

Abstract
Many common cancers have a propensity to metastasize to bone. Although malignancies often go undetected in their native tissues, bone metastases produce excruciating pain that severely compromises patient quality of life. Cancer-induced bone pain (CIBP) is poorly managed with existing medications, and its multifaceted etiology remains to be fully elucidated. Novel analgesic targets arise as more is learned about this complex and distinct pain state. Over the past two decades, multiple animal models have been developed to study CIBP’s unique pathology and identify therapeutic targets. Here, we review animal models of CIBP and the mechanistic insights gained as these models evolve. Findings from immunocompromised and immunocompetent host systems are discussed separately to highlight the effect of model choice on outcome. Gaining an understanding of the unique neuromolecular profile of cancer pain through the use of appropriate animal models will aid in the development of more effective therapeutics for CIBP.
Keywords: metastatic bone disease, syngeneic tumor model, ASIC, NGF, oxidative stress, cytokines, inflammation
Introduction
The pain associated with cancer bone metastasis is debilitating and difficult to manage clinically. Many common cancers (eg, breast, prostate, and lung cancers) go undetected in their native tissues but cause excruciating pain upon metastasis to bone.1,2 Cancer-induced bone pain (CIBP) is a growing health concern, as it is both increasingly common and inadequately managed with current standard-of-care therapeutics.3 The World Cancer Report 2014 prepared by International Agency for Research on Cancer estimates that the annual number of cancer cases worldwide will rise from 14 million in 2012 to 22 million by 2032. A significant portion of these patients will experience pain. Cancer pain of all types is reported to be suffered by 30%–50% of all cancer patients4 and 75%–90% of advanced, late-stage cancer patients.5 Of several categories of pain, metastatic CIBP is the most common type of pain reported.6
Two classifications of CIBP have been outlined based on the experiences of these patients: ongoing pain and breakthrough pain. Ongoing pain is dull in character, persistent in presentation, and progressive in intensity.7 Breakthrough pain is a transient, debilitating exacerbation of sharp pain sensations that “breakthrough” the analgesic regimen designed to control a patient’s ongoing pain.8 Breakthrough pain can be associated with movement of the afflicted limb or occurs spontaneously in the absence of a precipitating event.3,6 Pain intensity varies among cancer patients and is related to an individual’s pain sensitivity, the type of cancer, and the tumor location.9,10
Current management of CIBP largely revolves around the World Health Organization’s (WHO) guidelines for cancer pain relief.11 These guidelines outline a treatment progression from non-opioid analgesics through strong opioids with adjuvant supplementation (eg, bisphosphonates, local radiotherapy) to treat progressively worsening pain.12 Unfortunately, many of these therapies are associated with severe dose-limiting side effects that further compromise quality of life of patients.13 Nonsteroidal anti-inflammatory drugs and acetaminophen, used to treat minor cancer pain, are associated with adverse gastrointestinal and renal effects.14 Opioids, used to combat severe cancer pain, are associated with nausea, constipation, sedation, cognitive effects, and respiratory depression and carry an abuse potential.14 Additionally, chronic morphine is associated preclinically with enhanced bone loss and increased (twofold) spontaneous fracture rate.15
The development of dose-limiting side effects, combined with tumor progression, limits analgesic efficacy in nearly 42% of cancer pain patients.16 Thus, clinical management of CIBP would be improved by the identification and development of innovative agents with analgesic efficacy and a more favorable side effect profile. While the etiology of CIBP remains to be fully elucidated, increasing evidence suggests that CIBP is uniquely complex and is accompanied by neurochemical changes distinct from other chronic pain pathologies (eg, neuropathic pain, inflammatory pain).1 Tumors within the bone medullary space activate primary afferent fibers, alter osteoblast/osteoclast balance, and induce a pronounced inflammatory infiltrate.5 A number of animal models have been developed to study CIBP’s unique pathology, what might drive these types of pain and to identify molecular targets, with the end goal of finding novel efficacious analgesics for this devastating pain state.
Animal Models of CIBP
Prior to 1999, there existed two strategies for generating in vivo models of solid tumor-induced bone destruction.17 The first strategy involved injecting tumor cells into the left ventricle of the heart (ie, via intracardiac injection) in rodents. These cells then spread to multiple sites around the body, including the bone marrow. Tumor cell proliferation in the bone marrow results in the formation of a solid tumor within the intramedullary space and destruction of surrounding bone.18,19 This model replicates the clinical observation that many common cancers (ie, breast, prostate, and lung cancers) have a propensity to metastasize to bone.1 Unfortunately, it is difficult to study CIBP using this strategy because of high inter-animal variability in the site and size of metastases;17 the extent of tumor development in any part of the animal, including the bone, is uncontrolled. The second strategy involved direct injection of cancer cells into the intramedullary space of the mouse tibia or femur.17 This strategy had a distinct advantage over intracardiac injection in that bone metastasis is ensured. However, the wet, bony surface of the femur distal end head precluded sealing of the injection site with conventional sealing agents. The unsealed injection hole in the bone head resulted in a large and highly variable extraskeletal tumor mass. Tumor that had escaped the bone and invaded surrounding soft tissue confounded results in CIBP studies as this mass (1) interfered with assessment of pain-related behaviors and (2) destroyed nerves resulting in neuropathic pain.20
The first controlled animal model of CIBP was reported in 1999. In this syngeneic model, fibrosarcoma cells (NCTC 2472) are implanted directly into the mouse femur.21,22 Critically, by plugging the injection site with dental amalgam (Fig. 1), tumor cells are confined to the marrow space until expansion of the tumor and cancer-induced bone degradation releases the cancer from the femur. The initial tumor proliferates within the femur intramedullary space and initially does not invade adjacent soft tissues. This model allows for reproducible tumor development within the femur and assessment of CIBP-related behaviors of the afflicted limb while limiting the confounds of soft tissue invasion and secondary metastases. Furthermore, intraosseous injection permits simultaneous evaluation of bone pain-related behaviors, tumor growth, local and central neurochemical changes, and bone remodeling.23 This strategy of arthrotomy followed by tumor cell inoculation and injection site plugging was adapted for use in a rat model of bone cancer pain in 2002. In this model, Sprague-Dawley MRMT-1 mammary gland carcinoma cells are implanted into the tibia of a female Sprague-Dawley rat, and the injection site is sealed with dental amalgam.24 To date, the use of arthrotomy and direct bone injection followed by injection site plugging has generated tens of distinct animal models of CIBP by varying rodent species and/or strain, site of implantation (eg, femur, tibia, humerus, calcaneus), tumor cell type (eg, fibrosarcoma, mammary adenocarcinoma, prostate adenocarcinoma), and tumor cell species of origin (eg, human being, mouse, rat) (Table 1).
Figure 1.
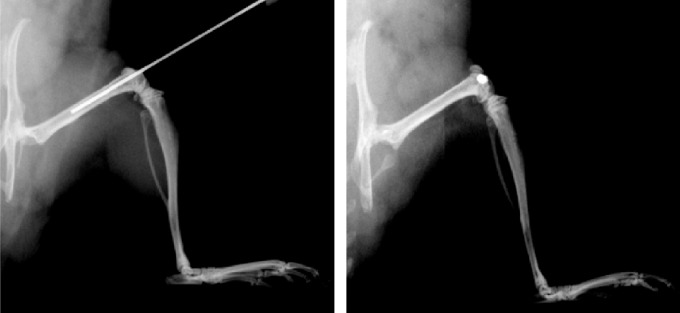
Arthrotomy followed by tumor cell inoculation and injection site plugging. Radiographs demonstrating placement of an injection needle into a mouse femur intramedullary space (left) and sealing of the injections site with dental amalgam (right). This figure was originally published in: King TE, Pawar SC, Majuta L, et al. The Role of Alpha 6 Integrin in Prostate Cancer Migration and Bone Pain in a Novel Xenograft Model. Cordes N, ed. PLoS ONE. 2008;3(10):e3535. doi:10.1371/journal.pone.0003535. It is reused here under the terms of the Creative Commons Attribution (CC BY) license.
Table 1.
Selected animal models of CIBP.
| SPECIES AND STRAIN | TUMOR CELL LINE | TUMOR CELL TISSUE OF ORIGIN | INJECTION | SYNGENEIC | REFERENCES |
|---|---|---|---|---|---|
| Mouse | |||||
| BALB/c | 66.1 | Mammary Pad—adenocarcinoma (Murine) | Femur | X | 101 |
| BALB/cJ | 4T1 | Mammary Pad—adenocarcinoma (Murine) | Femur | X | 165 |
| B6C3/F1 | NCTC 2472 | Mesenchyme—Osteosarcoma (Murine) | Humerus | X | 166 |
| C57BL/6 | Lewis Lung | Lung—Carcinoma (Murine) | Femur | X | 167 |
| C57BI/6J | MC57G | Fibrosarcoma (Murine) | Femur | X | 27 |
| CDF1 | Colon-26 | Colon—adenocarcinoma (murine) | Intracardiac | X | 41 |
| C3H/He | NCTC 2472 | Mesenchyme—Osteosarcoma (Murine) | Humerus | X | 166 |
| Calcaneus | X | 168 | |||
| C3H/HeN | NCTC 2472 | Mesenchyme—Osteosarcoma (Murine) | Femur | X | 165, 169 |
| C3H/HeNCr | NCTC 2472 | Mesenchyme—Osteosarcoma (Murine) | Calcaneus | X | 170 |
| C3H/HeJ | NCTC 2472 | Mesenchyme—Osteosarcoma (Murine) | Femur | X | 77, 78, 105 |
| Tibia | X | 171 | |||
| C3H/SCID | 4T1 | Mammary Pad—adenocarcinoma (Murine) | Femur | 68 | |
| SCID | PC3N | Prostate—carcinoma (Human) | Femur | 172 | |
| Nude nu/nu | ACE-1 | Prostate—adenocarcinoma (Canine) | Intracardiac | 173 | |
| Femur | 68, 71, 75 | ||||
| MDA-MB-231 | Breast—adenocarcinoma (Human) | Intracardiac | 41, 56 | ||
| Mammary Pad | 45 | ||||
| Femur | 87 | ||||
| MDA-MB-231-TXSA | Breast—adenocarcinoma, OPG overexpressing (Human) | Tibia | 42 | ||
| Rat | |||||
| Sprague-Dawley | MRMT1 | Mammary Pad—gland carcinoma cells (Rat) | Tibia | X | 24 |
| Walker 256 | Mammary Gland—carcinoma (Rat) | Tibia | X | 82, 110, 111, 114, 174 | |
| Copenhagen | AT-3 | Prostate—adenocarcinoma (Rat) | Tibia | X | 175 |
| Wistar | MRMT1 | Mammary Pad—gland carcinoma cells (Rat) | Tibia | 81 | |
Selection of host species
CIBP has been modeled in many domestic animals, including dogs, cats, and rodents;25 rodent models predominate preclinical studies. The decision to use rats versus mice is largely dictated by three factors: ease of use, recapitulation of human pathophysiology, and technologies available.26 Rats offer larger bones than mice, which facilitates intramedullary implantation of tumor cells. Similarities to human presentation (ie, hormone responsiveness and premalignant stages) make rats an excellent model organism for studying breast cancer.26 However, the technologies allowing for knock-out or knock-in of a protein of interest in rats has lagged behind the development of such techniques in mice. Thus, mouse models are superior in this nature (ie, more amenable to genetic manipulation). Transgenic animals that exhibit over- or underexpression of a protein of interest are especially helpful for investigating mechanism-based research questions.27 Therefore, the selection of host species, in addition to status of immune system, must be considered when choosing a model for CIBP.
Selection of host immunocompetency
CIBP models can be categorized according to the immune competency of the host. Models using both immunocompromised and immunocompetent animal hosts are employed to study CIBP pathology. Advantages and disadvantages accompany each model, with appropriate model choice depending on question(s) under investigation. For example, human neoplasms can be studied in immune-deficient mice and rats. These studies require nude (athymic) or severe combined immunodeficient (SCID) rodents that are T- and B-cell deficient to allow for engraftment and growth of the human tumor xenograft.28 In addition to compromised adaptive immune systems in these animals, there are changes in innate immunity, including increased natural killer (NK) cell activity and tumoricidal macrophages.28 Non-syngeneic, immunocompromised host models, therefore, provide an in vivo system in which human cancer cells can be passaged within a bone microenvironment and produce tumors with greater histopathological similarity to those encountered in human beings.29 This opportunity allows for both the study of genetic changes necessary for the survival of tumor cells within the bone microenvironment and changes in the bone microenvironment induced by engraftment of human cancer cells.29 Non-syngeneic models with signaling incompatibilities may preclude ligands of the host species from interacting with tumor receptors of the donor species and vice versa.29 The downfall of using such rodents is the inability of the immune system to play its natural role in contributing to the proliferative, inflammatory, and painful processes.
