

Abstract
Neuropeptide Y (NPY) is expressed in mammalian retina but the location and potential modulatory effects of NPY receptor activation remain largely unknown. Retinal ganglion cell (RGC) death is a hallmark of several retinal degenerative diseases, particularly glaucoma. Using purified RGCs and ex vivo rat retinal preparations, we have measured RGC intracellular free calcium concentration ([Ca2+]i) and RGC spiking activity, respectively. We found that NPY attenuated the increase in the [Ca2+]i triggered by glutamate mainly via Y1 receptor activation. Moreover, (Leu31, Pro34)−NPY, a Y1/Y5 receptor agonist, increased the initial burst response of OFF-type RGCs, although no effect was observed on RGC spontaneous spiking activity. The Y1 receptor activation was also able to directly modulate RGC responses by attenuating the NMDA-induced increase in RGC spiking activity. These results suggest that Y1 receptor activation, at the level of inner or outer plexiform layers, leads to modulation of RGC receptive field properties. Using in vitro cultures of rat retinal explants exposed to NMDA, we found that NPY pretreatment prevented NMDA-induced cell death. However, in an animal model of retinal ischemia-reperfusion injury, pretreatment with NPY or (Leu31, Pro34)−NPY was not able to prevent apoptosis or rescue RGCs. In conclusion, we found modulatory effects of NPY application that for the first time were detected at the level of RGCs. However, further studies are needed to evaluate whether NPY neuroprotective actions detected in retinal explants can be translated into animal models of retinal degenerative diseases.
Keywords: calcium imaging, electrophysiology, neuromodulation, neuropeptide y, retinal explants, retinal ganglion cells
Introduction
Neuropeptide Y (NPY) is widely distributed in the central and peripheral nervous systems (Silva et al., 2005). This peptide belongs to a family of highly conserved peptides which also includes pancreatic polypeptide (PP) and peptide YY (Michel et al., 1998). During the last three decades, NPY has been associated with a multitude of physiological functions such as energy homeostasis (Sousa-Ferreira et al., 2011; Chambers and Woods, 2012), stress response (Hirsch and Zukowska, 2012), circadian rhythm (Yannielli and Harrington, 2004), bone physiology (Lee and Herzog, 2009), neurogenesis (Malva et al., 2012), and immune cell regulation (Dimitrijevic and Stanojevic, 2013). NPY, peptide YY, and PP activate seven transmembrane G protein-coupled receptors (GPCRs) named NPY receptors. All these receptors bind to Gi/G0 proteins, which results in inhibition of adenylyl cyclase. Other signaling pathways are also regulated, such as Ca2+ channels and G protein-coupled inwardly rectifying potassium channels (Sun and Miller, 1999), phospholipase C (Perney and Miller, 1989), and phosphoinositide 3-kinase (Rosmaninho-Salgado et al., 2012). In humans, only four NPY receptors were cloned and known to be functionally active, Y1, Y2, Y4, and Y5 (Babilon et al., 2013). These receptors have been implicated as potential neuroprotective therapeutic targets because they can prevent neuronal cell death induced by excitotoxic insults (Silva et al., 2003; Xapelli et al., 2007).
In the retina, NPY presence has been demonstrated in different species. However, the localization of the various NPY receptors in retinal cells has been scarcely investigated, and its function in retinal physiology remains largely unknown (Santos-Carvalho et al., 2014). Previous works from our laboratory have shown that NPY exerts neuroprotective action against different toxic insults. Particularly, in primary rat mixed retinal cell cultures, NPY pretreatment prevented increased cell death induced by both 3,4-methylenedioxy-N-methylamphetamine and glutamate (Alvaro et al., 2008b; Santos-Carvalho et al., 2013a). In an animal model of excitotoxicity-induced retinal injury, intravitreal administration of NPY inhibited both the increase in cell death and retinal ganglion cell (RGC) loss induced by glutamate (Santos-Carvalho et al., 2013a). In addition, we and others presented evidence suggesting a neuromodulatory role of NPY in the retina. For example, NPY application regulates neurotransmitter release in the rabbit and chicken retinas (Bruun and Ehinger, 1993). Also, NPY attenuates depolarization-induced increase in [Ca2+]i in primary mixed retinal cell cultures (Alvaro et al., 2009). Moreover, NPY decreases depolarization-dependent Ca2+ influx into bipolar cells via activation of Y2 receptors (D’Angelo and Brecha, 2004), and in retinas with selective ablation of NPY-expressing amacrine cells, an alteration in the receptive field properties of RGCs was reported, although a direct effect of NPY on RGCs was not demonstrated (Sinclair et al., 2004). These results suggest that NPY-induced modulation of visual circuitry might result in changes of RGC spiking activity. Therefore, in this study, we investigated the NPY modulatory effects at RGC level using a purified rat RGC culture and an ex vivo rat retinal preparation. In addition, since RGCs are lost in retinal degenerative diseases such as glaucoma, we also evaluated the neuroprotective potential of NPY against excitotoxic or ischemia-reperfusion (I-R) injuries.
Material and Methods
Animals
Wistar rats, 8 to 10 weeks old, were obtained from Charles River, France. Long Evans rats, 8 to 10 weeks old, were obtained from Charles River for RGC purification experiments and from Janvier Labs, Le Genest Saint Isle, France, for multielectrode array (MEA) experiments. Animals were provided with standard rodent diet and water ad libitum and kept on a 12 h light/12 h dark cycle. All procedures involving the animals were in agreement with the guidelines on the ethical use of animals from the European Community Council Directive 2010/63/EU.
Drugs
NPY and NPY receptor agonists: (Leu31, Pro34)−NPY, NPY13–36, and (Gly1, Ser3,22, Gln4,34, Thr6, Arg19, Tyr21, Ala23,31, Aib32)-PP ((Gly1,…Aib32)-PP) were all obtained from Bachem, Switzerland. NPY receptor antagonists: BIBP 3226, BIBO 3304, BIIE 0246, and L-152,804 were obtained from Tocris Bioscience, UK. The other used reagents were obtained from Sigma-Aldrich, USA, unless stated otherwise.
RGC Purification
Purified RGCs were obtained from the retinas of either 3 to 4 days old pups or 8 to 10 weeks old Wistar or Long Evans rats by a sequential immunopanning procedure yielding around 99% purity, as previously described (Barres et al., 1988), with some modifications, as follows. Rats were killed by decapitation or cervical dislocation, the eyes enucleated, and the retinas digested for 30 min at 37℃ in 16.5 U/mL papain (Worthington Biochemical, USA), 1.65 mM L-cysteine, and 124 U/mL deoxyribonuclease I (DNase I). The cell suspension was mechanically dissociated in 1.5 mg/mL ovomucoid (Roche, Switzerland), 1.5 mg/mL bovine serum albumin (BSA), and 124 U/mL DNase I in EBSS. The cell suspension was further triturated in 1.5 mg/mL ovomucoid, 1.5 mg/mL BSA, 124 U/mL DNase I, and 1:125 (v:v) rabbit anti-rat macrophage antiserum (Accurate Chemical, USA). After centrifugation for 11 min at 190 g at room temperature (RT), cells were resuspended in 10 mg/mL ovomucoid and 10 mg/mL BSA, and then centrifuged again for 10 min, at 190 g, at RT. Cells were resuspended in 0.2 mg/mL BSA and 5 µg/mL insulin. Cell suspension was plated in a goat anti-rabbit IgG (Rockland Immunochemicals, USA) coated dish. After 30 min at RT, nonadherent cells were removed to a second dish. After 30 min at RT, nonadherent cells were removed to a dish coated with goat anti-mouse IgM (Rockland Immunochemicals) and mouse anti-rat Thy1.1 hybridoma supernatant of T11D7e cell line (TIB-103, ATCC, USA). After 30 min, the nonadherent cells were removed, and RGCs were detached with a 0.125% trypsin solution. Trypsinization was stopped with 30% FBS (Gibco, Life Technologies, USA) in Neurobasal-A (Gibco). After final centrifugation for 10 min at 190 g, at RT, RGCs were resuspended. For cell culturing, RGCs were resuspended in Neurobasal-A medium containing 1 × B27 supplement (Gibco), 5 µg/mL insulin, 1 mM sodium pyruvate (Gibco), 1 × Sato/Bottenstein supplement (which includes 100 µg/mL transferrin, 100 µg/mL BSA, 16 µg/mL putrescine, 62 ng/mL progesterone, and 40 ng/mL sodium selenite), 40 ng/mL triiodo-L-thyronine, 2 mM L-glutamine, 5 mg/mL N-acetylcysteine, 100 µM inosine, 20 ng/mL ciliary neurotrophic factor and 25 ng/mL brain-derived neurotrophic factor (both from Peprotech, USA), 5 µM forskolin, 10 ng/mL basic fibroblast growth factor (Gibco), and 50 µg/mL gentamicin (Gibco) and were plated at a density of 460 cells/mm2 on 12 mm glass coverslips coated with 10 µg/mL poly-D-lysine and 10 µg/mL laminin. Cells were cultured for 16 to 48 h at 37℃ in a humidified environment of 5% CO2. For RNA extraction, the cell pellet was lysed in TRIzol reagent, as further described below. Regarding reverse-transcription polymerase chain reaction (RT-PCR), we extracted RNA immediately after the isolation of RGCs, and so we did not analyze mRNA expression from cultured purified RGCs. However, in RGC cultures, we did evaluate their purity by immunocytochemistry. The percentage of Brn3a+/DAPI+ cells varied between 70 and 98%. We also analyzed the mRNA expression of GFAP (marker of astrocytes and Muller cells) and CD11b (marker of microglia/macrophages) in some RGC isolations, and we did not find expression of these markers (data not shown).
RT-PCR
Total RNA was isolated from RGCs using TRIzol reagent (Ambion, Life Technologies, USA). Subsequently, cDNA first strand synthesis was performed from 2 µg DNase-treated RNA using random primers and SuperScript II Reverse Transcriptase (Invitrogen, Life Technologies, USA). The resulting cDNA (0.5 µL) was used for amplification of respective targets with AmpliTaq Gold DNA polymerase (Applied Biosystems, Life Technologies, USA), 200 nM of primer and 2 mM MgCl2, in a Veriti thermal cycler (Applied Biosystems). Reactions were performed as follows: denaturation for 3 min at 95℃; followed by 40 cycles each consisting of 30 s at 95℃, 30 s at annealing temperature, and 30 s at 72℃; and final extension at 72℃ for 5 min). The following primers were used (indicated as forward, reverse, annealing temperature, and product size): NPY, AGAGATCCAGCCCTGAGACA, TTTCATTTCCCATCACCACA, 57℃, 110 bp; Y1, ACGTTCGCTTGAAAAGGAGA, CATGACGTTGATTCGTTTGG, 57℃, 89 bp; Y2, CAGTTTTGTGCCATTTGGTG, AGGAAGCTGATTTGCTTGGA, 60℃, 142 bp; Y4, ATCTCATGGCCTCCCTTTCT, TCTCAACGCTGTAGGTGGTG, 57℃, 141 bp; Y5, ATACAGCTGCTGCTCGGAAT, GATTGCCCATAAAGCCAAGA, 57℃, 126 bp. Reaction specificity was confirmed by running nontemplate control for each primer set. Equal volumes of PCR products were separated on a 1.5% agarose gel. The gel images were digitally acquired in a Gel/ChemiDoc (Bio-Rad Laboratories, USA), and the level of gene transcription was evaluated and categorized as detected or not detected.
