

Abstract
The remediation of toxic metals from water with high concentrations of salt has been an emerging area for membrane separation. Cost-effective nanomaterials such as iron and iron oxide nanoparticles have been widely used in reductive and oxidative degradation of toxic organics. Similar procedures can be used for redox transformations of metal species (e.g. metal oxyanions to elemental metal), and/or adsorption of species on iron oxide surface. In this study, iron-functionalized membranes were developed for reduction and adsorption of selenium from coal-fired power plant scrubber water. Iron-functionalized membranes have advantages over iron suspension as the membrane prevents particle aggregation and dissolution. Both lab-scale and full-scale membranes were prepared first by coating polyvinylidene fluoride (PVDF) membranes with polyacrylic acid (PAA), followed by ion exchange of ferrous ions and subsequent reduction to zero-valent iron nanoparticles. Water permeability of membrane decreased as the percent PAA functionalization increased, and the highest ion exchange capacity (IEC) was obtained at 20% PAA with highly pH responsive pores. Although high concentrations of sulfate and chloride in scrubber water decreased the reaction rate of selenium reduction, this was shown to be overcome by integration of nanofiltration (NF) and iron-functionalized membranes, and selenium concentration below 10 μg/L was achieved.
Keywords: Surface modification, Polyelectrolyte, Nanoparticles, Nanocomposite membrane
Graphical abstract
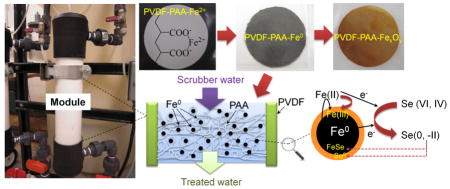
1. Introduction
Various water sources may contain both toxic heavy metals and chlorinated organic compounds (COCs). Those contaminants are either recalcitrant to environmental degradation, or exist at high concentrations of total dissolved solids (TDS, such as, CaCl2, NaCl, etc.), which brings further complications in treatment.
Membrane technologies allow the separation of solutes in water based on size exclusion, charge exclusion and diffusivity/solubility difference. Commercial membranes such as reserve osmosis (RO) and nanofiltration (NF) membranes have been extensively used for water softening and desalination [1]. However, RO and NF membranes are not effective in rejecting organic pollutants such as trichloroethylene (TCE) and carbon tetrachloride, which have high solubility in membrane synthesis materials. Metal ions can be mostly retained by dense membranes, but concentrated metals in the retentate need further treatment. Non-membrane technologies existing for water remediation are either energy or capital intensive, such as physical and biological treatment. In recent years, chemical remediation especially reductive and oxidative treatment with iron and iron oxide nanomaterials has been an emerging area for selective removal of various trace contaminants from natural and waste water matrices.
Iron nanoparticles made by borohydride or polyphenol reduction are most commonly used materials in environmental remediation [2–5]. With a second metal coating such as Pd and Ni, more rapid and complete reduction of organic and inorganic pollutants can be achieved [6–8]. Alternatively, toxic organics can be destructed by oxidation of hydroxyl radicals (OH•) from iron oxide catalyzed hydrogen peroxide decomposition [9–13]. However, the direct injection use of iron is complicated by the aggregation and dissolution of nanoparticles, and the use of engineered nanomaterials has already raised public concerns about their potential toxicity [14–16].
One approach to utilize the high reactivity of iron nanoparticles but overcome the aggregation and dissolution issues is to synthesize nanoparticles through ion exchange or adsorption of precursor ions on porous substrates such as silica [17], activated carbon/carbon nanotubes [18], and polyelectrolyte pore-filled membranes [19–20], followed by reduction. The functional groups on those substrates can eliminate the aggregation during the particle formation and release issues to environment during the reaction, as well as allowing the control of particles size [21–22].
We recently reported the development of full-scale polyacrylic acid (PAA) functionalized polyvinylidene fluoride (PVDF) membrane with iron/iron oxide nanoparticles for combined reductive and oxidative degradation of polychlorinated biphenyls (PCBs) in groundwater [23–24]. The same redox chemistry and membrane approach can be used for selenium removal from coal-fired power plant scrubber water, as iron has been reported to reduce selenate (VI) and selenite (IV) to elemental selenium (Se0) and selenide (-II) [25–27]. Like those on toxic organic degradation, many of these studies were conducted in iron particle suspension without the use of stabilizers.
The scrubber water is generated in the wet scrubber during the flue gas desulfurization (FGD) process. This process is primarily designed for the forced oxidation (using excess air) of sulfur dioxide (SO2) to sulfates, and subsequent precipitation with the addition of limestone (CaCO3) slurry. It has co-benefit removals of volatile metals as well as other acid gases. As the limestone dissolves, it reacts with the sulfates and gypsum is precipitated and removed. The remaining scrubber liquor is saturated with gypsum, and also contains excess halides from the dissolution of other acid gases (HCl). TDS in this water varies from 15,000 to 50,000 mg/L, depending on the type of coal used as well as the upstream flue gas treatment unit operations. In addition to major ions such as calcium, magnesium, chloride, and sulfate, volatile metals such as selenium, arsenic and mercury are also present in trace amount. Selenium is an essential element in human and animal nutrition [26], but may be toxic at higher levels due to its interaction with body tissue. Therefore, the control of selenium release to environment from mining, oil refineries, manufacturing and agriculture drainage has been a critical issue [28–29]. This concern over selenium eco-toxicity has also driven its inclusion in proposed Effluent Limitation Guidelines for coal-fired power plants by the US EPA.
The main aim of this study is to understand the effects of surface functionalization on membrane transport and reactivity in toxic metal removal. The specific aims are: (i) to quantify the relationship of percent PAA functionalization in membrane pores to water permeability, responsive behavior and ion exchange capacity of membranes, (ii) to determine the reactivity of iron-functionalized membranes for selenium removal in different water matrices, and (iii) to evaluate the performance of full-scale functionalized membranes for power plan water applications.
2. Experimental
2.1. Materials
All chemicals used in the lab-scale membrane study were reagent grade without further purification. Sodium borohydride and selenium powder were purchased from Sigma-Aldrich. Sodium hydroxide, sulfuric acid, hydrochloric acid, nitric acid (trace metal grade), iron and selenium reference standard (1000 mg/L), and ferrous chloride tetrahydrate were obtained from Fisher Scientific. Acrylic acid, ammonium persulfate, N,N′-methylenebisacrylamide (NNMA), ethanol, and sodium selenite were purchased from Acros Organics. Sodium selenate was purchased from Alfa Aesar.
Commercial PVDF400HE and full-scale PVDF400HE-PAA (functionalized) microfiltration membranes were made in Nanostone/Sepro Membranes Inc., Oceanside, CA. The membrane thickness is 200±5 μm, including PVDF (70±5 μm) and nonwoven fabric backing (125±5 μm). The average pore size from bubble point tests is 420 nm for non-functionalized membrane and 124 nm for functionalized membrane. The spiral wound module of PVDF400HE-PAA has an effective area of 0.465 m2. The full-scale PV400B-PAA and spongy PVDF-PAA membranes were also studied, and they have different fabric backing and PVDF layer in thickness and pore structure. Commercial DVPP14250 PVDF membranes without fabric backing (hydrophilized, diameter: 142 mm, thickness: 125 μm, pore size: 0.65 μm, porosity: 70 %) were obtained from EMD Millipore. All solutions were prepared with Milli-Q ultrapure deionized (DI) water (18.2 MΩ·cm at 25 °C). Deoxygenated water was obtained by purging nitrogen gas into DI water for 30 min.
2.2. Functionalized membrane synthesis and characterization
PVDF membranes were functionalized via in situ polymerization method reported in [23]. The polymerization solution contains the monomer acrylic acid (AA, 0.9–4.9 M), initiator ammonium persulfate (1 mol% of AA), cross-linker NNMA (1 mol% of AA), and deoxygenated water.
Iron precursors (e.g. Fe2+) were immobilized in PAA functionalized PVDF membranes through two-step ion exchange process, followed by borohydride reduction to form iron nanoparticles. The iron loading in membranes can be increased by repeated ion exchange and reduction. The amount of iron immobilized and ion exchange capacity (IEC) of membrane were determined by concentration change and acid digestion [9]. Through the oxidative treatment of iron with air or dilute hydrogen peroxide, iron oxides (e.g. ferrihydrite and magnetite) were formed as a passive oxide layer above iron core [9, 23]. The whole particle will be converted to iron oxide if enough oxidants are provided.