Syngeneic and autochthonous immunocompetent host models better represent the microenvironment built around tumor and immune cells, as immune cells are critical drivers of tumor proliferation, bone remodeling, and overall bone pain. These tumor–host systems allow a tumor to develop successfully in animals with fully intact immune systems, resulting in a more complete picture of tumor–immune interaction. Missing, however, is the question of whether the animal cancer cell line behaves, as would a human cancer cell line. Note that immunocompetent and immunocompromised are terms used here to describe characteristics of the host animal, while syngeneic and non-syngeneic are terms that describe the relationship between the implanted tumor cell line and the host animal. Immunological incompatibility usually precludes the use of an immunocompetent host in non-syngenic models; implanted tumor cells in these models are xenografts or allografts, originating from a different species or rodent strain. Syngeneic models use tumor cell lines originating from the same rodent strain as the host, thus permitting for the use of an immunocompetent host animal.
No CIBP model to date fully recapitulates all interactions between cancer cells and the host tissue. Immunocom-promised host models are especially lacking in this regard, as the artificially modulated immune system results in a dramatically different bone-tumor microenvironment. Even in immunocompetent hosts, however, intracardiac and skeletal injection models fail to account for certain biologic modifications that metastasizing tumor cells undergo since these cells do not originate from primary tumors growing in orthotopic sites.29,30 While models involving autochthonous tumor formation allow for the study of metastasis from orthotopic sites, they do not provide a robust skeletal metastasis phenotype.17 Additionally, both syngeneic and autochthonous animal models limit study to nonhuman tumors and/or tumor cell lines. Differences between rodent/canine and human tumors in terms of biochemistry and xenobiotic response have not been thoroughly documented but are described in the literature.31
Pathophysiology Underlying CIBP
Here, we review mechanistic findings from animal models of CIBP (Table 2). Reports from immunocompromised and immunocompetent host models are presented separately to demonstrate the effect of model choice on outcome, highlighting advantages and disadvantages of each. We do not include a comprehensive overview of all scientific literature employing animal models of CIBP but have selected articles that best illustrate the use of animal models to elucidate CIBP mechanisms.
Table 2.
CIBP therapeutic targets identified by use of animal models.
| TARGET | ABBREVIATION | CANCER | MODEL HOST SPECIES | REFERENCES |
|---|---|---|---|---|
| Bone remodeling | ||||
| Osteoprotegerin | OPG | Breast, Prostate | Mouse | 41–43, 50 |
| Transforming growth factor-β | TGFβ | Breast | Mouse | 56, 57 |
| Parathyroid hormone-related protein | PTHrP | Breast | Mouse | 56 |
| Receptor activator of nuclear factor κB ligand | RANKL | Breast, Prostate | Mouse | 41–43, 50 |
| Farnesyl pyrophosphate synthase | FPP synthase | Breast | Mouse | 44–46, 176, 177 |
| Cathepsin K | CKI | Breast | Mouse | 178 |
| Neurotrophins | ||||
| Nerve growth factor | NGF | Breast, Prostate, Sarcoma | Mouse | 68, 71, 75–77 |
| Brain-derived neurotrophic factor | BDNF | Breast | Rat | 81, 82 |
| Tropomyosin receptor kinase A,B | TrkA,B | Sarcoma | Mouse | 79 |
| Oxidative and nitrosative stress | ||||
| Cystine/Glutamate Antiporter System xc− | System xc− | Breast | Mouse | 87 |
| Inflammatory mediators | ||||
| Tumor necrosis factor-α | TNFα | Breast, Lung, Sarcoma | Mouse | 27, 101–105 |
| Interleukin-6 | IL-6 | Sarcoma | Mouse | 102, 103 |
| Interleukin-1β/receptor IL-1R | IL-1β/IL-1R | Sarcoma | Mouse | 102, 103 |
| Monocyte Chemoattractant Protein-1/receptor CCR2 | MCP-1, CCL2/CCr2 | Breast | Rat | 114 |
| Macrophage Inflammatory Protein-1α | MIP-1α, CCL3 | Breast | Mouse | 101 |
| Fractalkine/fractalkine receptor | CXCL1/CX3CR1 | Breast | Rat | 110, 111 |
| P38 mitogen-activated protein kinases | P38 MAPK | Breast | Rat | 111 |
| Cannabinoid receptor 2 | CB2 | Breast | Mouse | 101 |
| Neurochemical changes and sensitization | ||||
| Acid-sensing ion channel 3 | ASIC3 | Breast | Rat | 132 |
| Acid-sensing ion channel 1a/1b (ASIC1a/1b) | ASIC1a/1b | Breast | Rat | 47 |
| Transient receptor potential vanilloid type 1 | TRPV1 | Sarcoma Breast, | Mice | 121 |
| Sarcoma | Rat | 124, 126, 127 | ||
| P2X purinoceptor 3 | P2X3 | Breast | Rat | 136, 137 |
| MicroRNAs miR-1a-3p and miR-483-3p | Sarcoma | Mice | 142 | |
Bone remodeling
Several studies in both immuno-compromised and immunocompetent host systems link pathological bone remodeling and CIBP. Osteoclasts mediate bone resorption, in part, by secreting protons, thereby making the adjacent microenvironment acidic.32 Acidosis is a well-known cause of pain, as extracellular protons sensitize primary afferent neurons.33–35 Osteoclasts, the body’s principal bone-resorbing cell, are required for cancer-induced bone destruction.36 Both osteolytic (bone-destroying) and osteoblastic (bone-forming) cancers are characterized by osteoclast proliferation and hypertrophy.37
Osteoprotegerin (OPG), a member of the tumor necrosis factor receptor superfamily, plays a critical role in bone remodeling and osteoclastogenesis.38 OPG is a secreted decoy receptor for receptor activator of nuclear factor κB ligand (RANKL). OPG binding prevents RANKL from associating with receptor activator of nuclear factor κB (RANK). RANK receptor is expressed by both osteoclasts and pre-osteoclasts and is essential for their differentiation, activation, and survival.39,40 By preventing RANKL–RANK binding on osteoclast and osteoclast precursors, OPG decreases osteoclast activation and function.
Here, we highlight one example of this phenomenon. Intracardiac or intramammary pad injection of the human breast cancer line MDA-MB-231 in nude mice results in osteolytic metastases. Repeated OPG prophylactic treatment reduced radiographic osteolytic lesions, eradicated tumor-associated osteoclasts, and reduced skeletal tumor burden by 75% in this model.41 Transplantation of MDA-MB-231-TXSA breast cancer cells (MDA-MB-231) engineered to overexpress full-length human OPG into the tibia of nude mice reduced cancer-associated osteolysis and attenuated tumor growth.42 These findings were associated with a reduction in the number of osteoclasts lining the bone surface and an increase in bone volume. Positive OPG effects seem to be limited to the bone; OPG overexpression resulted in a significant increase in the incidence of pulmonary metastasis.42 In the SCID mice, OPG inhibited human prostate cell line C4-2B intratibial growth and cancer-induced osteoclastogeneis43 again suggesting local effects in the bone.
Bisphosphonates, a class of selective osteoclast inhibitors, have been shown to reduce osteolysis and tumor burden in nude mouse models of breast cancer bone metastasis. In MDA-231, innoculated nude mice repeated subcutaneous administration of the bisphosphonates ibandronate or risedronate in both prophylactic and therapeutic treatment paradigms induced apoptosis in osteoclasts, inhibited the progression of osteolytic bone metastases as assessed by radiographic analysis, reduced MDA-231 tumor burden selectively in the bone, and prolonged survival.44–46 While CIBP-related pain behaviors were not assessed in the above studies, such work on cancer-induced bone wasting set the stage for examining the effect of OPG and bisphosphonates on pain in immunocompetent host systems.
In a syngeneic rat model of CIBP in which MRMT-1 mammary pad tumor cells are inoculated into the tibia of female Sprague-Dawley rats, tumor proximity upregulated acid-sensing ion channel 1a/b (ASIC1a/b) expression in the primary sensory neuron.47 The bisphosphonate, zoledronic acid, attenuated cancer-induced hyperalgesia, spinal cord c-Fos elevation, and dorsal root ganglia (DRG) ASIC1a/b expression.47 Similarly, in a syngeneic murine model of CIBP in which 2472 osteolytic sarcoma cells are inoculated into the femur of adult male C3H/HeJ mice, the bisphosphonate ibandronate reduced ongoing and movement-evoked cancer pain-related behaviors, neurochemical markers of central sensitization, tumor burden, and tumor-induced bone destruction.48 In this same model, 50% of the ongoing and movement-evoked pain was blocked with elimination of osteoclast activity with OPG at the first sign of bone destruction, while the tumor grew unabated.22 Critically, because significant skeletal destruction is present in 2% of patients at the time of initial diagnosis of breast cancer and in 30% of patients with recurring breast cancer,49 OPG-induced elimination of osteoclast activity assuaged pain in animals with advanced tumor-induced bone destruction.50 In these animals, OPG halted further bone destruction, reduced ongoing and movement-evoked pain, and reversed several aspects of the neurochemical reorganization of the spinal cord.50 While OPG clearly reduced advanced bone cancer-related pain, there was also a component of the pain that continued despite nearly complete inhibition of osteoclast-mediated bone resorption, mirroring clinical findings with zoledronic acid or denosumab treatment,51 and highlighting the multifaceted nature of this pain state.
Following intracardiac injection of the murine colon adenocarcinoma Colon-26 cells, CDF1 mice develop aggressive metastases to bone and to other organs within 10–14 days.41 In these animals, systemic OPG treatment from the time of tumor cell inoculation decreased the number and area of radiographically evident lytic bone lesions and skeletal tumor burden.41 Tumor burden changes refer to a reduction in average tumor size but not change in the number of tumor nests. This finding suggests that part of the positive benefit of OPG in CIBP may be a result of retarded tumor growth, in addition to inhibition of osteolysis. Taken together, findings from immunocompromised host models illustrate the pathological bone remodeling that occurs following cancer bone metastasis and those from immunocompetent host models provide strong evidence for a role of this remodeling in CIBP.
Transforming growth factor-beta (TGFβ) is a bone-derived growth factor that has been implicated in metastatic bone disease and is released during tumor-induced osteolysis.52 Mineralized bone contains growth factors, one of the most abundant of these being the cytokine TGFβ.53 TGFβ, released from the matrix during bone resorption, can promote tumor cell proliferation54 and the release of osteolytic factors, including parathyroid hormone-related protein (PTHrP), interleukin-11 (IL-11), and vascular endothelial growth factor (VEGF), that further stimulate aberrant osteoclast activity.53 In this way, TGFβ drives the vicious feed-forward cycle of tumor growth and bone destruction characteristic of osteolytic bone metastases.55 Modulating TGFβ levels in the bone-tumor microenvironment, blocking TGFβ signaling or disrupting PTHrP, IL-11, or VEGF production, and/or release may prove valuable strategies in the treatment of bone metastases. Indeed, TGFβ signaling through both Smad and p38 MAPK pathways enhanced PTHrP production by MDA-MB-231 cells.56 Following intracardiac injection of MDA-MB-231 cells in nude mice, a PTHrP neutralizing antibody reduced bone-tumor burden, osteolytic lesion area, and osteolytic lesion number.56 Additionally, blockade of TGFβ signaling with the TFGβ receptor I kinase inhibitor LY2109761 in this same model decreased intramedullary tumor burden as determined by histological staining.57 The putative contribution of TGFβ signaling to CIBP has yet to be investigated.
Emerging data suggest that microRNA (miRNA) dysregulation may contribute to a number of pathologies, including cancer bone metastasis and cancer-induced osteolysis. MiRNAs are short noncoding RNA sequences that bind and prevent translation of target RNAs, thereby repressing protein expression.58 Ell and colleagues59 recently reported that tumor-conditioned media elicited changes in osteoclast miRNA expression. Critically, cancer-induced increases in circulating miRNAs correlated with tumor burden, suggesting that miRNA may be a novel biomarker for bone metastasis. Additionally, systemic administration of cancer-repressed miRNA inhibited osteoclasts in vivo and reduced breast cancer (MDA-MB-231) bone metastasis following intracardiac injection in nude mice.59 Further research is warranted to investigate osteoclast miRNA aberrations as pain biomarkers and/or CIBP therapeutic targets.
