMSAs of WT RctB (A to E) or RctB ΔC159 (F to J) with pseudo-origins depicted above the gels. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively.
Truncation of the 12-mer array to include only four 12-mers resulted in a different pattern of binding than was observed with the longer probes. The affinity of both proteins for their first binding reaction to the 12×4 probe was similar to that seen with the 12×6 fragment (compare Kds in Table 1); however, unlike for WT RctB, subsequent interactions for RctB ΔC159 occurred less readily, so that maximal probe shifting occurred with ~4- to 16-fold higher concentrations of protein (red arrows in Fig. 3B and C and 5D and I). This result suggests that the RctB-DNA complex forms less readily or is less stable when fewer binding sites are present if the protein’s cooperativity is reduced. These effects become even more apparent for both proteins when RctB interacts with a two-12-mer probe (Table 1; Fig. 5E and J).
Individual 12-mers within the oriCII array are not equivalent.
In the transformation assay, methylation site mutations within single 12-mers in the array reduced but did not abolish the capacity of the origin constructs to support replication, suggesting that no individual 12-mer makes an essential contribution to the functionality of the array. Consistent with these results, WT RctB and RctB ΔC159 bound to the corresponding probes (12-1, 12-2, 12-3, 12-4, 12-5, 12-6) with affinities similar to or slightly lower than those they had for the WT array (Table 1; Fig. 3B and C and 6; see Fig. S5 in the supplemental material). These binding experiments did not provide a clear explanation of why some mutations had a larger effect upon RctB ΔC159-based replication; no marked variation was observed in the truncated protein’s pattern of binding to the set of probes, and there was no precise correspondence between binding affinity and replication capacity (Table 1; Fig. 3C, 4, and 6G to L; see Fig. S5C in the supplemental material). However, an apparent correspondence between replication capacity and RctB binding was observed for the constructs with mutations in multiple 12-mers (12-135, 12-246, and 12-12456). Both WT RctB and RctB ΔC159 bound to these probes with markedly reduced affinity, suggesting that contiguous methylated 12-mers are important for high-affinity RctB binding to the origin (Table 1; see Fig. S5B and D and S6 in the supplemental material). WT RctB’s cooperative binding to these probes was slightly reduced as well, as shown by the appearance of weak intermediate species in the gels (see Fig. S6B to D, green arrows).
FIG 6 .
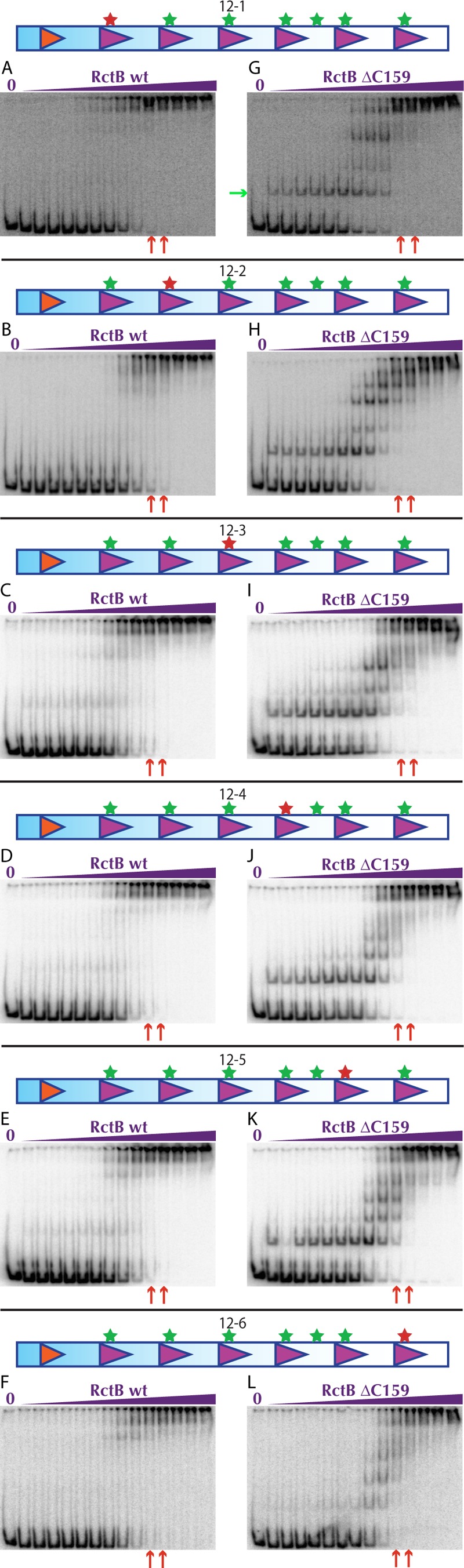
Individual 12-mer changes modestly reduce RctB binding affinity Representative EMSAs of RctB wt (A-F) or ΔC159 (G-L) with pseudo-origins depicted above gels. The concentrations of RctB ranged from 0.000006125-409.6 nM in a 4-fold dilution series. Red arrows below the gels indicate RctB wt and ΔC159 concentrations where near maximal binding is seen with the methylated 12×6 probe in Fig. 3B and 3C respectively.