The surface and cross-sectional morphology of functionalized membranes was obtained in scanning electron microscopy (SEM, Hitachi S4300). Membrane porosity was then estimated by ratio of pore area to total membrane area based on SEM images of membrane top surface. Transmission electron microscopy (TEM, JEOL 2010F) was used to study the base particle properties. The crystal structure of iron/iron oxide was studied by using the selected area electron diffraction (SAED) in TEM. To understand the iron surface transformation and composition change, line scanning and elemental mapping were conducted on single particles using either energy dispersive X-ray spectroscopy (EDS, Oxford INCA) or electron energy loss spectroscopy (EELS, Gatan GIF 2000) in scanning TEM mode. The composition of membrane top layer was studied with X-ray photoelectron spectroscopy (K-Alpha, Thermo-Scientific). Depth profile analysis with ion beam etching was also performed to prove PAA functionalization in membrane pores.
2.3. Water permeability and pH responsive behavior study
Water permeability of PVDF-PAA membranes was evaluated by using a lab-scale stainless steel pressure cell (Sepa ST, GE Osmonics, effective membrane area is 13.2 cm2) in dead-end mode (Fig. 1A) or a full-scale setup in cross-flow mode (Fig. 1B). Water flux was measured at three different pressures with three replicates at each pressure, and the average values were shown here. For pH responsive experiments, DI water at different pH was passed through the membrane until the permeate pH was identical to feed pH. Water permeability was determined at this equilibrium.
Fig. 1.

Schematic diagrams of lab-scale flat-sheet membrane (A) and full-scale functionalized membrane module (B) evaluation systems. 1. feed tank; 2. cooling coil; 3. nitrogen gas tank (to deoxygenate water); 4. pump; 5. globe valve; 6. pressure gauge; 7. spiral wound membrane module; 8. flowmeter.
2.4. Selenium removal by iro ron nanoparticles
Selenium removal was performed with iron nanoparticle suspension, and with iron-functionalized membranes in both batch and convective modes. The effects of TDS on selenium reduction with iron were studied in different water matrices, including DI water, synthetic water containing salts (e.g. CaCl2, NaCl, Na2SO4 and MgSO4), and real scrubber water. The scrubber water contained selenium between 0.5 and 2 mg/L, while selenium concentrations in DI water and synthetic water were made as 2 mg/L. Temperature experiments were conducted in an incubator shaker at 40 °C. In convective mode, feed solution was passed through membranes by applying the pressure in cell with compressed nitrogen.
2.5. Cation/anion analysis
The concentration of total dissolved iron was measured by atomic absorption spectrometer (SpectrAA 200 fast sequential). Cations such as calcium, magnesium and sodium in scrubber water were quantified by inductively coupled plasma optical emission spectrometry (ICP-OES, Varian VISTA-PRO) after acidification with nitric acid (1%). The wavelengths selected for analysis were 318.127 nm (Ca), 285.213 nm (Mg), and 568.821 nm (Na), respectively. Yttrium chloride (1 mg/L) was used as an internal standard. Selenium concentrations above 50 μg/L were analyzed by ICP-OES at the wavelength of 196.026 nm after acidification. Selenium concentrations below 50 μg/L were determined by graphite furnace atomic absorption spectrometer (GFAAS, Varian 880Z) after digesting the samples at 110 °C for 2 h. To account for interferences of dissolved salts in the samples, selenium standards were made with synthetic salt water similar to water matrix in samples. Anions including chloride (0.4 to 200 mg/L), sulfate (0.4 to 200 mg/L), nitrate (0.2 to 100 mg/L as N) and nitrite (0.2 to 100 mg/L as N) were analyzed by DIONEX IC25 ion chromatograph (column: IonPac AS18 4×250 mm) with Na2CO3/NaHCO3 buffer solution as the mobile phase (1 mL/min, 2000 psi). For scrubber water used in combined nanofiltration and functionalized membrane treatment, selenium speciation analysis was performed by ion chromatography inductively coupled plasma collision reaction cell mass spectrometry (IC-ICP-CRC-MS). Total elemental analyses were performed via inductively coupled plasma dynamic reaction cell mass spectrometry (ICP-DRC-MS) in Applied Speciation and Consulting, WA.
2.6. Full-scale functionalized membrane module study
The membranes in the spiral wound module were already functionalized by PAA, and thus can be used for ion exchange directly. To load iron nanoparticles in this module, NaCl solution (50 mM, pH 10) was passed through the module for 30 min to convert the carboxyl groups of PAA to Na+ form, followed by passing FeCl2 solution (3.57 mM, pH 4.9–5.2) through the module. After removing the residual solution from the system, NaBH4 (12 mM) was passed through the module for 15 min. To load more iron, repeated ion exchange and reduction were conducted. To prevent iron corrosion, the membrane module was stored in Fe2+/ascorbic acid solution (3.57 mM, pH 3.5) after reduction.
3. Results and discussion
3.1 Functionalized membrane surface characterization
To prove that PVDF membrane pores were successfully functionalized with PAA, XPS depth profile analysis was conducted from membrane top surface to pore region. The atomic percent (N, O, C and F) of functionalized membrane is shown in Fig. 2A. On the top surface (etch time 0 s), F and O content were 2.5% and 31.9% respectively, compared to 42.5% and 2.7% for the pristine PVDF400HE membrane. As the ion beam etching started, F content increased quickly while O content declined as approaching the pore region. The depth profile results clearly show that there was a thin layer of PAA above PVDF membrane. High resolution C1s spectra (Fig. 2B) further proved that the top surface was covered with PAA as evidenced by C-O (286.7 eV) and C=O (289.0 eV) peaks from carboxyl groups with a tiny C-F2 peak near 290.8 eV. The intensity of C-O and C=O peaks declined quickly from 0 s to 200 s (Fig. 2C) with the rise of F content to 8.75%. The spectra at 300 s (Fig. 2D) and 490 s (Fig. 2E) were identical with the exactly same elemental composition, suggesting the ion beam penetrated the PAA layer and reached the uniformly functionalized pore region.
Fig. 2.

XPS depth profile analysis of functionalized membrane (PVDF400HE-PAA, 12.5% membrane weight gain). (A) Atomic percent profile; High resolution C1s spectra at etch time (B) t=0 s, (C) t=200 s, (D) t=300 s, (E) t=490 s. Ion beam energy: 200 eV (high), etch speed: 10 s/level, 0.07nm/s as Ta2O5. Number 1 and 2 show the regions of membrane top surface and pore, respectively. XPS spectra of PVDF400HE and full-scale functionalized membrane were shown in (F) and (G).
An ideal functionalized membrane will have PAA in pores instead of on the top surface. However, PAA formation near the pore mouth always happened due to the residual polymerization solution from dip coating. This thin layer reduced the membrane permeability significantly. During the manufacturing of full-scale functionalized membranes, the polymerization solution was pulled through PVDF membranes by applying vacuum from the backing side, followed by surface cleaning with air knife. This improvement indeed removed the accumulated polymerization solution above pores effectively. XPS spectrum of full-scale functionalized membrane (Fig. 2G) shows similar C-F2 and C-N peaks as PVDF membrane substrate (Fig. 2F), since little PAA was formed on top of PVDF membrane. C-N peak could be from the cross-linker or additives during PVDF membrane formation such as polyvinylpyrrolidone (PVP). C=O peak was stronger in functionalized membrane due to PAA formation inside.
As expected, the full-scale functionalized membrane had higher F/C ratio from PVDF substrate and lower O/C ratio from PAA coating than lab-scale one (Table 1). The main carbon source of functionalized membrane included PVDF (F/C=1/1), PAA (O/C=2/3), and PVP (N/O/C=1:1:6). The carbon balance of full-scale PVDF400HE-PAA was close to commercial PVDF400HE and DVPP14250, showing the successful scale-up of functionalized membranes without blocking the pores. For iron/iron oxide immobilized membranes, iron can exist in multiple valence states, depending on the oxidation time and water pH. If Fe(OH)x is the overall formula of reacted particle, the value of x could vary from 0 (pure iron) to 3 (fully oxidized iron). With the carbon balance assumption, x fell between 1.5 and 3, suggesting iron was partially oxidized after selenium removal.
Table 1.
XPS surface analysis results of pristine PVDF, PAA functionalized PVDF (PVDF400HE-PAA) and iron/iron oxide immobilized PVDF-PAA membranes (PVDF-PAA-Fe/Fe(OH)x).
| Membranes | Atomic Ratio | Carbon Balance | Value of x in Fe(OH)x | |||
|---|---|---|---|---|---|---|
| F/C | O/C | N/C | Fe/C | |||
| PVDF400HE (Ultura) | 0.79 | 0.05 | 0.02 | N/A | 0.93 | N/A |
| PVDF400HE-PAA (Ultura) | 0.53 | 0.11 | 0.03 | N/A | 0.83 | N/A |
| DVPP 14250 (Millipore) | 0.50 | 0.26 | 0.005 | N/A | 0.88 | N/A |
| PVDF400HE-PAAa | 0.04 | 0.51 | 0.02 | N/A | 0.88 | N/A |
| PVDF400HE-PAA-Fe after Se reduction in DIUF watera | 0.22 | 0.65 | 0.04 | 0.16 | 0.68 (x=3)/1.39 (x=0) | 1.66 |
| PVDF400HE-PAA-Fe after Se reduction in synthetic water with CaCl2 (75 mM)b | 0.19 | 2.66 | 0.05 | 0.89 | 0.37 (x=3)/4.39 (x=0) | 2.53 |
| PVDF400HE-PAA-Fe after Se reduction in synthetic water with MgSO4 (30 mM)b | 0.21 | 2.62 | 0.03 | 1.03 | 0 (x=3)/4.28 (x=0) | 2.12 |
12.5% membrane weight gain;
20.7% membrane weight gain.
x values were obtained by assuming the carbon balance. DVPP 14250 (Millipore) is hydrophilized PVDF membrane with cross-linked polyacrylate coating. The coating was considered as PAA here for carbon balance calculation.
