

Abstract
Background
Recently, postnatal stem cells from dental papilla with neural crest origin have been considered as one of potent stem cell sources in regenerative medicine regarding their multi-differentiation capacity and relatively easy access. However, almost human oral tissues have been reported to be infected by mycoplasma which gives rise to oral cavity in teeth, and mycoplasma contamination of ex-vivo cultured stem cells from such dental tissues and its effect on stem cell culture has received little attention.
Results
In this study, mycoplama contamination was evaluated with stem cells from apical papilla which were isolated from human third molar and premolars from various aged patients undergoing orthodontic therapy. The ex-vivo expanded stem cells from apical papilla were found to express stem cell markers such as Stro-1, CD44, nestin and CD133, but mycoplama contamination was detected in almost all cell cultures of the tested 20 samples, which was confirmed by mycoplasma-specific gene expression and fluorescence staining. Such contaminated mycoplasma could be successfully eliminated using elimination kit, and proliferation test showed decreased proliferation activity in mycoplasma-contaminated cells. After elimination of contaminated mycoplasma, stem cells from apical papilla showed osteogenic and neural lineage differentiation under certain culture conditions.
Conclusion
Our study proposes that the evaluation of mycoplasma contamination and elimination process might be required in the use of stem cells from apical papilla for their potent applications to tissue engineering and regenerative medicine.
Keywords: Dental papilla, Stem cell, Mycoplasma, Proliferation, Differentiation, Osteogenic, Neural
Background
Tooth morphogenesis is known to be initiated from the cellular interactions between ectoderm-derived oral epithelial cells and neural crest-derived mesenchymes [1,2], and stem cell researches to generate osteoblasts and neurons have been done extensively using stem cells from dental tissues with such neural crest-derived ectomesenchyme origin [3-5]. To dates, various postnatal stem cells have been successfully isolated from the collected dental tissues such as dental pulp, periodontal ligament and dental papilla via non-invasive procurement process [3,6,7]. During tooth development, dentin and pulp tissue are known to be developed from dental papilla, which dental papilla is evident to have a pool of stem cells with high regeneration ability [8,9], and the majority of stem cell within dental papilla has been reported to be of neural crest-derived ectomesenchyme origin [10]. From these developmental origin, relatively easy accessibility of stem cell source from discarded tooth and possible autologous implantation via cryopreservation in dental stem cell bank, dental papilla has been suggested as potent stem cell source in regenerative medicine. For instance, in the field of tissue engineering and regenerative medicine, bioengineered tooth with the correct tooth structure was developed using stem cells from molar tooth germ tissue [11], in vivo bone formation could be generated via transplantation of tissue engineered bone tissue formed by cryopreserved dental stem cells [12,13], and the generation of functional neurons were reported to be from dental stem cell under neural inductive cues [14].
However, in spite of high applicability of stem cells from dental tissues in tissue engineering and regenerative medicine, mycoplasma contamination of the primary cultured stem cells from dental tissues where is easily infected by oral bacteria has been overlooked, and the evaluation and removal of infected mycoplasma in dental stem cells need to be considered, regarding biological safety in dental stem cells applications to regenerative medicine. It was reported that almost human oral tissue was frequently found to be infected small microorganism such as mycoplasma [15], and many kinds of bacteria give rise to oral cavity in teeth, and mycoplasma, the smallest and simplest self-replicating organisms, is known as one of major bacteria found in oral cavity [16]. Postnatal stem cells from infected dental tissues are evident to be infected by mycoplasma, and such mycoplama infection in dental tissues might influence dental tissue-derived stem cell’s behavior including cell proliferation. It is well known that mycoplasma infection influenced cell proliferation, chromosomal aberration in cells and induced immunological reactions [17,18]. Hence, in this study, postnatal stem cells were primarily isolated and cultured from apical papilla of the third molar and premolar teeth from various aged patients undergoing orthodontic therapy, and mycoplasma contamination was evaluated for each isolated stem cells from apical papilla (hSCAPs) of human teeth. The cell proliferation capacity was also tested with mycoplasma-infected and -eliminated hSCAPs, and then 2D and 3D osteogenic and neural differentiation capacities of mycoplasma-eliminated hSCAPs were evaluated for the potent application to bone and neural tissue engineering (Figure 1 I).