Immunofluorescence Labeling
For immunocytochemistry in purified RGC cultures, cells were fixed with 4% paraformaldehyde (PFA) for 20 min at RT and permeabilized with 1% Triton X-100 for 5 min. Unspecific binding was prevented with 3% BSA and 0.2% Tween 20 blocking solution for 60 min at RT. Cells were incubated with primary antibodies diluted in blocking solution for 90 min at RT as follows: rabbit anti-NPY (1:1000, Sigma-Aldrich), sheep anti-Y1 (1:500, AbD Serotec, Bio-Rad Laboratories, USA), rabbit anti-Y2 (1:500, Alomone Labs, Israel), rabbit anti-Y4 (1:25, Alomone Labs), rabbit anti-Y5 (1:250, Alomone Labs), or mouse anti-Brn3a (1:25, Merck Millipore, USA). Cells were incubated with the secondary antibodies diluted in blocking solution for 60 min at RT as follows: Alexa-Fluor 568 goat anti-mouse IgG (1:200), Alexa-Fluor 488 goat anti-rabbit IgG (1:200), or Alexa-Fluor 488 donkey anti-sheep IgG (1:200, all from Molecular Probes, Life Technologies, USA). The nuclei were stained with DAPI (2.5 µg/mL, Molecular Probes). Images were acquired in a laser scanning confocal microscope LSM 710 (Zeiss, Germany) using a 40 × numerical aperture (NA) = 1.3 objective lens, magnification 400×.
For immunohistochemistry in retinal sections, adult Wistar rats were transcardially perfused with 250 mL PBS followed by 250 mL 4% PFA at RT, under deep anesthesia induced by 90 mg/kg ketamine (Imalgene™) and 10 mg/kg xylazine (Rompun™). Then, the eyes were enucleated and fixed with PFA for 60 min. The cornea and lens were removed, and the eye cup was further fixed for 60 min in 4% PFA. Eye cups were transferred sequentially to 15% and 30% (w/v) sucrose for at least 120 min each. The eye cup was embedded in a mixture 1:1 of 30% sucrose and cryomatrix embedding resin (Thermo Fisher Scientific, USA), and stored at −80℃. Retinal sections, 10 µm thickness, were obtained in a cryostat and collected on SuperFrost Plus glass slides (Menzel-Glaser, Thermo Fisher Scientific) and stored at −20℃. Retinal sections were air dried for at least 45 min at RT and were fixed with acetone for 10 min at −20℃ and permeabilized in 0.25% Triton X-100 for 30 min at RT. In the case of Y1, Y2, Y4, and Y5 receptors, the fixation and permeabilization were replaced by an antigen retrieval step with 10 mM sodium citrate, pH 6.0, for 30 min at 95℃. The sections were permeabilized in 0.25% Triton X-100 for 30 min at RT, and blocked in 1% BSA and 10% goat serum for 30 min at RT. Sections were incubated overnight at 4℃ with primary antibodies diluted in 1% BSA in PBS: rabbit anti-NPY (1:10000), rabbit anti-Y1 (1:100) (Immunostar, USA), rabbit anti-Y2 (1:2000), rabbit anti-Y4 (1:200), rabbit anti-Y5 (1:2000), mouse anti-Brn3a (1:500), or mouse anti-vimentin (1:500; Lab Vision, USA). Sections were incubated for 60 min at RT with the corresponding secondary antibodies diluted in 1% BSA in PBS: Alexa-Fluor 488 goat anti-mouse IgG (1:500) or Alexa-Fluor 568 goat anti-rabbit IgG (1:500). The nuclei were stained with DAPI (2.5 µg/mL). The specificity of anti-NPY and anti-NPY receptor antibodies has been confirmed by the corresponding manufacturer. Images were acquired in a laser scanning confocal microscope LSM 710 using a 20 × NA = 0.8 objective lens, magnification 200×.
NPY-Stimulated [35S]GTPγS Functional Binding
Eyes of 8 to 10 weeks old male Wistar rats were enucleated and frozen in dry ice. Retinal sections (18 µm thickness) were obtained in a cryostat, collected onto SuperFrost Plus glass slides and stored at −80℃ until further processing. Sections were air dried for 30 min at RT and then rehydrated in assay buffer A (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4) for 10 min at RT. Sections were preincubated in assay buffer B (assay buffer A + 0.2 mM dithiothreitol, 1 µM 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), 0.5% w/v BSA, and 2 mM GDP) for 15 min at RT to shift all G proteins into the inactive state. Subsequently, incubation of retinal slices was performed in assay buffer B + 50 pM [35S] guanosine 5’-[γ-thio]triphosphate (GTPγS; 1250 Ci/mmol; PerkinElmer, USA) for 60 min at RT with 1 to 10 µM NPY. In each experiment, basal binding was determined by incubation without NPY receptor ligands but with assay buffer B + 50 pM [35S]GTPγS (1250 Ci/mmol). Specificity was confirmed by adding a combination of the NPY receptor antagonists: 10 µM BIBO 3304 for Y1; 10 µM BIIE 0246 for Y2; 100 µM L-152,804 for Y5. For antagonistic studies, NPY receptor antagonists were also added to the preincubation buffer B. Incubation was terminated by two washes of 5 min each in ice cold 50 mM Tris-HCl buffer, pH 7.4, followed by a final wash in deionized water. Sections were air dried at RT and exposed to Kodak BioMax MR films (Carestream Health, Rochester, USA) together with 14C-microscales (Amersham, GE Healthcare, Little Chalfont, UK) for 5 days at −20℃. The films were developed in Kodak GBX developer, and the optical densities of retinal slices measured using ImageJ software (http://imagej.nih.gov/ij). The values obtained were converted to estimated nCi/g tissue using 14C-microscales.
Fura-2 Intracellular Ca2+ Imaging in Purified RGCs
Purified RGCs cultured for 1 or 2 DIVs were used to assess the changes in [Ca2+]i using the Ca2+ dye Fura-2-AM, with a fluorescence microscope Axiovert 200 (Zeiss) coupled to a perfusion system. RGCs were loaded with 5 µM Fura-2-AM in the presence of 0.02% Pluronic F-127 (both Molecular Probes, Life Technologies, USA) for 45 min at 37℃, in HBSS Mg2+-free solution (in mM: 138 NaCl, 5.3 KCl, 0.34 Na2HPO4, 0.44 KH2PO4, 2.6 CaCl2, 5.6 glucose, 15 HEPES, 4.17 NaHCO3, pH 7.4) supplemented with 0.1% fatty acid-free BSA. Under continuous perfusion (2.9 ± 0.1 mL/min) with HBSS Mg2+-free solution, RGCs were exposed to glutamate for 30 s, and all glutamate stimuli included 10 µM glycine, a coagonist of N-methyl-D-aspartic acid (NMDA) glutamate receptor, as previously described (Hartwick et al., 2004). RGCs were alternately excited at 340 and 380 nm, and a ratio of fluorescence intensity (340 nm/380 nm) was calculated for each individual cell by Metafluor software (Molecular Devices, Sunnyvale, USA). Fura-2 ratios (R) were converted to [Ca2+]i in separate calibration experiments using the following formula: [Ca2+]i = [Kd(F0/FS)][R – Rmin)/(Rmax – R)], with Kd for Fura-2 of 224 nM, and where F0/FS is the ratio of fluorescence intensity at 380 nm excitation in Ca2+-free solution over the intensity in solution with saturated Ca2+ levels (Hartwick et al., 2004). The minimum value for the Fura-2 ratio (Rmin) was obtained using Ca2+-free HBSS and 1 µM ionomycin, after which the cells were perfused with Mg2+-free HBSS and 1 µM ionomycin in order to calculate the maximum value for Fura-2 ratio (Rmax).
Ex Vivo MEA Recordings
Long Evans rats (8 weeks old) were dark-adapted for at least 12 h and killed by CO2 inhalation and quick cervical dislocation under dim red light. Eyes were enucleated and placed in oxygenated Ames’ medium at RT. Square pieces of retina (1–2 mm2) were placed into the recording chamber, with the ganglion cell layer (GCL) facing the MEA60 biochip electrode array. The electrode array was composed of 60 titanium nitride electrodes, 10 µm diameter each, disposed in a 8 × 8 layout with 100 µm interelectrode spacing (Multi Channel Systems—MCS, GmbH, Germany). Retinas were held in the center of the electrode array using a piece of polycarbonate membrane covered by a U-shaped platinum ring with a nylon mesh. During recording sessions, retinas were continuously perfused with Ames’ medium equilibrated with 95% O2 and 5% CO2, pH 7.4, at a flow rate of 1.3 mL/min. Retinas were maintained at 34 to 37℃ through a TC01 heating pad (MCS) of the recording system. In order to obtain stable recordings, each session started 60 min after placing the retina in MEA recording chamber. MEA recordings were conducted using MEA60 setup (MCS). The analog extracellular neuronal signals from 60 channels were AC amplified (×1000–1200), band-pass filtered (200–3000 Hz), sampled at 20 to 30 kHz, and saved in PC-compatible computer for subsequent off-line analysis. RGC spiking activity was monitored during recording sessions using MC_Rack software (MCS). Light-induced responses were evaluated under dark conditions. To elicit light responses in the RGCs, white light stimulation blocks produced from light-emitting diodes driven by a stimulus generator STG-1008 (MCS) were applied. The light-emitting diodes were positioned 5 mm below the transparent MEA chamber and used to generate full-field stimuli in the photopic range (5.0 cd/m2). Stimuli consisted of 10 consecutive stimulus blocks with 5 s light followed by 10 s dark each. The recordings were subsequently subjected to off-line spike sorting and analysis using Spike2 (Cambridge Electronic Design, UK). Waveforms were isolated using a combination of template matching and cluster cutting. The spontaneous activity was calculated for each RGC as spiking rate (Hz). To detect changes in spontaneous activity induced by light, the raster and peri-stimulus time histograms were generated from 10 stimulus blocks using 50 ms bin widths. The onset of ON- and OFF-type RGC responses were defined as an increase in spike number higher than two standard deviations than the prestimulus frequency over at least three consecutive bins. The initial burst responses to both light onset (ON-type RGCs) and light offset (OFF-type RGCs) were quantified over a 50 ms bin width. Latency was defined as the time delay between light onset or offset (ON- or OFF-type RGCs, respectively) and the RGC light response as defined above, when aligned in raster plots for nine consecutive light stimuli.
Culture of Retinal Explants and Cell Death Evaluation
Wistar rats (8–10 weeks old) were killed by cervical dislocation. Retinas were flat-mounted onto 30 mm diameter culture plate hydrophilic polytetrafluoroethylene inserts with a 0.4 µm pore size (Millicell, Merck Millipore), with the GCL side facing upward. The retinal explants were cultured in Neurobasal-A media containing 1 × B27 supplement, 2 mM L-glutamine, and 50 µg/mL gentamicin, and maintained for 4 DIVs in a humidified environment of 5% CO2 at 37℃. To induce cell death, cultured retinal explants were exposed to 300 µM NMDA at DIV2.