Neurotrophins
Neurotrophins are a family of four structurally related polypeptides in mammals: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4/5.60 Through actions at tropomyosin receptor kinases (Trks) and p75 neurotrophin receptor (p75), these factors regulate survival, development, and activity of subsets of sensory and sympathetic neurons.1 Critically, NGF plays a significant role in the generation of pain and hyperalgesia in a variety of acute and chronic pain states in rodents.61 NGF expression is enhanced in injured and inflamed tissues62 as a result of mast cell degranulation,63 macrophage and neutrophil activation,64 and possible fibroblast and/or Schwann cell release.62 Secreted NGF directly activates and/or sensitizes primary afferent neurons that express the NGF receptors such as tropomyosin receptor kinase A (TrkA) and/or p75.65 Of note, NGF may play a role in the survival and proliferation of some tumor types, including prostate cancer,66 and establish mitogenic and antiapoptotic effects in several human breast cancer cell lines (eg, MCF-7, T47-D, BT-20, MDA-MB-231).67
Given that the majority of nerve fibers that innervate the bone expresses neurotrophin receptors trkA and p7568,69 and that trkA is selectively expressed by nociceptive DRG neurons,70 neurotrophins are attractive analgesic targets in CIBP. The first evidence of the utility of a neurotrophin-targeting agent in CIBP came in 2005 when it was reported that a blocking antibody to NGF attenuated both early- and late-stage bone cancer pain.68 Anti-NGF therapy produced a reduction in pain-related behaviors greater than or equivalent to 10 or 30 mg/kg morphine sulfate in nude mice inoculated intrafemorally with the canine prostate carcinoma cell line ACE-1.68 Behavioral changes were not associated with reduction in tumor-induced bone remodeling, osteoblast proliferation, osteoclastogenesis, or tumor growth. However, 26 days post tumor cell inoculation in this model, sprouting of calcitonin gene-related peptide (CGRP+) and neurofilament 200 kDa (NF200+) sensory nerve fibers, and the formation of neuroma-like features in the periosteum are evident.71
Ectopic sprouting and/or pathological remodeling of sensory nerve fibers drives pain in several other difficult to manage human pain states, such as injury-induced neuromas72 and complex regional pain syndrome (where the most common precipitating event is bone fracture).73,74 A growing body of evidence suggests that NGF-induced aberrant nerve growth and reorganization contributes to CIBP. TrkA, the receptor for NGF, is expressed in fibers that undergo cancer-induced sprouting. Ectopic sprouting is a dynamic process wherein nerve fibers degenerate as parent cancer cell colonies undergo necrosis and new sprouting occurs at the leading, viable edge of new daughter cancer cell colonies.75 Fibers innervating the tumor-bearing bone appear to undergo active pathological remodeling throughout the course of the disease. Preventative anti-NGF therapy, when started at the first sign of pain and bone remodeling, blocked ectopic sprouting and attenuated cancer pain.75
Similar findings have been documented with the human breast cancer cell line MDA-MB-231-BO. Nerve sprouting [CGRP+/TrkA+/growth associated protein-43 (GAP43+)] and neuroma formation were documented following intrafemoral inoculation of MDA-MB-231-BO cells in nude mice.76 As with prostate cancer-induced neuronal remodeling, treatment with an anti-NGF sequestering antibody at the onset of pain attenuated both ectopic sprouting and CIBP. Taken together, these data from immunocompromised host models suggest that NGF may contribute to CIBP by both direct activation and/or sensitization of primary afferent nociceptors, as well as through induction of ectopic sprouting.
Similar to findings in immunocompromised host systems, following intrafemoral inoculation of osteolytic murine sarcoma cells (NCTC 2472), male C3He/HeJ mice display pathological sprouting and neuroma formation in the periosteum that is likely driven by NGF. Sequestration of NGF using an anti-NGF antibody largely blocked this pathological reorganization when administered early in the disease course (days 8–12 post tumor cell inoculation) but not at a later stage of disease progression (day 18).77 In accordance, early but not late administration of NGF sequestering therapy reduced cancer-related pain behaviors.77 These findings deviate from those in nude mice where both preventive and late administration of anti-NGF therapy attenuated tumor-induced nerve sprouting, neuroma formation, and cancer pain.75 In both cases, however, ectopic sprouting and neuroma formation was blocked by anti-NGF therapy, suggesting that prevention of pathological reorganization inhibits the development of severe cancer pain. Anti-NGF treatment also reduced cancer-induced peripheral and central neurochemical changes.78
In male C3He/HeJ mice with intrafemoral NCTC 2472 sarcoma inoculation, oral administration of the small molecule Trk inhibitor ARRY-470 (with affinity for TrkA, TrkB, and TrkC receptors) significantly attenuated CIBP-related behaviors and tumor-induced remodeling of sensory nerve fibers.79 No change in tumor growth or tumor-induced bone remodeling was found. As seen with anti-NGF therapy in this model, early (initiated day 6 post tumor cell inoculation), but not late (day 18 post tumor cell injection) administration of ARRY-470 had positive effects on CIBP and ectopic sprouting.79
Blockade of Trk receptors prevents downstream signaling not only from NGF but also from other neurotrophins. The efficacy of ARRY-470 may be attributable to the inhibition of a number of factors in addition to NGF and changes in neuronal excitability independent of sprouting. Recently, a second member from the neurotrophin family has been implicated in CIBP: BDNF. BDNF, through interaction with TrkB and p75 receptors, modulates N-methyl-d-aspartate (NMDA) receptor activity and synaptic transmission.80 BDNF expression is upregulated in L3 DRG following intratibial inoculation of the rat mammary gland carcinoma cell line MRMT-1 in male Wistar rats.81 In a similar rat model of CIBP in which mammary gland carcinoma Walker 256 cells are inoculated into the tibia of female Sprague-Dawley rats, BDNF-induced NMDA receptor activation in the spinal cord or DRG contributed to central sensitization and behavioral hypersensitivity. Intrathecal BDNF siRNA prevented CIBP at an early stage of tumor growth and attenuated, but did not completely block, established CIBP.82 By altering the primary or secondary neuron’s response to the excitatory neurotransmitter glutamate, BDNF may contribute to peripheral and/or central sensitization in CIBP.
Taken together, these data suggest that neurotrophins may contribute to prostate, breast, and sarcoma CIBP through initiation of aberrant nerve sprouting, neuroma formation, and dysregulation of glutamatergic neurotransmission. Notably, early intervention with neurotrophin-targeting agents has been successful at attenuating pain in both immunocompromised and immunocompetent host systems, whereas late-stage antineurotrophic therapy is ineffective (ie, anti-NGF therapy, Trk inhibition) or less effective (ie, anti-BDNF therapy) in syngeneic models. This dynamic discrepancy of anti-neurotrophin efficacy between immune-compromised versus immune-intact model system may be attributed to the source of neurotrophins driving ectopic sprouting of sympathetic and nociceptive primary afferents or sensitization. For example, variation in the efficacy of neurotrophin-targeting agents in late-stage metastatic bone disease between immunocompromised and immunocompetent host models may stem from changes in neurotrophin production and release in immunocompromised animals secondary to loss of NGF and BDNF secreting T-lymphocytes. Neurotrophin differences may also reflect nuances of distinct tumor cell lines. Some sarcoma and prostate cancer cell lines produce small but significant amounts of NGF,78,83 suggesting that the tumor may be a source of this factor. However, the canine prostate cell line ACE-1 that does not express any identifiable NGF mRNA does induce CIBP and anti-NGF therapy sensitive nerve sprouting.68 These data, together with literature illustrating that macrophages, neutrophils, endothelial cells, T-lymphocytes, and fibroblasts express significant levels of NGF, suggest that most NGF produced in cancers may arise from tumor-associated inflammatory, immune, and stromal cells, as well as some cancer cells themselves.77,84,85 BDNF is also produced and secreted by several types of neuronal cells and immune cells (eg, T-lymphocytes, B-lymphocytes, monocytes, and hematogenic precursors).86 However, the cellular sources of BDNF are not fully mapped, and the differences observed with anti-NGF at early-/late-stage CIBP may extend to other members of the neurotrophin family.
Oxidative and nitrosative stress
Oxidative stress, an established hallmark of tumor burden and known contributor to numerous chronic pain states, has a central role in CIBP.13 The pronociceptive actions of oxidative and nitrosative stress in CIBP are hypothesized to be a function of (1) tumor-derived nitrating species altering the response to glutamate by primary afferent neurons and (2) the release of glutamate itself as an algogenic substance by cancer cells.1,13,87 Sources of oxidative and nitrosative stress in the tumor-bone microenvironment include tumor-associated immune cells (eg, tumor-associated macrophages, tumor-associated neutrophils) and cancer cells themselves. In immunocompromised host animal model systems, the neoplasm may be the major source of reactive oxygen and nitrogen species (ROS/RNS). Tumor cells produce ROS/RNS, as a result of the inefficient use of glucose (Warburg effect), leading to reduced antioxidant capacity and excessive generation of reactive species as byproducts of respiration.88
In addition to promoting glutamatergic neurotransmission, ROS/RNS may also drive tumor cell glutamate release. Several tumor cell lines, including C3L5 (mouse breast cancer), B16F1 (mouse melanoma), B16F10 (mouse melanoma), MAT-LyLu (rat prostate cancer), CNS-1 (rat astrocytoma), MDA-MB-231 (human breast cancer), and MCF-7 (human breast cancer), have been shown to release glutamate via the oxidative-stress inducible cystine-glutamate antiporter system xc−.89 A member of the heteromeric amino acid transporter family90, system xc−’s major physiological role is to support two pathways for which intracellular cysteine is the rate-limiting step: the synthesis of the anti-oxidant molecular glutathione and the completion of the cystine-cysteine redox loop.91
System xc− helps to maintain cellular redox balance and is upregulated in response to oxidative challenge.91 Rapid proliferation and tumor burden make survival in the bone-tumor microenvironment just such an oxidative challenge. High expression levels of system xc− in tumor cells are thought to be maintained in this environment and, consequently, tumor cell glutamate release is enhanced.92
Tumor-derived glutamate may stimulate nociceptors by activating NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, and metabotropic-type glutamate receptors on peripheral endings,1 thus leading to the persistent nociceptive state found in CIBP. Furthermore, because of glutamate’s role in bone homeostasis, elevated glutamate may lead to further dysregulation of bone metabolism. Extracellular glutamate released from bone-tumor cells induces osteoclastogenesis and modulates osteoblast function.93
The validity of system xc− as an analgesic target in CIBP is bolstered by recent evidence that repeated administration of the anti-inflammatory agent and established system xc− inhibitor sulfasalazine attenuates pain-related behaviors in a murine model of CIBP.87 Systemic sulfasalazine treatment reduced nociceptive behaviors and extended time until onset of pain but did not alter bone integrity following intrafemoral inoculation of MDA-MB-231 human breast adenocarcinoma cell line in female nude mice.87 The potential disease-modifying effects (ie, inhibition of bone wasting) of system xc− targeting agents have yet to be fully investigated. Further research is needed to evaluate the repurposing of sulfasalazine for CIBP.