3.2. Functionalized membrane properties
3.2.1. Membrane permeability with weight gain
The weight gain of functionalized membrane is defined as the ratio of dry weight difference through in-situ polymerization to the weight of pristine PVDF membrane.
| (1) |
where W0 and W′ are the weight of membrane before and after functionalization, respectively. It should be noted that the weight of backing was also included in W0 since the control experiment shows that PAA was also formed in the nonwoven backing due to its porous structure. The weight gain of PVDF400HE membrane increased linearly with the concentrations of monomer in polymerization solution (Fig. 3A). Meanwhile water permeability of membrane dropped rapidly. From 2.2% to 36.3% PAA, water permeability declined from 1220 L/(m2·h·bar) to 1.9 L/(m2·h·bar) at pH 3. It was mainly due to the decrease of membrane pore volume or effective pore size.
Fig. 3.
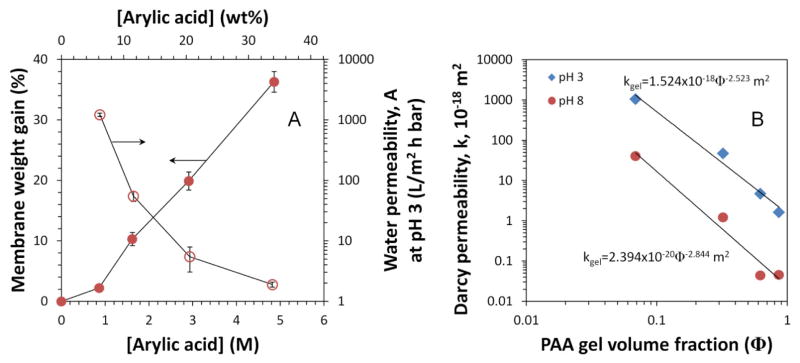
(A) Weight gain and water permeability of functionalized membranes (PVDF400HE-PAA) with concentration of acrylic acid in polymerization; (B) Darcy permeability of pore-filled membrane as a function of polymer gel volume fraction at pH 3 and 8. Water permeability of PVDF400HE is 7512±125 L/(m2·h·bar).
The hydrodynamic permeability of functionalized membrane (km, m2) is dependent on the physical properties of cross-linked PAA gel formed in membrane pores. With km obtained from experiments, Darcy permeability of gel (kgel, m2) can be calculated as,
| (2) |
where εs and τs are the porosity and tortuosity of substrate PVDF membrane. The surface porosity of PVDF400HE membrane is 50±5%, which was confirmed by SEM image of membrane top surface. The cross section image showed the bulk porosity is quite similar to surface porosity. τs can be obtained from Kozeny constant,
| (3) |
where KS is assumed as 5 for PVDF400HE membrane, and K*=2 with the assumption of parallel capillary-type pores of circular cross-section [30–31]. KS has been reported as a function of porosity based on the experimental data [32]. For a substrate membrane with the porosity between 0.5 and 0.8, KS varies slightly between 4.0 and 6.5. For spongy membranes, the porosity is usually greater than 0.8, and KS increases exponentially as the porosity increases. The flow mechanism is also transformed from capillary flow to free flow around cylinders. km can be calculated from the following equations,
| (4) |
| (5) |
where Jw is the water flux through the functionalized membrane, Q is the water volume flow rate, Am is the membrane area (13.2 cm2), d is the thickness of PVDF layer (70±5 μm) measured with cross-sectional images obtained in SEM, η is the water viscosity (0.89 mPa·s at 25 °C), and ΔP is the transmembrane pressure.
The PAA gel volume fraction of functionalized membranes (Φ) is defined as the ratio of gel volume (Vgel) to total membrane pore volume (Vm). It can be estimated by
| (6) |
where ρgel is the density of PAA hydrogel in membrane pores. The volume and weight of PAA gel obtained in aqueous polymerization were measured, and the density was calculated to be 1.21±0.10 g/cm3, which was close to reported value of uncross-linked PAA (1.37 g/cm3) [33]. The gel volume fraction of membranes is proportional to the weight gain from polymerization, increasing from 0.069 (2.2%) to 0.862 (36.3%). The gel Darcy permeability obtained at pH 3 and 8 (Fig. 3B) can be fit in a power law similar to previous studies [30–31], with a correlation coefficient R2=0.981 (kgel=1.524×10−18 φ −2.523m2) at pH 3 and R2=0.965 (kgel = 2.394×10−20φ−2.844m2) at pH 8. The exponent on Φ represents the rate of permeability change with the gel volume fraction. When pH increased from 3 to 8 with the same gel, the exponent varied within 12.7%, proving the consistency of PAA gel and no significant change of substrate PVDF membrane structure with water pH.
Darcy permeability of PAA gel synthesized in aqueous phase decreased faster with the increase of gel volume than previous study [34], which used the organic phase polymerization. In the presence of organic solvent, PAA coils might be formed more uniformly in membrane pores with higher weight gain and less space between polymer chains for gel to swell. The aqueous polymerization used here might cause greater gel heterogeneity, resulting in a larger exponent of Φ. Generally, free radical polymerization of acrylic acid in aqueous phase has a higher propagation rate than that in organic phase. Water can enhance the reactivity of monomer and free radicals by solvation [35–36]. Looser polymer coils are formed in water, and these polymer coils facilitate greater water and monomer transport, resulting in higher concentration of monomer near their active growing center. Additionally, hydrogen bonding might also enhance the polymerization by creating complexes between monomers, oligomers, and water [37].
While the gel formed in aqueous phase may not be as uniform as that made in organic phase, one advantage of aqueous phase polymerization is that membranes may be more precisely tuned for desired water transport based on more precisely controlled PAA loading. The use of water as the solvent will also reduce the emission of organic vapor during the thermal treatment. PVDF400HE membrane used here also has a thinner PVDF layer (70±5 μm) with highly porous nonwoven backing, and this composite structure enhances the water permeability of membrane. However, after normalized with membrane thickness, gel permeability at Φ=0.1 is still enhanced by 29 times at pH 8, compared to gel synthesized at cross-linking degree of 4 mol% in 1-propanol (50 wt%) [34]. It should be noted that higher cross-linking degree should give higher water permeability due to the formation of gel with greater heterogeneity.
Darcy permeability of gel at pH 3 is about 63 times higher than that at pH 8 when Φ=1, while this ratio decreased to 30 times when Φ=0.1. This significant difference indicates that the gel permeability was also related to the swelling of PAA with environmental pH, which can be the control factor when Φ>0.1.
3.2.2. pH responsive behavior and its applications
PAA is well-known for its pH responsive (swelling/deswelling) behavior due to the reversible protonation and deprotonation of carboxyl groups with water pH variation. The acid dissociation constant (pKa) of PAA is usually around 4.2–4.9. At very low pH, carboxyl groups are in compact state without ionization. With pH increasing, carboxyl groups start the deprotonation accompanied by ion exchange with cations in water. The mesh size of PAA hydrogel also increased with increasing pH, resulting in higher swelling ratio. Our gel swelling study shows that PAA synthesized through thermal polymerization had a weight gain around 194% at pH 1.9 and 1676% at pH 9.3 [38]. With PAA incorporated in membrane pores as a “smart” valve, one can manipulate the water permeability of membranes by adjusting the water pH [10]. Although we have reported the pH responsive behavior of full-scale functionalized membranes, the optimization of monomer solution has not been studied.