For TdT-mediated dUTP nick-end labeling (TUNEL) assay, the manufacturer’s instructions were followed (Promega, USA). Briefly, retinal explants were fixed in 4% PFA for 15 min at RT and permeabilized with 20 µg/mL proteinase K for 15 min at RT followed by further fixation in 4% PFA for 5 min. Retinal explants were incubated with the recombinant TdT enzyme and nucleotide mix containing dUTP conjugated to fluorescein at 37℃ for 60 min. The reaction was stopped by incubating the retinal explants in saline-citrate buffer for 15 min at RT. The nuclei were stained with DAPI (2.5 µg/mL). At least 12 images of GCL per retinal explant (three images per quadrant) were acquired in a laser scanning confocal microscope LSM 710 using a 40 x NA = 1.3 objective lens, magnification 400×. TUNEL-positive cells localized in GCL were quantified, since this layer is highly affected in diseases like glaucoma, and the values were then normalized to the value obtained in nonNMDA treated retinal explant (control), in each independent experiment.
For propidium iodide (PI) incorporation assay, cultured retinal explants were incubated with 2 µM PI for 180 min at DIV2 and at DIV4. Since the acquisition of images was performed with live retinal explants using fluorescence microscopy (DM IRE2, Leica, Germany) with a 10 × NA = 0.22 objective lens, magnification 100×, we quantified PI-positive cells across all retinal layers. Images comprising the four quadrants of retinal explant were acquired, and the PI-positive cells were counted at DIV2 (before NMDA treatment and at DIV4). The extent of cell death was expressed as the ratio between PI-positive cells at DIV4 and DIV2.
Intravitreal Injections and Retinal I-R Injury
Wistar rats (8–10 weeks old) were anesthetized by 2.5% isoflurane inhalation using a gas-anesthetizing system (VetEquip, USA). Then, 4 mg/mL oxybuprocaine (Laboratórios Edol, Portugal) anesthetic was applied topically to the eyes and the pupils dilated with 10 mg/mL tropicamide (Laboratórios Edol). Intravitreal injection of 5 µL containing 10 µg (2.34 nmol) NPY, 10 µg (2.36 nmol; Leu31, Pro34)−NPY, or sterile saline solution was performed using a 10 µL Hamilton syringe (Hamilton, USA) with a 30-gauge needle, in both eyes, 120 min before the induction of retinal I-R injury. Fusidic acid (10 mg/g, Leo Pharmaceutical, Denmark) ointment was applied in the conjunctival sac after the intravitreal injections. Retinal I-R injury was induced in one eye by elevating the intraocular pressure (IOP) to 80 mmHg for 60 min. IOP was measured with a tonometer (Tonolab, Icare, Finland). The anterior chamber of one eye was cannulated with a 30-gauge needled connected to a reservoir infusing sterile saline solution. The contralateral eye was taken as the control eye. The IOP was raised by elevating the reservoir to a height of 1.8 m. Retinal ischemia was confirmed by whitening of the iris and loss of the red reflex. In order to avoid corneal opacity, 2% methocel™ (Dávi II, Portugal) was applied to both eyes. Reperfusion was confirmed by returning of red color to the iris and the red reflex to the eye fundus. Hypothermia was avoided using warmed blankets covering the animals during all the procedure. After removing the needle from the anterior chamber, the animals were observed until full recovery which occurred within 5 min. Animals were killed after 24 h of reperfusion. There were no animals excluded from the study.
Electroretinogram Recordings
Electroretinograms (ERGs) were recorded before the onset of I-R injury (baseline) and after 24 h of reperfusion. After dark adaptation for at least 12 h, the animals were anesthetized with 90 mg/kg ketamine and 10 mg/kg xylazine (i.p). Then, 4 mg/mL oxybuprocaine anesthetic was applied topically to the eyes and the pupils dilated with 10 mg/mL tropicamide under dim red light illumination. The body temperature was maintained with a heating pad set to 37℃. Using a Ganzfeld stimulator, white light flashes (0.0095–9.49 cd-s/m2) were applied under scotopic and photopic conditions (in the latter case, after 16 min of light adaptation to a white background, 25 cd/m2). ERGs were recorded with a corneal gold wire electrode, a reference electrode at the head and a ground electrode in the tail of the animal. A band width of 1 to 300 Hz and sampling rate of 3.4 kHz were used for acquisition (Roland Consult GmbH, Germany). The scotopic a-wave and b-wave, and photopic b-wave, were evaluated. Off-line digital filter was applied on b-wave (high frequency cutoff of 50 Hz) with the RETIport software (Roland Consult GmbH).
Brn3a Labeling and TUNEL Assay in Retinal Sections
The animals subjected to I-R injury were used for Brn3a labeling by immunohistochemistry with antibody mouse anti-Brn3a (1:500, Merck Millipore, USA), using the same protocol as for retinal sections. After this procedure, a TUNEL assay was performed following manufacturer’s instructions (Promega). Briefly, sections were permeabilized with 20 µg/mL proteinase K for 10 min at RT and incubated with the recombinant TdT enzyme and nucleotide mix containing dUTP conjugated to fluorescein at 37℃ for 60 min. The reaction was stopped by immersing the slides in saline-citrate buffer for 15 min at RT. The nuclei were stained with DAPI (2.5 µg/mL). Images of retinal sections were acquired in a fluorescence microscope. The Brn3a- and TUNEL-positive cells were counted, and results were expressed per mm of GCL length. Images were acquired using a fluorescence microscope (DM IRE2, Leica) with a 20 × NA = 0.3 objective lens, magnification 200×.
Statistical Analysis
Statistical analysis was performed with Prism 5 (GraphPad, USA) using one-way analysis of variance followed by Bonferroni’s test. When distribution normality was not achieved, one-way Kruskal-Wallis test was used followed by Dunn’s test, as indicated in figure legends. P values less than 0.05 were taken as significant. All values are presented as mean ± SEM.
Results
Expression of NPY and NPY Receptors in RGCs
The presence of NPY and NPY receptors (Y1, Y2, Y4, and Y5) was assessed in purified rat RGCs. Total RNA was extracted immediately after purification. To assess the presence of mRNA encoding for NPY and NPY receptors (Y1, Y2, Y4, and Y5) by RT-PCR, we used cDNA from RGCs isolated from the retinas of P3 to P4 pups and 8 weeks old adult rats of two different strains: Wistar and Long Evans (Figure 1(a)). We found that mRNA for both NPY and NPY receptors (Y1, Y2, Y4, and Y5) was detected in purified RGCs from both rat strains and at both ages.
Figure 1.

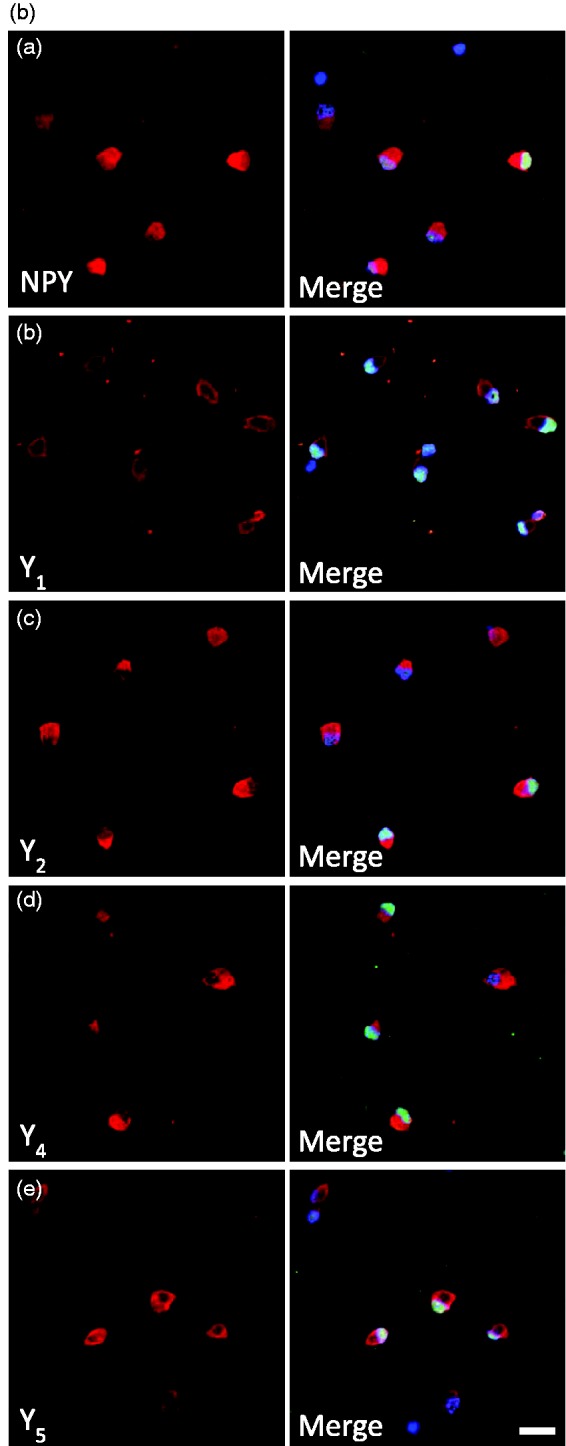

Expression of NPY and NPY receptors in RGCs. (a) Detection of NPY and NPY receptors (Y1, Y2, Y4, and Y5) mRNA expression by RT-PCR in purified RGCs. RGCs were isolated from the retinas of either pups or young adult Long Evans and Wistar rats. DNA ladder. (b) NPY and NPY receptor immunoreactivity (ir) in a purified RGC culture obtained from Wistar pups (a–e, red). RGCs were stained with antiBrn3a (RGC marker, green) and nuclei with DAPI (blue). Scale bar: 20 µm. (c) NPY-ir and NPY receptor-ir in retinal slices obtained from young adult Wistar rats (a–e, red). RGCs were stained with the RGC marker Brn3a (green) and nuclei with DAPI (blue). NPY-ir was mainly detected in strata 1, 3, and 5 of IPL (arrows; a). Y1-ir was detected in distal and proximal INL, strata 2 and 4 of IPL, and RGCs (arrows; b). Y2-ir was detected in INL (c). Y4-ir was detected in INL and GCL (d). and Y5-ir was detected in Müller cells (e). (d) Y5-ir (red) is colocalized with Müller cells (green) in retinal slices of young adult Wistar rats. Müller cells were identified by vimentin-ir (green). Nuclei were stained with DAPI (blue). GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Scale bar: 50 µm.
To assess the NPY and NPY receptor expressions at protein level, we also evaluated their immunoreactivity (ir) in purified RGCs from P3 to P4 Wistar rats cultured overnight. We found NPY-ir in RGCs with colocalization with the RGC marker Brn3a (Figure 1(ba)). Similar to NPY-ir, immunoreactivity for Y1, Y2, Y4, and Y5 receptors was also detected in RGCs (Figure 1(bb–e)). In addition, we assessed the localization of NPY and NPY receptors in retinal sections of adult Wistar rats. NPY-ir was detected in ramified dendrites in strata 1, 3, and 5 of inner plexiform layer (IPL; Figure 1(ca)), and cell bodies of GCL and inner nuclear layer (INL). Similar observations were reported previously, identifying these cells as amacrine cells (Oh et al., 2002). Y1 receptor-ir was detected in cell bodies in the distal and proximal INL (Figure 1(cb)), arrows). A previous study has identified these cells as horizontal and amacrine cells (D’Angelo et al., 2002). In accordance, we also detected Y1-ir processes ramifying in strata 2 and 4 of IPL. In addition, we found that some Brn3a-ir RGCs colocalized with Y1. We also detected, for the first time, Y2 receptor-ir in stratum 1 of IPL and in cell bodies in proximal INL (Figure 1(cc)). Y4 receptor-ir was localized in cell bodies of GCL, and proximal and distal INL (Figure 1(cd)). The cell bodies in GCL that were immunoreactive for Y4 receptor were both RGC (colocalized with Brn3a, yellow arrow) and nonBrn3a-positive cells, likely displaced amacrine cells (green arrow). Concerning the Y5 receptor, immunoreactivity was detected in Müller cells (Figure 1(ce)). Müller cell identity was confirmed by colocalization with vimentin (Figure 1(d)). This is the first time the localization of Y4 and Y5 receptor was assessed in rat retinal sections. The lack of clear immunoreactivity for NPY, and for Y2 and Y5 receptors in RGCs in retinal sections of adult rat, while it could be found in cultured RGCs from P3 to P4 rats, may be explained by decreased expression in adulthood.