In vitro mutagenesis and BLAST sequence comparisons reveal a novel domain in the AT-rich region important for oriCII-based replication.
Besides the site-directed approach for dissecting oriCII features required for its function (Fig. 4), we also developed a high-throughput open-ended approach to identify oriCII nucleotides and elements that are necessary for this sequence to act as an origin. Our deep sequencing-based strategy (OriSeq), which is akin to Mut-seq (28), is based on negative selection against oriCII mutations that reduce its capacity to act as an origin. Using error-prone PCR with the oriCII-min as a template, we generated an extensive library of mutant oriCII sequences. The library was ligated to a kanamycin resistance cassette and then transformed into an E. coli strain expressing RctB from a plasmid, similar to the transformation assay. Origins from the resulting colonies were purified and sequenced, and the locations of mutations were compiled in order to identify sites that were underrepresented, which were expected to be important or essential for oriCII-based replication. The findings from the screening are outlined in Fig. 7A. Green vertical lines show the sites and relative abundances of mutations that have higher-than-average mutation frequencies, while the red vertical lines represent nucleotides with lower-than-average mutation frequencies. Figure 7A clearly demonstrates that the mutation frequencies at all six 12-mers and the DnaA box were below the average mutation frequency per base across the entire library (Fig. 7, blue boxes). Since these elements are known to be important for oriCII function, this result provides strong evidence for the validity of our strategy. In addition to these sites, Fig. 7 also shows that a section of the AT-rich region that contains three Dam methylation sites was underrepresented in the library (Fig. 7, orange box), suggesting that this region has a previously unappreciated functional significance.
FIG 7.
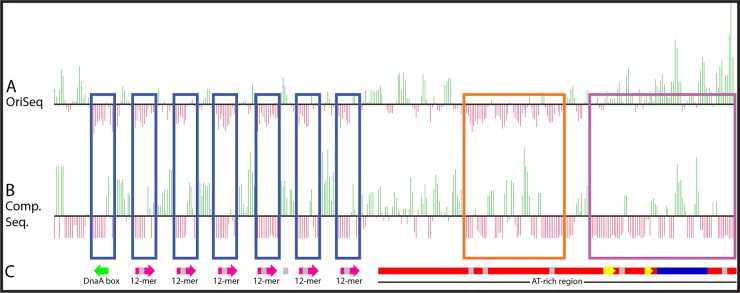
Origin-Seq and comparative sequence analysis reveal a novel element in the AT-rich region important for oriCII function (A) OriSeq results. The black line represents the average mutation frequency per base pair averaged across the entire oriCII fragment that was subjected to error-prone PCR. The green lines above the black bar signify bases that had a higher mutation frequency than average and are therefore less important, while the red bars indicate bases that had a lower mutation frequency and are therefore more important to oriCII function. (B) Sequence comparisons of vibrio chrII putative origins. The minimum functional oriCII sequence from V. cholerae was compared to 28 sequences of other vibrio species genomes using BLASTN. The black line represents the average mutation frequency per base. The green lines are bases that had a higher mutation frequency and are therefore less important, while the red lines are bases that had a lower mutation frequency and are therefore more important to oriCII function. (C) Schematic of known elements within the oriCII minimum functional sequence. The green arrow indicates the DnaA box, pink arrows indicate 12-mers, the red rectangle indicates the AT-rich region, gray squares indicate GATC Dam methylation sites, yellow arrows indicate putative -35 and -10 boxes of the RctB promoter, and the blue rectangle indicates the 29-mer.
We hypothesized that comparisons of the oriCII-min region from all sequenced vibrios would yield information regarding the evolutionary constraints on the sequence of oriCII and provide a means to corroborate our experimental OriSeq findings. There is a remarkably good correlation between the pattern of sequence conservation (Fig. 7B) and the OriSeq results (Fig. 7A). The DnaA box, the six 12-mer RctB binding sites, and a section of the AT-rich region overlapping the three GATC sites all showed more conservation than surrounding sequences among the vibrio species we queried (Fig. 7). The conservation of these sequences through evolutionary time provides powerful validation of the results garnered from the in vitro mutagenesis.