The fraction of ionized carboxyl groups (degree of dissociation) with water pH can be estimated by,
| (7) |
where [COO−] and [COOH] are concentrations of ionized and unionized carboxyl groups, respectively. They can be calculated from Henderson-Hasselbach equation,
| (8) |
At pH 3, only 3.1% of carboxyl groups were ionized (assume pKa=4.5), while they are fully deprotonated (99.97%) at pH 8. Water flux ratio (Jw, pH=3/Jw, pH=8) increased as the amount of carboxyl groups increased. However, as the PAA loading in membrane pores approaches the maximum, the water permeability at all pH values is minimized. For example, the water flux ratio reached as high as 92±21 with 0.49 mmol carboxyl groups (19.9% weight gain) incorporated in functionalized membranes. However, even at PH=3 (most compact state), water permeability was only 1.9 L/(m2·h·bar) with 0.89 mmol carboxyl groups (36.3% weight gain). This high PAA loading resulted in some swollen gel being excluded from pores, and anchored at the pore mouth with low stability. Some entangled gel was also observed on top of membranes. While the gel homogeneity generally increases with increasing pH and consequent gel swelling, the reduced space between polymer segments and pore volume limited the gel swelling at higher weight gain (36.3%).
Similar pH valve effect of PAA functionalized membranes was also reported by Hu et.al, with water flux ratio up to 112 [34]. At the same gel volume fraction (Φ<0.1), higher cross-linking degree created more heterogeneity and higher elastic modulus in polymer segments, resulting in higher water permeability and swelling ratio. In our study, the gel volume fraction was mainly higher than 0.1 (0.069<Φ<0.862), indicating the formation of less heterogeneous polymer network via overlapping and interpenetration.
The flux modulation of three full-scale functionalized membranes with pH proves the successful scale-up of PAA functionalized PVDF membrane (Fig. 4 inset). As expected, the spongy PVDF membrane allowed more PAA incorporated inside with the same functionalization procedure due to its larger pore volume.
Fig. 4.

Water flux ratio (at pH=3 and pH=8) and ion exchange capacity (IEC) with total amount of carboxyl groups from functionalization. Three full-scale membranes (Spongy PVDF-PAA, PVDF400HE-PAA, and PV400B-PAA) are shown in the inset, where the amount of carboxyl groups is calculated from ion exchange studies.
3.2.3. Ion exchange capacity of functionalized membranes
During the ion exchange process, the deporotonated carboxyl groups in PAA chains chelate with Fe2+ (molar ratio 2:1). Fe2+ can also be absorbed by carboxyl groups (molar ratio 1:1) through electrostatic binding. Ion exchange capacity of functionalized membranes varied from 0.60 to 7.57 meq/g PVDF, depending on the amount of carboxyl groups in membranes (Fig. 4). The swelling of polymer led to the overlapping and interpenetration of polymer chains, and the diffusivity of ions in swelling gel was much lower than that in bulk. It explains why carboxyl groups were not fully utilized during ion exchange. The obtained IEC values were about 30% less than calculated values based on the membrane weight gain.
Higher amount of carboxyl groups in pores did not necessarily result in higher IEC. As the amount of PAA increased, some swollen gel came out of pores and lost the stability, as evidenced by ion exchange results of membrane with a high loading of PAA (0.89 mmol) (Fig. 4). For full-scale functionalized membrane synthesis, the weight gain between 10% and 20% provided the advantages of a high swelling ratio and ion exchange capacity.
3.2.4. Nanoparticle size control
PAA acted as a stabilizer of iron precursor in nanoparticle synthesis, and also controlled the particle size. With PAA incorporated in membrane pores, both the rates of nucleation and particle growth were retarded. The aggregate formation during the particle growth can be effectively reduced as evidenced by SEM images (Fig. S1). A higher molar ratio of carboxyl groups to ferrous ions resulted in smaller particle size and higher specific surface area. Although the nanoparticle size control by lab-scale functionalized membranes has been reported in [22], the same property has not been studied for full-scale functionalized membranes. Additionally, the full-scale membranes were synthesized in aqueous phase without using any organic solvent. This property suggests the potential of using the functionalized membrane as a stabilizer to manipulate the nanoparticles reactivity via size control [21–22].
3.2.5. Membrane stability
PVDF is well-known for its robust mechanical strength. The mechanical properties of PVDF membranes prepared via the phase inversion method have also been reported [39]. The surface modification with cross-linked polyelectrolytes and inorganic particles appears primarily inside pores without attacking PVDF chains. Therefore, the overall mechanical performance (such as tensile strength and elongation) of functionalized membranes should be similar to PVDF membranes. In addition, the fabric backing will also enhance the membrane stability.
Our previous study also shows that iron/iron oxide nanoparticles immobilized in the cross-linked polymer matrix have improved stability, while those unsupported in aqueous phase or loosely supported on top of membrane tend to aggregate or become dislodged [9, 23]. The morphology of particles may change during iron oxidation.
3.3. Selenium oxyanion reactions
With iron as an electron donor, selenate may be reduced to selenite and elemental selenium (Se0). In reducing environment, iron selenide formation has also been reported [25–26, 40–41]. The possible reactions of selenium oxyanions with iron as well as their standard oxidation-reduction potentials (E0) are shown below.
| (9) |
| (10) |
| (11) |
Ferrous ions released from iron surfaces can precipitate as hydroxides containing Fe(II) at near neutral pH. This increasing oxide layer creates more resistance for reactants to transfer from bulk solution to iron surfaces. Eventually, iron will be consumed completely. Low pH will benefit the reduction by minimizing the formation of iron hydroxide [42].
3.3.1. Selenium removal in solution and effects of competing anions
Because iron is in excess, selenium oxyanion reduction is assumed to follow a pseudo-first-order law (d[Se]/dt = −kobs,Se [Se]), where [Se] is the selenium concentration, and kobs,Se (observed reaction rate) is related to the particle loading (ρm) and specific surface area (αS). It is used here to compare the reactivity of same iron nanoparticles in different water matrix. In aqueous phase nanoparticle synthesis, the average particle size obtained is 20.0±0.2 nm based on the BET specific surface area result (37.8 m2/g). The same feed solution (2 mg/L as Se) was used in DI and synthetic water experiments. The concentration of selenate in DI water (pH 4.5) decreased to less than 5 μg/L within 10 minutes in the presence of iron (0.5 g/L). Iron nanoparticles maintained the reactivity with five cycle treatment, and kobs,Se in DI water (32.8 h−1) is 160 times higher than that obtained in real scrubber water (0.204 h−1).
Based on the speciation results, selenium in scrubber water existed in the form of selenate (49.9%), selenite (48.3%) and methylseleninic acid (MeSe, 1.8%). The scrubber water mainly contains cations such as Ca2+ (90±10 mM), Mg2+ (30±3 mM), and Na+ (2.8±1.5 mM), and anions such as Cl− (200±12 mM) and SO42− (30±5 mM). Sulfate has very similar chemical properties as selenate, and could lower the rate of selenate removal via competing adsorption on iron surfaces [43]. It should be noted that the sulfate concentration in scrubber water is much higher than selenium (molar ratio=1000–1500:1), and thus its interference has to be considered. Point of zero charge (PZC) of iron/iron oxide nanoparticles is between 7.5 and 7.8. At low pH (4.5), the particle surface is positively charged, and thus the adsorption capacity of anions is enhanced.
The experimental result in Na2SO4 solution (Fig. 5) further proved that sulfate indeed inhabited the removal of selenate [8], and this effect was enhanced at a higher concentration (Table 2). A similar trend was observed when studying the effect of chloride with NaCl solution. Compared to sulfate, chloride had limited inhibition on the removal of selenate below 75 mM (kobs,Se=13.4 h−1 at 75 mM). The rate constant decreased by 4 times (kobs=3.20 h−1) after increasing its concentration to 150 mM. Similar results were reported by Mondal et al [8], showing the selenate removal rate was greatly reduced by competing anions such as chloride at 10 g/L and sulfate at 2.5 g/L.
Fig. 5.

Selenium removal with iron nanoparticle suspension in batch mode. [Fe]0=0.50±0.02 g/L; [Se]0=1.3±0.1 mg/L in scrubber water, and 2.0±0.2 mg/L in synthetic NaCl and Na2SO4 solutions. pH of feed solution was adjusted to 4.5. Dash lines show the fit of pseudo-first-order law.
Table 2.
Observed rate constant (kobs,Se, h−1) of selenium oxyanion removal with iron nanoparticle suspension in different water matrix. [Fe]0=0.5±0.02 g/L. Specific surface area: 37.8 m2/g.
| Water matrix | kobs,Se (h−1) |
|---|---|
| DI water | 32.8 |
| Scrubber Water | 0.204 |
| NaCl (75 mM/150 mM) | 13.4/3.20 |
| Na2SO4 (30 mM/75 mM) | 1.14/0.626 |
In addition to competitive adsorption, sulfate and chloride lowered kobs due to enhanced corrosion of iron, by forming an oxide layer and increasing the solution pH. After deoxygenated water washing, the reactivity of iron from selenate removal (100 mM Cl−) was partially rejuvenated, with kobs,Se=4.09 h−1 in NaCl solution and kobs,Se=8.69 h−1 in DI water after washing (0.50±0.02 g/L iron). The increase of treatment time caused the rapid decline of reactivity in DI water (kobs,Se=0.618 h−1 in the second cycle and kobs,Se=0.473 h−1 in the third cycle). Those results provided the evidence that both anion adsorption and corrosion contributed to the slow removal of selenium oxyanions in scrubber water. Additionally, other ions existed in water such as NO3− (1.5±0.6 mM) and Ni2+ (6.7 μM) could also lower the reactivity of iron through reduction and/or adsorption [29, 43–44].
