

Abstract
Drug-related hepatotoxicity is the leading cause of acute liver failure, and hepatic problems are responsible for a significant number of liver transplantations and deaths worldwide. Calcium has been associated with various metabolic processes that lead to cell death and apoptosis, and increased cytosolic Ca2+ has been implicated in hepatotoxicity. This study was designed to investigate the effects of calcium channel blockers (CCBs) on isoniazid-rifampicin, zidovudine and erythromycin-induced hepatotoxicity in rats. Treatment groups comprised control, hepatotoxicant, hepatotoxicant along with each of silymarin, nifedipine, verapamil and diltiazem at subclinical, clinical and supraclinical doses. A day to the end of treatment for each model, rats were subjected to the hexobarbitone-induced hypnosis test. On the last days of treatment, blood samples were collected and serum was analyzed for relevant biochemical parameters. Animals were sacrificed after blood collection and livers were harvested, and samples obtained for in vivo antioxidant indices assay and histopathology. The hepatotoxicants significantly increased serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase (ALP), as well as duration of sleep in the hypnosis test. These drugs significantly reduced the hepatic levels of reduced glutathione (GSH), superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) and increased the level of malondialdehyde (MDA). The CCBs at the various doses significantly reversed the effects of isoniazid-rifampicin, zidovudine and erythromycin. The results obtained in this study suggest that the CCBs possess hepatoprotective activity in drug-induced hepatotoxicity and may be beneficial at the subclinical and clinical doses.
Keywords: calcium channel blockers, drug-induced hepatotoxicity, isoniazid, rifampicin, zidovudine, erythromycin
Introduction
The liver is a vital organ involved in detoxification, protein synthesis and production of biochemicals essential for the process of digestion (e.g. bile). It plays a major role in metabolism, glycogen storage, decomposition of red blood cells, plasma protein synthesis, hormone production and detoxification. In the process of performing its essential role in metabolism and detoxification, especially as regards exogenous agents, liver damage may occur. Common causes of liver damage include xenobiotics, alcohol consumption, malnutrition, infection, anemia and medications (Mroueh et al., 2004[34]). Liver toxicity induced by chemicals is a well recognized problem (Kalantari and Valizadeh, 2000[25]). According to Satoh (1991[50]), after acute exposure, lipid accumulation in the hepatocytes, cellular necrosis or hepatobiliary dysfunction usually occurs, whereas cirrhosis or neoplastic changes are considered to be the results of chronic exposure. Navarro and Senior (2006[35]) defined hepatotoxicity as injury to the liver that is associated with impaired liver function caused by exposure to drugs or noninfectious agents.
Hepatitis is one of the most prevalent diseases in the world and drug related hepatotoxicity is the leading cause of acute liver failure among patients referred for liver transplantation (Lee, 2005[29]; Wang et al., 2008[59]). Hepatic problems are responsible for a significant number of liver transplantations and deaths recorded worldwide and available therapeutic options are very limited (Akindele et al., 2010[5]). In most cases of drug induced hepatotoxicity, there is no effective treatment other than cessation of drug use and general supportive care with the possible use of N-acetylcysteine after acetaminophen overdose and intravenous carnitine for valproate induced mitochondrial injury (Navarro and Senior, 2006[35]; Bohan et al., 2001[9]; Polson and Lee, 2005[44]). Silymarin, a flavonolignan from “milk thistle” (Silybum marianum) plant has clinical applications in alcoholic liver diseases, liver cirrhosis, Amanita mushroom poisoning, viral hepatitis, toxic and drug-induced liver disease and in diabetic patients, with non-traditional use in the protection of other organs in addition to the liver also being advocated (Pradhan and Girish, 2006[45]). Due to the wide range of drugs that can cause hepatotoxicity (including antibiotic, antitubercular, antiviral, anticonvulsant, analgesic (non-steroidal anti-inflammatory drugs [NSAIDs] and acetaminophen), antihypertensive, antipsychotic, antidepressant and oral contraceptive agents) and the increasing incidence and fatality of drug-induced hepatotoxicity, efforts are ongoing worldwide to discover new drugs, especially from natural products, or find relevant uses for old orthodox drugs.
Exaggerated calcium influx into cells is an important signal that may lead to cell death and a number of hepatotoxic effects of drugs is associated with increase in intracellular calcium (Thomas and Reed, 1989[55]; Nicotera et al., 1992[36]; Farghali et al., 2000[15]). Kristian and Siesjo (1998[27]) reported that calcium as a triggering factor has also been associated with ischemic cell death. According to Nicotera et al. (1992[36]) and Gasbarrini et al. (1992[18]), a number of hepatocyte injuries is associated with an elevation of intracellular calcium and that calcium signaling plays a pivotal role in the cascade of events that leads to cell injury. Based on these facts, calcium antagonism has been explored in the management of hepatotoxicity.
In respect of profiling hepatoprotective actions of calcium channel blockers (CCBs), Farghali et al. (2000[15]) investigated the effects of CCBs on tert-butyl hydroperoxide (TBH) induced liver injury using isolated perfused rat hepatocytes. The authors concluded that CCBs (verapamil and diltiazem) exhibited hepatoprotective properties, in hepatocyte oxidative injury, accompanied by a decrease in ATPase activity, which suggests normalization of Ca2+i after TBH intoxication. Sippel et al. (1993[52]) demonstrated the modulatory action of some CCBs on impaired hepatocyte calcium homeostasis induced with diamidinothionaphthene (DAMTP). Landon et al. (1986[28]) measured calcium in liver homogenates and scored tissues for centrilobular necrosis 24 h after administration of hepatotoxic doses of several chemicals in vivo. Carbon tetrachloride, chloroform, dimethylnitrosamine, thioacetamide and acetaminophen all caused massive increase in hepatocellular calcium content and produced pericentral necrosis. Administration of calcium entry blockers (nifedipine, verapamil or chlorpromazine) 1 h before or 7 h after the hepatotoxins significantly attenuated increase in cellular calcium content and necrosis, thus demonstrating protective effect against chemical injury in the liver in vivo. Kalantari and Valizadeh (2000[25]) investigated the protective effect of nifedipine against acetaminophen overdose (700 mg/kg) induced hepatotoxicity in mice. In the study, peak hepatoprotective activity was produced by nifedipine at the dose of 500 mg/kg.
The aim of this study was to investigate the effect of subclinical, clinical and supraclinical doses of CCBs (nifedipine, verapamil and diltiazem) on isoniazid-rifampicin combination, azidothymidine (zidovudine) and erythromycin-induced hepatotoxicity in rats.
Materials and Methods
Drugs and chemicals
Zidovudine (Aurobindo Pharma, Hyderabad, India), rifampicin (Mekophar Pharmaceutical, Ho Chi Minh City, Vietnam), isoniazid (Pfizer Pharmaceuticals, Lagos, Nigeria), erythromycin stearate (Aurochem Pharmaceuticals Pvt Ltd, Mumbai, India), verapamil (APS/Berk Pharmaceutical Ltd, Eastbourne, England), nifedipine (Gemini Pharmaceuticals, Lagos, Nigeria), diltiazem (Sanofi-Aventis, Milan, Italy), silymarin and hexobarbital (Sigma-Aldrich, St Louis, USA).