NPY Stimulates Functional Binding in Retinal Slices
We performed [35S]GTPγS-binding assay (Christensen et al., 2006) to assess the functional activity of NPY receptors in retinal slices from adult Wistar rats (Figure 2). [35S]GTPγS-binding assay is able to evaluate the activation of GPCRs taking advantage of a radiolabeled nonhydrolyzable GTP analog—[35S]GTPγS. Upon activation of the GPCR by a receptor agonist, Gα binds [35S]GTPγS allowing the measurement of the amount of radiolabeled GTP bound to the cell membrane using an autoradiography film. After incubation with 10 µM NPY for 60 min (n = 10 animals), we detected an increase in [35S]GTPγS binding in inner retinal layers comparing to basal conditions (n = 10)—nonstimulated retinal slices; Figure 2(a) and (b). The small size of the rat retinal tissue and the resolution of autoradiography films made it difficult to identify the exact part of the inner retinal layers responsible for the increased NPY-stimulated [35S]GTPγS functional binding. However, when comparing the hematoxylin-eosin staining of native retinal slices with autoradiography film pictures of the same retinal slices, we found that the retinal layers displaying increased [35S]GTPγS binding upon NPY stimulation corresponded to the GCL and IPL (Figure 2(c)). It is also of note that an intense binding signal corresponding to the photoreceptor layer was detected both under basal and NPY-stimulated conditions (Figure 2(aa) and (ab), white arrows). This likely represents the high amount of G proteins in the photoreceptor outer segments, mainly transducin (Arshavsky et al., 2002). Moreover, NPY did not induce a statistically significant increase in binding signal in the photoreceptor layer (data not shown). Since 1 µM NPY (n = 2, not used for statistical tests) was not sufficient to increase [35S]GTPγS binding in retinal sections, we speculated that low levels of NPY receptors were functionally active in frozen retinal sections, thus requiring increased concentrations of NPY. A cocktail of NPY receptor antagonists (10 µM BIBO 3304—Y1 receptor antagonist; 10 µM BIIE 0246 for Y2; 100 µM L-152,804 for Y5) was used to evaluate the selectivity of NPY-stimulated [35S]GTPγS binding (Figure 2(ac) and (b)). The blockade of Y1, Y2, and Y5 receptors (n = 8) prevented the increased [35S]GTPγS binding induced by NPY, confirming the selectivity of the signal corresponding to the GCL and IPL. To assess the nonspecific binding, a competitive control with nonradioactive GTPγS was used, which exhibit no clear binding signal in the autoradiography film (Figure 2(ad)).
Figure 2.

NPY stimulates [35S]GTPγS functional binding in retinal sections. (a) Examples of retinal sections from autoradiography films using [35S]GTPγS-binding assay. (b) Quantification of [35S]GTPγS binding in inner retinal layers. Incubation with 10 µM NPY increased [35S]GTPγS binding in inner retinal layers (ab, black arrow) compared with basal binding (aa). (c) [35S]GTPγS binding signal (green) merged with hematoxylin-eosin staining (red). The green represents the [35S]GTPγS binding signal as in Figure 2ab, where the black was converted to green in order to be better visualized in the merged picture with hematoxylin-eosin staining which is in red. Note the increased signal in IPL (black arrow). An intense signal was found in the photoreceptor layer (white arrows) both under basal and NPY-stimulated binding conditions that may represent the high amount of G proteins in photoreceptor outer segments, mainly transducin. The blockade of Y1, Y2, and Y5 receptors prevented the increased [35S]GTPγS binding induced by NPY (ac). NSB refers to nonspecific binding, a competitive control with nonradioactive GTPγS (ad). Antagonists used: Y1, BIBO 3304 (10 µM); Y2, BIIE 0246 (10 µM); Y5, L-152,804 (100 µM). Bar: 2 mm. Data are presented as mean ± SEM of n = 2 to 10 independent experiments. *p < .05, compared with basal; ##p < .01, compared with 10 µM NPY. One-way analysis of variance followed by Bonferroni’s test.
NPY Attenuates Glutamate-Induced [Ca2+]i Increase in Purified RGCs
We aimed to evaluate the potential modulatory effect of NPY directly on RGCs. For this purpose, we cultured purified RGCs obtained from retinas of P3 to P4 rat pups and performed intracellular free calcium concentration ([Ca2+]i) measurements using Fura-2 calcium indicator. As previously described (Hartwick et al., 2004), we stimulated RGCs with increasing glutamate concentrations (from 1 to 1000 µM) for 30 s, including also 10 µM glycine (Figure 3(a)). After testing these glutamate concentrations, we used 30 µM glutamate in the following experiments since this concentration induced a submaximal increase in [Ca2+]i that was rapidly reversible. The ratios between emission of Fura-2 when excited by light at 340 nm and 380 nm wavelengths were quantified in cell bodies of RGCs as indicative of [Ca2+]i (Figure 3(b)). In calibration experiments, we converted Fura-2 ratio to [Ca2+]i (see methods section). We obtained basal values of 54 ± 6 nM, while after stimulation with 30 µM glutamate for 30 s, a peak [Ca2+]i of 733 ± 59 nM was found (data not shown). After the first glutamate stimulus, different drugs were applied for 10 min: NPY (1 µM, n = 6 independent RGC cultures), the Y1/Y5 agonist (Leu31, Pro34)−NPY (1 µM, n = 8), the Y2 agonist NPY13–36 (300 nM, n = 5), and the Y5 agonist (Gly1,…Aib32)-PP (1 µM, n = 5), or a drug-free solution (control, n = 10), followed by a second glutamate stimulus coapplied with the drug (Figure 3(c)). The increase above basal Fura-2 ratio was quantified for each stimulus as a Delta value. When evaluating the average [Ca2+]i responses of the cell population analyzed, a small decrease in Delta 2/Delta 1 ratios was found for NPY (0.83 ± 0.04) and (Leu31, Pro34)−NPY (0.81 ± 0.05), compared with control (0.93 ± 0.02) (Figure 3(d)). In fact, a small significant change may be hard to detect in the overall population, as can be observed in scatter plots showing individual RGCs from an independent experiment (Figure 3(e)). Hence, we quantified the percentage of RGCs where the Delta 2/Delta 1 ratio was lower than 0.9 (Figure 3(f)). The application of NPY or (Leu31, Pro34)−NPY for 10 min significantly increased the percentage of cells with Delta 2/Delta 1 ratio below 0.9 (77.7 ± 10.3% or 68.5 ± 8.3%, respectively) compared with control (32.7 ± 8.4%). Since the Y5 receptor agonist alone was not able to affect the RGC response to glutamate, it is likely that the effect of NPY or (Leu31, Pro34) − NPY might occur via Y1 receptor activation.
Figure 3.

NPY attenuates glutamate-induced [Ca2+]i increase in purified RGCs. (a) [Ca2+]i trace from a cultured RGC illustrating the response of RGCs to increasing concentrations of glutamate (10–1000 µM). (b) Example of cultured RGCs. Bright field image (ba) and pseudocolor representation of Fura-2 ratio on basal condition (bb) and after stimulation with 30 µM glutamate (bc) are shown. (c) [Ca2+]i traces showing RGC responses to two consecutive glutamate stimuli (30 µM) for 30 s each stimulus. After the first glutamate stimulus, 1 µM NPY or a drug-free solution (control) was applied to RGCs during 10 min followed by a second glutamate stimulus. For illustration, two experiments (control and NPY) are shown. The increase above basal Fura-2 ratio (340 nm/380 nm) was quantified for each stimulus as a Delta value. (d) The Delta 2 (second stimulus)/Delta 1 (first stimulus) ratios are presented for different drug applications for 10 min: 1 µM NPY, Figure 3. Continued. 1 µM (Leu31, Pro34) − NPY, 300 nM NPY13–36, 1 µM (Gly1, . . . Aib32)-PP, or a drug-free solution (control). A small decrease in Delta 2/Delta 1 ratios may be apparent in RGCs exposed to NPY and (Leu31, Pro34) − NPY. (e) Scatter plots for two populations of RGCs from the same cell culture showing the dispersion of Delta 2/Delta 1 ratio values among cells. Each point represents an individual RGC. The effect of NPY is evident in some RGCs. Cells were treated with a drug-free solution (control) or NPY. Note that in NPY-treated population, a small downward shift may be observed. Dashed line indicates 0.9 ratio value. (f) Percentage of cells presenting Delta 2/Delta 1 ratio below 0.9. The application of NPY or (Leu31, Pro34) − NPY for 10 min increased the percentage of cells with Delta 2/Delta 1 ratio below 0.9. Data are presented as mean ± SEM of n = 5 to 10 independent experiments. *p < .05, compared with control. Kruskal-Wallis followed by Dunn’s test.
RGC Spiking Activity Modulation via Y1 Receptor Activation
Since NPY is able to modulate neuronal activity in various brain regions (Silva et al., 2002; Benarroch, 2009), we hypothesized whether application of NPY to ex vivo retinas could directly modulate RGC spiking activity. For this purpose, we used a MEA system, which allows simultaneous recording spiking activity from various RGCs. After spike sorting based on template matching, the spiking rate for individual RGCs was quantified. In the first set of experiments, we recorded RGC spontaneous activity (no stimulus) before (baseline) and after the application of NPY (1 µM, n = 4 retinas), the Y1/Y5 agonist (Leu31, Pro34) − NPY (1 µM, n = 3), the Y2 agonist NPY13–36 (1 µM, n = 3), or a drug-free solution (control, n = 3) for 10 min under continuous perfusion (Figure 4(a)). In addition, a period of washout up to 60 min was included. We found a small decrease in RGC spiking rate over time reaching 79.5 ± 7.2% of baseline in control at 60 min of washout. However, exposure to NPY or NPY receptor agonists caused no effect since RGCs presented a decrease in spiking rate similar to control.
Figure 4.

The Y1 and Y5 receptor agonist (Leu31, Pro34) − NPY increases the response of OFF-type RGCs to light offset. (a) RGC spontaneous spiking rate quantification after the application of 1 µM NPY, 1 µM (Leu31, Pro34) − NPY, 1 µM NPY13–36, or a drug-free solution (control) for 10 min and upon 60 min of washout was recorded in ex vivo retinas using a MEA. A decrease in RGC spiking rate was observed over time, though no effects were found for drug treatments. (b) Examples of peri-stimulus time histograms and raster plots for ON-type RGC and OFF-type RGC responses are shown for seven consecutive stimulus blocks. White rectangles indicate duration of light period. (c) Quantification of initial burst to light onset of ON-type RGCs after application of the same drug treatments as in (a). No effect was found for the different drug treatments compared with control. (d) Quantification of initial burst to light offset of OFF-type RGCs after application of the same drug treatments as in (a). The application of 1 µM (Leu31, Pro34) − NPY for 10 min was able to increase the magnitude of OFF-type response compared with control. All data were normalized to the values obtained before drug application (baseline). Data are presented as mean ± SEM of n = 3 to 4 independent experiments. *p < .05, compared with control. Kruskal-Wallis followed by Dunn’s test.