3.3.2. Selenium removal in functionalized membranes
The high specific surface area of iron/iron oxide nanoparticles (20–300 m2/g depending on the particle size) enhanced the removal of toxic metals and organics through an adsorption-reduction-desorption process [6], but the particle aggregation due to ferromagnetic nature of iron, and the toxicity from iron leaching also need be evaluated before applying nanotechnology to real water treatment [14–16]. PAA functionalized PVDF membrane provides a tunable platform for metal capture and nanoparticle in-situ synthesis, resulting in non-aggregated particles with uniform size [22]. By applying the convective flow operation, the conversion of the model compound is directly related to its residence time in membranes bulk phase. Water is forced to flow through membranes under pressure, and thus the diffusion resistance from bulk solution to membrane pore area can be neglected. Additionally, there is less separation need when using membrane supported nanoparticles.
The residence time (τ) is defined as the ratio of membrane pore volume to water volume flow rate,
| (12) |
where mFe and ρFe are the mass and density of iron nanoparticles in functionalized membranes. With a residence time of 10 s (Fig. 6), 91.4% of selenate was removed in DI water at 25.2±1.9 L/(m2·h·bar), while the conversion was 21.8% in scrubber water with a residence time of 8.4 s at 15.3±0.4 L/(m2·h·bar) (Fig. S2). As discussed, the interference of salts and dissolved metal ions in scrubber water lowered the reactivity of iron. To recover the reactivity, either low TDS water (to reduce the competing adsorption) or more reactive iron (to compensate the reactivity loss with nitrate and dissolved metal ions) is required.
Fig. 6.

Selenium removal with iron functionalized membranes in different water matrix under convective flow condition. [Se]0=1.3±0.1 mg/L in scrubber water, [Fe]0=8.4 mg (20.7 % weight gain), [TDS]0=12 g/L ([SO42−]0=11.7 mM or 1120 mg/L); [Se]0=52.3 μg/L in NF permeate-1 (positively charged NF membrane), [Fe]0=9.9 mg (23 % weight gain), [TDS]0=2.4 g/L ([SO42−]0=292 μM or 28 mg/L); [Se]0=11.0 μg/L in NF permeate-2 (negatively charged NF membrane), [Fe]0=9.2 mg (23 % weight gain), [TDS]0=0.92 g/L ([SO42−]0=0.094 μM or 9 mg/L); [Se]0=2.0±0.2 mg/L in DI water, [Fe]0=4.0 mg (12.5 % weight gain). pH of feed solution: 4.5–5.5.
3.3.3. Integration of nanofiltration (NF) and functionalized membranes
By applying NF membrane, divalent cations/anions (including selenium) were retained by charge exclusion, and counter ions associated with those ions were also retained due to charge balance (within 5% difference). The rejection of total TDS and selenium were 94.0±1.5% and 97.9±0.9%. It should be noted that chloride rejections in NF membrane will be completely different, depending on which cation (Ca2+ or Na+) is acting as the counter ion. Divalent Ca2+ has a higher electrical potential than Na+, resulting in higher rejection of CaCl2. The scrubber water mainly contains Ca2+ (90±10 mM) and Mg2+ (30±3 mM) as cations, with a small amount of Na+ (2.8±1.5 mM). Therefore, NF membrane is a better choice here to remove TDS, considering its higher water permeability and lower energy consumption than RO membrane.
Two types of nanofiltration membranes with different surface charge (NF3A and PNF2A) were used, and the rejections of sulfate and chloride were 99.47±0.41% and 93.03±1.90%, respectively. The NF permeate had much less sulfate (0.07±0.05 mM) and chloride (13.1±3.6 mM), which didn’t affect the removal of residual selenium oxyanions (0.16±0.07 μM). The molar ratio of sulfate to selenium in the NF permeate was as low as 265:1. With the addition of NF membrane prior to treatment with iron-functionalized membrane, the conversion of selenium via iron reduction was 77.5% with a residence time of 8 s (Fig. 6). The major cation (e.g. calcium and magnesium) and anion (e.g. chloride and sulfate) concentrations hardly changed when passing through functionalized membranes as they don’t generally react with iron. With this combined technology, selenium concentration in the final permeate was below 10 μg/L, which met U.S. EPA’s drinking water standard.
It should be noted that the selenium concentration in scrubber water may vary from 0.6 mg/L to 2 mg/L, depending on the coal composition and flue gas treatment operations. Additional iron particle loading or increased residence time (multiple membranes in series) may afford lower final permeate concentrations. The selenium conversion in the functionalized membranes is expected to increase and achieve the effluent limitation. This platform also benefited the physical adsorption and chemical reduction of some other toxic metals such as arsenic, nickel and mercury as well as nitrate. Arsenic concentration as low as 1 μg/L were achieved (Table 3).
Table 3.
Selenium and other toxic compound removal from coal-fired power plant scrubber water via combined treatment with nanofiltraion and iron functionalized membranes.
| Se (μg/L) | As (μg/L) | Ni (μg/L) | Hg (μg/L) | NO3− (mg/L) | |
|---|---|---|---|---|---|
| Scrubber water-Feed | 634 | 4.7 | 391 | 0.17 | 51.8 |
| NF membrane treatment-Permeate | 9–17 | 0.7–0.9 | 22.2–41.7 | 0.05–0.07 | 3.1–16.4 |
| NF membrane followed by iron functionalized membrane treatment-Permeate | 1.9–9.4 | 0.5 | 7.8–16.3 | N/A | 2.7–4.3 |
3.3.4. Effect of temperature
One possible solution to enhancing the reactivity of iron in scrubber water is to conduct the selenium removal at elevated temperatures. Mondal et al. reported that selenium removal increased by 22% when temperature rose from 25 °C to 65 °C [8]. The scrubber water from FGD process contains some waste heat, which could be a benefit for selenium removal. Our study shows the first-order rate constant of iron-functionalized membranes increased by 3.8 times in DI water (Fig. 7A), and 2.7 times in scrubber water at 40 °C (Fig. 7B). The activation energy of iron in functionalized membranes was calculated to be 56.95 kJ/mol in DI water and 42.95 kJ/mol in scrubber water by Arrhenius equation and rate constants obtained (Table S1). The low activation energy in scrubber water can be due to the acceleration of iron corrosion by salts. The frequency factor in DI water (1.052×1010 h−1) was three magnitudes higher than that in scrubber water (0.935×107 h−1). It again shows the competing adsorption of anions significantly inhabited the selenium removal.
Fig. 7.

Temperature effect on selenium removal from DI water (A) and scrubber water (B) with iron nanoparticle suspension and iron functionalized membranes in batch mode. Dash lines show the fit of pseudo-first-order law. [Fe]0=0.50±0.02 g/L (nanoparticles) and 0.47±0.04 g/L (membranes); [Se]0=1.3±0.1 mg/L in scrubber water, and 2.0±0.2 mg/L in DI water. pH of feed solution was adjusted to 4.5. T=22 °C or 40 °C.
3.3.5. Effect of membrane surface charge
Iron-functionalized membranes show slightly higher reactivity in scrubber water but much lower reactivity in DI water than nanoparticles in suspension (Fig. 7). Due to the incorporation of PAA, the functionalized membrane pores were negatively charged at pH 4.5 or above in DI water. Meanwhile, selenium oxyanions mainly existed as SeO42− (pKa2=1.9) and SeO42−/HSeO3− (pKa1=2.9 and pKa2=7.3) [45]. The repulsive electrostatic force between ionized carboxyl groups and selenium oxyanions might retard the selenium transport through membrane pores, especially inside the polymer domain where iron was immobilized, and thus delay the selenium removal. This assumption was proved by experimental results obtained in scrubber water, where membrane supported iron had better reactivity than iron suspension. Divalent cations (e.g. Ca2+/Mg2+) present in scrubber water immediately formed chelates with carboxyl groups (in Na+ form), neutralizing the negative charge in membrane pores. The cations adsorbed by PAA could also facilitate the diffusion of selenium oxyanions through the membrane. Additionally, the aggregation of unsupported nanoparticles (see TEM results in 3.6) could also increase the particle size and lower the specific surface area, resulting in lower reaction rate in suspension.