Animals
The animals used in this study, 4-6 weeks old male and female albino rats weighing between 150-200 g, were obtained from the Laboratory Animal Center of the College of Medicine, University of Lagos, Nigeria. The animals were housed in polypropylene cages and kept in standard environmental conditions having access to standard rodent feed (Livestock Feeds PLC, Lagos, Nigeria) and clean water ad libitum. Rats were acclimatized for two weeks before the experimental procedures began.
The experimental procedures were carried out in compliance with the United States National Academy of Sciences Guide for the Care and Use of Laboratory Animals (Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011[10]).
Experimental design
Isoniazid-rifampicin-induced hepatotoxicity model
Rats were randomly allotted to 12 different treatment groups (n = 5) as shown in Table 1(Tab. 1).
Table 1. Treatment groups in the isoniazid-rifampicin-induced hepatotoxicity model.

Treatment was carried out for 21 days. The hepatoxicants and therapeutic interventions were given concurrently (Balakrishnan et al., 2010[7]).
The clinical dose was calculated as the average of the range of doses used for the indications of the CCBs. The subclinical dose was 1/5th of the clinical dose while the supraclinical dose was 5 times the clinical dose.
Zidovudine-induced hepatotoxicity model
Treatment groups in this test are as outlined in Table 1(Tab. 1) except that zidovudine 800 mg/kg p.o. was used in place of isoniazid-rifampicin combination to induce hepatotoxicity. Administration of drugs was carried out for 28 days (Tikoo et al., 2008[56]).
Erythromycin-induced hepatotoxicity model
In this model erythromycin stearate 100 mg/kg p.o. was used to induce hepatotoxicity and the treatment period in respect of the groups earlier outlined in Table 1(Tab. 1) was 14 days (Abdel-Hameid, 2007[1]).
Hexobarbitone-induced hypnosis test
A day before the last administration in respect of the models (20th, 27th and 13th day for the isoniazid-rifampicin, zidovudine and erythromycin models respectively), rats were subjected to the hexobarbitone-induced hypnosis test for the indirect assessment of the level of cytochrome P450 metabolizing enzyme activity. Each rat in the different groups received hexobarbitone 60 mg/kg i.p. Rats were then placed on their backs in separate observation chambers and the duration of loss of the righting reflex (starting at the time of hexobarbitone injection until they regained their righting reflexes) were recorded. The recovery of righting reflex was confirmed if the animal, when gently placed on its back again, rights itself within one minute (Vogel and Vogel, 1997[58]; Akindele and Adeyemi, 2010[4]). The duration of sleep (min) was then recorded for each animal.
Assessment of parameters
Two hours after the administration of drugs on the last day of treatment for each of the models (21st, 28th and 14th day for the isoniazid-rifampicin, zidovudine and erythromycin models respectively), blood samples were collected from each of the rats by retro-orbital artery bleeding into plain sample bottles. Serum separated out by centrifugation at 3000 rpm at 25 °C for 15 min was used for the estimation of relevant biochemical parameters. After blood collection, the animals were sacrificed by cervical dislocation and laparatomized for the harvest of liver. A slice of approximately 1/3rd of the liver of each rat was processed for estimation of in vivo antioxidant indices while the remaining 2/3rd was fixed in 10 % formo-saline for histopathological assessment.
Biochemical analysis
Levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), total cholesterol, total bilirubin (TBILI), albumin and total protein were estimated using Roche and Cobas commercial kits and Roche/Hitachi 904 automated analyzer.
Measurement of antioxidant indices
Measurement of antioxidant indices in liver samples including catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPx), reduced glutathione (GSH) and malondialdehyde (MDA) was done according to established procedures (Soon and Tan, 2002[53]; Habbu et al., 2008[23]) Total protein was determined using the Biuret method (Gornall et al., 1949[22]).
Histopathological assessment
The various tissues obtained from experimental animals fixed in 10 % formo-saline were dehydrated in graded alcohol, embedded in paraffin, and cut into 4-5 μm thick sections. Hematoxylin-eosin was used to stain the sections for photomicroscopic assessment using a Model N-400ME photomicroscope (CEL-TECH Diagnostics, Hamburg, Germany). Slides were examined using the × 40, × 100, and × 400 objectives (Galigher and Kozloff, 1971[17]; Habbu et al., 2008[23]).
Statistical analysis
Results are expressed as mean ± SEM. Data were analyzed using One-way ANOVA followed by Dunnett's multiple comparison test using GraphPad Prism 5 (GraphPad Software Inc., CA, USA). Values were considered significant when P < 0.05.
Results
Isoniazid-rifampicin-induced hepatotoxicity model
Biochemical parameters
Isoniazid-rifampicin significantly (P < 0.001) increased the level of AST, ALT and ALP while significantly (P < 0.001) reducing the level of total protein and albumin (Table 2(Tab. 2)). Silymarin significantly (P < 0.05, 0.001) reduced the level of AST and increased the level of total protein compared with isoniazid-rifampicin alone treated group of animals. Nifedipine at all doses administered (subclinical, 0.17 mg/kg p.o.; clinical, 0.86 mg/ kg p.o.; and supraclinical, 4.23 mg/kg p.o.) significantly (P < 0.05, 0.01, 0.001) reduced the level of AST and total bilirubin while increasing the level of total protein and albumin compared with the isoniazid-rifampicin group, with peak effect observed at the clinical dose. Verapamil at the clinical (4.29 mg/kg, p.o.) and supraclinical (21.43 mg/kg, p.o.) doses significantly (P < 0.05) reduced the level of AST, ALT and ALP while increasing the level of total protein and direct bilirubin compared with isoniazid-rifampicin group. There was an increase in total cholesterol at the subclinical dose (0.86 mg/kg, p.o.) when compared to the hepatotoxicant group. Diltiazem intervention significantly (P < 0.05) reduced the levels of AST, ALT and ALP while increasing the level of total protein compared with isoniazid-rifampicin alone treated group, with effects being most pronounced at the clinical (3.43 mg/kg, p.o.) and supraclinical (17.13 mg/kg, p.o.) doses (Table 2(Tab. 2)).
Table 2. Effect of calcium channel blockers (nifedipine, verapamil and diltiazem) on serum biochemical parameters in isoniazid-rifampicin combination-induced hepatotoxicity in rats.

In vivo antioxidant indices
Isoniazid-rifampicin significantly (P < 0.05, 0.01) reduced the levels of GSH, SOD, CAT and total protein with increase in the level of MDA when compared to rats of the control group (Table 3(Tab. 3)). Silymarin significantly (P < 0.05, 0.01) increased the level of GSH and reduced the level of MDA relative to the isoniazid-rifampicin only treated group. Co-administration of the antitubercular drugs with nifedipine significantly (P < 0.05 0.01) reduced the level of MDA, at all the treatment doses, compared to the group of rats receiving isoniazid-rifampicin alone. However, peak effect was observed at the supraclinical dose (4.29 mg/kg, p.o.). At this dose, nifedipine also significantly (P < 0.01) reversed the reduction in GSH level induced by isoniazid-rifampicin. Verapamil and diltiazem significantly (P < 0.001) reduced the level of MDA compared with isoniazid-rifampicin only treated group, with peak effect produced at the clinical doses of 4.29 mg/kg and 3.43 mg/kg respectively (Table 3(Tab. 3)).