While the role of oxidative and/or nitrosative stress in CIBP has not been explored in an immunocompetent host model, extensive investigation is warranted. Tumor-associated immune cells (eg, tumor-associated macrophages, tumor-associated neutrophils) are likely critical contributors to the oxidative state of the bone-tumor microenvironment; production of ROS/RNS is a characteristic of activated myeloid cells.94 As such, oxidative and nitrosative stress would be expected to be significantly elevated in immunocompetent host systems, making the study of oxidative and nitrosative-stress-related CIBP pathology in these models critical. Elevated production of reactive species from both immune and tumor cells may lead to increased nitration-associated dysregulation of glutamate recycling and enhanced glutamatergic neurotransmission as well as exaggerated tumor cell glutamate release, as compared to that in immunocompromised hosts. The increasing number of redox-active pharmacological tools will facilitate the study of oxidative/nitrosative stress in the context of CIBP pathophysiology.95,96
Inflammatory mediators
Cytokines are small protein mediators that are involved in the communication of immune and immune-related cells and play a critical role in regulation of the inflammatory response.97 Subtypes of cytokines, including pro-inflammatory cytokines and chemokines (chemotactic cytokines), contribute to both adaptive and pathologic inflammatory pain as well as chronic neuropathic pain.98 Additionally, these mediators (eg, IL-1β, TNFα, IL-6, and TGFβ) act at their respective receptors on osteoclasts, which are derived from monocyte/macrophage hematopoietic lineage, to stimulate osteoclastogenesis and alter bone metabolism.99 Given their ability to modulate pain neurotransmission centrally and peripherally and stimulate bone resorption, upregulation of cytokines in the bone microenvironment likely plays a profound role in both peripheral and central components of CIBP.1
Pro-inflammatory cytokines and chemokines that contribute to CIBP include TNFα, IL-6, IL-1β, TGFβ, MCP-1, and MIP-1A. Tumor necrosis factor-α (TNFα) is released from cells as a soluble cytokine after being cleaved from its membrane-bound precursor protein (tmTNF) by TNF-α-converting enzyme. Both the soluble and transmembrane forms are biologically active and bring about their effects by binding to two transmembrane receptors: TNF receptor 1 (TNFR1) or TNF receptor 2 (TNFR2). TNFα is produced by immune and nonimmune cells, including macrophages, mast cells, granulocytes, T-lymphocytes, NK cells, fibroblasts, neurons, smooth muscle cells, and some tumor cells.100 At low concentrations in tissues, TNFα is an effective sentinel molecule, initiating host defense against invading microbes and the production of other inflammatory mediators. At high concentrations, however, TNFα can lead to excess inflammation and tissue damage.100
Dysregulation of TNFα occurs in several inflammatory diseases, including rheumatoid arthritis, Crohn’s disease, ankylosing spondylitis, psoriatic arthritis, plaque psoriasis, ulcerative colitis, and, recently, CIBP.100,101 In disease, TNFα expression is elevated because of inappropriate activation of both the innate and adaptive immune responses. Indeed, postmammary pad adenocarcinoma (66.1) inoculation, femur TNFα levels are significantly elevated (threefold) over those in nontumor-bearing female Balb/c mice.101 Additionally, TNFα protein levels are upregulated in tumor site homogenates and in the spinal cord in syngeneic mouse models of fibrosarcoma in the calcaneus102 and tibial bone.103 Critical to CIBP, TNFα promotes nociception both indirectly by increasing prostanoids and sympathetic amines and directly via activation of receptors on nociceptive fibers.98 TNFα can also sensitize the nociceptor-specific heat transducer ion channel transient receptor potential vanilloid 1 (TRPV1) channels via p38/MAP kinase (mitogen-activated protein) and protein kinase C (PKC) pathways to promote pain.104
Given the role of TNFα in driving disease pathology, the removal of excess TNFα from sites of inflammation (ie, the bone-tumor microenvironment) has become a therapeutic goal.100 In a murine osteosarcoma CIBP model in which NCTC 3472 cells are inoculated into the femur intramedullary space of male C3H/HeJ mice, repeated intraperitoneal injection of the TNFα suppressing agent thalidomide attenuated CIBP-related mechanical allodynia and thermal hyperalgesia.105 Systemic antagonism of TNFα with the soluble receptor etanercept, marketed as Enbrel®, reduced tactile hypersensitivity, spontaneous, and ambulatory pain in fibro-sarcoma tumor-bearing C57Bl/6J male mice.27 Likewise, in transgenic C57Bl/6J male mice deficient in both TNFR1 and TNFR2 (TNFR1−/−, TNFR2−/−), the development of tactile hypersensitivity following tumor cell injection was completely inhibited and spinal astrogliosis was markedly reduced, despite increased tumor growth.27 In a soft tissue model of cancer pain, TNFα enhanced DRG expression of TRPV1 and increased the amplitudes of capsaicin (TRPV1 agonist) and heat-activated ionic currents via p38/MAP kinase and PKC.104 Deletion of TNFR2 (TNFR2−/−) but not TNFR1 (TNFR1−/−) attenuated heat hyperalgesia and prevented TRPV1 upregulation in tumor-bearing mice, suggesting a role for TNFR2 in TNFα TRPV1 regulation.104 Thus, enhanced CIBP in the presence of increased tumor burden may be in part related to interactions between TNFα and TRPV1 expression in primary afferent fibers.
Interleukins, IL-1β and IL-6, also play a role in the development of CIBP in animal models. IL-1β and IL-6 are released from infiltrating immune cells (eg, macrophages, monocytes)106 and cancer cells themselves.107 These cytokines promote nociception by increasing the expression of other pronociceptive factors such as prostaglandins via upregulation of COX-2108,109 and NGF109 as well as increasing the production and release of other cytokines.101 As with TNFα, IL-1β and IL-6 protein levels are elevated in the bone-tumor microenvironment in a syngeneic model of breast cancer in the femur and in the bone-tumor microenvironment and spinal cord of syngeneic models of fibrosarcoma in the calcaneus102 and tibial bone.103 A role for peripheral IL-1β is bolstered by the observation that systemic, but not intrathecal, administration of the IL-1 receptor antagonist anakinra attenuated tibial osteosarcoma-induced mechanical hyperalgesia.103
Chemokines, including fractalkine (CX3CL1), MCP-1, MIP-1A, and the growth factor TGFβ, are also implicated in CIBP. Fractalkine and its receptor CX3CR1 mediate spinal cord neuron-to-microglia activation. Spinal, microglial CX3CR1 expression is elevated in the Sprague-Dawley— Walker 256 model of CIBP. In these same rats, a CX3CR1 neutralizing antibody both delayed the onset of ambulatory pain and mechanical allodynia and attenuated established mechanical sensitization.110,111 Blockade of CX3CR1 suppressed pain behavior and spinal microglial activation while reducing expression of p38 MAPK.111 These findings suggest that CX3CR1 may promote pain in the spinal cord through a mechanism involving p38 MAPK and microglial activation.
The ability of the bone-derived growth factor TGFβ to promote the vicious cycle of tumor growth and bone destruction52 may be cancer and/or environment dependent. Ex vivo coculture of mouse femur bone and tumor cells confirmed that bone is a major contributor of TGFβ112 and demonstrated that bone and sarcoma cells synergistically enhanced monocyte chemoattractant protein (MCP-1) secretion, a regulator of monocyte/macrophage migration and infiltration.112,113 The coculture of bone with breast carcinoma cells, in contrast, resulted in a reduction in bone TGFβ and MCP-1 release.112 This ex vivo study is, however, at odds with in vivo data; tibial implantation of a mammary gland carcinoma cell line in rats increased spinal MCP-1 and its receptor CCR2.114 Intrathecal administration of recombinant MCP-1-induced mechanical hypersensitivity in naïve rats that were neutralized by an anti-MCP-1 antibody.114
Our laboratory has demonstrated a significant increase in a panel of cytokines and chemokines, including TNFα, IL-6, MIP-1A, and MCP-1, in the mouse femur bone-tumor microenvironment 14 days postinoculation with the spontaneously occurring murine mammary adenocarcinoma cell line 66.1.101 Upregulation of inflammatory mediators, cytokine/chemokine release, bone-tumor progression, and pain were all attenuated by systemic administration of a cannabinoid-2 receptor (CB2) agonist.101 The well-documented broad anti-inflammatory effects of CB2 make it an attractive novel therapeutic target in CIBP. Taken together, these data suggest that inflammatory mediators have a profound effect on metastatic bone pain, the role of which can only be properly studied in fully immunocompetent host model systems.
Neurochemical changes and sensitization
A mounting body of evidence from syngeneic animal models suggests that the DRG and spinal dorsal horn undergo significant alteration in response to CIBP. These changes constitute the unique “neurochemical signature” of CIBP: a constellation of changes distinct from that of other chronic pain states.21 Persistent inflammatory pain, such as that modeled by hindpaw injection of complete Freund’s adjuvant, is associated with spinal increases in substance P, substance P receptor, CGRP, and PKCγ2. In rodent models of neuropathic pain (ie, sciatic nerve transaction, L5 spinal nerve ligation) substance P and CGRP are decreased and galanin and neuropeptide Y increased in both spinal cord and primary afferent neurons.2 In contrast, in the NCTC 2472—C3H/HeJ murine model of osteolytic sarcoma—there are no spinal or DRG (L4) changes in substance P, CGRP, galanin or neuropeptide Y2. CIBP is characterized spinally (ipsilateral to cancer-bearing limb) by massive astrocyte hypertrophy without neuronal loss, increased neuronal expression of c-Fos, internalization of substance P receptor, and increased number of dynorphin-positive neurons in lamina III–VI.2,115
In addition to these spinal alterations, CIBP is associated with a marked increase in sensory afferent expression of multiple acid-sensing ion channels, including TRPV1 and ASICs. This finding is especially critical given the established pronociceptive roles of these channels116 and the acidity of the bone-tumor microenvironment.117,118 Note that osteoclasts secrete protons to maintain the acidic microenvironment (pH < 5.5) needed for bone resorption.119 Other sources of protons in the bone-tumor microenvironment include tumor cells, considered to be because of lactate secretion from anaerobic glycolysis and inflammatory cells.117
TRPV1 is found in small C and Aδ sensory fibers and colocalized with TrKA in DRG.120 Critical to CIBP, TRPV1 is expressed in a significant portion of sensory neurons innervating tumor-bearing bone and TRPV1 expression is maintained during disease progression.121 It is activated by low pH (<6), capsaicin, resiniferatoxin (a capsaicin analog), noxious heat (>43°C), voltage, and endovanilloids.122,123 In the Walker 256 mammary gland carcinoma tibial injection Sprague-Dawley CIBP model, ipsilateral expression of TRPV1 and capsaicin-evoked inward currents in DRG (L4–6) neurons were significantly elevated above those in sham animals.124 Additionally, blockade of TRPV1 with the selective antagonist JNJ-17203212125 in 2472 osteolytic sarcoma-bearing C3H/HeJ mice attenuated both tumor-induced ongoing nocifensive behaviors and movement-evoked allodynia.121 JNJ-17203212’s antihyperalgesic effect was evident in TRPV1 wild-type (+/+) and TRPV1 heterozygous mutant (+/−) mice but was occluded in TRPV1 homozygous mutant (−/−) mice, which displayed reduced ongoing and evoked pain-related behaviors as compared to wild-type mice, suggesting that a component of CIBP is TRPV1 mediated. Of note, certain sarcoma cell lines (ie, rhabdomyosarcoma CRL1598 and osteosarcoma CRL 1543) produce a soluble, lipophilic factor that activates peripheral nociceptors via TRPV1.126 Neuronal activation as evidenced by CGRP release and calcium imaging was blocked by the TRPV1 inhibitor I-RTX.126,127 These data suggest that TRPV1 activation in CIBP may be secondary to both the acidic tumor environment and tumor-derived agonists.
ASICs are voltage-insensitive proton-gated cation channels in the degenerin–epithelial Na+ channel superfamily. The ASICs family consists of at least seven subtypes (ie, ASIC1a, ASIC1b, ASIC1b2, ASIC2a, ASIC2b, ASIC3, ASIC4) encoded by four genes.128 These channels, which form homomeric and heteromeric complexes, are widely expressed in the nervous system,129 including in DRG,130 and can participate in tissue acidosis-induced nociception.131
ASIC3 is capable of detecting a narrow range of acidic pH (7.3–6.7) and is expressed abundantly in DRG.132 The threshold pH for activation of ASIC3 is dependent on nociceptor co-expression of other ASIC channels.133 Following inoculation of Walker 256 mammary gland carcinoma cells into the tibial medullary cavity of Sprague-Dawley rats, ASIC3 mRNA and protein was upregulated in ipsilateral L4/5 DRG neurons, as compared to sham animals.132 In the closely related MRMT-1 (mammary gland carcinoma) tibial injection Sprague-Dawley rat model, L4/5 DRG ASIC1a/1b expression was elevated.47 At odds with the aforementioned findings, L4/5 DRG expression of ASIC3 and TRPV1 was unchanged in this model.47 The contradictory findings regarding disease regulation of ASIC3 and TRPV1 are surprising, given that the models utilized the same host (female Sprague-Dawley rats) and mammary gland carcinoma cells (Walker 256 or MRMT-1). The dynamic expression of acid-sensing ion channels in CIBP would support their involvement in CIBP development and/or maintenance.
In addition to modulation of acid-sensing ion channels, CIBP pathology is associated in syngeneic rodent models with robust primary afferent upregulation of P2X3 receptors. P2X receptors, activated by extracellular adenosine triphosphate (ATP), belong to the ligand-gated ion channel family and allow for Na+ and Ca2+ influx. Of the seven P2X receptors (P2X1–7) identified to date,134 P2X3 is highly expressed by small- and medium-diameter nociceptive DRG neurons and may have a significant role in pain processing.135 In the Walker 256 Wistar rat intratibial injection model, P2X3 receptor mRNA and protein were upregulated (~50%) in ipsilateral L4/5 DRG neurons in rats with bone cancer.136 Intrathecal or local injection of the P2X3 receptor antagonist A-317491 significantly attenuated cancer-induced pain behaviors.136 In the MRMT-1 Sprague-Dawley rat intratibial injection model, there was a functional upregulation of P2X3 in ipsilateral L4/5 DRGs137 that were mediated by the neuronal calcium sensor protein visinin-like protein-1 (VILIP-1). In this model, the orally available P2X3 and P2X2/3 antagonist AF-353 reversed cancer-induced pain behaviors. In vivo electrophysiology and in vitro ATP tumor cell release studies suggest that AF-353 may have dual actions at peripheral and central terminals.138 Enhanced functional expression of P2X3 may contribute to DRG hyperexcitability and CIBP.