Figure 1.
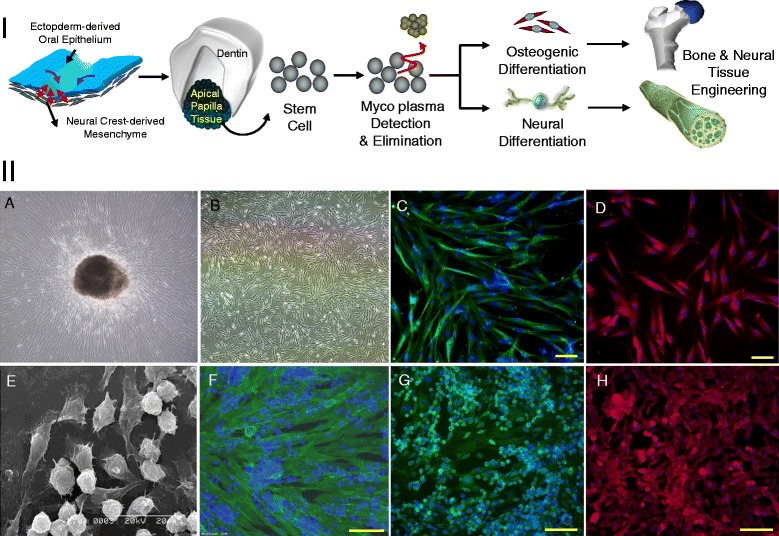
Schematic illustration of the applications of hSCAPs to bone and neural tissue engineering and characterization of primary cultured hSCAPs. I. Schematic illustration of bone and neural differentiation of mycoplasma eliminated hSCAPs for bone and neural tissue engineering. II. Morphology and immunocytochemical images of the primary cultured stem cells from apical papilla (hSCAPs). A. Cell outgrowth from apical papilla tissue fragment. B. Expanded hSCAPs. C. Stro-1(green) and nuclear staining (DAPI; blue). D. CD44 (red) and nuclear staining (DAPI; blue). E. SEM image of the expanded hSCAPs in the presence of NGF, FGF2 and LIF. F. CD44 (green) and nuclear staining (DAPI; blue) of the expanded hSCAPs in the presence of NGF, FGF2 and LIF. G. Nestin (green) and nuclear staining (DAPI; blue) of the expanded hSCAPs in the presence of NGF, FGF2 and LIF. H. CD133 (red) and nuclear staining (DAPI; blue) the expanded hSCAPs in the presence of NGF, FGF2 and LIF.
Method
Primary culture of Stem Cells from Apical Papilla (hSCAPs) of the human premolar and third molar teeth
Briefly, human dental papilla tissue was procured from discarded 6 ~ 24 aged donor’s premolar and third molar teeth with informed consent of patients undergoing routine extractions at the Dental Clinic of School of Dentistry in Kyung Hee University, under approved guidelines set by the Kyung Hee University and School of Dentisty Human Subjects Research Committees (IRB# KHUSD 0908–01). The extracted teeth were directly stored in alpha-MEM (Lonza) containing 1% penicillin/streptomycin (P/S, Lonza), and the teeth were used to procure papilla tissue within 2 hours after teeth extraction. Dental apical papilla tissues were extracted from the premolar and third molar teeth, and were minced with a scalpel. The fragmented papilla tissues were washed with phosphate buffered saline (PBS: Gibco) three times, and the minced tissues were allowed to attach on T25 tissue culture flasks in basic culture medium consisting of alpha-MEM, 10% fetal bovine serum (FBS, lonza), and 1% penicillin/streptomycin. Cultures were fed every 2 days and passaged by treatment with 0.25% trypsin/EDTA. Cell Cultures were grown at 37°C in a humidified atmosphere containing 5% CO2.