Table 3. Effect of nifedipine, verapamil and diltiazem on in vivo antioxidant indices in liver samples in isoniazid-rifampicin combination-induced hepatotoxicity in rats.

Hexobarbitone-induced hypnosis test
Isoniazid-rifampicin combination significantly (P < 0.01) increased the duration of sleep compared with the control group. Silymarin and nifedipine at the supraclinical dose (4.29 mg/kg, p.o.) significantly reduced the duration of sleep compared with the isoniazid-rifampicin alone treated group (Table 4(Tab. 4)). Verapamil and diltiazem at the various doses used did not elicit any significant effect (P > 0.05) on duration of sleep compared to the hepatotoxicant group.
Table 4. Effect of nifedipine, verapamil and diltiazem on duration of sleep in hexobarbitone-induced hypnosis test in isoniazid-rifampicin combination-induced hepatotoxicity in rats.

Zidovudine-induced hepatotoxicity model
Biochemical parameters
Significant (P < 0.01, 0.001) increase in the levels of AST, ALT, ALP and total bilirubin but significant (P < 0.01) reduction in the level of total protein were observed in the zidovudine treated rats compared to control (Table 5(Tab. 5)). Silymarin significantly (P < 0.05, 0.01) reversed the effect of zidovudine on ALP and total bilirubin. Nifedipine at the clinical dose (0.86 mg/kg, p.o.) and supraclinical dose (4.29 mg/kg, p.o.) produced significant (P < 0.05, 0.01) reduction in the levels of AST and ALP compared to the zidovudine treated group. Verapamil at the supraclinical dose (21.43 mg/kg, p.o.) significantly (P < 0.05, 0.01) reduced the level of AST and increased the level of total protein relative to the zidovudine only group. In respect of diltiazem, there was significant (P < 0.05, 0.01, 0.001) reduction in the levels of AST and total bilirubin while the level of total protein was significantly (P < 0.05, 0.01) increased compared to the zidovudine treated rats, at all doses used. However, peak protective effect was elicited at the subclinical dose, 0.68 mg/kg, p.o. (Table 5(Tab. 5)).
Table 5. Effect of calcium channel blockers (nifedipine, verapamil and diltiazem) on serum biochemical parameters in zidovudine-induced hepatotoxicity in rats.

In vivo antioxidant indices
Zidovudine (800 mg/kg, p.o.) caused significant (P < 0.01) reductions in the levels of GSH, SOD, CAT, GPx and total protein in liver samples but the level of MDA was significantly (P < 0.001) increased compared to control (Table 6(Tab. 6)). Silymarin (50 mg/kg, p.o.) significantly (P < 0.05, 0.01, 0.001) reversed the effect of zidovudine on these parameters especially as it relates to GSH, CAT and MDA. Nifedipine at the subclinical dose (0.17 mg/kg, p.o.), clinical dose (0.86 mg/kg, p.o.) and supraclinical dose (4.23 mg/kg, p.o.) reversed the elevation in the levels of MDA produced by zidovudine, with peak protective effect observed at the clinical dose. However, the level of CAT observed with nifedipine at the clinical and supraclinical doses were significantly (P < 0.05, 0.01) lower when compared with the zidovudine only treated rats. Verapamil at all doses (subclinical, 0.86 mg/ kg; clinical, 4.29 mg/kg; and supraclinical, 21.43 mg/kg, p.o.) significantly (P < 0.01, 0.001) reversed the elevation of MDA level produced by zidovudine. Verapamil also significantly (P< 0.05) increased the levels of SOD at the clinical dose but there was a significant (P < 0.01) reduction in CAT level at the supraclinical dose compared to zidovudine. Diltiazem at the supraclinical dose (4.23 mg/kg, p.o.) significantly (P < 0.05, 0.01) increased zidovudine diminished level of GSH, with a reversal of the increased level of MDA observed with zidovudine (Table 6(Tab. 6)).
Table 6. Effect of nifedipine, verapamil and diltiazem on in vivo antioxidant indices in liver samples in zidovudine-induced hepatotoxicity in rats.

Hexobarbitone-induced hypnosis test
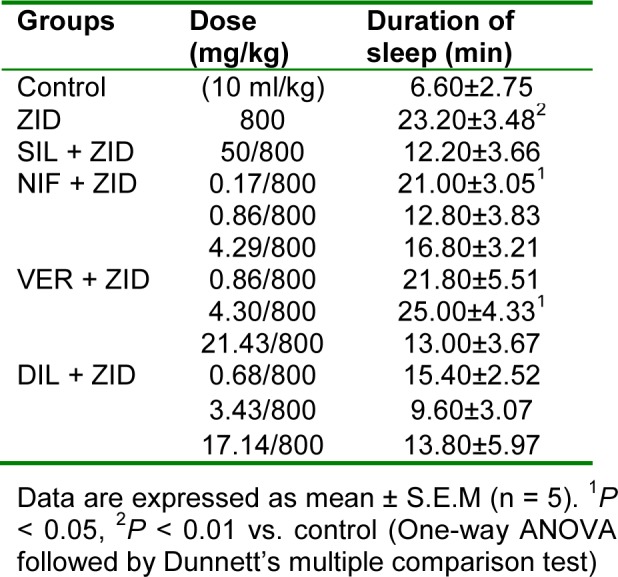
Significant (P < 0.01) increase in the duration of sleep was observed in the zidovudine treated group of rats compared to the control group. Silymarin and the CCBs at all doses used did not significantly (P > 0.05) affect the duration of sleep compared to the zidovudine only group (Table 7(Tab. 7)).
Table 7. Effect of nifedipine, verapamil and diltiazem on duration of sleep in hexobarbitone-induced hypnosis test in zidovudine-induced hepatotoxicity in rats.
Erythromycin-induced hepatotoxicity model
Biochemical parameters
As shown in Table 8(Tab. 8), erythromycin caused a significant (P < 0.01, 0.001) increase in serum ALT and AST levels when compared to the control group. Silymarin produced a decrease in AST and ALT levels compared to the erythromycin group but the effect was only significant (P < 0.01) in respect of AST. Treatment with the CCBs reduced the elevated levels of AST and ALT but the effect was only significant (P < 0.05) in respect of AST. Nifedipine administered at the subclinical dose of 0.17 mg/kg produced the most pronounced effect with the effect of verapamil at the clinical dose of 4.30 mg/kg also being significant (P < 0.05). Erythromycin also caused a significant (P < 0.001) increase in serum ALP level compared to the control group. Silymarin and the CCBs produced significant reductions (P < 0.001) in ALP level compared to erythromycin. Diltiazem at the clinical dose of 3.43 mg/kg produced the most pronounced effect. Erythromycin had no significant effect (P > 0.05) on total protein compared to the control. Silymarin produced a significant (P < 0.05) reduction in the level of total protein compared to erythromycin. The CCBs did not produce any significant effect (P > 0.05) on total protein compared to the erythromycin group (Table 8(Tab. 8)). Erythromycin produced a significant (P < 0.01) increase in albumin level when compared to control. Silymarin produced a significant (P < 0.05) reduction in the level of albumin compared to erythromycin. The CCBs did not generally produce a significant effect on serum albumin compared to the erythromycin group except in the case of verapamil at the clinical dose of 4.30 mg/kg in which there was a significant (P < 0.001) reduction (Table 8(Tab. 8)). In respect of total bilirubin, direct bilirubin and cholesterol, there was no significant difference in comparing erythromycin with control and in comparing silymarin and the CCBs with erythromycin except in the case of verapamil at the subclinical dose of 0.86 mg/kg in which a significant (P < 0.001) increase in total bilirubin was produced compared to erythromycin (Table 8(Tab. 8)).