Other neurochemical changes in primary afferent neurons in response to CIBP include upregulation of activating transcription factor (ATF3)139 and downregulation of µ-opioid receptors.140 Increased ATF3 immunoreactive DRG neurons in cancer-bearing animals suggest tumor-induced injury of nerve fibers. A reduction in µ-opioid receptors in NCTC 2472 sarcoma-bearing mice may explain the observation that higher doses of morphine are needed to produce analgesia in bone cancer, as compared with those required for pain management in nonmalignant inflammatory conditions.140
As with other chronic pain pathologies,141 CIBP is associated with a unique miRNA expression “signature” in primary sensory afferents.142 Injection of fibrosarcoma cells into the calcaneus bone of C3H/HeNCrl mice resulted in perturbation of levels of 57 miRNAs (26 upregulated, 31 downregulated) in L3–4 DRGs. Reversing cancer-induced miRNA upregulation of miR-1a-3p, which regulates chloride channel 3 (Ccln3), or downregulation of miR-483-3p attenuated tumor-associated pain and validated miRNA as therapeutic targets in CIBP.142
In sum, neurochemical reorganization in CIBP promotes nociceptive transmission and induces central sensitization. Elucidating the mechanisms through which CIBP elicits these alterations will provide insight into CIBP pathophysiology and reveal novel therapeutic targets.
Methodological Considerations
Much has been discovered about the mechanisms underlying the development and maintenance of CIBP with the refinement of preclinical models. However, the translational impact of animal models on clinical pain research could be enhanced by improving methodological quality.143 Results from a systematic review of 150 publications describing 38 different models of bone cancer pain suggest that there is room yet to improve the rigger of CIBP scientific investigations. Currie and colleagues found reported methodological quality to be low, with only 31% of included publications reporting blinded assessment of outcome and only 11% reporting random allocation to group. No publication reported a sample size calculation. Additionally, studies that included measures to reduce bias (eg, blinded behavior assessment) reported smaller differences in pain-related behaviors between tumor-bearing and control animals. Of concern, articles that contained a conflict of interest statement reported larger differences in behavioral outcomes between tumor-bearing and control animals.143
As there are sex differences in pain perception, sex selection in animal models is a critical consideration. In human beings, women are more likely to be diagnosed with a chronic pain-related disorder (eg, migraine, fibromyalgia, irritable bowel syndrome),144 show lower mean pain threshold and pain tolerance in response to thermal stimuli,145 report greater unpleasantness with pain,145 and exhibit greater responsiveness to µ- and κ-opioid analgesics,145–147 as compared to men. In rodents, females exhibit increased pain sensitivity to chemical, heat, and electrical stimuli144 and blunted responsiveness to µ-opioid agonists,148 as compared to males. Of note, pain sensitivity is related to menstrual cycle in women149 and estrous cycle in rodents.150 Sex selection for animal models of CIBP should be determined by tumor tissue of origin and epidemiological data on the relative disease burden in men and women. For bone-seeking tumors that arise in both men and women (eg, lung cancer with male:female incidence ratio of 12:10),151 studies of CIBP pathophysiology should include both male and female animals.
Increasing evidence suggests that the sex of the experimenter may affect behavioral findings, particularly responses to noxious stimuli. A recent study reported that male experimenters trigger stress-induced analgesia, with mice displaying elevated plasma corticosterone levels, decreased facial grimacing, and increased tail and paw withdrawal latencies in response to thermal stimuli.152 Based on these findings, consistency of the experimenter throughout the study may be critical.
Another methodological concern involves the volume of tumor cell suspensions inoculated into murine femurs in some models. The volume contained in the intramedullary capacity of mouse bones is relatively small and, as such, the injection of large volumes of tumor cell suspensions is inappropriate. Tumor cell suspensions that are not fully contained in the bone intramedullary space can form masses in the surrounding soft tissue and result in distal secondary metastases, both of which confound pain and biochemical assessments.20 In murine models of bone cancer involving intrafemoral tumor cell injection, reported injection volumes range from 5 to 30 µL.27,101,153,154 In C57BL/6 wild-type male mice (20–25 g), the anatomical capacity of the femur is 9.5 µL.155 For comparison, the intramedullary capacity of female Sprague-Dawley (225–275 g) femora is estimated to be 510 ± 10 µL and tibias 380 ± 10 µL.156 While the volume of the mouse femur intramedullary space varies depending on strain, age, weight, and sex of the animal, single injections of greater than 10 µL into a mouse femur should be avoided because of leakage.155 Thus, care is advised when selecting a CIBP model and designing experiments.
Conclusions
In the past two decades, animal models have revealed CIBP to be a truly multifaceted disorder. Immunocompetent host models have the great advantage of allowing for study of the interaction between an intact immune system and tumor within the marrow space. A growing body of work suggests that interaction is especially critical; recall the profound contribution of inflammatory mediators as sensitizing agents, modulators of bone metabolism and initiators of neurochemical changes to CIBP. Contributing mechanisms include sensitization of nociceptors by bone cell-derived (eg, protons), tumor cell-derived (eg, protons, nitrating species, glutamate) and immune cell-derived (eg, cytokines, chemokines, neurotrophins) algogenic products, periosteal neuroma formation, and peripheral and central neurochemical changes. The identification of such therapeutic targets has led to the development of pharmacological agents with validated preclinical efficacy against CIBP (Fig. 2). The power of CIBP animal models to predict clinical efficacy has yet to be determined but will likely be related to both their ability to recapitulate human CIBP pathology and the scientific rigger of the studies in which they are included.
Figure 2.
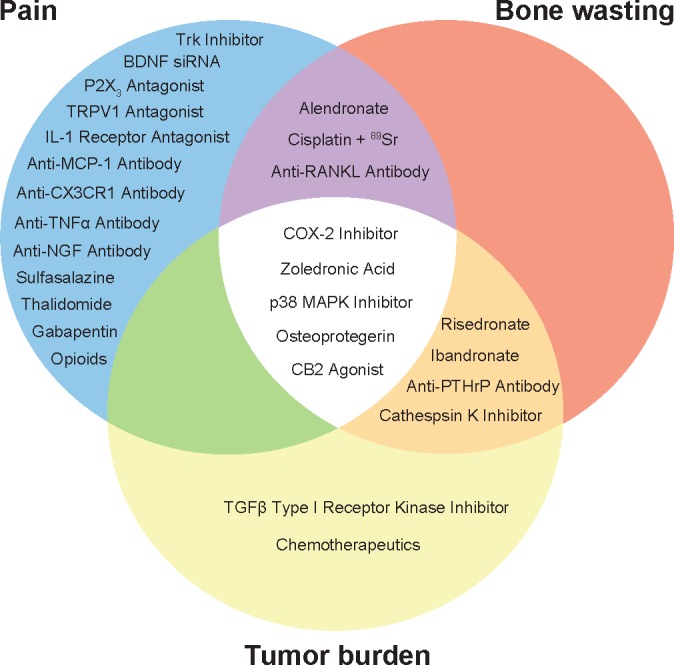
Selected pharmacological interventions with positive effects in animal models of CIBP: Trk inhibitor (ARRY-470);79 BDNF siRNA;82 P2X3 antagonist (AF-353);138 TRPV1 antagonist (JNJ-17203212);125 IL-1 receptor antagonist (anakinra);103 anti-MCP-1 antibody;114 anti-CX3CR1 antibody;110,111 anti-TNFα antibody (etanercept);27 anti-NGF antibody;75,77 sulfasalzine;87 thalidomide;105 gabapentin;179 opioids (morphine, oxycodone);180,181 alendronate;177 cisplatin + 89SR;182 anti-RANKL antibody (denosumab);183 Cox-2 inhibitor (MF tricyclic);20 zoledronic acid;176 p38 MAPK inhibitor (SB203580);184 osteoprotegerin;41,42 CB2 agonist (JWH015);101 ibandronate;44,45 risedronate;44,46 anti-PTHrP antibody;56 cathepsin K inhibitor;178 TGFβ type I receptor kinase inhibitor (LY2109761);57 and chemotherapeutics. Trk inhibitor, tropomyosin receptor kinase inhibitor; BDNF siRNA, brain-derived neurotrophic factor small interfering RNA; P2X3 antagonist, P2X purinoceptor 3 antagonist; TRPV1 antagonist, transient receptor potential vanilloid type 1 antagonist; IL-1 receptor antagonist, interleukin-1 receptor antagonist; anti-MCP-1 antibody, anti-monocyte chemoattractant protein-1 antibody; anti-CX3CR1 antibody, anti-fractalkine receptor antibody; anti-TNFα antibody, antitumor necrosis factor-α antibody; anti-NGF antibody, anti-nerve growth factor antibody; anti-RANKL antibody, anti-receptor activator of nuclear factor κB ligand antibody; CB2 agonist, cannabinoid receptor 2 agonist; anti-PTHrP antibody, anti-parathyroid hormone related protein antibody.
Future Directions
It may be possible to improve translation between animal and clinical pain research by increasing the scientific rigger of preclinical studies and ensuring that selected animal models exemplify human pathology. Measures are currently being taken in human beings with advanced stages of bone cancer, including markers for bone remodeling157 and imaging.158 As more is learned about the human condition, these markers should be applied to animal models to assess model validity. Perhaps notes can be taken in this regard from work on oral cancer pain. Much effort has been dedicated in this field to the development of animal models that recapitulate natural cancer development and its resulting functional impairment. For example, cancer pain-induced gnawing dysfunction, the rodent correlate of a clinically relevant impairment, is being assessed following carcinogen (4-nitroquinoline 1-oxide)-induced oral malignancy in fully immunocompetent mice.159,160 More “natural” models of CIBP may be implanted as we learn more about the mechanisms of bone metastasis.
It is fitting that a number of distinct CIBP animal models exist, as CIBP sufferers are a diverse group of patients, having different types of cancer and metastases in diverse, and at times multiple bones.161,162 Pain etiology and analgesic drug responsiveness may vary with tumor type and host bone and between patients with similar cancer types.14,163
Given the multifaceted nature of CIBP, its management is likely to involve polypharmacy or agents with polypharmacology.14 The more the aspects of human CIBP recapitulated in the animal model, the greater the likelihood of identifying agents with clinical efficacy in this pain state. Adding an additional layer of complication, analgesic agents in this patient population will likely be coadministered with chemotherapeutic agents (known to produce their own form of neuropathy), radiological interventions, and other adjunct therapies. Such adjunct therapies may alter the immune competency of the patient (eg, chemotherapy)164 and modulate the efficacy of immune-targeting CIBP interventions (eg, cytokine/chemokine neutralizing antibodies). The compatibility of analgesic agents with other therapeutics and therapeutic goals is essential and should be considered preclinically. For example, while anti-TNFα therapy attenuates CIBP, the associated increase in tumor growth27 is at odds with the therapeutic goal of reducing tumor burden and prolonging survival.
Pain, bone wasting, and tumor burden are not entirely independent, and many pharmacological interventions with preclinical efficacy address multiple aspects of CIBP pathology (Fig. 2). While animal models have begun to shed light on the mechanisms underlying CIBP and allowed for the identification of novel analgesic drug targets, much work remains. A more comprehensive understanding of the unique neuromolecular profile of cancer pain will aid in the development of more effective therapeutics for CIBP.