Morphological and immunocytochemical characterization of hSCAPs
1 × 104 cell/cm2 of hSCAPs were seeded on 24 well plate (Corning) and cultured for 2 days. The attached cells were fixed with 3.7% formaldehyde (Sigma) for 20 mins at room temperature, and fixed cells were washed with PBS. The cells were permeabilized with 0.2% Triton X-100 (Sigma) treatment for 20 mins, and, after brief washing with PBS, were treated with 4% bovine serum albumin (BSA: Sigma) to block non-specific binding of antibodies at 4°C for overnight. After blocking, the cells were incubated with primary antibodies such as 1:200-diluted mouse-anti-nestin (Abcam) antibody, 1:200-diluted mouse-anti-Stro-1 (Abcam) antibody, and 1:200-diluted mouse-anti-CD44 (Abcam), and 1:200-diluted mouse-anti-CD133 (Abcam) at 4°C for overnight. After reaction with primary antibodies, the cells were washed gently with PBS three times, and then incubated with secondary antibodies such as 1:2000-diluted alexa 488 (goat anti-mouse IgG, Invitrogen), 1:2000-diluted alexa 596 (goat anti-mouse IgG, Invitrogen) for 1 hr at room temperature under dark condition. Finally, after brief washing with PBS three times, the cells were counter-stained with DAPI, and mounted. The stained cells were observed under an inverted fluorescence microscope (Olympus, IX-72).
Detection of mycoplasma contamination
Mycoplasma contamination of primary cultured hSCAPs was evaluated by mycoplasma specific staining method and polymerase chain reaction (PCR) method.
For mycoplasma staining, cell suspension was prepared by trypsin-EDTA treatment for 5 minutes and subsequent neutralization with culture media. Mycoplama staining was processed with prepared hSCAPs suspension and Mycoplasma Detection Kit according to manufacturer’s instruction. Briefly explaining, 95 μl of cell suspension (1 × 105 cell/ml) was mixed with 5 μl of MycoFluor™ Mycoplasma Detection Kit (Invitrogen) in micro-centrifuge tube, and the mixed cell suspension was incubated for 30 mins at 37°C in a 5% CO2 incubator. After incubation, the stained sample was observed under an inverted fluorescence microscope (Olympus, IX-72).
To evaluate mycoplasma contamination in gene level, PCR was processed with BioMycoX Mycoplasma PCR Detection Kit (Cells-safe) according to manufacturer’s instruction. Briefly, after 24 hrs of culture, 1 ml of culture media was collected, and centrifuged at 3000 rpm for 5 mins. After centrifuge, supernatant was transferred to microcentrifuge tube, and was centrifuged at 13,000 rpm for 10 mins. After centrifuge, mycoplasma pellet was collected, and suspended with 100 μl of dH2O, and then boiled at 98°C for 10 mins. 50 μl of supernatant was transferred into new PCR tubes. For PCR reaction, supernatant was mixed with 5 μl PCR template, 2 μl primer mix (Cells-safe), 2 μl dH2O and 10 μl 2X PCR premix (Cells-safe). PCR reactions were performed as following conditions: pre-denaturation process at 95°C for 5 mins with 1 cycle, denaturation process at 94°C for 30 secs with 35 cycles, annealing process at 55°C for 30 secs with 35 cycles, and extension process at 72°C for 30 secs with 35 cycles. Finally, all PCR products were loaded on 0.7% argarose gel containing ethidium bromide at concentration of 0.5 μg/ml.
Mycoplasma elimination
Mycoplasma elimination of contaminated hSCAPs was processed using BioMycoX® Mycoplasma Elimination Kit (Cells-safe). After detection of mycoplasma contamination, 5 × 106 of the mycoplasma-infected hSCAPs was seeded on a T75 flask and cultured in culture media at 37°C in a 5% CO2 incubator. When the confluence of cells reached at 70 ~ 80%, the cells were trypsinized, neutralized with culture media, and then centrifuged at 1500 rpm for 5 mins. After centrifuged, cell pellet was collected, and then suspend with culture media containing 5% FBS. 2.5 × 106 cells/ml was mixed with 200 μl of BioMycoX® Reagent 1, and total volume was adjusted to 10 ml with culture media. The cell suspension was transferred to a T75 flask, and incubated for 3 days without media change at 37°C in a 5% CO2 incubator. After 3 days of incubation, attached cells were trypsinized, and then centrifuged at 1500 rpm for 5 mins. After centrifuge, cell pellet was collected and suspended with culture media containing 10% FBS. Cell suspension was mixed with 200 μl of BioMycoX® Reagent 2, and total volume was adjusted to 10 ml with culture media. The cells were incubated for 2 days without media change at 37°C in a 5% CO2 incubator, which treatment with BioMycoX® Reagent 2 was repeated two times.