In one region (outlined by the purple box in Fig. 7), there is a discrepancy between the OriSeq results, where the nucleotides were generally mutable (green), and the conservation analysis, where these nucleotides were conserved (red). This region corresponds to the rctB promoter, a cis-acting sequence required for the expression of rctB, an essential gene. The conservation of this locus, seen in a comparative sequence analysis, is expected and likely reflects genuine selective pressure for the maintenance of this sequence. However, in our in vitro OriSeq experiment, we expressed RctB in trans from an exogenous plasmid, rather than from the endogenous locus; thus, under these particular conditions, the rctB promoter was dispensable for oriCII function.
We engineered additional oriCII constructs to confirm that the three GATC sites in the AT-rich region are important for replication in the transformation assay. When all three GATC sites were changed to GTAC, preventing their methylation, no colonies were recovered from the transformation, suggesting that methylation of these sites is essential for oriCII replication (Fig. 4). RctB did not bind to probes corresponding to this region (see Fig. S7 in the supplemental material; Fig. 7, orange box). Although we cannot exclude the possibility that RctB could bind to these sequences if additional adjacent sequences (e.g., the 12×6 array) were present, it is also possible that methylation of these sites contributes to oriCII replication through different means. For example, methylation of these GATC sites may modulate the unwinding of the AT-rich region, as previously reported for oriC (29).
DISCUSSION
The genomes of all species of the family Vibrionaceae are divided between two circular chromosomes, and the factors that control initiation of chrII replication, oriCII and RctB, are highly conserved in this family (4–8). oriCII-min, the minimum cis-acting sequence required for initiation of replication of V. cholerae chrII, was known to consist of an array of six 12-mer binding sites for the initiator, RctB, as well as an adjacent DnaA-binding site and an AT-rich sequence. Here, we used both targeted and random mutagenesis to finely map the oriCII-min structural features and sequences required for RctB-mediated DNA replication. Collectively, our findings reveal the remarkable evolutionary honing of the architecture and motifs that constitute oriCII-min—most of the extensive set of mutant forms of oriCII we tested were incapable of serving as templates for initiation of replication.
In general, we observed a correlation between the extent of WT RctB binding to variants of oriCII-min and the replicative capacity of these mutated origins of replication. Changes that markedly reduced RctB binding, so that >4-fold elevated levels of protein were required for occupancy of all RctB binding sites (e.g., 11×6, 11+1×6, 12-135, 12-246, 12-12456) also prevented recovery of plasmids dependent upon the mutant origins. In contrast, most changes that did not markedly reduce WT RctB binding, such as disruption of the GATC motif within an individual 12-mer, at least partially preserved origin function. The exception to this pattern was the reversal of the 12×6 array, which permitted WT binding levels but not replication. However, this mutation likely prevents interactions that span the array and adjacent sequences and thereby prevents replication. It is likely that replicative failures due to the absence of sequences outside the 12×6 array (e.g., in Delta-DnaA, Rev-DnaA, Delta-AT, etc.) also do not reflect changes in RctB binding; however, this was not assessed in our study. The correlation between binding and replicative capacity was less consistent for RctB ΔC159-based replication than for WT RctB-based replication; it is not clear from the binding assays why a subset of mutations within 12-mer GATC sites (12-1, 12-2, 12-5) abolish replication while others do not. Overall, our results suggest that RctB binding is likely to be a limiting factor in oriCII-mediated replication, as has generally been observed for other replication initiator proteins (30, 31). Fine calibration of initiator binding (which is likely aided by RctB’s previously described transcription autorepression, as well as by binding to low-affinity sites within the inc region) presumably allows cells to avoid the toxicity associated with replication over initiation (32–36).
Our results also provide insight into the nature of RctB binding sites and how small variations in them influence RctB binding and oriCII functionality. We observed that RctB’s affinity for 12-mer consensus sequences (found in oriCII-min and in the inc region) consistently exceeds that for 11-mer consensus sequences (naturally present in the inc region of oriCII). This difference in affinity was observed both when using individual binding motifs, where it might have reflected differences in flanking sequences, and when using oriCII-min derivatives in which 12-mers were compared with 11-mers in the same sequence context. Since the inc region is thought to negatively regulate oriCII, the apparent lower affinity for 11-mers than for 12-mers was unexpected (8, 14, 18). However, it is possible that the single 12-mer in the inc region serves as a nucleation site for RctB binding to adjacent 11-mers, potentially enabling the formation of a high-affinity RctB-inc complex that restricts the cell’s capacity for initiation.