3.3.6. Iron recapture by functionalized membranes
Iron oxidation in selenium removal and water corrosion can release ferrous ions into water, which are further oxidized to ferric ions in the presence of oxygen. Both ferrous and ferric ions precipitate as hydroxides at high pH. In DI water at 40 °C, iron loss with functionalized membranes (0.20±0.17%) was significantly lower than iron suspension result (5.4±0.6%) (Fig. 8). The carboxyl groups of PAA formed chelates with Na+ first, and then with Fe2+ during the ion exchange. After NaBH4 reduction, the carboxyl groups were back to Na+ form, which could capture Fe2+ again. By taking advantage of this property, iron loading continued to grow with repeated ion exchange and reduction. However, with more iron immobilized in polymer network, the entry of Fe2+ into the carboxyl groups became more difficult, and IEC of membrane decreased by 43±5% after eight cycles. While in scrubber water, iron-functionalized membrane lost the recapture ability since the carboxyl groups of PAA also had strong binding affinity for Ca2+ and Mg2+. With high TDS and an elevated temperature (40 °C), the dissolved iron percent also increased to 10.2±0.5% for iron-functionalized membrane, and 8.7±1.9% for iron suspension.
Fig. 8.

Dissolved iron percent in solutions during selenium removal with iron nanoparticle (NP) suspension and iron functionalized membranes in batch mode at 40 °C. [Fe]0=0.47±0.04 g/L (membranes); [Se]0=1.3±0.1 mg/L in scrubber water, and 2.0±0.2 mg/L in DIUF water. pH of feed solution was adjusted to 4.5.
3.4. Iron nanoparticle synthesis and selenium removal in spiral wound module
The water flux of spiral wound functionalized membrane module (0.465 m2) varied between pH 4 and 9 with a transition pH at around 6 (Fig. 9A), suggesting the successful pore functionalization. With repeated ion exchange and NaBH4 reduction, 680±9 mg Fe was immobilized in this module. Synthetic solution with both selenate and selenite (same fraction as in scrubber water) was passed through iron-functionalized membrane module convectively. With water flux of 110.4 L/(m2·h) and a residence time of 1.2 s, the conversion of selenium was 0.47 in the permeate after running it for 1 min. It decreased rapidly from 1 min to 5 min, and maintained around 0.12 after 15 min (Fig. 9B). The total volume of permeate was 17 L, and 27.6% of selenium was removed from synthetic solution, giving a minimum reduction capacity of 14.6±1.1 mg Se/g Fe. Feed concentration was measured during test, and the difference was within 3.3%. It should be noted that iron loading in the module (0.15 mg/cm2) was half of that in lab-scale DI water study (0.30 mg/cm2), resulting in a lower conversion. Iron corrosion was also faster in the module test since the selenium solution was exposed to air during the circulation.
Fig. 9.
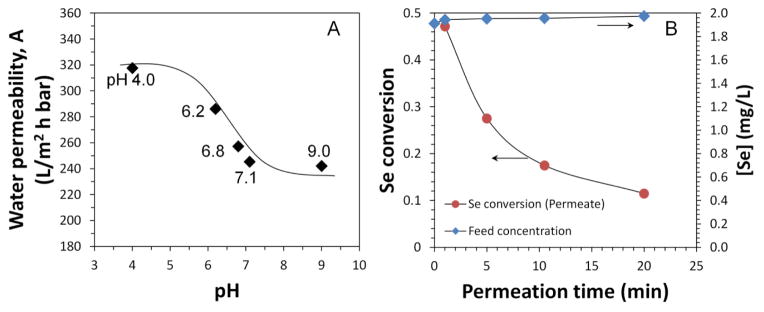
Full-scale PVDF400HE-PAA membrane module water flux and reactivity in selenium oxyanion removal. (A) Water permeability with pH at 25 °C; (B) Selenium removal results by passing synthetic selenium solution through iron immobilized module convectively, [Fe]0=0.68 g, [Se(VI)]0=1.00±0.05 mg/L, [Se(IV)]0=1.00±0.05 mg/L (2.0±0.1 mg/L in total) in synthetic feed solution (pH=6.2), V=18 L, Jw=110.4 L/(m2·h) and τ=1.2 s. The retentate flow rate was 22.5 L/h, and the total volume of permeate was 17 L. Effective membrane area: 0.465 m2. In pH responsive study, water flux was measured after being stabilized for 15 min. Water flux was already normalized with temperature due to viscosity change.
3.5. Membrane regeneration
The maximum reduction capacity obtained in membrane convective experiments was 32.1±2.0 mg Se/g Fe, and a significant amount of iron was not utilized during the selenium removal. As the thickness of iron oxide shell increases, neither electrons nor selenium oxyanions may transfer through this layer without iron surface regeneration. By acid treatment to dissolve the iron oxide, the membrane reactivity should be at least partially restored. For long-term treatment, the used membrane modules can be replaced by new modules considering the toxicity of selenium deposited on iron/iron oxide surface. Selenium can also be recycled via the post treatment such as digestion and purification for further use in electronics industry.
3.6. Reduction and adsorption mechanism
The reduction of selenate by iron forms the elemental selenium (Se0) and iron selenide deposited on iron surface, as evidenced by X-ray absorption near edge structure (XANES) and extended x-ray absorption fine structure (EXAFS) spectroscopy [41]. The release of ferrous ions due to electron transfer in selenium reduction resulted in the formation of an iron oxide layer above iron, which may contain ferrous/ferric hydroxides, lepidocrocite, maghemite and magnetite, depending on oxidation conditions (aerobic/anaerobic) and pH of solution [25–26, 46]. The Gibbs free energy change (ΔG<0) and experimental results both shows that iron oxides containing Fe(II) such as green rust, ferrous hydroxide, mackinawite, siderite and magnetite could also achieve the transition from selenate/selenite to Se0 [40–41, 46–48]. Additionally, iron oxides such as ferrihydrite have been considered as good adsorbents for selenium removal, forming inner- and/or outer-sphere surface complexes [49–50]. Therefore, selenium removal by iron nanoparticles involves both reduction and adsorption mechanism.
XPS spectra of iron after selenium removal shows both selenate (166.26 eV, 53.7 %) and Se0 (161.07 eV, 46.3 %) existed on iron surface (Fig. 10). The oxidation of Se0 deposited on iron surface was much slower compared to that of iron [51]. Therefore, selenate was mainly from iron oxide adsorption. Three main feature peaks including 2p3/2 (710.6 eV), satellite 2p3/2 (719.3 eV), and 2p1/2 (724.0 eV) are shown in Fe 2p spectra (Fig. 10), suggesting even the freshly made iron had an iron oxide layer [23, 52]. Iron nanoparticles were only partially oxidized after selenium removal as evidenced by Fe0 peak at 706.6 eV.
Fig. 10.

XPS spectra of iron nanoparticles after selenate removal. The left graph shows Se 3p spectra of (A) elemental selenium (Se0), (B) sodium selenite, (C) sodium selenate, and (D) iron nanoparticles (0.5 g/L) after mixing with sodium selenate ([Se]0=50 mg/L) for 1 h in DI water. The right graph shows Fe 2p spectra of (E) iron oxide (Fe2O3), (F) same as (D), and (G) freshly made iron (Fe0). Se 3p spectra was used here due to an overlap between Se 3d and Fe 3p.
TEM image shows the morphology of iron/iron oxide nanoparticles after reduction of selenate (Fig. 11A). SAED patterns were similar to that of magnetite, suggesting that iron oxide layer contained mostly magnetite (Fig. 11B). Similar results were reported by Olegario et al. from XRD studies of nano Fe0 oxidation during reaction with selenate [25]. High resolution image provides more details about the core-shell structure of partially oxidized iron, which contains an iron core (diameter: 15±2 nm) and iron oxide shell (thickness: 10±2 nm) (Fig. 11C). The oxide layer was made of polycrystalline magnetite with size around 5 nm. Those iron oxides had high specific surface area, which enhanced the selenium adsorption. EDS line scan of particles in STEM mode also shows that outside particles had higher O/Fe ratio than those in core, and they were stacked on top of each other, forming a highly porous oxide layer (Fig. 11D, E). The intensity of Se peak scan was associated with that of iron peak, since the main source of selenium on iron surface was Se0 from iron reduction. Even though iron was oxidized, the ratio of Fe/Se should have no significant change if there was no selenium dislodging from iron oxide surfaces.
Fig. 11.

TEM images of iron nanoparticles after selenate removal in DI water (five cycle reaction). (A) Bright field image; (B) Selected area electron diffraction (SAED); (C) High-resolution TEM of iron/iron oxide core-shell structure; (D) Dark field image in STEM mode; (E) STEM-EDS line scan spectrum (step size: 1.5 nm, pixel time: 30s). For each cycle, the reaction time was 10 min. [Fe]0=0.50±0.02 g/L, [Se]0=2.0±0.2 mg/L (made by Na2SeO4), and pH was adjusted to 4.5. The dash lines separate the core and shell of particles. Blue/red/green lines indicate Fe/Se/O, respectively.