Footnotes
ACADEMIC EDITOR: Marc D. Basson, Editor in Chief
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 945 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Wrote the first draft of the manuscript: LMS, TML, TWV. Contributed to the writing of the manuscript: LMS, TML, TWV. Agree with manuscript results and conclusions: LMS, TML, TWV. Jointly developed the structure and arguments for the paper: LMS, TML, TWV. Made critical revisions and approved final version: LMS, TML, TWV. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Lozano-Ondoua AN, Symons-Liguori AM, Vanderah TW. Cancer-induced bone pain: mechanisms and models. Neurosci Lett. 2013;557(pt A):52–59. doi: 10.1016/j.neulet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honore P, Rogers SD, Schwei MJ, et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98(3):585–598. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- 3.Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695–1700. doi: 10.1016/S0140-6736(99)01310-0. [DOI] [PubMed] [Google Scholar]
- 4.Kumar SP. Cancer pain: a critical review of mechanism-based classification and physical therapy management in palliative care. Indian J Palliat Care. 2011;17(2):116–126. doi: 10.4103/0973-1075.84532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabino MA, Mantyh PW. Pathophysiology of bone cancer pain. J Support Oncol. 2005;3(1):15–24. [PubMed] [Google Scholar]
- 6.Mercadante S. Malignant bone pain: pathophysiology and treatment. Pain. 1997;69(1–2):1–18. doi: 10.1016/s0304-3959(96)03267-8. [DOI] [PubMed] [Google Scholar]
- 7.Kerba M, Wu JS, Duan Q, Hagen NA, Bennett MI. Neuropathic pain features in patients with bone metastases referred for palliative radiotherapy. J Clin Oncol. 2010;28(33):4892–4897. doi: 10.1200/JCO.2010.28.6559. [DOI] [PubMed] [Google Scholar]
- 8.Portenoy RK, Hagen NA. Breakthrough pain: definition, prevalence and characteristics. Pain. 1990;41(3):273–281. doi: 10.1016/0304-3959(90)90004-W. [DOI] [PubMed] [Google Scholar]
- 9.Foley KM. Controlling the pain of cancer. Sci Am. 1996;275(3):164–165. doi: 10.1038/scientificamerican0996-164. [DOI] [PubMed] [Google Scholar]
- 10.Foley KM. Advances in cancer pain. Arch Neurol. 1999;56(4):413–417. doi: 10.1001/archneur.56.4.413. [DOI] [PubMed] [Google Scholar]
- 11.WHO . Cancer Pain Relief. Geneva, Switzerland: World Health Organization; 1998. [Google Scholar]
- 12.Urch C. The pathophysiology of cancer-induced bone pain: current understanding. Palliat Med. 2004;18(4):267–274. doi: 10.1191/0269216304pm887ra. [DOI] [PubMed] [Google Scholar]
- 13.Ungard RG, Seidlitz EP, Singh G. Oxidative stress and cancer pain. Can J Physiol Pharmacol. 2013;91(1):31–37. doi: 10.1139/cjpp-2012-0298. [DOI] [PubMed] [Google Scholar]
- 14.Nersesyan H, Slavin KV. Current approach to cancer pain management: availability and implications of different treatment options. Ther Clin Risk Manag. 2007;3(3):381–400. [PMC free article] [PubMed] [Google Scholar]
- 15.King T, Vardanyan A, Majuta L, et al. Morphine treatment accelerates sarcoma-induced bone pain, bone loss, and spontaneous fracture in a murine model of bone cancer. Pain. 2007;132(1–2):154–168. doi: 10.1016/j.pain.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, Schouten HC, van Kleef M, Patijn J. High prevalence of pain in patients with cancer in a large population-based study in The Netherlands. Pain. 2007;132(3):312–320. doi: 10.1016/j.pain.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 17.Mantyh P. Bone cancer pain: causes, consequences, and therapeutic opportunities. Pain. 2013;154(suppl 1):S54–S62. doi: 10.1016/j.pain.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 18.Arguello F, Baggs RB, Frantz CN. A murine model of experimental metastasis to bone and bone marrow. Cancer Res. 1988;48(23):6876–6881. [PubMed] [Google Scholar]
- 19.Yoneda T, Sasaki A, Mundy GR. Osteolytic bone metastasis in breast cancer. Breast Cancer Res Treat. 1994;32(1):73–84. doi: 10.1007/BF00666208. [DOI] [PubMed] [Google Scholar]
- 20.Sabino MA, Ghilardi JR, Jongen JL, et al. Simultaneous reduction in cancer pain, bone destruction, and tumor growth by selective inhibition of cyclooxygen-ase-2. Cancer Res. 2002;62(24):7343–7349. [PubMed] [Google Scholar]
- 21.Schwei MJ, Honore P, Rogers SD, et al. Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. J Neurosci. 1999;19(24):10886–10897. doi: 10.1523/JNEUROSCI.19-24-10886.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honore P, Luger NM, Sabino MA, et al. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med. 2000;6(5):521–528. doi: 10.1038/74999. [DOI] [PubMed] [Google Scholar]
- 23.Goblirsch MJ, Zwolak P, Clohisy DR. Advances in understanding bone cancer pain. J Cell Biochem. 2005;96(4):682–688. doi: 10.1002/jcb.20589. [DOI] [PubMed] [Google Scholar]
- 24.Medhurst SJ, Walker K, Bowes M, et al. A rat model of bone cancer pain. Pain. 2002;96(1–2):129–140. doi: 10.1016/s0304-3959(01)00437-7. [DOI] [PubMed] [Google Scholar]
- 25.Pacharinsak C, Beitz A. Animal models of cancer pain. Comp Med. 2008;58(3):220–233. [PMC free article] [PubMed] [Google Scholar]
- 26.Iannaccone PM, Jacob HJ. Rats! Dis Model Mech. 2009;2(5–6):206–210. doi: 10.1242/dmm.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geis C, Graulich M, Wissmann A, et al. Evoked pain behavior and spinal glia activation is dependent on tumor necrosis factor receptor 1 and 2 in a mouse model of bone cancer pain. Neuroscience. 2010;169(1):463–474. doi: 10.1016/j.neuroscience.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 28.Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007;170(3):793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein RH, Weinberg RA, Rosenblatt M. Of mice and (wo)men: mouse models of breast cancer metastasis to bone. J Bone Miner Res. 2010;25(3):431–436. doi: 10.1002/jbmr.68. [DOI] [PubMed] [Google Scholar]
- 30.Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–U554. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 31.de Jong M, Maina T. Of mice and humans: are they the same? Implications in cancer translational research. J Nucl Med. 2010;51(4):501–504. doi: 10.2967/jnumed.109.065706. [DOI] [PubMed] [Google Scholar]
- 32.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289(5484):1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 33.Bevan S, Geppetti P. Protons: small stimulants of capsaicin-sensitive sensory nerves. Trends Neurosci. 1994;17(12):509–512. doi: 10.1016/0166-2236(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 34.Krishtal OA, Pidoplichko VI. A receptor for protons in the nerve cell membrane. Neuroscience. 1980;5(12):2325–2327. doi: 10.1016/0306-4522(80)90149-9. [DOI] [PubMed] [Google Scholar]
- 35.Reeh PW, Steen KH. Tissue acidosis in nociception and pain. Prog Brain Res. 1996;113:143–151. doi: 10.1016/s0079-6123(08)61085-7. [DOI] [PubMed] [Google Scholar]
- 36.Clohisy DR, Ogilvie CM, Carpenter RJ, Ramnaraine ML. Localized, tumor-associated osteolysis involves the recruitment and activation of osteoclasts. J Orthop Res. 1996;14(1):2–6. doi: 10.1002/jor.1100140103. [DOI] [PubMed] [Google Scholar]
- 37.Mantyh PW, Clohisy DR, Koltzenburg M, Hunt SP. Molecular mechanisms of cancer pain. Nat Rev Cancer. 2002;2(3):201–209. doi: 10.1038/nrc747. [DOI] [PubMed] [Google Scholar]
- 38.Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 39.Hsu H, Lacey DL, Dunstan CR, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96(7):3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Sarosi I, Yan XQ, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97(4):1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morony S, Capparelli C, Sarosi I, Lacey DL, Dunstan CR, Kostenuik PJ. Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res. 2001;61(11):4432–4436. [PubMed] [Google Scholar]
- 42.Zinonos I, Luo KW, Labrinidis A, et al. Pharmacologic inhibition of bone resorption prevents cancer-induced osteolysis but enhances soft tissue metastasis in a mouse model of osteolytic breast cancer. Int J Oncol. 2014;45(2):532–540. doi: 10.3892/ijo.2014.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Dai J, Qi Y, et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107(10):1235–1244. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoneda T, Michigami T, Yi B, Williams PJ, Niewolna M, Hiraga T. Actions of bisphosphonate on bone metastasis in animal models of breast carcinoma. Cancer. 2000;88(12 suppl):2979–2988. doi: 10.1002/1097-0142(20000615)88:12+<2979::aid-cncr13>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 45.Hiraga T, Williams PJ, Mundy GR, Yoneda T. The bisphosphonate ibandronate promotes apoptosis in MDA-MB-231 human breast cancer cells in bone metastases. Cancer Res. 2001;61(11):4418–4424. [PubMed] [Google Scholar]
- 46.Sasaki A, Boyce BF, Story B, et al. Bisphosphonate risedronate reduces metastatic human breast cancer burden in bone in nude mice. Cancer Res. 1995;55(16):3551–3557. [PubMed] [Google Scholar]
- 47.Nagae M, Hiraga T, Yoneda T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J Bone Miner Metab. 2007;25(2):99–104. doi: 10.1007/s00774-006-0734-8. [DOI] [PubMed] [Google Scholar]
- 48.Halvorson KG, Sevcik MA, Ghilardi JR, et al. Intravenous ibandronate rapidly reduces pain, neurochemical indices of central sensitization, tumor burden, and skeletal destruction in a mouse model of bone cancer. J Pain Symptom Manage. 2008;36(3):289–303. doi: 10.1016/j.jpainsymman.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahan J, Seligson D. Orthopedic management of skeletal metastases. In: Donegan W, Spratt J, editors. Cancer of the Breast. 4th ed. Philadelphia: W. B. Saunders; 1995. pp. 682–695. [Google Scholar]
- 50.Luger NM, Honore P, Sabino MA, et al. Osteoprotegerin diminishes advanced bone cancer pain. Cancer Res. 2001;61(10):4038–4047. [PubMed] [Google Scholar]
- 51.Stopeck AT, Lipton A, Body JJ, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol. 2010;28(35):5132–5139. doi: 10.1200/JCO.2010.29.7101. [DOI] [PubMed] [Google Scholar]
- 52.Xu Q, Zhang XM, Duan KZ, et al. Peripheral TGF-beta 1 signaling is a critical event in bone cancer-induced hyperalgesia in rodents. J Neurosci. 2013;33(49):19099–19111. doi: 10.1523/JNEUROSCI.4852-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delany AM, Canalis E. Growth factors and bone. In: Derek L, Carolyn B, editors. Growth Factors and Cytokines in Health and Disease. Vol. 3. Greenwich, CT: JAI Press; 1997. pp. 127–155. [Google Scholar]
- 54.Sato S, Futakuchi M, Ogawa K, et al. Transforming growth factor beta derived from bone matrix promotes cell proliferation of prostate cancer and osteoclast activation-associated osteolysis in the bone microenvironment. Cancer Sci. 2008;99(2):316–323. doi: 10.1111/j.1349-7006.2007.00690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buijs JT, Stayrook KR, Guise TA. The role of TGF-beta in bone metastasis: novel therapeutic perspectives. Bonekey Rep. 2012;1:96. doi: 10.1038/bonekey.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kakonen SM, Selander KS, Chirgwin JM, et al. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J Biol Chem. 2002;277(27):24571–24578. doi: 10.1074/jbc.M202561200. [DOI] [PubMed] [Google Scholar]
- 57.Korpal M, Yan J, Lu X, Xu SW, Lerit DA, Kang YB. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2009;15(8):960–U169. doi: 10.1038/nm.1943. [DOI] [PubMed] [Google Scholar]
- 58.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ell B, Mercatali L, Ibrahim T, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell. 2013;24(4):542–556. doi: 10.1016/j.ccr.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skaper SD. The neurotrophin family of neurotrophic factors: an overview. Methods Mol Biol. 2012;846:1–12. doi: 10.1007/978-1-61779-536-7_1. [DOI] [PubMed] [Google Scholar]
- 61.Shu XQ, Mendell LM. Neurotrophins and hyperalgesia. Proc Natl Acad Sci U S A. 1999;96(14):7693–7696. doi: 10.1073/pnas.96.14.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McMahon SB. NGF as a mediator of inflammatory pain. Philos Trans R Soc Lond B Biol Sci. 1996;351(1338):431–440. doi: 10.1098/rstb.1996.0039. [DOI] [PubMed] [Google Scholar]
- 63.Forsythe P, Bienenstock J. The mast cell-nerve functional unit: a key component of physiologic and pathophysiologic responses. Chem Immunol Allergy. 2012;98:196–221. doi: 10.1159/000336523. [DOI] [PubMed] [Google Scholar]
- 64.Susaki Y, Shimizu S, Katakura K, et al. Functional properties of murine macrophages promoted by nerve growth factor. Blood. 1996;88(12):4630–4637. [PubMed] [Google Scholar]