Changes that disrupted individual 12-mers—either via targeted mutation of GATC sites or via random mutagenesis of oriCII-min sequences—also impaired the origin’s replicative capacity. The lack of replication with the 11-mer substitutions could be a result of RctB binding to this motif with an affinity lower than that for a 12-mer. It is possible that RctB has different affinities for the various 12-mers in oriCII-min and that high-affinity 12-mers provide critical nucleation points for the formation of a competent RctB-oriCII initiation complex, as has been observed with DnaA and oriC (37). Overall, our results suggest that the sequence of oriCII-min has evolved such that RctB’s affinity for it is just sufficient to permit replication, since disruption of a single high-affinity binding site reduces replication. Similar findings have been obtained in studies of E. coli replication in which low-affinity DnaA binding sites have replaced high-affinity sites (35, 38).
Binding of WT RctB to target sites within the oriCII-min probe was highly cooperative, so that most of the probe was either unbound or fully shifted in EMSAs. Despite its reduced affinity for 11-mers, RctB binding of oriCII-min probes containing 11-mers was also highly cooperative. The fact that the 11-mer replacements principally impair the initial step of RctB-probe interaction, rather than subsequent binding events, suggests that the existence of an RctB-DNA complex influences later binding more strongly than the precise sequence of unoccupied target sites. Such a model is generally consistent with our observations using WT RctB and probes lacking one or more methylation sites, with which cooperative binding was also maintained. It is possible that cooperativity reflects changes in the DNA structure due to RctB binding that facilitate subsequent interactions or lessen the dissociation of complexes. Cooperative binding may be facilitated by RctB’s previously described capacity to dimerize; although the structures of RctB-DNA complexes have not been described, it is possible that RctB dimers might be able to simultaneously occupy two target sites. Cooperativity might also result from interactions between RctB molecules that stabilize protein-DNA complexes, which could potentially occur between monomeric or dimeric molecules. In some other replicative systems, dimeric initiator protein molecules are inactive for replication (39–42), but it is not clear from our data whether this is true in V. cholerae as well. However, our analyses do indicate that a reduction in cooperative binding (as observed with RctB ΔC159, which is also thought to remain monomeric) does not abrogate the protein’s replicative capacity.
Our analyses also revealed previously unrecognized determinants of replication within oriCII-min’s AT-rich region. It appears that this region does not contribute to replication simply via its relatively low melting temperature, which is presumed to aid in the formation of an open complex, since it could not be replaced by the AT-rich region from oriCI. The findings from our undirected mutational (OriSeq) experiment were consistent with comparisons of oriCII sequences in a variety of vibrios (Fig. 7), as well as with the results obtained with our site-directed mutant forms of oriCII. Together, these findings strongly support the idea that specific sequences in this region are important for replication: many sites are resistant to modification. In particular, we found that disruption of three target sites for Dam methyltransferase (GATC) prevented oriCII-min-based replication. These sites may assist in unwinding the DNA duplex, since it has been shown that nucleotides with methyl adducts, specifically, the 6-methyladenine generated by Dam methylase, lead to greater destabilization of double-stranded DNA oligomers than their unmethylated counterparts (29, 43, 44). Methylation may also affect the topology of the oriCII-min region by altering its interactions with DNA-binding proteins other than RctB, such as IHF (45). GATC methylation has been shown to increase oriC binding by IHF, as well as enhance the bending of an oriC fragment in the absence of protein (46, 47). Finally, these GATC sites may facilitate the binding of replisome components, such as the DnaB helicase, or the as-yet-unidentified V. cholerae helicase loader, thereby licensing replication.
Overall, the results from our OriSeq analysis highlight the utility of this approach in identifying sequence regions that are critical for function, since there was remarkable congruence between the key elements defined in our targeted analysis and underrepresented mutation sites following undirected mutagenesis. Thus, the OriSeq approach is likely to be widely applicable for defining critical bases in cis-acting sequences.
MATERIALS AND METHODS
Plasmids and strains.
The plasmids used in this study are listed in Table S1 in the supplemental material. The primers used in this study are listed in Table S2 in the supplemental material. For specific strain construction details, see Text S1 in the supplemental material.
Protein purification.
WT RctB-6His and RctB ΔC159-6His were purified as described before (8, 14).
Transformation efficiency assay.
The transformation efficiency assay was performed as previously described (14). For a detailed explanation of the protocol, see Text S1 in the supplemental material.
OriSeq.