In order to evaluate the effect of NPY on RGC light responses, ex vivo retinas were exposed to light stimuli in the photopic range (5.0 cd/m2), while RGC spiking activity was recorded with the MEA system. The light stimulation protocol consisted of 10 consecutive stimulation blocks composed of 5 s light followed by 10 s dark. RGC light responses were classified as ON- or OFF-type. An ON-type RGC light response was considered if increased spiking activity was detected at light onset, and an OFF-type RGC light response was considered when increased spiking activity was detected at light offset in each stimulation block. In Figure 4(b), examples of ON- and OFF-type responses are presented. The peri-stimulus time histograms were generated from RGC responses to nine consecutive stimulation blocks and used to calculate the initial burst response to light onset or offset, as well as to estimate the latency of RGC light response. The first RGC light response to a series of 10 stimulation blocks was always omitted from the quantification since it was harder to discriminate it due to the robust spike distortion within burst discharge during light presentation. Thus, in order to evaluate the potential modulatory effect of NPY on RGC light responses, the recordings were performed before (baseline) and after the application of NPY (1 µM, n = 4 retinas), the Y1/Y5 agonist (Leu31, Pro34) − NPY (1 µM, n = 4), the Y2 agonist NPY13–36, (1 µM, n = 4), or a drug-free solution (control, n = 3) for 10 min under continuous perfusion (Figure 4(c) and (d)). Regarding ON-type light response, no apparent effect was found on the initial burst of spiking activity to light onset for the listed above drugs (Figure 4(c)). On the contrary, the application of 1 µM (Leu31, Pro34) − NPY for 10 min was able to induce a small, but statistically significant, increase to 112.4 ± 2.8% of baseline on the initial burst of spiking activity triggered by ceasing light in OFF-type RGCs (Figure 4(d)), when compared with control (100.3 ± 1.1% of baseline), while no statistically significant effect was found after the application of 1 µM NPY or 1 µM NPY13–36. This result suggests that Y1 or Y5 activation may be sufficient to modulate the OFF-type RGC light response directly by changing the receptive field properties or acting upstream of RGCs in the vertical pathway of visual information, namely at the level of glutamatergic bipolar or photoreceptor cells. When considering the latency of RGC light responses, no effect was found in both ON- or OFF-type RGC responses after the different drug treatments used (data not shown).
We also evaluated the potential modulatory effect of NPY application on RGC spiking activity upon glutamate receptor activation. In this experimental set, no light stimulation was applied. Glutamate is easily cleared within the retina by glutamate uptake (Higgs and Lukasiewicz, 1999), and therefore, we applied NMDA directly to ex vivo retinas in order to induce synaptic excitation of RGCs, since NMDA receptors are abundantly expressed in RGCs (Shen et al., 2006). The application of NMDA (30 µM) for 5 min induced an acute increase in RGC spiking activity, which was reversed by 10 min of washout (Figure 5(a)). After washout of the first NMDA stimulus, different drugs were applied for 10 min under continuous perfusion: NPY (1 µM, n = 8 retinas), the Y1/Y5 agonist (Leu31, Pro34) − NPY (1 µM, n = 8), the Y2 agonist NPY13–36 (1 µM, n = 6), the Y5 agonist (Gly1,…Aib32)-PP (1 µM, n = 5), or a drug-free solution (control, n = 10). After 10 min of drug treatment, a second NMDA stimulus was coapplied, followed by 10 min of washout. The Delta value represents the difference between the maximum spiking frequency (or spiking rate) during NMDA application and the spiking frequency obtained before NMDA exposure. Subsequently, the Delta 2 (second stimulus)/Delta 1 (first stimulus) ratios were calculated for each individual RGC. We found that application of (Leu31, Pro34) − NPY was able to decrease the Delta 2/Delta 1 ratio to 0.67 ± 0.07, when compared with control, 0.96 ± 0.04 (Figure 5(b)), indicating that activation of Y1 or Y5 receptor was sufficient to attenuate the NMDA-stimulated RGC spiking activity. For the other drug treatments, no statistically significant alterations were detected compared with control. The effect of (Leu31, Pro34) − NPY was demonstrated to be mediated by Y1 receptor activation since the coapplication of Y1 receptor antagonist BIBP 3226 (1 µM, n = 3) and (Leu31, Pro34) − NPY was able to prevent the agonist effect (Figure 5(b)). Moreover, the application of the Y5 agonist alone did not affect the RGC response to NMDA, suggesting that the effect found for (Leu31, Pro34) − NPY is not mediated by Y5 receptor activation. Focusing on the RGCs that presented more pronounced differences between responses to first and second NMDA stimulus, we calculated the percentage of RGCs presenting Delta 2/Delta 1 ratio values below 0.9 for each independent experiment (Figure 5(c)), thus, overcoming the masking effect associated with overall population mean calculation. As expected, the application of (Leu31, Pro34) − NPY increased the percentage of cells (79.0 ± 6.9% of the cells analyzed) with Delta 2/Delta 1 ratio below 0.9, when compared with control (40.8 ± 5.0% of the cells analyzed; Figure 5(c)). Also, the Y1 receptor antagonist, BIBP 3226, was able to prevent the effect of (Leu31, Pro34) − NPY, confirming the involvement of Y1 receptor activation. No statistically significant difference was detected for the other drugs. We hypothesized that a possible opposite effect of Y2 activation might be responsible for counteracting the effect of Y1 activation by NPY. In this regard, the application of Y2 receptor agonist NPY13–36 resulted in a Delta 2/Delta 1 ratio of 1.2 ± 0.1 (Figure 5(b)), and the percentage of cells with Delta 2/Delta 1 ratio below 0.9 was 24.8 ± 7.2% (Figure 5(c)), both values apparently in opposite direction of Y1 receptor activation, although no statistically significant difference was detected.
Figure 5.

Y1 receptor activation decreases NMDA-stimulated RGC spiking activity. (a) Representative recordings of RGC spiking activity in ex vivo retina using MEA. Note the increase in spiking rate upon 30 µM NMDA. After washout of first NMDA stimulus, 1 µM (Leu31, Pro34) − NPY was applied for 10 min followed by a second NMDA stimulus. (b) The Delta 2 (second stimulus)/Delta 1 (first stimulus) ratios are presented for different drug applications, for 10 min, between NMDA stimuli: 1 µM NPY, 1 µM (Leu31, Pro34) − NPY, 1 µM NPY13–36, 1 µM (Gly1, . . . Aib32)-PP, or a drug-free solution (control). The application of (Leu31, Pro34) − NPY was able to reduce the NMDA-stimulated RGC spiking activity. This effect was blocked by the Y1 receptor antagonist BIBP 3226 (1 µM). (c) Percentage of RGCs presenting Delta 2/Delta 1 ratio below 0.9. The application of (Leu31, Pro34) − NPY significantly increased the percentage of cells with Delta 2/Delta 1 ratio below 0.9. BIBP 3226 was able to block the effect of (Leu31, Pro34) − NPY confirming the involvement of Y1 receptor activation. Data are presented as mean ± SEM of n = 3 to 10 independent experiments. **p < .01, compared with control; #p < .05, compared with (Leu31, Pro34) − NPY. Kruskal-Wallis followed by Dunn’s test.
NPY Reduces NMDA-Induced Cell Death in the GCL in Retinal Explants via Y1 and Y5 Receptor Activation
NPY exerts neuroprotective actions in various brain regions, and also in the retina (Alvaro et al., 2008b; Santos-Carvalho et al., 2013a), suggesting that NPY receptors might be possible therapeutic targets for retinal degenerative diseases such as glaucoma. Therefore, in addition to the NPY modulatory effect on RGC activity detected in ex vivo retinas, we also evaluated whether NPY could be able to prevent retinal cell death induced by an excitotoxic insult. The excitotoxic insult was induced in retinal explants (cultured for four DIV) exposed to 300 µM NMDA for 48 h. Apoptotic retinal cell death was assessed by TUNEL assay, and necrotic or late apoptotic cell death was assessed by PI incorporation assay (Figure 6). Different drug treatments were applied to assess the potential protective effect of NPY receptor activation. Thus, NPY (1 µM, n = 6 retinas for TUNEL, n = 9 retinas for PI), the Y1/Y5 agonist (Leu31, Pro34) − NPY (1 µM, n = 6 for TUNEL, n = 7 for PI), the Y2 agonist NPY13–36, (300 nM, n = 6 for TUNEL, n = 11 for PI), the Y5 agonist (Gly1,…Aib32)-PP (1 µM, n = 6 for TUNEL, n = 6 for PI), or a drug-free solution (control, n = 11 for TUNEL, n = 10 for PI), were applied at DIV1 and DIV2, respectively 24 h and 60 min before NMDA treatment.
Figure 6.

NPY reduces NMDA-induced cell death in the GCL of retinal explants via Y1 and Y5 receptor activation. Retinal explants were exposed to 300 µM NMDA for 48 h to induce an excitotoxic insult. The agonists, 1 µM NPY, 1 µM (Leu31, Pro34) − NPY, 300 nM NPY13–36, and 1 µM (Gly1, . . . Aib32)-PP, were applied 24 h and 60 min before NMDA application. All the antagonists: 1 µM BIBP 3226, 1 µM BIIE 0246, and 1 µM L-152,804 were applied 30 min before the agonist. (a) Representative images of TUNEL-positive cells (green) in the GCL of control (aa) or NMDA-treated (ab-af) retinal explants. Nuclei were stained with DAPI (blue). Retinal explants were pretreated with different drugs 24 h before exposure to NMDA. Scale bar: 100 µm. (b) Quantification of TUNEL-positive cells. NMDA exposure induced a significant increase in the number of TUNEL-positive cells in the GCL compared with control. Pretreatment with 1 µM NPY or 1 µM (Leu31, Pro34) − NPY (Y1/Y5 receptor agonist), 24 h and 60 min before NMDA, was able to reduce the increase in the number of TUNEL-positive cells induced by NMDA. Pretreatment with Y1 or Y5 receptor antagonist (1 µM BIBP 3226 or 1 µM L-152,804, respectively), when applied 30 min before NPY, blocked its protective effect. The effect of (Leu31, Pro34) − NPY was blocked by BIBP 3226. (c) NMDA exposure induced a significant increase in the number of PI-positive cells in retinal explants. Data are presented as the ratio between PI-positive cells at DIV4 and DIV2. NPY or (Leu31, Pro34) − NPY was able to reduce the increase in the number of PI-positive cells induced by NMDA. BIBP 3226 or L-152,804 blocked the protective effect of NPY, and BIBP 3226 blocked the protective effect of (Leu31, Pro34) − NPY. The application of 1 µM (Gly1, . . . Aib32)-PP was able to reduce the increase in PI-positive cells induced by NMDA, and this effect was blocked by L-152,804. Together, the results from TUNEL and PI assays suggest that NPY is able to protect retinal cells from an excitotoxic insult through the activation of Y1 and Y5 receptors. Data are presented as mean ± SEM of n = 5 to 11 independent experiments. ***p < .001, compared with control; ##p < .01, ###p < .001, compared with NMDA; §p < .05, §§p < .01, §§§p < .001, compared with NMDA + NPY, or NMDA + (Leu31, Pro34) − NPY, or NMDA + (Gly1, . . . Aib32)-PP. Kruskal-Wallis followed by Dunn’s test.