Table 8. Effect of calcium channel blockers (nifedipine, verapamil and diltiazem) on serum biochemical parameters in erythromycin-induced hepatotoxicity in rats.

In vivo antioxidant indices
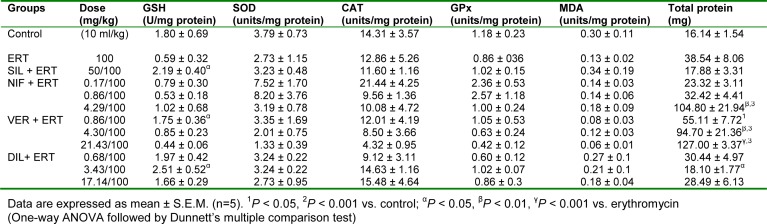
Erythromycin reduced the level of GSH compared to control but this effect was not significant (P > 0.05). Silymarin, verapamil at the subclinical dose of 0.86 mg/kg, and diltiazem at the clinical dose of 3.43 mg/kg reversed the effect of erythromycin by significantly (P < 0.05) increasing the level of GSH. Nifedipine at all the doses used, verapamil at 4.30 and 17.40 mg/kg, and diltiazem at 0.68 and 17.14 mg/kg did not produce any significant effect on GSH level compared to erythromycin (Table 9(Tab. 9)). Erythromycin did not produce any significant effect (P > 0.05) on SOD, CAT, GPx, and MDA compared to control. Silymarin and the CCBs also did not produce any significant (P > 0.05) effect on these parameters relative to erythromycin (Table 9(Tab. 9)). Erythromycin increased the level of total protein in liver samples but this effect was not significant (P > 0.05) compared to the control. Compared to erythromycin, silymarin reduced the level of total protein but this effect was also not significant (P > 0.05). Nifedipine at the supraclinical dose of 4.29 mg/kg and verapamil at the clinical and supraclinical doses of 4.30 mg/kg and 17.30 mg/kg respectively, significantly (P < 0.01, 0.001) increased the total protein level compared to erythromycin. However, diltiazem at the clinical dose of 3.43 mg/kg significantly (P < 0.05) reduced the total protein level compared to erythromycin (Table 9(Tab. 9)).
Table 9. Effect of nifedipine, verapamil and diltiazem on in vivo antioxidant indices in liver samples in erythromycin-induced hepatotoxicity in rats.
Hexobarbitone-induced hypnosis test
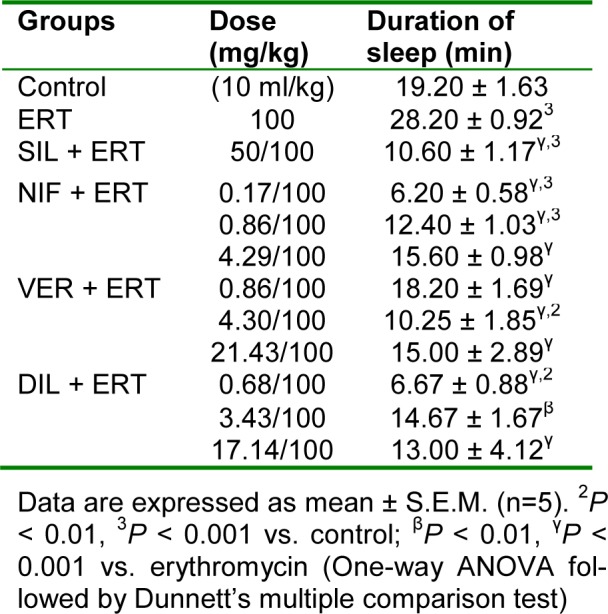
In relation to control, erythromycin significantly (P < 0.001) increased the duration of sleep. Silymarin and the CCBs at all doses used significantly (P < 0.001) reduced the duration of sleep compared to erythromycin. The effect of the CCBs in preserving the metabolizing effect of cytochrome P450 enzymes on hexobarbitone was most pronounced for nifedipine at the subclinical dose of 0.17 mg/kg, for verapamil at the clinical dose of 4.30 mg/kg, and for diltiazem at the subclinical dose of 0.68 mg/kg (Table 10(Tab. 10)).
Table 10. Effect of nifedipine, verapamil and diltiazem on duration of sleep in hexobarbitone-induced hypnosis test in erythromycin-induced hepatotoxicity in rats.
Histopathological assessment
Hepatotoxicants representative samples presented with cytoplasmic inclusions with frosted appearance of hepatocytes due to intracellular accumulation of material, and periportal hemorrhage with the observation of presence of blood around the portal tracts. Control, silymarin, and CCBs intervention representative samples generally presented as normal liver with hepatocytes arranged as plates radiating away from the central vein to the portal tracts. Representative photomicrographs are presented as Figures 1A-F(Fig. 1).
Figure 1. Representative photomicrographs of liver samples. A. control group (normal saline 10 ml/kg, p.o.) showing normal liver architecture; B. isoniazid-rifampicin group (100 mg/kg each, p.o.) showing cytoplasmic inclusions; C. Silymarin (50 mg/kg, p.o.) + isoniazid-rifampicin (100 mg/kg each, p.o.) group showing normal liver; D. Nifedipine (clinical dose; 0.86 mg/kg, p.o.) + isoniazid-rifampicin (100 mg/kg each, p.o.) group showing normal liver; E. Zidovudine (800 mg/kg, p.o.) group showing periportal hemorrhage; F. Verapamil (clinical dose; 4.30 mg/kg, p.o.) + zidovudine (800 mg/kg, p.o.) group showing normal liver (×400).

Discussion
Drug-induced liver injury is a potential complication of nearly every prescribed medication and many fatal and near-fatal drug reactions occur each year (Kaplowitz, 1992[26]; Zimmerman and Maddrey, 1993[61]; Farrell, 1994[16]; Lee, 1995[29]). According to Pandit et al. (2012[41]), more than 900 drugs, toxins and herbs have been reported to cause liver injury, and drug-induced liver injury is responsible for 5 % of all hospital admissions and 50 % of all acute liver failures. More than 75 % of cases of idiosyncratic drug reactions result in liver transplantation or death (Ostapowicz et al., 2002[39]). Drug-induced liver injury is reported to be the most common reason for drug withdrawal from the market (Pandit et al., 2012[41]). With increasing morbidity and mortality arising from drug-induced liver injury, a lot of attention is being devoted worldwide to understanding the mechanistic basis of drug-induced hepatotoxicity, strategy for management, and search for drugs (complementary or orthodox; natural or synthetic; old or new) that may ameliorate or reverse drug-induced liver injury by chemically or therapeutically diverse agents. Among the numerous drugs reputed to induce hepatotoxicity are the antitubercular drugs isoniazid and rifampicin, antiviral drug zidovudine, and antibiotic erythromycin.