Proliferation assay of mycoplasma-infected and -eliminated hSCAPs
Mycoplasma-infected hSCAPs and mycoplasma-eliminated hSCAPs were seeded at cell density of 1 × 104 cells on 96 well plate and incubated for 24 hrs at 37°C in a 5% CO2 incubator. The BrdU incorporation based cell proliferation assay was processed with BrdU assay kit (Merck) according to manufacturer’s instruction. Briefly, after 24 hrs of incubation, 1:2000 diluted BrdU working solution was added into hSCAPs culture media, and the cells were incubated for 3 hrs. The BrdU incorporated cells were treated with 200 μl of fixative/denaturing solution for 30 mins at room temperature. After reaction, fixative/denaturing solution was aspirated, and the cells were incubated with 1:100 diluted mouse anti-BrdU in dilution buffer solution for 1 hr at room temperature. After washing with washing buffer three times, the cells were reacted with 100 μl goat anti-mouse IgG HRO conjugate for 30 mins at room temperature, and then washed with washing buffer three times. Finally, the cells were reacted with 100 μl of substrate solution in the dark at room temperature for 15 mins, and subsequently 100 μl of stop solution was added to each well. The content of incoporated BrdU in cellular DNA was measured using a spectrophotometric plate reader at dual wavelengths of 450–540 nm (or 450–595).
2D and 3D osteogenic differentiation of mycoplasma-eliminated hSCAPs
Prior to osteogenic differentiation, primary cultured hSCAPs were expanded in α-MEM supplemented with 15% FBS, and 1% penicillin/streptomycin. For osteogenic differentiation, hSCAPs was seeded on culture plated at cell density of 1 × 105/cm2 and cultured in osteogenic medium; α-MEM supplemented with 15% FBS, and 1% penicillin/streptomycin containing 50 μg/ml ascorbic acid (Sigma) in PBS, 1 μM dexamethasone (Sigma) in PBS and 10 mM β-glycerophosphate (Sigma) in PBS. The cultures were maintained at 37°C in a 5% CO2 humidified incubator and fed every 2 days up to 20 days. After 20 days of osteogenic culture, the differentiated cells were fixed with 3.7% formaldehyde for 1 min or 20 mins at room temperature, and fixed cells were washed with PBS. The fixed cells were stained with phenotypic alkaline phosphatase (ALP) staining kit (Chemicon) and alizarin red-S (Sigma) staining solution according to manufacturer’s instruction.
In addition, for 3D osteogenic differentiation, hSCAPs were suspended in 1.1% (w/v) alginic acid (Sigma) and 0.1% (v/v) porcine gelatin (Sigma) solution (all dissolved in PBS, pH 7.4), as described before [19]. Briefly, the cell-gel solution was passed through a peristaltic pump (EYELA) and dropped using a 25-gauge needle into sterile alginate gelation solution composing of 100 mM CaCl2 (Sigma), 10 mM HEPES (Sigma), and 0.01% (v/v) Tween (Sigma) at pH 7.4 with stirring. Approximately 10,000 cells were encapsulated in each alginate hydrogel. The hydrogels remained in gently stirred CaCl2 solution for 6–10 mins and were then washed with PBS. The hSCAPs containing hydrogels were transferred to in 10 ml vessels of HARV bioreactors (Synthecon) and the vessels was rotated at 25 rpm. Subsequently, 3D osteogenic differentiation was induced using the same osteogenic medium described above. The bioprocess is illustrated in Figure 2 II. After 3D osteogenic differentiation, the sections of hydrogels were stained with alizarin red-S staining solution according to manufacturer’s instruction.
Figure 2.