Exposure of retinal explants to NMDA for 48 h increased the number of TUNEL-positive cells in the GCL, up to 16.3 ± 1.3 times higher than in control (Figure 6(b)). There was also an increase in the DIV4/DIV2 ratio for PI-positive cells compared with control, namely 3.4 ± 0.2 in NMDA treated explants versus 1.3 ± 0.1 in control (Figure 6(c)). Pretreatment with NPY was able to reduce the increase in the number of TUNEL-positive cells induced by NMDA (4.8 ± 0.5 times higher than in control). NPY pretreatment was also able to reduce the increase in DIV4/DIV2 ratio for PI-positive cells (1.4 ± 0.2; Figure 6(c)). The Y1 antagonist BIBP 3226 (1 µM, n = 5 for TUNEL, n = 7 for PI) or the Y5 antagonist L-152,804 (1 µM, n = 5 for TUNEL, n = 9 for PI), when applied 30 min before NPY, was able to block its protective effect both on TUNEL and PI assays, indicating the involvement of Y1 and Y5 receptors on the neuroprotective effects of NPY. The protective effect of Y1 receptor activation was confirmed also by the application of (Leu31, Pro34) − NPY, which was able to reduce the NMDA-induced increase in TUNEL- and PI-positive cells. Again, the Y1 antagonist BIBP 3226 (n = 6 for TUNEL, n = 5 for PI) blocked this effect. Concerning the Y5 receptor activation, the use of (Gly1, . . . Aib32)-PP reduced the number of TUNEL-positive cells comparing to NMDA-treated explants, though no statistically significant difference was detected. Accordingly, Y5 receptor activation was able to reduce the number of PI-positive cells compared with NMDA condition. This later effect was blocked by L-152,804 (n = 5 for TUNEL, n = 5 for PI), thus confirming the protective effect of Y5 receptor activation. The Y2 receptor was not involved in the protective action of NPY since the Y2 antagonist BIIE 0246 (1 µM, n = 5 for TUNEL, n = 8 for PI) did not block the neuroprotective effect of NPY. Also, NPY13–36 did not reduce the increase in TUNEL- and PI-positive cells induced by NMDA in retinal explants.
Intravitreal Administration of NPY or (Leu31, Pro34) − NPY Does Not Prevent Retinal Cell Death Induced by I-R Injury
In a previous study from our group, pretreatment with NPY before the intravitreal injection of glutamate was shown to afford neuroprotection to RGCs (Santos-Carvalho et al., 2013a). Here, we explored the potential neuroprotective action of NPY in an animal model of retinal damage induced by I-R injury (Figure 7). Saline (n = 5 animals), 10 µg (2.34 nmol) NPY (n = 6), or 10 µg (2.36 nmol; Leu31, Pro34) − NPY (Y1/Y5 receptor agonist, n = 5) were intravitreally injected 2 h prior to I-R injury. Retinal cell death was assessed by TUNEL assay. I-R injury induced an increase in the number of TUNEL-positive cells (31.8 ± 6.6) per mm of retinal section length across all the retinal nuclear layers compared with the retina of the contralateral eye (0.6 ± 0.3 TUNEL-positive cells; Figure 7(b)). This increase was not prevented by pretreatment with NPY or (Leu31, Pro34) − NPY. RGC survival was evaluated by counting the number of Brn3a-immunoreactive cells, a RGC marker Brn3a (Nadal-Nicolas et al., 2012). We found that I-R injury decreased the number of Brn3a-positive RGCs per mm of retinal section length to 12.0 ± 2.5 RGCs compared with the contralateral eye (26.0 ± 4.5 Brn3a-positive cells), and again, this effect was not prevented by NPY or (Leu31, Pro34) − NPY administration (Figure 7(c)). We also performed ERGs in order to assess the functional effect of I-R injury on retinal light responses and a potential protective effect of NPY. Scotopic and photopic ERGs were recorded before the onset of I-R injury (baseline) and after 24 h of reperfusion (Figure 7(d)). We found that I-R injury induced a clear decrease in scotopic and photopic b-wave amplitudes but not in scotopic a-wave amplitude (data not shown). Similarly to results on TUNEL and Brn3a assays, the NPY or (Leu31, Pro34) − NPY administration before I-R injury did not prevent the reduction in ERG b-wave amplitudes. Together, these results indicate that, contrarily to the protective action against an excitotoxic injury in retinal explants, NPY or (Leu31, Pro34) − NPY pretreatment was not able to prevent retinal I-R injury within 24 h of reperfusion.
Figure 7.

Intravitreal administration of NPY or (Leu31, Pro34) − NPY did not prevent cell death induced by retinal I-R injury at 24 h of reperfusion. Retinal ischemia was induced for 60 min followed by 24 h of reperfusion. Ischemia was induced in one eye, and the contralateral eye was taken as an internal control. (a) Representative images of retinal sections showing TUNEL-positive cells (green) and Brn3a-positive RGCs (red). Nuclei were stained with DAPI (blue). Saline (aa, ad), 10 µg (2.34 nmol) NPY (ab, ae), or 10 µg (2.36 nmol; Leu31, Pro34) − NPY (ac, af) were intravitreally injected 2 h before the onset of I-R injury. GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Scale bar: 50 µm. (b) TUNEL-positive cells are Figure 7. Continued. expressed per mm of section length. I-R injury induced an increase in the number of TUNEL-positive cells. NPY or (Leu31, Pro34) − NPY administration was not able to reduce the number of TUNEL-positive cells. (c) Brn3a-positive RGCs were expressed per mm of section length. NPY or (Leu31, Pro34) − NPY administration was not able to prevent the reduction in Brn3a-positive RGC number induced by I-R injury. (d) Examples of scotopic ERG traces for saline (da, dd), NPY-treated (db, de), or (Leu31, Pro34) − NPY-treated eyes (dc, df). ERG recordings were performed before (baseline) and 24 h after I-R injury. Note that in the injured eye the b-wave was reduced (dd). NPY or (Leu31, Pro34) − NPY administration did not prevent the reduction in b-wave (de, df). Data are presented as mean ± SEM of n = 5 to 6 independent experiments. *p < .05, **p < .01, compared with contralateral eye. Kruskal-Wallis followed by Dunn’s test.
Discussion
The presence of mRNA encoding NPY and NPY receptors (Y1, Y2, Y4, and Y5) in the rat retina has been demonstrated in previous studies (D’Angelo and Brecha, 2004; Alvaro et al., 2007). We here present evidence that NPY and NPY receptors are specifically present in freshly isolated RGCs (Figure 1). NPY-ir in the retina has been evaluated in different species (Santos-Carvalho et al., 2014). In human retina, NPY-ir was found in a subset of amacrine cells and RGCs, with processes extending mainly in the IPL (Tornqvist and Ehinger, 1988; Straznicky and Hiscock, 1989). In rat retina, NPY-ir was reported to localize to cell bodies of amacrine cells in INL and displaced amacrine cells in GCL, and to colocalize mainly with GABAergic neurons. The corresponding cell processes extend and ramify mainly in strata 1, 3, and 5 of IPL (Oh et al., 2002). We confirmed these observations using retinal slices of adult rats (Figure 1). In particular, we found NPY-ir in a purified culture of RGCs obtained from P3 to P4 pups. Previously, we also suggested the presence of NPY-ir in different retinal cell types in primary cultures (Alvaro et al., 2007). Regarding NPY receptor localization, only a few studies addressed this issue. Y1 receptor-ir was detected in glial cells of diseased human retina and in horizontal and amacrine cells of rat retina (Canto Soler et al., 2002; D’Angelo et al., 2002), and we have previously detected immunoreactivity for Y1 and Y2 receptors in neurons and glial cells in cultured retinal cells (Santos-Carvalho et al., 2013b). Moreover, we and other authors have provided functional evidence for the presence of Y1, Y2, Y4, and Y5 receptors in retinal cells (Bruun et al., 1994; D’Angelo and Brecha, 2004; Milenkovic et al., 2004; Alvaro et al., 2008a; Alvaro et al., 2009; Santos-Carvalho et al., 2013a). Regarding purified RGC cultures, we detected immunoreactivity for NPY and Y1, Y2, Y4, and Y5 receptors in Brn3a-positive RGCs. In retinal sections, Y1-ir was found in INL and strata 2 and 4 of IPL as previously reported (D’Angelo et al., 2002). In addition, we detected Y1-ir RGCs (Figure 8), which is in agreement with the labeling found in purified RGC cultures. We described, for the first time, the localization of Y2 receptor-ir in stratum 1 of IPL and in cell bodies in proximal INL. We also detected for the first time Y4 and Y5 receptor-ir in retinal sections. Y4 receptor-ir was localized to cell bodies of GCL, and proximal and distal INL. The cell bodies in GCL that were immunoreactive for Y4 receptor were both RGC (colocalized with Brn3a) and nonBrn3a-positive cells, likely displaced amacrine cells. Concerning the Y5 receptor, immunoreactivity was detected in Müller cells. The lack of clear immunoreactivity for NPY, and for Y2 and Y5 receptors in RGCs in retinal sections of adult rat, while it could be found in cultured RGCs from P3 to P4 rats, may be explained by decreased expression in adulthood or related with cell culture conditions, which might favor the expression of NPY and NPY receptors. We also found functional active NPY receptors in the inner retina using [35S]GTPγS-binding assay (Figure 2). Binding signal detected in the photoreceptor layer might represent the high amount of G proteins in photoreceptor outer segments, mainly transducin (Arshavsky et al., 2002).
Figure 8.

NPY receptor activation leads to RGC modulation and increases cell survival in the inner retina. (a) NPY receptor location in the different layers of the adult rat retina. (b) The activation of Y1 receptor leads to decreased Ca2+ influx in RGCs, and also increased OFF RGC response, potentially by inhibiting ON-driven surround antagonism in OFF-center RGCs. The activation of Y1 in RGCs and amacrine cells and Y5 in Müller cells leads to increased cell survival in the inner retina. GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer.
NPY has been associated with inhibitory actions after electrical or chemical stimulation of excitatory synaptic transmission (El Bahh et al., 2002; Tu et al., 2006; Kovac et al., 2011). In fact, NPY was reported to decrease depolarization-evoked increase in [Ca2+]i by inhibiting voltage-dependent Ca2+ channels. This effect was reported in a variety of neuronal cell types such as dorsal root ganglion neurons (Bleakman et al., 1991), hippocampal neurons (Bleakman et al., 1992), submandibular ganglion neurons (Endoh et al., 2012), and hypothalamic arcuate nucleus neurons (Sun and Miller, 1999). Moreover, the activation of Y1 receptors has been associated with inhibition of neurotransmitter release (Bitran et al., 1999; Hastings et al., 2004; Wang, 2005), though the opposite behavior can be found depending on the neurotransmitter or cell type (Hastings et al., 2001). In the retina, Y2 receptor activation attenuated depolarization-evoked Ca2+ influx in rod bipolar cell terminals (D’Angelo and Brecha, 2004). In addition, previous work in our laboratory has shown that NPY application to retinal cell cultures was able to reduce depolarization-evoked [Ca2+]i increase via activation of Y1 and Y5 receptors (Alvaro et al., 2009). Thus, the present study was aimed at evaluating the effect of direct application of NPY or NPY receptor agonists on [Ca2+]i changes in RGCs, using a purified RGC culture.