The oriCII mutant library was generated by taking the following steps. oriCII-min DNA fragments were generated by mutational PCR with the GeneMorph II EZclone domain mutagenesis kit (Agilent) on pYB199-oriCII-wt and primers pseudo-ori mut 5′ Xma fuse and pseudo-ori mut 3′ Xho fuse. We used 50 ng of pYB199 as the template and did 50 amplification rounds in an attempt to obtain ~10 to 15 mutations per kb. The kanamycin resistance cassette was PCR amplified from pYB199 with PfuUltra II Fusion HS DNA polymerase (Agilent Technologies) and primers kan 5′ Xma fuse and kan 3′ Xho fuse. The PCR fragments were purified with a Qiagen PCR purification kit and ligated by Gibson assembly (48). The assembled mutant library was transformed into electrocompetent E. coli DH5α cells harboring either pYB285 or pYB296 made as described above. Transformants were grown in Super Optimal Broth with 0.4% Glucose for 1 h at 37°C with shaking and then plated onto LB 1% agar plates containing chloramphenicol (20 µg/ml), kanamycin (50 µg/ml), and isopropyl-β-d-thiogalactopyranoside (100 µM) and placed at 37°C overnight. A second round of Gibson assembly and transformation was done, and colonies from both experiments were pooled based on strain. WT RctB(pYB285) had approximately 50,000 and 44,000 colonies, for a total of 94,000. RctB ΔC159(pYB296) had approximately 38,000 and 45,000 colonies, for a total of 83,000.
Plasmid DNA was prepared using 1/8 of each strain pool and a Qiagen Miniprep kit. The entirety of both Minipreps was digested with BstAPI, BssHII, and NaeI (NEB). The digests were run on 1.5% agarose gels, and the 531-bp fragment was excised and purified with a Qiagen gel purification kit and cleaned with GE Illustra MicroSpin G-50 columns. The mutant libraries were then prepared for sequencing with the Nextera XT DNA sample preparation kit and indices from the Nextera XT Index kit (Illumina). The number of reads per microliter of each library was quantitated by quantitative PCR against a standard curve, and the amount of DNA required to obtain 12 million reads for each strain was sequenced with a 600-cycle MiSeq Reagent kit v3 and a MiSeq sequencer (Illumina).
The reads were trimmed and evaluated for quality score with CLC Genomics Workbench (CLCbio) and then mapped to the WT oriCII-min sequence of V. cholerae (bases 514 to 887 of oriCII, Fig. 1) with the Bowtie aligner (49). A custom python script was used to analyze the numbers of mismatches and total reads for each base, resulting in the average mutation frequency at each base. The mutation frequency at every base was averaged to obtain the average mutation frequency per base across all of the oriCII-min fragments queried. A comparison of each individual base mutation frequency versus the average mutation frequency was done, and this analysis was visualized with Artemis, release 15.0.0 (Wellcome Trust Sanger Institute).
Comparative sequence analysis.
The oriCII-min (bases 514 to 887 of oriCII, Fig. 1) sequence of V. cholerae was used to perform a standard nucleotide BLAST search of the NCBI website against Vibrionaceae optimized for more dissimilar sequences (discontinuous MegaBLAST). This resulted in a comparison against 28 Vibrionaceae species (see Table S2 in the supplemental material). The total number of times an individual base was present, as well as the total number of mismatches for that base across the 28 species, was analyzed with a custom python script. The mutation frequency at every base was averaged to obtain the average mutation frequency per base across all of the oriCII-min fragments queried. A comparison of each individual base mutation frequency with the average mutation frequency was done, and this analysis was visualized with Artemis, release 15.0.0 (Wellcome Trust Sanger Institute).
SUPPLEMENTAL MATERIAL
Supplemental Material and Methods. Download
RctB binding to unmethylated Inc 11 and Inc 12 is greatly reduced. (A) Schematic of the unmethylated probes used in the EMSAs shown in panels B to E. (B to E) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 (D and E) with unmethylated Inc 12 (B and D) and Inc 11 (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
RctB binding to Inc 12 GTAC is greatly reduced. (A) Schematic of the probes used in the EMSAs shown in panels B to E. (B to E) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 (D and E) with Inc 12 (B and D) and Inc 12-GTAC (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
Sequences of individual 12-mers in oriCII-min. Download
Twelve-mer substitutions and deletions reduce RctB binding affinity and/or cooperativity. (A) Schematics of the probes used in EMSAs. (B) Representative binding curves of WT RctB with pseudo-origins. (C) Representative binding curves of RctB ΔC159 with pseudo-origins. (D) Representative binding curves of WT RctB and RctB ΔC159 with pseudo-origins. Download
Multiple mutations in the oriCII-min 12×6 array reduce WT RctB and RctB ΔC159 affinity more than individual mutations do. Representative binding curves of WT RctB (B and C) and RctB ΔC159 (C and D) with the pseudo-origins depicted above. Download
Multiple 12-mer changes reduce RctB affinity and cooperativity. Representative EMSAs of WT RctB (B to D) and RctB ΔC159 (E to G) with pseudo-origins depicted above the gels. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
RctB binding to AT-rich region. (A) Schematic of the probes used in the EMSAs shown. (B to D) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 with AT-rich WT (B and D) and AT-rich GTAC mutant (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
Plasmids and primers used in this study.