- 65.Mendell LM, Albers KM, Davis BM. Neurotrophins, nociceptors, and pain. Microsc Res Tech. 1999;45(4–5):252–261. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<252::AID-JEMT9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 66.Djakiew D. Dysregulated expression of growth factors and their receptors in the development of prostate cancer. Prostate. 2000;42(2):150–160. doi: 10.1002/(sici)1097-0045(20000201)42:2<150::aid-pros10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 67.Descamps S, Toillon RA, Adriaenssens E, et al. Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J Biol Chem. 2001;276(21):17864–17870. doi: 10.1074/jbc.M010499200. [DOI] [PubMed] [Google Scholar]
- 68.Halvorson KG, Kubota K, Sevcik MA, et al. A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer Res. 2005;65(20):9426–9435. doi: 10.1158/0008-5472.CAN-05-0826. [DOI] [PubMed] [Google Scholar]
- 69.Castañeda-Corral G, Jimenez-Andrade JM, Bloom AP, et al. The majority of myelinated and unmyelinated sensory nerve fibers that innervate bone express the tropomyosin receptor kinase A. Neuroscience. 2011;178:196–207. doi: 10.1016/j.neuroscience.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang X, Djouhri L, McMullan S, et al. trkA is expressed in nociceptive neurons and influences electrophysiological properties via Nav1.8 expression in rapidly conducting nociceptors. J Neurosci. 2005;25(19):4868–4878. doi: 10.1523/JNEUROSCI.0249-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jimenez-Andrade JM, Bloom AP, Stake JI, et al. Pathological sprouting of adult nociceptors in chronic prostate cancer-induced bone pain. J Neurosci. 2010;30(44):14649–14656. doi: 10.1523/JNEUROSCI.3300-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. 2008;64(6):644–653. doi: 10.1002/ana.21527. [DOI] [PubMed] [Google Scholar]
- 73.Janig W, Baron R. Complex regional pain syndrome: mystery explained? Lancet Neurol. 2003;2(11):687–697. doi: 10.1016/s1474-4422(03)00557-x. [DOI] [PubMed] [Google Scholar]
- 74.de Mos M, de Bruijn AG, Huygen FJ, Dieleman JP, Stricker BH, Sturkenboom MC. The incidence of complex regional pain syndrome: a population-based study. Pain. 2007;129(1–2):12–20. doi: 10.1016/j.pain.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 75.Jimenez-Andrade JM, Ghilardi JR, Castaneda-Corral G, Kuskowski MA, Mantyh PW. Preventive or late administration of anti-NGF therapy attenuates tumor-induced nerve sprouting, neuroma formation, and cancer pain. Pain. 2011;152(11):2564–2574. doi: 10.1016/j.pain.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bloom AP, Jimenez-Andrade JM, Taylor RN, et al. Breast cancer-induced bone remodeling, skeletal pain, and sprouting of sensory nerve fibers. J Pain. 2011;12(6):698–711. doi: 10.1016/j.jpain.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mantyh WG, Jimenez-Andrade JM, Stake JI, et al. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience. 2010;171(2):588–598. doi: 10.1016/j.neuroscience.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sevcik MA, Ghilardi JR, Peters CM, et al. Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain. 2005;115(1–2):128–141. doi: 10.1016/j.pain.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 79.Ghilardi JR, Freeman KT, Jimenez-Andrade JM, et al. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Mol Pain. 2010;6:87. doi: 10.1186/1744-8069-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kerr BJ, Bradbury EJ, Bennett DL, et al. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19(12):5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tomotsuka N, Kaku R, Obata N, et al. Up-regulation of brain-derived neurotrophic factor in the dorsal root ganglion of the rat bone cancer pain model. J Pain Res. 2014;7:415–423. doi: 10.2147/JPR.S63527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang LN, Yang JP, Ji FH, et al. Brain-derived neurotrophic factor modulates N-methyl-d-aspartate receptor activation in a rat model of cancer-induced bone pain. J Neurosci Res. 2012;90(6):1249–1260. doi: 10.1002/jnr.22815. [DOI] [PubMed] [Google Scholar]
- 83.Cohen S, Levi-Montalcini R, Hamburger VA. Nerve growth-stimulating factor isolated from sarcom as 37 and 180. Proc Natl Acad Sci U S A. 1954;40(10):1014–1018. doi: 10.1073/pnas.40.10.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lindsay TH, Jonas BM, Sevcik MA, et al. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain. 2005;119(1–3):233–246. doi: 10.1016/j.pain.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 85.Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- 86.Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999;189(5):865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ungard RG, Seidlitz EP, Singh G. Inhibition of breast cancer-cell glutamate release with sulfasalazine limits cancer-induced bone pain. Pain. 2014;155(1):28–36. doi: 10.1016/j.pain.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 88.El Sayed SM, Mahmoud AA, El Sawy SA, et al. Warburg effect increases steady-state ROS condition in cancer cells through decreasing their antioxidant capacities (anticancer effects of 3-bromopyruvate through antagonizing Warburg effect) Med Hypotheses. 2013;81(5):866–870. doi: 10.1016/j.mehy.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 89.Sharma MK, Seidlitz EP, Singh G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun. 2010;391(1):91–95. doi: 10.1016/j.bbrc.2009.10.168. [DOI] [PubMed] [Google Scholar]
- 90.Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 2004;447(5):532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 91.Lewerenz J, Hewett SJ, Huang Y, et al. The cystine/glutamate antiporter system x(c)(−) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18(5):522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nashed MG, Balenko MD, Singh G. Cancer-induced oxidative stress and pain. Curr Pain Headache Rep. 2014;18(1):384. doi: 10.1007/s11916-013-0384-1. [DOI] [PubMed] [Google Scholar]
- 93.Seidlitz EP, Sharma MK, Singh G. Extracellular glutamate alters mature osteoclast and osteoblast functions. Can J Physiol Pharmacol. 2010;88(9):929–936. doi: 10.1139/y10-070. [DOI] [PubMed] [Google Scholar]
- 94.Corzo CA, Cotter MJ, Cheng P, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Slosky LM, Vanderah TW. Therapeutic potential of peroxynitrite decomposition catalysts: a patent review. Expert Opin Ther Pat. 2015;25(4):443–466. doi: 10.1517/13543776.2014.1000862. [DOI] [PubMed] [Google Scholar]
- 96.Batinic-Haberle I, Tovmasyan A, Roberts ERH, Vujaskovic Z, Leong KW, Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal. 2014;20(15):2372–2415. doi: 10.1089/ars.2012.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arai KI, Lee F, Miyajima A, Miyatake S, Arai N, Yokota T. Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem. 1990;59:783–836. doi: 10.1146/annurev.bi.59.070190.004031. [DOI] [PubMed] [Google Scholar]
- 98.Verri WA, Jr, Cunha TM, Parada CA, Poole S, Cunha FQ, Ferreira SH. Hypernociceptive role of cytokines and chemokines: targets for analgesic drug development? Pharmacol Ther. 2006;112(1):116–138. doi: 10.1016/j.pharmthera.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 99.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 100.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117(2):244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 101.Lozano-Ondoua AN, Hanlon KE, Symons-Liguori AM, et al. Disease modification of breast cancer-induced bone remodeling by cannabinoid 2 receptor agonists. J Bone Miner Res. 2013;28(1):92–107. doi: 10.1002/jbmr.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wacnik PW, Eikmeier LJ, Simone DA, Wilcox GL, Beitz AJ. Nociceptive characteristics of tumor necrosis factor-alpha in naive and tumor-bearing mice. Neuroscience. 2005;132(2):479–491. doi: 10.1016/j.neuroscience.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 103.Baamonde A, Curto-Reyes V, Juarez L, Meana A, Hidalgo A, Menendez L. Antihyperalgesic effects induced by the IL-1 receptor antagonist anakinra and increased IL-1beta levels in inflamed and osteosarcoma-bearing mice. Life Sci. 2007;81(8):673–682. doi: 10.1016/j.lfs.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 104.Constantin CE, Mair N, Sailer CA, et al. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J Neurosci. 2008;28(19):5072–5081. doi: 10.1523/JNEUROSCI.4476-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gu X, Zheng Y, Ren B, et al. Intraperitoneal injection of thalidomide attenuates bone cancer pain and decreases spinal tumor necrosis factor-alpha expression in a mouse model. Mol Pain. 2010;6:64. doi: 10.1186/1744-8069-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Massi P, Vaccani A, Parolaro D. Cannabinoids, immune system and cytokine network. Curr Pharm Des. 2006;12(24):3135–3146. doi: 10.2174/138161206777947425. [DOI] [PubMed] [Google Scholar]
- 107.Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431(7007):405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 108.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 109.Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410(6827):471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 110.Yin Q, Cheng W, Cheng MY, Fan SZ, Shen W. Intrathecal injection of anti-CX3CR1 neutralizing antibody delayed and attenuated pain facilitation in rat tibial bone cancer pain model. Behav Pharmacol. 2010;21(7):595–601. doi: 10.1097/FBP.0b013e32833e7e2a. [DOI] [PubMed] [Google Scholar]
- 111.Hu JH, Yang JP, Liu L, et al. Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res. 2012;1465:1–9. doi: 10.1016/j.brainres.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 112.Schiller KR, Zillhardt MR, Alley J, Borjesson DL, Beitz AJ, Mauro LJ. Secretion of MCP-1 and other paracrine factors in a novel tumor-bone coculture model. BMC Cancer. 2009;9:45. doi: 10.1186/1471-2407-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu JH, Zheng XY, Yang JP, Wang LN, Ji FH. Involvement of spinal monocyte chemoattractant protein-1 (MCP-1) in cancer-induced bone pain in rats. Neurosci Lett. 2012;517(1):60–63. doi: 10.1016/j.neulet.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 115.Clohisy DR, Mantyh PW. Bone cancer pain. Clin Orthop Relat Res. 2003;415:S279–S288. doi: 10.1097/01.blo.0000093059.96273.56. [DOI] [PubMed] [Google Scholar]
- 116.Szallasi A, Sheta M. Targeting TRPV1 for pain relief: limits, losers and laurels. Expert Opin Investig Drugs. 2012;21(9):1351–1369. doi: 10.1517/13543784.2012.704021. [DOI] [PubMed] [Google Scholar]
- 117.Yoneda T, Hata K, Nakanishi M, et al. Involvement of acidic microenvironment in the pathophysiology of cancer-associated bone pain. Bone. 2011;48(1):100–105. doi: 10.1016/j.bone.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 118.Yoneda T, Hiasa M, Nagata Y, Okui T, White F. Contribution of acidic extracellular microenvironment of cancer-colonized bone to bone pain. Biochim Biophys Acta. 2015 Feb 14; doi: 10.1016/j.bbamem.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kato Y, Ozawa S, Miyamoto C, et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013;13(1):89. doi: 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Palazzo E, Luongo L, de Novellis V, Rossi F, Marabese I, Maione S. Transient receptor potential vanilloid type 1 and pain development. Curr Opin Pharmacol. 2012;12(1):9–17. doi: 10.1016/j.coph.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 121.Ghilardi JR, Röhrich H, Lindsay TH, et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25(12):3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 123.Tominaga M, Caterina MJ, Malmberg AB, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21(3):531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 124.Pan HL, Zhang YQ, Zhao ZQ. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCepsilon pathway in dorsal root ganglion neurons. Mol Pain. 2010;6:85. doi: 10.1186/1744-8069-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Swanson DM, Dubin AE, Shah C, et al. Identification and biological evaluation of 4-(3-trifluoromethylpyridin-2-yl)piperazine-1-carboxylic acid (5-triflu-oromethylpyridin-2-yl)amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist. J Med Chem. 2005;48(6):1857–1872. doi: 10.1021/jm0495071. [DOI] [PubMed] [Google Scholar]
- 126.Lautner MA, Ruparel SB, Patil MJ, Hargreaves KM. In vitro sarcoma cells release a lipophilic substance that activates the pain transduction system via TRPV1. Ann Surg Oncol. 2011;18(3):866–871. doi: 10.1245/s10434-010-1328-1. [DOI] [PubMed] [Google Scholar]
- 127.Seabrook GR, Sutton KG, Jarolimek W, et al. Functional properties of the high-affinity TRPV1 (VR1) vanilloid receptor antagonist (4-hydroxy-5-iodo-3- methoxyphenylacetate ester) iodo-resiniferatoxin. J Pharmacol Exp Ther. 2002;303(3):1052–1060. doi: 10.1124/jpet.102.040394. [DOI] [PubMed] [Google Scholar]
- 128.Waldmann R, Lazdunski M. H(+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8(3):418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- 129.Lingueglia E. Acid-sensing ion channels in sensory perception. J Biol Chem. 2007;282(24):17325–17329. doi: 10.1074/jbc.R700011200. [DOI] [PubMed] [Google Scholar]