Glutamate is the major retinal excitatory neurotransmitter mediating the transmission of visual information from bipolar cells to RGCs. In this experiment, 30 µM glutamate plus 10 µM glycine was used to stimulate Ca2+ influx into cultured RGCs by activating both NMDA and nonNMDA glutamate receptors and indirectly voltage-dependent calcium channels, as previously described (Hartwick et al., 2004). However, under these conditions, Ca2+ influx was found to be primarily mediated by NMDA glutamate receptors (Hartwick et al., 2008). We now present new evidence showing an inhibitory effect of NPY on the glutamate-evoked increase in [Ca2+]i in purified RGCs through Y1 receptor activation (Figure 3). This result suggests that NPY, released from amacrine cells in the rat retina (Oh et al., 2002), may activate postsynaptic Y1 receptors on RGCs inhibiting glutamate receptors or voltage-dependent calcium channels, thus modulating the effect of glutamate released from bipolar cells (Figure 8). The specific activation of Y2 or Y5 receptors did not evoke any significant change in [Ca2+]i in purified RGCs (Figure 3(f)), although these receptors were expressed in similar purified RGC cultures (Figure 1(b)). This may result from the presence of nonfunctional Y2 and Y5 receptors in these cultures.
We further explored the modulatory potential of NPY on RGC spiking activity using a MEA technique (Meister et al., 1994). Although application of NPY or NPY receptor agonists tested did not affect the spontaneous spiking activity, a small but statistically significant increase was found at the initial burst response of OFF-type RGCs upon the application of Y1 and Y5 receptor agonist (Leu31, Pro34) − NPY (Figures 4 and 8). We speculate that the modulatory effects of NPY receptor activation may be exerted within the circuitry generating center-surround organization within the receptive field of OFF-type RGCs at the level of amacrine cell-RGCs (Sinclair et al., 2004). A decreased contribution of ON bipolar cell signal to this circuitry might not be responsible for this effect since activation of Y2 receptors, but neither Y1 nor Y5, was reported to inhibit rod bipolar cells (D’Angelo and Brecha, 2004). Another possibility is that the modulatory mechanism may reside at the level of horizontal cell circuitry in OPL, since it contributes to center-surround organization (Wassle and Boycott, 1991), and the Y1 receptor localizes to rat horizontal cells (D’Angelo et al., 2002).
In another experimental paradigm, ex vivo rat retinal preparations were exposed to NMDA in order to induce an excitation of RGCs. We chose NMDA instead of glutamate since in intact retina even high concentrations of glutamate have no effect on [Ca2+]i in RGCs (Hartwick et al., 2004), due to the presence of glutamate uptake systems (Thoreson and Witkovsky, 1999). We clearly show for the first time that Y1 receptor activation is able to modulate directly RGC responses by attenuating the NMDA-induced increase in RGC spiking activity (Figure 5). A possible effect of NPY application cannot be excluded, but it might be masked by an effect of Y2 activation counteracting the effect of Y1 activation by NPY. These results suggest that the activation of Y1 receptors present on RGC dendrites is responsible for the modulatory effect observed. Nevertheless, the contribution of Y1 receptor expressing amacrine cell inputs onto RGC dendrites may also be important in this modulation (D’Angelo et al., 2002). Additional neuroanatomical and electrophysiological studies are needed to clarify the cellular contribution (RGCs vs. amacrine cells) and localization of Y1 receptor activation responsible for the alterations in RGC spiking activity, as well as whether Y1 receptor activation is associated with a particular RGC type or modulates a specific visual task (Gollisch and Meister, 2010).
NPY has been shown to prevent neuronal cell death, also in the retina, induced by excitotoxic insults (Silva et al., 2003; Xapelli et al., 2007; Santos-Carvalho et al., 2013a). To further address the potential neuroprotective role of NPY in the retina, we used an in vitro model, cultured retinal explants exposed to NMDA, as well as an animal model of retinal I-R injury. Both cultured retinal explants and the I-R model have been previously used to evaluate neuroprotective strategies targeting retinal neurons, specially RGCs (Lagreze et al., 1998; Goodyear and Levin, 2008; Peng et al., 2011; Zhang et al., 2012). We now found that NPY pretreatment was able to prevent NMDA-induced cell death in retinal explants, through the activation of Y1 and Y5 receptors (Figures 6 and 8). This result is in agreement with previous studies showing NPY neuroprotective effects under activation of Y1 or Y5 receptors (Silva et al., 2003; Xapelli et al., 2007; Smialowska et al., 2009), including in retinal neurons (Santos-Carvalho et al., 2013a). However, in I-R injury model, pretreatment with NPY or (Leu31, Pro34) − NPY was not able to prevent cell death or rescue RGCs (Figure 7). This observation contrasts with the protective effect of NPY when injected intravitreally before the intravitreal injection of glutamate, in a model of glutamate-induced injury in the retina (Santos-Carvalho et al., 2013a). In fact, there is a complex involvement of a series of events leading to cell injury triggered by I-R, compared with the exposure to glutamate injected into the vitreous. Those events include primarily the formation of free radicals during the early stage of reperfusion which overwhelms normal cellular antioxidant defense mechanisms, as well as inflammation and excitotoxicity (Osborne et al., 2004). Taking into account the protective effects we found using the glutamate intravitreal injection model as well as the protective effect in retinal explants, clearly indicating that NPY can be neuroprotective in the retina, it is likely that a protective effect could be obtained with a less severe insult. Therefore, further studies are needed in order to evaluate whether NPY neuroprotective effects detected in cultured retinal explants can be translated into animal models of retinal degenerative diseases.
Summary
In this study, we found that neuropeptide Y (NPY) and NPY receptors are present in retinal ganglion cells (RGCs). The activation of Y1 receptor modulates intracellular calcium levels and also spiking activity in RGCs.
Author contribution
J. M., F. E., D. B., B. K., D. P. D. W., S. P., and A. F. A. designed research; J. M., F. E., D. B., T. M., B. K., P. T., C. R. G, and A. R. S. performed research; J. M., C. C., M. C. B, D. P. D.W, S. P, A. R. S, and A. F. A analyzed data; and J. M., A. R. S., and A. F. A wrote the paper. All authors read and corrected the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fundação para a Ciência e a Tecnologia, Portugal [PTDC/SAU-NEU/099075/2008, PTDC/NEU-OSD/1113/2012, PEst-C/SAU/UI3282/2011-2013, PEst-C/SAU/LA0001/2011-2013, UID/NEU/04539/2013, SFRH/BD/44817/2008], COMPETE/FEDER, and AIBILI.
References
- Alvaro A. R., Martins J., Araujo I. M., Rosmaninho-Salgado J., Ambrosio A. F., Cavadas C. (2008a) Neuropeptide Y stimulates retinal neural cell proliferation—Involvement of nitric oxide. Journal of Neurochemistry 105: 2501–2510. [DOI] [PubMed] [Google Scholar]
- Alvaro A. R., Martins J., Costa A. C., Fernandes E., Carvalho F., Ambrosio A. F., Cavadas C. (2008b) Neuropeptide Y protects retinal neural cells against cell death induced by ecstasy. Neuroscience 152: 97–105. [DOI] [PubMed] [Google Scholar]
- Alvaro A. R., Rosmaninho-Salgado J., Ambrosio A. F., Cavadas C. (2009) Neuropeptide Y inhibits [Ca2+]i changes in rat retinal neurons through NPY Y1, Y4, and Y5 receptors. Journal of Neurochemistry 109: 1508–1515. [DOI] [PubMed] [Google Scholar]
- Alvaro A. R., Rosmaninho-Salgado J., Santiago A. R., Martins J., Aveleira C., Santos P. F., Cavadas C. (2007) NPY in rat retina is present in neurons, in endothelial cells and also in microglial and Muller cells. Neurochemistry International 50: 757–763. [DOI] [PubMed] [Google Scholar]
- Arshavsky V. Y., Lamb T. D., Pugh E. N., Jr (2002) G proteins and phototransduction. Annual Review of Physiology 64: 153–187. [DOI] [PubMed] [Google Scholar]
- Babilon S., Morl K., Beck-Sickinger A. G. (2013) Towards improved receptor targeting: Anterograde transport, internalization and postendocytic trafficking of neuropeptide Y receptors. Biological Chemistry 394: 921–936. [DOI] [PubMed] [Google Scholar]
- Barres B. A., Silverstein B. E., Corey D. P., Chun L. L. (1988) Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1: 791–803. [DOI] [PubMed] [Google Scholar]
- Benarroch E. E. (2009) Neuropeptide Y: Its multiple effects in the CNS and potential clinical significance. Neurology 72: 1016–1020. [DOI] [PubMed] [Google Scholar]
- Bitran M., Tapia W., Eugenin E., Orio P., Boric M. P. (1999) Neuropeptide Y induced inhibition of noradrenaline release in rat hypothalamus: Role of receptor subtype and nitric oxide. Brain Research 851: 87–93. [DOI] [PubMed] [Google Scholar]
- Bleakman D., Colmers W. F., Fournier A., Miller R. J. (1991) Neuropeptide Y inhibits Ca2+ influx into cultured dorsal root ganglion neurones of the rat via a Y2 receptor. British Journal of Pharmacology 103: 1781–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakman D., Harrison N. L., Colmers W. F., Miller R. J. (1992) Investigations into neuropeptide Y-mediated presynaptic inhibition in cultured hippocampal neurones of the rat. British Journal of Pharmacology 107: 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruun A., Edvinsson L., Ehinger B. (1994) Neuropeptide Y inhibits adenylyl cyclase activity in rabbit retina. Acta Ophthalmologica (Copenhagen) 72: 326–331. [DOI] [PubMed] [Google Scholar]
- Bruun A., Ehinger B. (1993) NPY-induced neurotransmitter release from the rabbit and chicken retina. Acta Ophthalmologica (Copenhagen) 71: 590–596. [DOI] [PubMed] [Google Scholar]
- Canto Soler M. V., Gallo J. E., Dodds R. A., Hokfelt T., Villar M. J., Suburo A. M. (2002) Y1 receptor of neuropeptide Y as a glial marker in proliferative vitreoretinopathy and diseased human retina. Glia 39: 320–324. [DOI] [PubMed] [Google Scholar]
- Chambers A. P., Woods S. C. (2012) The role of neuropeptide Y in energy homeostasis. Handbook of Experimental Pharmacology 209: 23–45. [DOI] [PubMed] [Google Scholar]
- Christensen D. Z., Olesen M. V., Kristiansen H., Mikkelsen J. D., Woldbye D. P. (2006) Unaltered neuropeptide Y (NPY)-stimulated [35S]GTPgammaS binding suggests a net increase in NPY signalling after repeated electroconvulsive seizures in mice. Journal of neuroscience research 84: 1282–1291. [DOI] [PubMed] [Google Scholar]