Vibrio species used for comparative sequence analysis.
Footnotes
Citation Gerding MA, Chao MC, Davis BM, Waldor MK. 2015. Molecular dissection of the essential features of the origin of replication of the second Vibrio cholerae chromosome. mBio 6(4):e00973-15. doi:10.1128/mBio.00973-15.
REFERENCES
- 1.Messer W. 2002. The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol Rev 26:355–374. doi: 10.1111/j.1574-6976.2002.tb00620.x. [DOI] [PubMed] [Google Scholar]
- 2.Fuller RS, Funnell BE, Kornberg A. 1984. The dnaA protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell 38:889–900. doi: 10.1016/0092-8674(84)90284-8. [DOI] [PubMed] [Google Scholar]
- 3.Bramhill D, Kornberg A. 1988. A model for initiation at origins of DNA replication. Cell 54:915–918. [DOI] [PubMed] [Google Scholar]
- 4.Trucksis M, Michalski J, Deng YK, Kaper JB. 1998. The Vibrio cholerae genome contains two unique circular chromosomes. Proc Natl Acad Sci U S A 95:14464–14469. doi: 10.1073/pnas.95.24.14464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaichi Y, Iida T, Park KS, Yamamoto K, Honda T. 1999. Physical and genetic map of the genome of Vibrio parahaemolyticus: presence of two chromosomes in Vibrio species. Mol Microbiol 31:1513–1521. [DOI] [PubMed] [Google Scholar]
- 6.Okada K, Iida T, Kita-Tsukamoto K, Honda T. 2005. Vibrios commonly possess two chromosomes. J Bacteriol 187:752–757. doi: 10.1128/JB.187.2.752-757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egan ES, Waldor MK. 2003. Distinct replication requirements for the two Vibrio cholerae chromosomes. Cell 114:521–530. doi: 10.1016/S0092-8674(03)00611-1. [DOI] [PubMed] [Google Scholar]
- 9.Duigou S, Knudsen KG, Skovgaard O, Egan ES, Løbner-Olesen A, Waldor MK. 2006. Independent control of replication initiation of the two Vibrio cholerae chromosomes by DnaA and RctB. J Bacteriol 188:6419–6424. doi: 10.1128/JB.00565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkova-Canova T, Srivastava P, Chattoraj DK. 2006. Transcriptional inactivation of a regulatory site for replication of Vibrio cholerae chromosome II. Proc Natl Acad Sci U S A 103:12051–12056. doi: 10.1073/pnas.0605120103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava P, Chattoraj DK. 2007. Selective chromosome amplification in Vibrio cholerae. Mol Microbiol 66:1016–1028. doi: 10.1111/j.1365-2958.2007.05973.x. [DOI] [PubMed] [Google Scholar]
- 12.Yamaichi Y, Fogel MA, McLeod SM, Hui MP, Waldor MK. 2007. Distinct centromere-like parS sites on the two chromosomes of Vibrio spp. J Bacteriol 189:5314–5324. doi: 10.1128/JB.00416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasmussen T, Jensen RB, Skovgaard O. 2007. The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J 26:3124–3131. doi: 10.1038/sj.emboj.7601747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaichi Y, Gerding MA, Davis BM, Waldor MK. 2011. Regulatory cross-talk links Vibrio cholerae chromosome II replication and segregation. PLoS Genet 7:e1002189–e1002189. doi: 10.1371/journal.pgen.1002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Val M-E, Skovgaard O, Ducos-Galand M, Bland MJ, Mazel D. 2012. Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet 8:e1002472. doi: 10.1371/journal.pgen.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baek JH, Chattoraj DK. 2014. Chromosome I controls chromosome II replication in Vibrio cholerae. PLoS Genet 10:e1004184. doi: 10.1371/journal.pgen.1004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demarre G, Chattoraj DK. 2010. DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet 6:e1000939. doi: 10.1371/journal.pgen.1000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkova-Canova T, Chattoraj DK. 2011. Transition from a plasmid to a chromosomal mode of replication entails additional regulators. Proc Natl Acad Sci U S A 108:6199–6204. doi: 10.1073/pnas.1013244108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal D, Venkova-Canova T, Srivastava P, Chattoraj DK. 2005. Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol 187:7167–7175. doi: 10.1128/JB.187.21.7167-7175.