- 130.Voilley N, de Weille J, Mamet J, Lazdunski M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci. 2001;21(20):8026–8033. doi: 10.1523/JNEUROSCI.21-20-08026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sluka KA, Price MP, Breese NA, Stucky CL, Wemmie JA, Welsh MJ. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain. 2003;106(3):229–239. doi: 10.1016/S0304-3959(03)00269-0. [DOI] [PubMed] [Google Scholar]
- 132.Qiu F, Wei XL, Zhang SZ, Yuan WX, Mi WD. Increased expression of acid-sensing ion channel 3 within dorsal root ganglia in a rat model of bone cancer pain. Neuroreport. 2014;25(12):887–893. doi: 10.1097/WNR.0000000000000182. [DOI] [PubMed] [Google Scholar]
- 133.Lingueglia E, de Weille JR, Bassilana F, et al. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem. 1997;272(47):29778–29783. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- 134.Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev. 2011;63(3):641–683. doi: 10.1124/pr.110.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chizh BA, Illes P. P2X receptors and nociception. Pharmacol Rev. 2001;53(4):553–568. [PubMed] [Google Scholar]
- 136.Wu JX, Xu MY, Miao XR, et al. Functional up-regulation of P2X3 receptors in dorsal root ganglion in a rat model of bone cancer pain. Eur J Pain. 2012;16(10):1378–1388. doi: 10.1002/j.1532-2149.2012.00149.x. [DOI] [PubMed] [Google Scholar]
- 137.Liu M, Yang H, Fang D, et al. Upregulation of P2X3 receptors by neuronal calcium sensor protein VILIP-1 in dorsal root ganglions contributes to the bone cancer pain in rats. Pain. 2013;154(9):1551–1568. doi: 10.1016/j.pain.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 138.Kaan TK, Yip PK, Patel S, et al. Systemic blockade of P2X3 and P2X2/3 receptors attenuates bone cancer pain behaviour in rats. Brain. 2010;133(9):2549–2564. doi: 10.1093/brain/awq194. [DOI] [PubMed] [Google Scholar]
- 139.Peters CM, Ghilardi JR, Keyser CP, et al. Tumor-induced injury of primary afferent sensory nerve fibers in bone cancer pain. Exp Neurol. 2005;193(1):85–100. doi: 10.1016/j.expneurol.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 140.Yamamoto J, Kawamata T, Niiyama Y, Omote K, Namiki A. Down-regulation of mu opioid receptor expression within distinct subpopulations of dorsal root ganglion neurons in a murine model of bone cancer pain. Neuroscience. 2008;151(3):843–853. doi: 10.1016/j.neuroscience.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 141.Kusuda R, Cadetti F, Ravanelli MI, et al. Differential expression of microRNAs in mouse pain models. Mol Pain. 2011;7:17. doi: 10.1186/1744-8069-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bali KK, Selvaraj D, Satagopam VP, Lu J, Schneider R, Kuner R. Genome-wide identification and functional analyses of microRNA signatures associated with cancer pain. EMBO Mol Med. 2013;5(11):1740–1758. doi: 10.1002/emmm.201302797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Currie GL, Delaney A, Bennett MI, et al. Animal models of bone cancer pain: systematic review and meta-analyses. Pain. 2013;154(6):917–926. doi: 10.1016/j.pain.2013.02.033. [DOI] [PubMed] [Google Scholar]
- 144.Wiesenfeld-Hallin Z. Sex differences in pain perception. Gend Med. 2005;2(3):137–145. doi: 10.1016/s1550-8579(05)80042-7. [DOI] [PubMed] [Google Scholar]
- 145.Wise EA, Price DD, Myers CD, Heft MW, Robinson ME. Gender role expectations of pain: relationship to experimental pain perception. Pain. 2002;96(3):335–342. doi: 10.1016/s0304-3959(01)00473-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gear RW, Miaskowski C, Gordon NC, Paul SM, Heller PH, Levine JD. Kappa-opioids produce significantly greater analgesia in women than in men. Nat Med. 1996;2(11):1248–1250. doi: 10.1038/nm1196-1248. [DOI] [PubMed] [Google Scholar]
- 147.Chia YY, Chow LH, Hung CC, Liu K, Ger LP, Wang PN. Gender and pain upon movement are associated with the requirements for postoperative patient-controlled IV analgesia: a prospective survey of 2,298 Chinese patients. Can J Anaesth. 2002;49(3):249–255. doi: 10.1007/BF03020523. [DOI] [PubMed] [Google Scholar]
- 148.Kest B, Sarton E, Dahan A. Gender differences in opioid-mediated analgesia: animal and human studies. Anesthesiology. 2000;93(2):539–547. doi: 10.1097/00000542-200008000-00034. [DOI] [PubMed] [Google Scholar]
- 149.Riley JL, III, Robinson ME, Wise EA, Price DD. A meta-analytic review of pain perception across the menstrual cycle. Pain. 1999;81(3):225–235. doi: 10.1016/S0304-3959(98)00258-9. [DOI] [PubMed] [Google Scholar]
- 150.Frye CA, Cuevas CA, Kanarek RB. Diet and estrous cycle influence pain sensitivity in rats. Pharmacol Biochem Behav. 1993;45(1):255–260. doi: 10.1016/0091-3057(93)90116-b. [DOI] [PubMed] [Google Scholar]
- 151.Cancer Research UK Lung Cancer Incidence by Sex and UK Region. 2015. [Accessed June 2, 2015]. Available from: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/lung-cancer/incidence.
- 152.Sorge RE, Martin LJ, Isbester KA, et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods. 2014;11(6):629–632. doi: 10.1038/nmeth.2935. [DOI] [PubMed] [Google Scholar]
- 153.Luo Y, Zhang LL, Ning L, He DL, Fan JH, Hou HL. Establishment of bone metastasis model of prostate cancer in nude mice by intratibia injection of human prostate cancer cell line Du145. Zhonghua Nan Ke Xue. 2006;12(2):133–136. [PubMed] [Google Scholar]
- 154.Luger NM, Sabino MA, Schwei MJ, et al. Efficacy of systemic morphine suggests a fundamental difference in the mechanisms that generate bone cancer vs. inflammatory pain. Pain. 2002;99(3):397–406. doi: 10.1016/S0304-3959(02)00102-1. [DOI] [PubMed] [Google Scholar]
- 155.Zilber S, Epstein NJ, Lee SW, et al. Mouse femoral intramedullary injection model: technique and microCT scan validation. J Biomed Mater Res B Appl Biomater. 2008;84B(1):286–290. doi: 10.1002/jbm.b.30872. [DOI] [PubMed] [Google Scholar]
- 156.Rosen HN, Chen V, Cittadini A, et al. Treatment with growth-hormone and Igf-I in growing rats increases bone-mineral content but not bone-mineral density. J Bone Miner Res. 1995;10(9):1352–1358. doi: 10.1002/jbmr.5650100912. [DOI] [PubMed] [Google Scholar]
- 157.Watts NB. Clinical utility of biochemical markers of bone remodeling. Clin Chem. 1999;45(8 pt 2):1359–1368. [PubMed] [Google Scholar]
- 158.Hamaoka T, Madewell JE, Podoloff DA, Hortobagyi GN, Ueno NT. Bone imaging in metastatic breast cancer. J Clin Oncol. 2004;22(14):2942–2953. doi: 10.1200/JCO.2004.08.181. [DOI] [PubMed] [Google Scholar]
- 159.Lam DK, Dang DM, Zhang JA, Dolan JC, Schmidt BL. Novel animal models of acute and chronic cancer pain: a pivotal role for PAR2. J Neurosci. 2012;32(41):14178–14183. doi: 10.1523/JNEUROSCI.2399-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dolan JC, Lam DK, Achdjian SH, Schmidt BL. The dolognawmeter: a novel instrument and assay to quantify nociception in rodent models of orofacial pain. J Neurosci Methods. 2010;187(2):207–215. doi: 10.1016/j.jneumeth.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20 pt 2):6243s–6249s. doi: 10.1158/1078-0432.CCR-06-0931. [DOI] [PubMed] [Google Scholar]
- 162.Coleman RE. Skeletal complications of malignancy. Cancer. 1997;80(8 suppl):1588–1594. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1588::aid-cncr9>3.3.co;2-z. [DOI] [PubMed] [Google Scholar]
- 163.Sabino MA, Luger NM, Mach DB, Rogers SD, Schwei MJ, Mantyh PW. Different tumors in bone each give rise to a distinct pattern of skeletal destruction, bone cancer-related pain behaviors and neurochemical changes in the central nervous system. Int J Cancer. 2003;104(5):550–558. doi: 10.1002/ijc.10999. [DOI] [PubMed] [Google Scholar]
- 164.Rasmussen L, Arvin A. Chemotherapy-induced immunosuppression. Environ Health Perspect. 1982;43(Feb):21–25. doi: 10.1289/ehp.824321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hansen RR, Nielsen CK, Nasser A, et al. P2X7 receptor-deficient mice are susceptible to bone cancer pain. Pain. 2011;152(8):1766–1776. doi: 10.1016/j.pain.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wacnik PW, Kehl LJ, Trempe TM, Ramnaraine ML, Beitz AJ, Wilcox GL. Tumor implantation in mouse humerus evokes movement-related hyperalgesia exceeding that evoked by intramuscular carrageenan. Pain. 2003;101(1–2):175–186. doi: 10.1016/s0304-3959(02)00312-3. [DOI] [PubMed] [Google Scholar]
- 167.Huang D, Huang XL, Yan XB, Wu LX, Wang MA. Establishment and evaluation of a bone cancer pain model. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2008;33(2):115–120. [PubMed] [Google Scholar]
- 168.Wacnik PW, Eikmeier LJ, Ruggles TR, et al. Functional interactions between tumor and peripheral nerve: morphology, algogen identification, and behavioral characterization of a new murine model of cancer pain. J Neurosci. 2001;21(23):9355–9366. doi: 10.1523/JNEUROSCI.21-23-09355.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hald A, Nedergaard S, Hansen RR, Ding M, Heegaard AM. Differential activation of spinal cord glial cells in murine models of neuropathic and cancer pain. Eur J Pain. 2009;13(2):138–145. doi: 10.1016/j.ejpain.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 170.Hamamoto DT, Giridharagopalan S, Simone DA. Acute and chronic administration of the cannabinoid receptor agonist CP 55,940 attenuates tumor-evoked hyperalgesia. Eur J Pharmacol. 2007;558(1–3):73–87. doi: 10.1016/j.ejphar.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Menéndez L, Lastra A, Fresno MF, et al. Initial thermal heat hypoalgesia and delayed hyperalgesia in a murine model of bone cancer pain. Brain Res. 2003;969(1–2):102–109. doi: 10.1016/s0006-8993(03)02284-4. [DOI] [PubMed] [Google Scholar]
- 172.King TE, Pawar SC, Majuta L, et al. The role of alpha 6 integrin in prostate cancer migration and bone pain in a novel xenograft model. PLoS One. 2008;3(10):e3535. doi: 10.1371/journal.pone.0003535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Thudi NK, Martin CK, Nadella MV, et al. Zoledronic acid decreased osteolysis but not bone metastasis in a nude mouse model of canine prostate cancer with mixed bone lesions. Prostate. 2008;68(10):1116–1125. doi: 10.1002/pros.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu S, Yang J, Wang L, et al. Tibia tumor-induced cancer pain involves spinal p38 mitogen-activated protein kinase activation via TLR4-dependent mechanisms. Brain Res. 2010;1346:213–223. doi: 10.1016/j.brainres.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 175.Zhang RX, Liu B, Wang L, et al. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118(1–3):125–136. doi: 10.1016/j.pain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 176.Walker K, Medhurst SJ, Kidd BL, et al. Disease modifying and anti-nociceptive effects of the bisphosphonate, zoledronic acid in a model of bone cancer pain. Pain. 2002;100(3):219–229. doi: 10.1016/S0304-3959(02)00040-4. [DOI] [PubMed] [Google Scholar]
- 177.Sevcik MA, Luger NM, Mach DB, et al. Bone cancer pain: the effects of the bisphosphonate alendronate on pain, skeletal remodeling, tumor growth and tumor necrosis. Pain. 2004;111(1–2):169–180. doi: 10.1016/j.pain.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 178.Le Gall C, Bellahcène A, Bonnelye E, et al. A cathepsin K inhibitor reduces breast cancer induced osteolysis and skeletal tumor burden. Cancer Res. 2007;67(20):9894–9902. doi: 10.1158/0008-5472.CAN-06-3940. [DOI] [PubMed] [Google Scholar]
- 179.Donovan-Rodriguez T, Dickenson AH, Urch CE. Gabapentin normalizes spinal neuronal responses that correlate with behavior in a rat model of cancer-induced bone pain. Anesthesiology. 2005;102(1):132–140. doi: 10.1097/00000542-200501000-00022. [DOI] [PubMed] [Google Scholar]
- 180.Kato A, Minami K, Ito H, et al. Oxycodone-induced analgesic effects in a bone cancer pain model in mice. Oncology. 2008;74(suppl 1):55–60. doi: 10.1159/000143220. [DOI] [PubMed] [Google Scholar]
- 181.Urch CE, Donovan-Rodriguez T, Gordon-Williams R, Bee LA, Dickenson AH. Efficacy of chronic morphine in a rat model of cancer-induced bone pain: behavior and in dorsal horn pathophysiology. J Pain. 2005;6(12):837–845. doi: 10.1016/j.jpain.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 182.Sciuto R, Festa A, Rea S, et al. Effects of low-dose cisplatin on 89Sr therapy for painful bone metastases from prostate cancer: a randomized clinical trial. J Nucl Med. 2002;43(1):79–86. [PubMed] [Google Scholar]
- 183.Steger GG, Bartsch R. Denosumab for the treatment of bone metastases in breast cancer: evidence and opinion. Ther Adv Med Oncol. 2011;3(5):233–243. doi: 10.1177/1758834011412656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sukhtankar D, Okun A, Chandramouli A, et al. Inhibition of p38-MAPK signaling pathway attenuates breast cancer induced bone pain and disease progression in a murine model of cancer-induced bone pain. Mol Pain. 2011;7:81. doi: 10.1186/1744-8069-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]