Isoniazid and rifampicin are first-line anti-tuberculosis chemotherapeutic agents that have been associated with hepatotoxicity. A major metabolic pathway of isoniazid is acetylation to acetylisoniazid and subsequent hydrolysis to acetylhydrazine and isonicotinic acid (Tostmann et al., 2008[57]). Acetylhydrazine is either hydrolyzed to hydrazine or acetylated to diacetylhydrazine (Ellard and Gammon, 1976[13]; Mitchell et al., 1976[33]). Hydrazine has been suggested as the toxic metabolite of isoniazid responsible for its hepatotoxicity (Gent et al., 1992[19]; Sarich et al., 1996[48]). Hydrazine is known to cause irreversible cellular damage (Tostmann et al., 2008[57]) and a study in rat liver microsomes showed that nitrogen-centered radicals are formed during hydrazine metabolism which possibly participates in the hepatotoxicity process (Noda et al., 1985[37]). Rifampicin undergoes desacetylation to desacetylrifampicin and hydrolysis to produce 3-formylrifampicin (Acocella and Conti, 1980[2]; Holdiness, 1984[24]). The mechanism of rifampicin-induced hepatotoxicity is unknown (Tostmann et al., 2008[57]) and there is no evidence for the presence of toxic metabolites (Westphal et al., 1994[60]). However, rifampicin is a potent inducer of hepatic CYP450 and induction of isoniazid hydrolase results in increased hydrazine production when rifampicin is combined with isoniazid resulting in higher toxicity (Blair et al., 1985[8]; Sarma et al., 1986[49]).
Zidovudine is a potent inhibitor of HIV replication and the first clinically approved drug for AIDS (Tikoo et al., 2008[56]). Several antiretrovirals, including zidovudine, have been reported to cause fatal acute hepatitis (Pandit et al., 2012[41]). Mechanisms that have been suggested for zidovudine-induced toxicity include alteration in liver mitochondrial DNA, alteration in oxidative phosphorylation coupling and changes in fine ultra-structure of liver mitochondrial (Tikoo et al., 2008[56]). Generation of reactive oxygen species (ROS), peroxide production and oxidative damage in mitochondria have been reported to play vital roles in zidovudine-induced toxicity (Majid et al., 1991[31]; Tikoo et al., 2008[56]).
Erythromycin is a commonly used antibiotic which induces severe liver injury and cardiovascular dysfunction both in humans and experimental animals at high doses (Adams, 1976[3]; Pari and Murugan, 2004[42]). As reported by Pessayre et al. (1985[43]), overdoses of erythromycin led to occurrence of hepatitis, hepatic dysfunction, jaundice and necrosis of hepatocytes. Erythromycin is metabolized to reactive nitrosoalkane derivatives, which may be further metabolized to nitroso radicals which could be responsible for the degradation of phospholipids in the liver (Pessayre et al., 1985[43]). The highly reactive nitrosoalkane derivative is normally detoxified by glutathione but excessive consumption of glutathione stores enables the reactive intermediate of erythromycin to destroy hepatic cells and other cells (Pessayre et al., 1985[43]).
Clinical chemistry variables considered useful in identifying liver toxicity include ALT, AST, sorbitol dehydrogenase (SDA), glutamate dehydrogenase (GLDH), total bile acids (TBA), lactate dehydrogenase (LDH), ornithine carbamyltransferase (OCT) and unconjugated bilirubin (UBILI) (hepatocellular toxicity); TBA, ALP, gamma glutamyltransferase (GGT), 5'-nucleotidase (5-NT) and TBILI (hepatobiliary toxicity); and glutamate dehydrogenase (GLDH), lactate and OCT (mitochondrial toxicity; EMEA, 2008[14]). According to EMEA (2008[14]), total protein, albumin, triglycerides, cholesterol, glucose and blood urea nitrogen, activated partial thromboplastin time (APTT) and prothrombin time (PT) can be used as supplementary tests for hepatic synthetic functions. Liver disease is implicated in cases of elevated levels of AST, ALT, LDH, total and direct bilirubin, ALP, gamma-glutamyltranspeptidase, 5-NT, prothrombin time and diminution in level of albumin, hepatic demethylation and galactose elimination (Cotran et al., 2005[11]).
Generally, hepatotoxicity is associated with diminished activity of cytochrome P450 metabolizing enzymes. The hexobarbitone-induced hypnosis test is an indirect method of assessing the activity of this class of enzymes. Hepatic damage resulting in reduced metabolizing enzyme activity will cause increase in the duration of sleep. For example, it has been reported that paracetamol and CCl4-induced hepatic injury decreased the activity of cytochrome P450 enzymes and the metabolic functional activity of hepatocytes, thus delaying barbiturate metabolism and excretion of phenobarbitone leading to prolongation of sleeping time (Girish et al., 2009[20]; Girish and Pradhan, 2012[ref:20]). According to these authors, pretreatment with hepatoprotective phytochemicals restored the phenobarbitone-induced sleeping time in a dose-related fashion.
CCBs disrupt the movement of Ca2+ through calcium channels and are indicated for the treatment of hypertension, angina pectoris and arrhythmias. Nifedipine, a dihydropyridine, possesses high vascular selectivity like other members of its class and it is primarily used to reduce systemic vascular resistance and arterial pressure. Members of the dihydropyridines class are primarily used to treat hypertension. Verapamil is a non-dihydropyridine (phenylalkylamine class) relatively selective for the myocardium and less effective as a systemic vasodilator drug. It is used in the treatment of angina and arrhythmias. Diltiazem is also non-dihydropyridine (benzothiazepine class) intermediate in action between nifedipine and verapamil. It has both cardiac depressant and vasodilator actions. Due to the association of hepatotoxicity with disruption in calcium homeostasis, which may result in the activation of many membrane damaging enzymes like ATPases, phospholipases, proteases and endonucleases, disruption of mitochondrial metabolism and ATP synthesis and damage of microfilaments used to support cell structure (Singh et al., 2011[51]), attempts have been made in the past to investigate the modulatory actions of CCBs in drug-induced hepatotoxicity using mostly in vitro models. This study was designed to investigate the effect of subclinical, clinical and supraclinical doses of CCBs (nifedipine, verapamil and diltiazem) on isoniazid-rifampicin, zidovudine and erythromycin-induced hepatotoxicity in rats.