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao MC, Pritchard JR, Zhang YJ, Rubin EJ, Livny J, Davis BM, Waldor MK. 2013. High-resolution definition of the Vibrio cholerae essential gene set with hidden Markov model-based analyses of transposon-insertion sequencing data. Nucleic Acids Res 41:9033–9048. doi: 10.1093/nar/gkt654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jha JK, Demarre G, Venkova-Canova T, Chattoraj DK. 2012. Replication regulation of Vibrio cholerae chromosome II involves initiator binding to the origin both as monomer and as dimer. Nucleic Acids Res 40:6026–6038. doi: 10.1093/nar/gks260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch B, Ma X, Løbner-Olesen A. 2012. rctB mutations that increase copy number of Vibrio cholerae oriCII in Escherichia coli. Plasmid 68:159–169. doi: 10.1016/j.plasmid.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Jha JK, Ghirlando R, Chattoraj DK. 2014. Initiator protein dimerization plays a key role in replication control of Vibrio cholerae chromosome 2. Nucleic Acids Res 42:10538–10549. doi: 10.1093/nar/gku771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konieczny I, Helinski DR. 1997. Helicase delivery and activation by DnaA and TrfA proteins during the initiation of replication of the broad host range plasmid RK2. J Biol Chem 272:33312–33318. doi: 10.1074/jbc.272.52.33312. [DOI] [PubMed] [Google Scholar]
- 25.Datta HJ, Khatri GS, Bastia D. 1999. Mechanism of recruitment of DnaB helicase to the replication origin of the plasmid pSC101. Proc Natl Acad Sci U S A 96:73–78. doi: 10.1073/pnas.96.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woelker B, Messer W. 1993. The structure of the initiation complex at the replication origin, oriC, of Escherichia coli. Nucleic Acids Res 21:5025–5033. doi: 10.1093/nar/21.22.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu J, Bramhill D, Thompson CM. 1994. Open complex formation by DnaA initiation protein at the Escherichia coli chromosomal origin requires the 13-mers precisely spaced relative to the 9-mers. Mol Microbiol 11:903–911. [DOI] [PubMed] [Google Scholar]
- 28.Robins WP, Faruque SM, Mekalanos JJ. 2013. Coupling mutagenesis and parallel deep sequencing to probe essential residues in a genome or gene. Proc Natl Acad Sci U S A 110:E848–E857. doi: 10.1073/pnas.1222538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaki H, Ohtsubo E, Nagai K, Maeda Y. 1988. The oriC unwinding by dam methylation in Escherichia coli. Nucleic Acids Res 16:5067–5073. doi: 10.1093/nar/16.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atlung T, Løbner-Olesen A, Hansen FG. 1987. Overproduction of DnaA protein stimulates initiation of chromosome and minichromosome replication in Escherichia coli. Mol Gen Genet 206:51–59. doi: 10.1007/BF00326535. [DOI] [PubMed] [Google Scholar]
- 31.Atlung T, Hansen FG. 1993. Three distinct chromosome replication states are induced by increasing concentrations of DnaA protein in Escherichia coli. J Bacteriol 175:6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun RE, O’Day K, Wright A. 1987. Cloning and characterization of dnaA(Cs), a mutation which leads to overinitiation of DNA replication in Escherichia coli K-12. J Bacteriol 169:3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernander R, Merryweather A, Nordström K. 1989. Overinitiation of replication of the Escherichia coli chromosome from an integrated runaway-replication derivative of plasmid R1. J Bacteriol 171:674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haugan K, Karunakaran P, Tøndervik A, Valla S. 1995. The host range of RK2 minimal replicon copy-up mutants is limited by species-specific differences in the maximum tolerable copy number. Plasmid 33:27–39. doi: 10.1006/plas.1995.1004. [DOI] [PubMed] [Google Scholar]
- 35.Grimwade JE, Torgue JJ-C, McGarry KC, Rozgaja T, Enloe ST, Leonard AC. 2007. Mutational analysis reveals Escherichia coli oriC interacts with both DnaA-ATP and DnaA-ADP during pre-RC assembly. Mol Microbiol 66:428–439. doi: 10.1111/j.1365-2958.2007.05930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duigou S, Yamaichi Y, Waldor MK. 2008. ATP negatively regulates the initiator protein of Vibrio cholerae chromosome II replication. Proc Natl Acad Sci U S A 105:10577–10582. doi: 10.1073/pnas.0803904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller DT, Grimwade JE, Betteridge T, Rozgaja T, Torgue JJ, Leonard AC. 2009. Bacterial origin recognition complexes direct assembly of higher-order DnaA oligomeric structures. Proc Natl Acad Sci U S A 106:18479–18484. doi: 10.1073/pnas.0909472106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langer U, Richter S, Roth A, Weigel C, Messer W. 1996. A comprehensive set of DnaA-box mutations in the replication origin, oriC, of Escherichia coli. Mol Microbiol 21:301–311. [DOI] [PubMed] [Google Scholar]