- D’Angelo I., Brecha N. C. (2004) Y2 receptor expression and inhibition of voltage-dependent Ca2+ influx into rod bipolar cell terminals. Neuroscience 125: 1039–1049. [DOI] [PubMed] [Google Scholar]
- D’Angelo I., Oh S. J., Chun M. H., Brecha N. C. (2002) Localization of neuropeptide Y1 receptor immunoreactivity in the rat retina and the synaptic connectivity of Y1 immunoreactive cells. Journal of Comparative Neurology 454: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrijevic M., Stanojevic S. (2013) The intriguing mission of neuropeptide Y in the immune system. Amino Acids 45: 41–53. [DOI] [PubMed] [Google Scholar]
- El Bahh B., Cao J. Q., Beck-Sickinger A. G., Colmers W. F. (2002) Blockade of neuropeptide Y(2) receptors and suppression of NPY’s anti-epileptic actions in the rat hippocampal slice by BIIE0246. British Journal of Pharmacology 136: 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh T., Nobushima H., Tazaki M. (2012) Neuropeptide Y modulates calcium channels in hamster submandibular ganglion neurons. Neuroscience Research 73: 275–281. [DOI] [PubMed] [Google Scholar]
- Gollisch T., Meister M. (2010) Eye smarter than scientists believed: Neural computations in circuits of the retina. Neuron 65: 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear E., Levin L. A. (2008) Model systems for experimental studies: Retinal ganglion cells in culture. Progress in Brain Research 173: 279–284. [DOI] [PubMed] [Google Scholar]
- Hartwick A. T., Hamilton C. M., Baldridge W. H. (2008) Glutamatergic calcium dynamics and deregulation of rat retinal ganglion cells. The Journal of Physiology 586: 3425–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwick A. T., Lalonde M. R., Barnes S., Baldridge W. H. (2004) Adenosine A1-receptor modulation of glutamate-induced calcium influx in rat retinal ganglion cells. Investigative Ophthalmology and Visual Science 45: 3740–3748. [DOI] [PubMed] [Google Scholar]
- Hastings J. A., McClure-Sharp J. M., Morris M. J. (2001) NPY Y1 receptors exert opposite effects on corticotropin releasing factor and noradrenaline overflow from the rat hypothalamus in vitro. Brain Research 890: 32–37. [DOI] [PubMed] [Google Scholar]
- Hastings J. A., Morris M. J., Lambert G., Lambert E., Esler M. (2004) NPY and NPY Y1 receptor effects on noradrenaline overflow from the rat brain in vitro. Regulatory Peptides 120: 107–112. [DOI] [PubMed] [Google Scholar]
- Higgs M. H., Lukasiewicz P. D. (1999) Glutamate uptake limits synaptic excitation of retinal ganglion cells. Journal of Neuroscience 19: 3691–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch D., Zukowska Z. (2012) NPY and stress 30 years later: The peripheral view. Cellular and Molecular Neurobiology 32: 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac S., Megalogeni M., Walker M. (2011) In vitro effects of neuropeptide Y in rat neocortical and hippocampal tissue. Neuroscience Letters 492: 43–46. [DOI] [PubMed] [Google Scholar]
- Lagreze W. A., Knorle R., Bach M., Feuerstein T. J. (1998) Memantine is neuroprotective in a rat model of pressure-induced retinal ischemia. Investigative Ophthalmology and Visual Science 39: 1063–1066. [PubMed] [Google Scholar]
- Lee N. J., Herzog H. (2009) NPY regulation of bone remodelling. Neuropeptides 43: 457–463. [DOI] [PubMed] [Google Scholar]
- Malva J. O., Xapelli S., Baptista S., Valero J., Agasse F., Ferreira R., Silva A. P. (2012) Multifaces of neuropeptide Y in the brain—Neuroprotection, neurogenesis and neuroinflammation. Neuropeptides 46: 299–308. [DOI] [PubMed] [Google Scholar]
- Meister M., Pine J., Baylor D. A. (1994) Multi-neuronal signals from the retina: Acquisition and analysis. Journal of Neuroscience Methods 51: 95–106. [DOI] [PubMed] [Google Scholar]
- Michel M. C., Beck-Sickinger A., Cox H., Doods H. N., Herzog H., Larhammar D, Westfall T. (1998) XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide, Y, peptide, YY, and pancreatic polypeptide receptors. Pharmacological Reviews 50: 143–150. [PubMed] [Google Scholar]
- Milenkovic I., Weick M., Wiedemann P., Reichenbach A., Bringmann A. (2004) Neuropeptide Y-evoked proliferation of retinal glial (Muller) cells. Graefe's Archive for Clinical and Experimental Ophthalmology 242: 944–950. [DOI] [PubMed] [Google Scholar]
- Nadal-Nicolas F. M., Jimenez-Lopez M., Salinas-Navarro M., Sobrado-Calvo P., Alburquerque-Bejar J. J., Vidal-Sanz M., Agudo-Barriuso M. (2012) Whole number, distribution and co-expression of brn3 transcription factors in retinal ganglion cells of adult albino and pigmented rats. PloS One 7: e49830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. J., D’Angelo I., Lee E. J., Chun M. H., Brecha N. C. (2002) Distribution and synaptic connectivity of neuropeptide Y-immunoreactive amacrine cells in the rat retina. Journal of Comparative Neurology 446: 219–234. [DOI] [PubMed] [Google Scholar]
- Osborne N. N., Casson R. J., Wood J. P., Chidlow G., Graham M., Melena J. (2004) Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Progress in Retinal and Eye Research 23: 91–147. [DOI] [PubMed] [Google Scholar]
- Peng P. H., Chao H. M., Juan S. H., Chen C. F., Liu J. H., Ko M. L. (2011) Pharmacological preconditioning by low dose cobalt protoporphyrin induces heme oxygenase-1 overexpression and alleviates retinal ischemia-reperfusion injury in rats. Current Eye Research 36: 238–246. [DOI] [PubMed] [Google Scholar]
- Perney T. M., Miller R. J. (1989) Two different G-proteins mediate neuropeptide Y and bradykinin-stimulated phospholipid breakdown in cultured rat sensory neurons. The Journal of Biological Chemistry 264: 7317–7327. [PubMed] [Google Scholar]
- Rosmaninho-Salgado J., Cortez V., Estrada M., Santana M. M., Goncalves A., Marques A. P., Cavadas C. (2012) Intracellular mechanisms coupled to NPY Y2 and Y5 receptor activation and lipid accumulation in murine adipocytes. Neuropeptides 46: 359–366. [DOI] [PubMed] [Google Scholar]
- Santos-Carvalho A., Alvaro A. R., Martins J., Ambrosio A. F., Cavadas C. (2014) Emerging novel roles of neuropeptide Y in the retina: From neuromodulation to neuroprotection. Progress in Neurobiology 112: 70–79. [DOI] [PubMed] [Google Scholar]
- Santos-Carvalho A., Aveleira C. A., Elvas F., Ambrosio A. F., Cavadas C. (2013b) Neuropeptide Y receptors Y1 and Y2 are present in neurons and glial cells in rat retinal cells in culture. Investigative Ophthalmology and Visual Science 54: 429–443. [DOI] [PubMed] [Google Scholar]
- Santos-Carvalho A., Elvas F., Alvaro A. R., Ambrosio A. F., Cavadas C. (2013a) Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Disease 4: e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Liu X. L., Yang X. L. (2006) N-methyl-D-aspartate receptors in the retina. Molecular Neurobiology 34: 163–179. [DOI] [PubMed] [Google Scholar]
- Silva A. P., Cavadas C., Grouzmann E. (2002) Neuropeptide Y and its receptors as potential therapeutic drug targets. Clinica Chimica Acta 326: 3–25. [DOI] [PubMed] [Google Scholar]
- Silva A. P., Pinheiro P. S., Carvalho A. P., Carvalho C. M., Jakobsen B., Zimmer J., Malva J. O. (2003) Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB Journal 17: 1118–1120. [DOI] [PubMed] [Google Scholar]
- Silva A. P., Xapelli S., Grouzmann E., Cavadas C. (2005) The putative neuroprotective role of neuropeptide Y in the central nervous system. Current drug targets. CNS and neurological disorders 4: 331–347. [DOI] [PubMed] [Google Scholar]
- Sinclair J. R., Jacobs A. L., Nirenberg S. (2004) Selective ablation of a class of amacrine cells alters spatial processing in the retina. The Journal of Neuroscience 24: 1459–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smialowska M., Domin H., Zieba B., Kozniewska E., Michalik R., Piotrowski P., Kajta M. (2009) Neuroprotective effects of neuropeptide Y–Y2 and Y5 receptor agonists in vitro and in vivo. Neuropeptides 43: 235–249. [DOI] [PubMed] [Google Scholar]
- Sousa-Ferreira L., Garrido M., Nascimento-Ferreira I., Nobrega C., Santos-Carvalho A., Alvaro A. R., Cavadas C. (2011) Moderate long-term modulation of neuropeptide Y in hypothalamic arcuate nucleus induces energy balance alterations in adult rats. PloS One 6: e22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straznicky C., Hiscock J. (1989) Neuropeptide Y-like immunoreactivity in neurons of the human retina. Vision Research 29: 1041–1048. [DOI] [PubMed] [Google Scholar]
- Sun L., Miller R. J. (1999) Multiple neuropeptide Y receptors regulate K+ and Ca2+ channels in acutely isolated neurons from the rat arcuate nucleus. Journal of Neurophysiology 81: 1391–1403. [DOI] [PubMed] [Google Scholar]
- Thoreson W. B., Witkovsky P. (1999) Glutamate receptors and circuits in the vertebrate retina. Progress in Retinal and Eye Research 18: 765–810. [DOI] [PubMed] [Google Scholar]
- Tornqvist K., Ehinger B. (1988) Peptide immunoreactive neurons in the human retina. Investigative Ophthalmology & Visual Science 29: 680–686. [PubMed] [Google Scholar]
- Tu B., Jiao Y., Herzog H., Nadler J. V. (2006) Neuropeptide Y regulates recurrent mossy fiber synaptic transmission less effectively in mice than in rats: Correlation with Y2 receptor plasticity. Neuroscience 143: 1085–1094. [DOI] [PubMed] [Google Scholar]
- Wang S. J. (2005) Activation of neuropeptide Y Y1 receptors inhibits glutamate release through reduction of voltage-dependent Ca2+ entry in the rat cerebral cortex nerve terminals: Suppression of this inhibitory effect by the protein kinase C-dependent facilitatory pathway. Neuroscience 134: 987–1000. [DOI] [PubMed] [Google Scholar]
- Wassle H., Boycott B. B. (1991) Functional architecture of the mammalian retina. Physiological Reviews 71: 447–480. [DOI] [PubMed] [Google Scholar]
- Xapelli S., Silva A. P., Ferreira R., Malva J. O. (2007) Neuropeptide Y can rescue neurons from cell death following the application of an excitotoxic insult with kainate in rat organotypic hippocampal slice cultures. Peptides 28: 288–294. [DOI] [PubMed] [Google Scholar]
- Yannielli P., Harrington M. E. (2004) Let there be “more” light: Enhancement of light actions on the circadian system through non-photic pathways. Progress in Neurobiology 74: 59–76. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Qin X., Zhao X., Tong N., Gong Y., Zhang W., Wu X. (2012) Valproic acid regulates antioxidant enzymes and prevents ischemia/reperfusion injury in the rat retina. Current Eye Research 37: 429–437. [DOI] [PubMed] [Google Scholar]