To eliminate the effect of dissolved oxygen on reduction, selenium removal was conducted in a mixture of ethanol and deoxygenated water (9:1, v/v) with freshly made iron nanoparticles (Fig. 12A) at high selenate concentration. After treatment, some needle-like materials with an amorphous structure was formed on iron surfaces (Fig. 12B). Some particles were even embedded in a cotton-like structure (Fig. 12C), which could be a mixture of iron corrosion products such as lepidocrocite and ferrous/ferric hydroxides [25–26]. EDS elemental analysis on those reacted particles shows the selenium content was 0.8 at% in Fig. 12B and 8.6 at% in Fig. 12C, with most selenium existing in iron oxides. The existence of iron selenide during reduction confirmed the deposition of Se0 on iron surfaces [25, 40–42]. Therefore, the diffusion of selenate through porous iron oxides and the electron transfer on iron surface could take place simultaneously. Ferrous ions are produced via reactions in Eq. (9)-(11) as well as the hydrogen generation with water [52]. Iron oxide precipitates containing Fe(II) can reduce selenate/selenite to Se0, which stays in oxide layer [40]. The dark field image clearly shows how iron oxide grew from iron surfaces. EELS mapping of a selected area in iron oxide layer gave the distribution of selenium, oxygen, and iron (Fig. 12D). In some area, Se existed with Fe only (marked with arrows). This was consistent with XPS results, proving the simultaneous removal of selenium by reduction and adsorption.
Fig. 12.

Iron oxide formation on iron nanoparticle surface during selenium removal (anaerobic). (A) Freshly made iron; (B) (C) iron/iron oxide after reaction; (D) STEM-EELS mapping of iron oxide. Red/blue/brown colors indicate Se/O/Fe, respectively. Sample preparation: fresh iron nanoparticles ([Fe]0=0.50±0.02 g/L) were mixed with Na2SeO4 solution for 24 h ([Se]0=100 mg/L, [Ethanol]:[Deoxygenated water]=9:1, v/v). After that, iron was washed with deoxygenated water and pure ethanol, and sonicated for 30 s. TEM grid (lacey carbon film on 300 mesh copper grids) was soaked in diluted solution for seconds and dried in N2 atmosphere before mounted in TEM sample holder.
4. Conclusions
PAA functionalized PVDF membranes were successfully developed for iron/iron oxide nanoparticle in-situ synthesis. The effects of membrane pore functionalization were studied on PAA gel permeability, pH responsive behavior, and ion exchange capacity. Membranes with weight gain around 20% show highly responsive pores with pH. PAA network also provided the benefits of reducing the particle agglomeration and dissolution, which enhanced the toxic metal (selenium) removal in scrubber water. At high salt concentrations, the selenium removal was significantly inhabited by competing adsorption and/or corrosion effect of sulfate and chloride on iron surface. However, the inherent reactivity of iron-functionalized membranes was demonstrated by lowering TDS concentration with NF membranes. By applying elevated temperature which might be available in full-scale power plant applications, the salt interference effects could also be significantly reduced although resulting in a faster oxidation of iron. For the first time, iron nanoparticles were in-situ synthesized in full-scale membrane module. Convective flow reaction results obtained from lab-scale and full-scale membranes demonstrated the potential of using this platform for industrial wastewater treatment. There is a great potential for improving and optimizing this treatment approach for selenium through the addition of secondary catalytic metals (such as nickel) for simultaneous hydrogenation, or through the improvement of reaction selectivity by modification of gel chemistry or iron surface.
Supplementary Material
Highlights.
Correlated the extent of functionalization with membrane transport and responsive behavior
Successful selenium removal from real scrubber water by integration of nanotechnology and membrane separation
Synthesized reactive nanoparticles in full-scale membrane module
Reduced dissolved iron through membrane recapture
Acknowledgments
This project has been supported by Southern Company in Birmingham, AL, National Institute of Environmental Health Sciences Superfund Research Program of the National Institutes of Health (NIH-NIEHS-SRP) under Award Number P42ES007380, and by NSF KY EPSCoR program. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH. XPS analysis was supported by NSF under Grant Number 0814194. We thank the significant contributions of Nanostone/Sepro Membranes, Inc. in Oceanside, CA for joint development of full-scale functionalized membranes. Joseph Papp was also supported by the NSF REU program. We also thank Mr. Xinjun Teng (formerly of Southern Company Services, Inc.) for his role in initiating and managing Southern’s contribution to this project in the early stages.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elimelech M, Phillip WA. The Future of Seawater Desalination: Energy, Technology, and the Environment. Science. 2011;333:712–717. doi: 10.1126/science.1200488. [DOI] [PubMed] [Google Scholar]
- 2.Zhang WX. Nanoscale iron particles for environmental remediation: An overview. J Nanopart Res. 2003;5:323–332. [Google Scholar]
- 3.He F, Zhao DY. Hydrodechlorination of trichloroethene using stabilized Fe-Pd nanoparticles: Reaction mechanism and effects of stabilizers, catalysts and reaction conditions. Appl Catal B-Environ. 2008;84:533–540. [Google Scholar]
- 4.Smuleac V, Varma R, Sikdar S, Bhattacharyya D. Green synthesis of Fe and Fe/Pd bimetallic nanoparticles in membranes for reductive degradation of chlorinated organics. J Membrane Sci. 2011;379:131–137. doi: 10.1016/j.memsci.2011.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson RL, Nurmi JT, Johnson GSO, Fan DM, Johnson RLO, Shi ZQ, Salter-Blanc AJ, Tratnyek PG, Lowry GV. Field-Scale Transport and Transformation of Carboxymethylcellulose-Stabilized Nano Zero-Valent Iron. Environ Sci Technol. 2013;47:1573–1580. doi: 10.1021/es304564q. [DOI] [PubMed] [Google Scholar]
- 6.Tee YH, Bachas L, Bhattacharyya D. Degradation of Trichloroethylene and Dichlorobiphenyls by Iron-Based Bimetallic Nanoparticles. J Phys Chem C. 2009;113:9454–9464. doi: 10.1021/jp809098z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang CB, Zhang WX. Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs. Environ Sci Technol. 1997;31:2154–2156. [Google Scholar]
- 8.Mondal K, Jegadeesan G, Lalvani SB. Removal of selenate by Fe and NiFe nanosized particles. Ind Eng Chem Res. 2004;43:4922–4934. [Google Scholar]
- 9.Gui M, Smuleac V, Ormsbee LE, Sedlak DL, Bhattacharyya D. Iron oxide nanoparticle synthesis in aqueous and membrane systems for oxidative degradation of trichloroethylene from water. J Nanopart Res. 2012;14:861–877. doi: 10.1007/s11051-012-0861-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis SR, Datta S, Gui M, Coker EL, Huggins FE, Daunert S, Bachas L, Bhattacharyya D. Reactive nanostructured membranes for water purification. Proc Natl Acad Sci U S A. 2011;108:8577–8582. doi: 10.1073/pnas.1101144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pignatello JJ, Oliveros E, MacKay A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Crit Rev Env Sci Tec. 2006;36:1–84. [Google Scholar]
- 12.Xue XF, Hanna K, Abdelmoula M, Deng NS. Adsorption and oxidation of PCP on the surface of magnetite: Kinetic experiments and spectroscopic investigations. Appl Catal B-Environ. 2009;89:432–440. [Google Scholar]
- 13.Greenlee LF, Torrey JD, Amaro RL, Shaw JM. Kinetics of Zero Valent Iron Nanoparticle Oxidation in Oxygenated Water. Environ Sci Technol. 2012;46:12913–12920. doi: 10.1021/es303037k. [DOI] [PubMed] [Google Scholar]