- 39.Giraldo R, Andreu JM, Díaz-Orejas R. 1998. Protein domains and conformational changes in the activation of RepA, a DNA replication initiator. EMBO J 17:4511–4526. doi: 10.1093/emboj/17.15.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krüger R, Konieczny I, Filutowicz M. 2001. Monomer/dimer ratios of replication protein modulate the DNA strand-opening in a replication origin. J Mol Biol 306:945–955. doi: 10.1006/jmbi.2000.4426. [DOI] [PubMed] [Google Scholar]
- 41.Kunnimalaiyaan S, Ross W, Rakowski SA, Filutowicz M. 2004. Binding modes of the initiator and inhibitor forms of the replication protein pi to the gamma ori iteron of plasmid R6K. J Biol Chem 279:41058–41066. doi: 10.1074/jbc.M403151200. [DOI] [PubMed] [Google Scholar]
- 42.Das N, Chattoraj DK. 2004. Origin pairing (’handcuffing’) and unpairing in the control of P1 plasmid replication. Mol Microbiol 54:836–849. doi: 10.1111/j.1365-2958.2004.04322.x. [DOI] [PubMed] [Google Scholar]
- 43.Engel JD, Hippel PH. 1974. Effects of methylation on the stability of nucleic acid conformations: studies at the monomer level. Biochemistry 13:4143–4158. [DOI] [PubMed] [Google Scholar]
- 44.Collins M, Myers RM. 1987. Alterations in DNA helix stability due to base modifications can be evaluated using denaturing gradient gel electrophoresis. J Mol Biol 198:737–744. doi: 10.1016/0022-2836(87)90214-2. [DOI] [PubMed] [Google Scholar]
- 45.Donczew R, Zakrzewska-Czerwińska J, Zawilak-Pawlik A. 2014. Beyond DnaA: the role of DNA topology and DNA methylation in bacterial replication initiation. J Mol Biol 426:2269–2282. doi: 10.1016/j.jmb.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Kimura T, Asai T, Imai M, Takanami M. 1989. Methylation strongly enhances DNA bending in the replication origin region of the Escherichia coli chromosome. Mol Gen Genet 219:69–74. doi: 10.1007/BF00261159. [DOI] [PubMed] [Google Scholar]
- 47.Polaczek P, Kwan K, Liberies DA, Campbell JL. 1997. Role of architectural elements in combinatorial regulation of initiation of DNA replication in Escherichia coli. Mol Microbiol 26:261–275. [DOI] [PubMed] [Google Scholar]
- 48.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 49.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material and Methods. Download
RctB binding to unmethylated Inc 11 and Inc 12 is greatly reduced. (A) Schematic of the unmethylated probes used in the EMSAs shown in panels B to E. (B to E) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 (D and E) with unmethylated Inc 12 (B and D) and Inc 11 (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
RctB binding to Inc 12 GTAC is greatly reduced. (A) Schematic of the probes used in the EMSAs shown in panels B to E. (B to E) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 (D and E) with Inc 12 (B and D) and Inc 12-GTAC (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
Sequences of individual 12-mers in oriCII-min. Download
Twelve-mer substitutions and deletions reduce RctB binding affinity and/or cooperativity. (A) Schematics of the probes used in EMSAs. (B) Representative binding curves of WT RctB with pseudo-origins. (C) Representative binding curves of RctB ΔC159 with pseudo-origins. (D) Representative binding curves of WT RctB and RctB ΔC159 with pseudo-origins. Download
Multiple mutations in the oriCII-min 12×6 array reduce WT RctB and RctB ΔC159 affinity more than individual mutations do. Representative binding curves of WT RctB (B and C) and RctB ΔC159 (C and D) with the pseudo-origins depicted above. Download
Multiple 12-mer changes reduce RctB affinity and cooperativity. Representative EMSAs of WT RctB (B to D) and RctB ΔC159 (E to G) with pseudo-origins depicted above the gels. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
RctB binding to AT-rich region. (A) Schematic of the probes used in the EMSAs shown. (B to D) Representative EMSAs of WT RctB (B and C) and RctB ΔC159 with AT-rich WT (B and D) and AT-rich GTAC mutant (C and E) probes. The concentrations of RctB ranged from 0.000025 to 1,638.4 nM in a 4-fold dilution series. Red arrows below the gels indicate WT RctB and RctB ΔC159 concentrations where near-maximal binding with the methylated 12×6 probe is shown in Fig. 3B and C, respectively. Download
Plasmids and primers used in this study.
Vibrio species used for comparative sequence analysis.