In this study, isoniazid-rifampicin, zidovudine and erythromycin significantly increased the levels of AST, ALT, ALP and total bilirubin while reducing the levels of total protein and albumin relative to control. Elevation in the level of AST and the liver specific ALT result from leakage from damaged tissues as a result of hepatocellular necrosis (Amacher, 2002[6]; Ozer et al., 2008[40]) while increase in ALP level is as a result of overproduction and release in blood due to hepatobiliary injury and cholestasis (Ramaiah, 2007[46]). Hepatobiliary injury and cholestasis cause reduced hepatic clearance and increase in the level of total bilirubin (Dufour et al., 2001[12]). Reduction in the level of albumin and total protein are consequence of decreased synthetic capacity as a result of hepatic dysfunction (Thapa and Walia, 2007[54]). The standard drug used, silymarin, significantly reduced the level of AST and increased the level of total protein in the isoniazid-rifampicin model; reduced the level of ALP and total bilirubin in zidovudine model; and reduced the level of AST and ALP in the erythromycin model, compared with the hepatotoxicant group. In the isoniazid-rifampicin model, the CCBs across the doses significantly reduced the level of AST, ALT, ALP and total bilirubin, while increasing the level of total protein and albumin relative to the hepatotoxicant. Effect was most prominent at the sub-clinical and clinical doses for nifedipine; supraclinical dose for verapamil; and clinical and supraclinical doses for diltiazem. In the zidovudine model, nifedipine most prominently reduced the AST and ALP level at the clinical dose; verapamil most prominently reduced AST and increased total protein at the supraclinical dose; while diltiazem at the subclinical dose most prominently reduced AST and total bilirubin while increasing total protein compared with the hepatotoxicant group. In the erythromycin model, nifedipine at the subclinical and supraclinical doses significantly reduced AST and ALP levels, respectively relative to the hepatotoxicant group. Verapamil across the doses significantly reduced the level of total bilirubin, AST and ALP, while for diltiazem there was significant reduction in ALP level at the clinical dose compared with the hepatotoxicant group. The action of the CCBs in reducing the level of liver enzymes and total bilirubin, while increasing the level of total protein and albumin suggests hepatoprotective activity in drug-induced liver injury.
In respect of the hexobarbitone-induced hypnosis test, all the hepatotoxic agents used significantly increased the duration of sleep compared to control suggesting reduction in the metabolism of the barbiturate as a result of inhibition of cytochrome P450 activity due to liver damage. Nifedipine at the supraclinical dose and silymarin significantly reduced the duration of sleep relative to the isoniazid-rifampicin only treated group. The effects of the CCBs and silymarin were not significant compared to the hepatotoxicant in the zidovudine model. In the erythromycin model, however, silymarin and the CCBs at all doses used significantly reduced the duration of sleep relative to the hepatotoxicant with effects being most prominent at the subclinical dose for nifedipine and diltiazem, and at the clinical dose for verapamil. The reduction in the duration of sleep by the CCBs relative to the hepatotoxicant groups suggest ability to protect the integrity of the liver and conserve the activity of the cytochrome P450 metabolizing enzymes.
Oxidative stress is implicated as a common pathologic mechanism contributing to the initiation and progression of hepatic damage in a variety of liver disorders (Girish et al., 2009[20]). Mammalian cells contain endogenous antioxidant enzymes, including superoxide dismutase (SOD), glutathione peroxidase and catalase (Saeed et al., 2005[47]). The initial step in the detoxification process is the dismutation of oxygen radicals to hydrogen peroxide by SOD. Catalase protects cells from oxidative stress of hydrogen peroxide by its cleavage to water and oxygen (Ookhtens and Kaplowitz, 1998[38]). Glutathione peroxidase facilitate the conjugation of hydrogen peroxide to glutathione (reduced) leading to the generation of water and oxidized glutathione (Saeed et al., 2005[47]; Akindele et al., 2010[5]). GSH is an important biomolecule against chemically induced toxicity and can participate in the elimination of reactive intermediaries by reduction of hydroperoxides in the presence of GSH dependent enzymes; it functions as a free radical scavenger, a generator of α-tocopherol and also an important role player in the maintenance of protein sulfhydryl group (Meister, 1984[32]; Girish et al., 2009[20]). Malondialdehyde is a useful index of lipid peroxidation being a major breakdown product of lipid peroxides. The engagement and overwhelming of antioxidant enzymes by free radicals result in the depletion of the antioxidant defenses and induction of lipid peroxidation evident in elevation of MDA level (Akindele et al., 2010[5]). Isoniazid-rifampicin significantly reduced the level of GSH, SOD and CAT while increasing the level of MDA relative to control. Silymarin significantly reduced the level of MDA compared with the hepatotoxicant group. Nifedipine at the supraclinical dose significantly increased the level of GSH relative to the isoniazid-rifampicin only treated group. All the CCBs at all doses used significantly reduced the level of MDA relative to the hepatotoxicant group with the most prominent effect produced at the supraclinical dose for nifedipine and the clinical dose for verapamil and diltiazem. Zidovudine significantly reduced all in vivo antioxidants and increased MDA compared with the control group. Silymarin significantly elevated the level of GSH and CAT while reducing MDA compared to zidovudine. Nifedipine at the clinical dose did not significantly affect the in vivo antioxidants except a significant reduction in CAT at the clinical and supraclinical doses relative to zidovudine. However, nifedipine at all doses used significantly reduced MDA level compared with zidovudine with the most prominent effect produced at the clinical dose. Verapamil at the clinical dose significantly increased the level of SOD but reduced CAT level at the supraclinical dose compared to the hepatotoxicant group. However, the drug at all doses used in this study significantly reduced MDA level relative to zidovudine with the most prominent effect produced at the supraclinical dose. Diltiazem at the supraclinical dose significantly increased GSH level but reduced GPx level relative to zidovudine with no significant effect on MDA. Erythromycin at the dose used in this study did not elicit significant changes in the level of antioxidants and MDA. Relative to erythromycin, however, silymarin, verapamil at the subclinical dose and diltiazem at the clinical dose all increased the level of GSH. The elevation of hepatotoxicants diminished in vivo antioxidants level and reduction in the level of MDA relative to isoniazid-rifampicin, zidovudine and erythromycin suggest that the CCBs counteracted lipid peroxidation and preserved the capacity of in vivo antioxidants in the liver.
Observations in the histopathological assessment of representative liver samples across the groups give further credence to the results obtained in the biochemical, antioxidant indices and hypnosis test determinations.
Although the CCBs used in this study demonstrated hepatoprotective activity at the supraclinical doses, it is suggested that the trial or use of this drugs in humans be restricted to the subclinical and clinical doses due to the side-effects associated with this class of drugs e.g. flushing, dizziness, headache, excessive hypotension, constipation, gingival overgrowth, edema, reflex tachycardia, excessive bradycardia, impaired electrical conduction/atrioventricular nodal block and depressed contractility.
Conclusion
The results obtained in this study suggest that the calcium channel blockers (nifedipine, verapamil and diltiazem) possess hepatoprotective activity in drug-induced hepatotoxicity and they may be beneficial at the sub-therapeutic and therapeutic doses in situations where stoppage of drug therapy as a result of induced hepatotoxicity may be critical to therapeutic outcome.
Acknowledgements
The authors express their gratitude to Mr. M. Chijioke (Department of Pharmacology) and Mr. S.O. Adenekan (Department of Biochemistry) of the Faculty of Basic Medical Sciences, College of Medicine, University of Lagos, Lagos, and Mr. S. Ogunnowo of the Chemical Pathology Department, Lagos University Teaching Hospital, Lagos, Nigeria, for technical assistance rendered in the course of this work.