- 14.Schug TT, Johnson AF, Balshaw DM, Garantziotis S, Walker NJ, Weis C, Nadadur SS, Birnbaum LS. ONE Nano: NIEHS’s Strategic Initiative on the Health and Safety Effects of Engineered Nanomaterials. Environ Health Perspect. 2013;121:410–414. doi: 10.1289/ehp.1206091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auffan M, Rose J, Bottero JY, Lowry GV, Jolivet JP, Wiesner MR. Towards a definition of inorganic nanoparticles from an environmental, health and safety perspective. Nat Nanotechnol. 2009;4:634–641. doi: 10.1038/nnano.2009.242. [DOI] [PubMed] [Google Scholar]
- 16.Lowry GV, Gregory KB, Apte SC, Lead JR. Transformations of Nanomaterials in the Environment. Environ Sci Technol. 2012;46:6893–6899. doi: 10.1021/es300839e. [DOI] [PubMed] [Google Scholar]
- 17.Zheng TH, Zhan JJ, He JB, Day C, Lu YF, Mcpherson GL, Piringer G, John VT. Reactivity characteristics of nanoscale zerovalent iron-silica composites for trichloroethylene remediation. Environ Sci Technol. 2008;42:4494–4499. doi: 10.1021/es702214x. [DOI] [PubMed] [Google Scholar]
- 18.Tseng HH, Su JG, Liang CJ. Synthesis of granular activated carbon/zero valent iron composites for simultaneous adsorption/dechlorination of trichloroethylene. J Hazard Mater. 2011;192:500–506. doi: 10.1016/j.jhazmat.2011.05.047. [DOI] [PubMed] [Google Scholar]
- 19.Kim H, Hong HJ, Lee YJ, Shin HJ, Yang JW. Degradation of trichloroethylene by zero-valent iron immobilized in cationic exchange membrane. Desalination. 2008;223:212–220. [Google Scholar]
- 20.Winnik FM, Morneau A, Mika AM, Childs RF, Roig A, Molins E, Ziolo RF. Polyacrylic acid pore-filled microporous membranes and their use in membrane-mediated synthesis of nanocrystalline ferrihydrite. Can J Chem. 1998;76:10–17. [Google Scholar]
- 21.Huang QG, Shi XY, Pinto RA, Petersen EJ, Weber WJ. Tunable Synthesis and Immobilization of Zero-Valent Iron Nanoparticles for Environmental Applications. Environ Sci Technol. 2008;42:8884–8889. doi: 10.1021/es8015588. [DOI] [PubMed] [Google Scholar]
- 22.Xu J, Bhattacharyya D. Membrane-based bimetallic nanoparticles for environmental remediation: Synthesis and reactive properties. Environ Prog. 2005;24:358–366. [Google Scholar]
- 23.Gui M, Ormsbee LE, Bhattacharyya D. Reactive Functionalized Membranes for Polychlorinated Biphenyl Degradation. Ind Eng Chem Res. 2013;52:10430–10440. doi: 10.1021/ie400507c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao L, Isner A, Waldrop K, Saad A, Takigawa D, Bhattacharyya D. Development of bench and full-scale temperature and pH responsive functionalized PVDF membranes with tunable properties. J Membrane Sci. 2014;457:39–49. doi: 10.1016/j.memsci.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olegario JT, Yee N, Miller M, Sczepaniak J, Manning B. Reduction of Se(VI) to Se(-II) by zerovalent iron nanoparticle suspensions. J Nanopart Res. 2010;12:2057–2068. [Google Scholar]
- 26.Yoon IH, Kim KW, Bang S, Kim MG. Reduction and adsorption mechanisms of selenate by zero-valent iron and related iron corrosion. Appl Catal B-Environ. 2011;104:185–192. [Google Scholar]
- 27.Huang YH, Peddi PK, Tang CL, Zeng H, Teng XJ. Hybrid zero-valent iron process for removing heavy metals and nitrate from flue-gas-desulfurization wastewater. Sep Purif Technol. 2013;118:690–698. [Google Scholar]
- 28.Lemly AD. Aquatic selenium pollution is a global environmental safety issue. Ecotox Environ Safe. 2004;59:44–56. doi: 10.1016/S0147-6513(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 29.Lalvani SB. Selenium Removal From Agricultural L Drainage Water: Lab Scale studies. Department of Water Resources; Sacramento, CA: 2004. [Google Scholar]
- 30.Kapur V, Charkoudian JC, Kessler SB, Anderson JL. Hydrodynamic permeability of hydrogels stabilized within porous membranes. Ind Eng Chem Res. 1996;35:3179–3185. [Google Scholar]
- 31.Mika AM, Childs RF. Calculation of the hydrodynamic permeability of gels and gel-filled microporous membranes. Ind Eng Chem Res. 2001;40:1694–1705. [Google Scholar]
- 32.Rushton A, Griffiths PVR. Filter media. In: Matteson MJ, Orr C, editors. Filtration: Principles and Practices. Marcel Dekker, Inc; New York: 1987. pp. 163–197. [Google Scholar]
- 33.Brandrup J, Immergut EH, Grulke EA. Polymer Handbook. Wiley-Interscience; 2003. pp. VII–198. [Google Scholar]
- 34.Hu K, Dickson JM. Development and characterization of poly(vinylidene fluoride)-poly (acrylic acid) pore-filled pH-sensitive membranes. J Membrane Sci. 2007;301:19–28. [Google Scholar]
- 35.Beuermann S. Propagation kinetics in free-radical polymerizations. Macromol Symp. 2002;182:31–42. doi: 10.1002/marc.200900131. [DOI] [PubMed] [Google Scholar]
- 36.Gromov VF, Galperina NI, Osmanov TO, Khomikovskii PM, Abkin AD. Effect of Solvent on Chain Propagation and Termination Reaction-Rates in Radical Polymerization. Eur Polym J. 1980;16:529–535. [Google Scholar]
- 37.Chapiro A, Mankowski Z. Influence of Solvents on Molecular Association and Kinetics of Acrylonitrile Polymerization - Supplementary Details on the Matrix Effect. Eur Polym J. 1981;17:457–472. [Google Scholar]
- 38.Hernandez S, Papp JK, Bhattacharyya D. Iron-Based Redox Polymerization of Acrylic Acid for Direct Synthesis of Hydrogel/Membranes and Metal Nanoparticles for Water Treatment. Ind Eng Chem Res. 2014;53:1130–1142. doi: 10.1021/ie403353g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu F, Hashim NA, Liu YT, Abed MRM, Li K. Progress in the production and modification of PVDF membranes. J Membrane Sci. 2011;375:1–27. [Google Scholar]
- 40.Myneni SCB, Tokunaga TK, Brown GE. Abiotic selenium redox transformations in the presence of Fe(II,III) oxides. Science. 1997;278:1106–1109. [Google Scholar]
- 41.Scheinost AC, Charlet L. Selenite reduction by mackinawite, magnetite and siderite: XAS characterization of nanosized redox products. Environ Sci Technol. 2008;42:1984–1989. doi: 10.1021/es071573f. [DOI] [PubMed] [Google Scholar]
- 42.Klas S, Kirk DW. Understanding the positive effects of low pH and limited aeration on selenate removal from water by elemental iron. Sep Purif Technol. 2013;116:222–229. [Google Scholar]
- 43.Zhang YQ, Wang JF, Amrhein C, Frankenberger WT. Removal of selenate from water by zerovalent iron. J Environ Qual. 2005;34:487–495. doi: 10.2134/jeq2005.0487. [DOI] [PubMed] [Google Scholar]
- 44.Su CM, Puls RW. Arsenate and arsenite removal by zerovalent iron: Effects of phosphate, silicate, carbonate, borate, sulfate, chromate, molybdate, and nitrate, relative to chloride. Environ Sci Technol. 2001;35:4562–4568. doi: 10.1021/es010768z. [DOI] [PubMed] [Google Scholar]
- 45.Goldberg S. Chemical Equilibrium and Reaction Modeling of Arsenic and Selenium in Soils. In: Magdi Selim H, editor. Dynamics and Bioavailability of Heavy Metals in the Rootzone. CRC Press; Boca Raton, FL: 2011. pp. 65–92. [Google Scholar]
- 46.Murphy AP. Removal of Selenate from Water by Chemical-Reduction. Ind Eng Chem Res. 1988;27:187–191. [Google Scholar]
- 47.Schellenger AEP, Larese-Casanova P. Oxygen Isotope Indicators of Selenate Reaction with Fe(II) and Fe(III) Hydroxides. Environ Sci Technol. 2013;47:6254–6262. doi: 10.1021/es4000033. [DOI] [PubMed] [Google Scholar]
- 48.Zingaro RA, Dufner DC, Murphy AP, Moody CD. Reduction of oxoselenium anions by iron(II) hydroxide. Environ Int. 1997;23:299–304. [Google Scholar]
- 49.Das S, Hendry MJ, Essilfie-Dughan J. Adsorption of selenate onto ferrihydrite, goethite, and lepidocrocite under neutral pH conditions. Appl Geochem. 2013;28:185–193. [Google Scholar]
- 50.Peak D, Sparks DL. Mechanisms of selenate adsorption on iron oxides and hydroxides. Environ Sci Technol. 2002;36:1460–1466. doi: 10.1021/es0156643. [DOI] [PubMed] [Google Scholar]
- 51.Qiu SR, Lai HF, Roberson MJ, Hunt ML, Amrhein C, Giancarlo LC, Flynn GW, Yarmoff JA. Removal of contaminants from aqueous solution by reaction with iron surfaces. Langmuir. 2000;16:2230–2236. [Google Scholar]
- 52.Sun YP, Li XQ, Cao JS, Zhang WX, Wang HP. Characterization of zero-valent iron nanoparticles. Adv Colloid Interfac. 2006;120:47–56. doi: 10.1016/j.cis.2006.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.