Declaration of interest
The authors declare that there is no conflict of interest in respect of this study. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- 1.Abdel-Hameid NH. Protective role of dimethyl diphenyl bicarboxylate (DDB) against erythromycin induced hepatotoxicity in male rats. Toxicol in Vitro. 2007;21:618–625. doi: 10.1016/j.tiv.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Acocella G, Conti R. Interaction of rifampicin with other drugs. Tubercle. 1980;61:171–177. doi: 10.1016/0041-3879(80)90007-0. [DOI] [PubMed] [Google Scholar]
- 3.Adams HR. Antibiotic-induced alterations of cardiovascular reactivity. Fed Proc. 1976;35:1148–1150. [PubMed] [Google Scholar]
- 4.Akindele AJ, Adeyemi OO. Anxiolytic and sedative effects of Byrsocarpus coccineus Schum. and Thonn. (Connaraceae) extract. Int J Appl Res Nat Prod. 2010;3:28–36. [Google Scholar]
- 5.Akindele AJ, Ezenwanebe KO, Anunobi CC, Adeyemi OO. Hepatoprotective and in vivo antioxidant effects of Byrsocarpus coccineus Schum. and Thonn. (Connaraceae) J Ethnopharmacol. 2010;129:46–52. doi: 10.1016/j.jep.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 6.Amacher DE. A toxicologist’s guide to biomarkers of hepatic response. Hum Exp Toxicol. 2002;21:253–262. doi: 10.1191/0960327102ht247oa. [DOI] [PubMed] [Google Scholar]
- 7.Balakrishnan BR, Sangameswaran B, Bhaskar VH. Effect of methanol extract of Cuscuta reflexa aerial parts on hepatotoxicity induced by antitubercular drugs in rats. Int J Appl Res Nat Prod. 2010;3:18–22. [Google Scholar]
- 8.Blair IA, Mansilla TR, Brodie MJ, Clare RA, Dollery CT, Timbrell JA, et al. Plasma hydrazine concentrations in man after isoniazid and hydralazine administration. Hum Toxicol. 1985;4:195–202. doi: 10.1177/096032718500400210. [DOI] [PubMed] [Google Scholar]
- 9.Bohan TP, Helton E, McDonald I, Konig S, Gazitt S, Sugimoto T, et al. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology. 2001;41:1405–1409. doi: 10.1212/wnl.56.10.1405. [DOI] [PubMed] [Google Scholar]
- 10.Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the care and use of laboratory animals. 8th. Washington, DC: National Academies Press; 2011. [Google Scholar]
- 11.Cotran RS, Kumar V, Fausto N, Nelso F, Robbins SL, Abbas AK. Robbins and Cotran Pathologic Basis of Disease. St. Louis, Mo: Elsevier Saunders; 2005. [Google Scholar]
- 12.Dufour DR, Lott JA, Nolte FS, Gretch DR, Koff RS, Seeff LB. Diagnosis and monitoring of hepatic injury. II. Recommendations for use of laboratory tests in screening, diagnosis and monitoring. Clin Chem. 2001;47:1133–1135. doi: 10.1093/clinchem/46.12.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellard GA, Gammon PT. Pharmacokinetics of isoniazid metabolism in man. J Pharmacokinet Biopharm. 1976;4:83–113. doi: 10.1007/BF01086149. [DOI] [PubMed] [Google Scholar]
- 14.EMAE (European Medicines Agency); Committee for Medicinal Products for Human Use (CHMP), editor Doc. Ref. EMEA/CHMP/SWP/150115/2006. London: Jan 24, 2008. Non-clinical guideline on drug-induced hepatotoxicity. [Google Scholar]
- 15.Farghali H, Kmonickova E, Lotkova H, Martinek J. Evaluation of calcium channel blockers as potential hepatoprotective agents in oxidative stress injury of perfused hepatocytes. Physiol Res. 2000;49:261–268. [PubMed] [Google Scholar]
- 16.Farrell GC. Drug-induced liver disease. Edinburgh, Scotland: Churchill Livingstone; 1994. [Google Scholar]
- 17.Galigher AE, Kozloff EN. Essentials of practical microtechniques. 2nd. Philadelphia, PA: Lea and Febiger; 1971. p. p 77. [Google Scholar]
- 18.Gasbarrini A, Borle AB, Farghali H, Francavilla A, van Thiel D. Fructose protects rat hepatocites from anoxic injury. J Biol Chem. 1992;267:5745–5752. [PubMed] [Google Scholar]
- 19.Gent WL, Seifart HI, Parkin DP, Donald PR, Lamprecht JH. Factors in hydrazine formation from isoniazid by paediatric and adult tuberculosis patients. Eur J Clin Pharmacol. 1992;43:131–136. doi: 10.1007/BF01740658. [DOI] [PubMed] [Google Scholar]
- 20.Girish C, Bidhan CK, Jayanthi S, Ramachandra Rao K, Rajesh B, Pradhan SC. Hepatoprotective activity of picroliv, curcumin and ellagic acid compared to silymarin on paracetamol induced liver toxicity in mice. Fund Clin Pharmacol. 2009;23:735–745. doi: 10.1111/j.1472-8206.2009.00722.x. [DOI] [PubMed] [Google Scholar]
- 21.Girish C, Pradhan SC. Hepatoprotective activities of picroliv, curcumin, and ellagic acid compared to silymarin on carbon-tetrachloride-induced liver toxicity in mice. J Pharmacol Pharmacother. 2012;3:149–155. doi: 10.4103/0976-500X.95515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- 23.Habbu PV, Shastry RA, Mahadevan KM, Joshi H, Das SK. Hepatoprotective and antioxidant effects of Argyreia speciosa in rats. Afr J Tradit Complement Altern Med. 2008;5:158–164. doi: 10.4314/ajtcam.v5i2.31268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holdiness MR. Clinical pharmacokinetics of the antituberculosis drugs. Clin Pharmacokinet. 1984;9:511–544. doi: 10.2165/00003088-198409060-00003. [DOI] [PubMed] [Google Scholar]
- 25.Kalantari H, Valizadeh M. Nifedipine in the treatment of liver toxicity induced by acetaminophen overdose in mice. Acta Med Iran. 2000;38:240–244. [Google Scholar]
- 26.Kaplowitz N. Drug metabolism and hepatotoxicity. In: Kaplowitz N, editor. Liver and biliary diseases. Baltimore: Williams and Wilkins; 1992. pp. 82–97. [Google Scholar]
- 27.Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- 28.Landon EJ, Naukam RJ, Sastry BVR. Effects of calcium channel blocking agents on calcium and centrilobular necrosis in the liver of rats treated with hepatotoxic agents. Biochem Pharmacol. 1986;30:697–705. doi: 10.1016/0006-2952(86)90369-2. [DOI] [PubMed] [Google Scholar]
- 29.Lee WM. Drug-induced acute liver failure in the United States. 2005: results from the U.S. Acute Liver Failure Study Group; FDA-PhRMA-AASLD Hepatotoxicity Steering Committee meeting 28 January 2005; [January 20, 2006]. Available from: http://www.fda.gov/cder/livertox/presentations2005/William Lee.ppt. [Google Scholar]
- 30.Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 1995;333:1118–1127. doi: 10.1056/NEJM199510263331706. [DOI] [PubMed] [Google Scholar]
- 31.Majid S, Khanduja KL, Gandhi RK, Kapur S, Sharma RR. Influence of ellagic acid on antioxidant defense system and lipid peroxidation in mice. Biochem Pharmacol. 1991;42:1441–1445. doi: 10.1016/0006-2952(91)90457-g. [DOI] [PubMed] [Google Scholar]
- 32.Meister A. New aspects of glutathione biochemistry and transport-selective alteration of glutathione metabolism. Nutr Rev. 1984;42:397–410. doi: 10.1111/j.1753-4887.1984.tb02277.x. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell JR, Zimmerman HJ, Ishak KG, Thorgeirsson UP, Timbrell JA, Snodgrass WR, et al. Isoniazid liver injury: clinical spectrum, pathology, and probable pathogenesis. Ann Intern Med. 1976;84:181–92. doi: 10.7326/0003-4819-84-2-181. [DOI] [PubMed] [Google Scholar]
- 34.Mroueh M, Saab Y, Riskallah R. Hepatoprotective activity of Centaurium erythraea on acetaminophen-induced hepatotoxicity in rats. Phytother Res. 2004;18:431–433. doi: 10.1002/ptr.1498. [DOI] [PubMed] [Google Scholar]
- 35.Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med. 2006;354:731–739. doi: 10.1056/NEJMra052270. [DOI] [PubMed] [Google Scholar]
- 36.Nicotera P, Bellomo G, Orrenius S. Calcium-mediated mechanisms in chemically induced cell death. Annu Rev Pharmacol Toxicol. 1992;32:449–470. doi: 10.1146/annurev.pa.32.040192.002313. [DOI] [PubMed] [Google Scholar]
- 37.Noda A, Noda H, Ohno K, Sendo T, Misaka A, Kanazawa Y, et al. Spin trapping of a free radical intermediate formed during microsomal metabolism of hydrazine. Biochem Biophys Res Commun. 1985;133:1086–91. doi: 10.1016/0006-291x(85)91247-1. [DOI] [PubMed] [Google Scholar]
- 38.Ookhtens M, Kaplowitz N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Semin Liver Dis. 1998;18:313–329. doi: 10.1055/s-2007-1007167. [DOI] [PubMed] [Google Scholar]
- 39.Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davron JT, Steven HB, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–54. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 40.Ozer J, Ratner M, Shaw M, Bailey W, Schomaker S. The current state of serum biomarkers of hepatotoxicity. Toxicology. 2008;245:194–205. doi: 10.1016/j.tox.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 41.Pandit A, Sachdeva T, Bafna P. Drug-induced hepatotoxicity: A review. J Appl Pharm Sci. 2012;02(05):233–243. [Google Scholar]
- 42.Pari L, Murugan P. Protective role of tetrahydrocurcumin against erythromycin estolate-induced hepatotoxicity. Pharmacol Res. 2004;49:481–486. doi: 10.1016/j.phrs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Pessayre D, Larrey D, Brentano FC, Benhamon JB. Drug interaction and hepatitis produced by some macrolide antibiotics. Journal Antimicrob Chemother. 1985;16:181–194. doi: 10.1093/jac/16.suppl_a.181. [DOI] [PubMed] [Google Scholar]
- 44.Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179–1197. doi: 10.1002/hep.20703. [DOI] [PubMed] [Google Scholar]
- 45.Pradhan SC, Girish C. Hepatoprotective herbal drug, silymarin from experimental pharmacology to clinical medicine. Indian J Med Res. 2006;124:491–504. [PubMed] [Google Scholar]
- 46.Ramaiah SK. A toxicologist guide to the diagnostic interpretation of hepatic biochemical parameters. Food Chem Toxicol. 2007;45:1551–1557. doi: 10.1016/j.fct.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 47.Saeed SA, Urfy MZS, Ali TM, Khimani FW, Gilani A. Antioxidants: their role in health and disease. Int J Pharmacol. 2005;1:226–233. [Google Scholar]
- 48.Sarich TC, Youssefi M, Zhou T, Adams SP, Wall RA, Wright JM. Role of hydrazine in the mechanism of isoniazid hepatotoxicity in rabbits. Arch Toxicol. 1996;70:835–840. doi: 10.1007/s002040050347. [DOI] [PubMed] [Google Scholar]
- 49.Sarma GR, Immanuel C, Kailasam S, Narayana AS, Venkatesan P. Rifampin-induced release of hydrazine from isoniazid. A possible cause of hepatitis during treatment of tuberculosis with regimens containing isoniazid and rifampin. Am Rev Respir Dis. 1986;133:1072–1075. doi: 10.1164/arrd.1986.133.6.1072. [DOI] [PubMed] [Google Scholar]
- 50.Satoh T. Recent advances in hepatotoxicity studies. Korean J Toxicol. 1991;7:113–28. [Google Scholar]
- 51.Singh A, Bhat TK, Sharma OP. Clinical biochemistry of hepatotoxicity. J Clinic Toxicol. 2011;S4:001. Available from: http://dx.doi.org/10.4172/2161-0495.S4-001. [Google Scholar]
- 52.Sippel H, Stauffert I, Estler CJ. Protective effect of various calcium antagonists against experimentally induced calcium overload in isolated hepatocytes. Biochem Pharmacol. 1993:1937–1944. doi: 10.1016/0006-2952(93)90634-9. [DOI] [PubMed] [Google Scholar]
- 53.Soon YY, Tan BKH. Evaluation of the hypoglycemic and antioxidant activities of Morinda officinalis in streptozocin-induced diabetic rats. Singapore Med J. 2002;43:77–85. [PubMed] [Google Scholar]
- 54.Thapa BR, Walia A. Liver function tests and their interpretation. Indian J Pediatr. 2007;74:663–671. doi: 10.1007/s12098-007-0118-7. [DOI] [PubMed] [Google Scholar]
- 55.Thomas CE, Reed DJ. Current status of calcium in hepatocellular injury. Hepatology. 1989;10:375–384. doi: 10.1002/hep.1840100322. [DOI] [PubMed] [Google Scholar]
- 56.Tikoo K, Tamta A, Ali IY, Gupta J, Gaikwad AB. Tannic acid prevents azidothymidine (AZT) induced hepatotoxicity and genotoxicity along with change in expression of PARG and histone H3 acetylation. Toxicol Lett. 2008;177:90–96. doi: 10.1016/j.toxlet.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 57.Tostmann A, Boeree MJ, Aarnoutse RE, de Lange Wiel CM, van der Ven Andre JAM, Dekhuijzen R. Antituberculosis drug-induced hepatotoxicity: Concise up-to-date review. J Gastroenterol Hepatol. 2008;23:192–202. doi: 10.1111/j.1440-1746.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- 58.Vogel HG, Vogel WH. Potentiation of hexobarbital sleeping time. In: Vogel HG, Vogel WH, editors. Drug discovery and evaluation; Berlin: Springer-Verlag; 1997. pp. 267–268. [Google Scholar]
- 59.Wang N, Li P, Wang Y, Peng W, Wu Z, Tan S, et al. Hepatoprotective effect of Hypericum japonicum extract and its fractions. J Ethnopharmacol. 2008;116:1–6. doi: 10.1016/j.jep.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 60.Westphal JF, Vetter D, Brogard JM. Hepatic side-effects of antibiotics. J Antimicrob Chemother. 1994;33:387–401. doi: 10.1093/jac/33.3.387. [DOI] [PubMed] [Google Scholar]
- 61.Zimmerman HJ, Maddrey WC. Toxic and drug-induced hepatitis. In: Schiff L, Schiff ER, editors. Diseases of the liver. Philadelphia, PA: Lippincott; 1993. pp. 707–783. [Google Scholar]