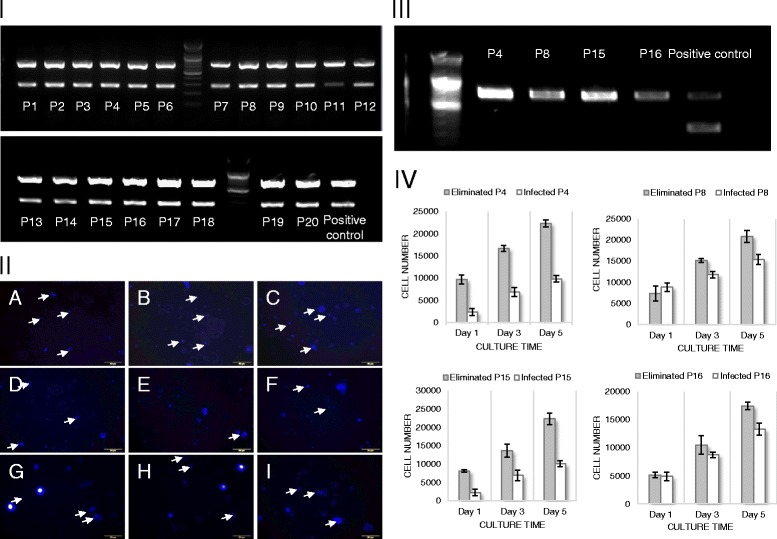
Mycoplasma-specific PCR analysis and fluorescence staining for primary cultured hSCAPs and proliferation activity. I. PCR analysis for 20 samples. II. fluorescence staining of mycoplasma in hSCAPs culture: arrow indicates positive staining of mycoplasma colony. III. mycoplasma-specific fluorescence staining of hSCAPs from selected 4 samples before and after mycoplasma elimination process. IV. proliferation assay for hSCAPs from selected 4 samples before and after mycoplasma elimination process.
2D and 3D neural differentiation of mycoplasma-eliminated hSCAPs
Prior to neural differentiation, primary cultured hSCAPs were expanded in culture medium consisting of DMEM/F12 mixture (1:1, with glucose, L-glutamine, HEPES Buffer, Lonza), 10% FBS, 20 ng/ml epithelial growth factor (EGF, Peprotech), 10 ng/ml basic fibroblast growth factor (FGF2, Peprotech), 1000 unit/ml LIF (Invitrogen) and 1% penicillin/streptomycin. For neural differentiation, hSCAPs was allowed to form neurospheres by plating non-adherent culture dishes in the presence of 100 ng/ml FGF2 in culture medium consisting of DMEM/F12 mixture, 10% FBS, 1000 unit/ml LIF (Invitrogen) and 1% penicillin/streptomycin for 3 days at 37°C in a 5% CO2 humidified incubator. The formed neurospheres were replated on laminin (10 μg/ml, Invitrogen)-coated culture dishes and were allowed to direct neural differentiation in neurobasal medium (Gibco) supplemented with 2% B27 (Invitrogen), 1X insulin-transferrin-sodium-selenite (ITS, BD science), 1% penicillin/streptomycin, 30 ng/ml of NGF, 20 ng/ml EGF and 10 ng/ml FGF2 for one week. Subsequently, hSCAPs were maintained in neuroinduction medium consisting of in neurobasal media (Gibco), 1% B27 supplement (Invitrogen), 1% non-essential amino acid (Gibco), 1 X ITS, 100 units/ml penicillin and 100 μg/ml streptomycin, 100 ng/ml GNDF (Peprotech) and 0.5 μM retinoic acid (Sigma). All of the media were replaced twice a week.
In addition, for 3D neural differentiation, hSCAPs which dissociated from neurospheres were mixed with matrigel (BD Bioscience), and the cell/matrigel solution and 1.1% (w/v) alginic acid solution were placed in separated syringes on syringe pump, and the solutions passed through double jet nodule and dropped into sterile alginate gelation solution composing of 100 mM CaCl2, 10 mM HEPES, and 0.01% (v/v) Tween at pH 7.4 with mild stirring. The hydrogels remained in gently stirred CaCl2 solution for 6–10 mins and were then washed with PBS. The bioprocess is illustrated in Figure 2 IIIE. The hSCAPs/matrigel were placed in inner side and alginate hydrogel were formed to be outer layer enveloping cell/matrigel. The 3D neural differentiation of hSCAPs containing hydrogels was induced by the culture under same culture condition described before. After 2D and 3D neural differentiation, the cells were fixed with 3.7% formaldehyde for 20 mins at room temperature, and neural differentiation was characterized by immunocytochemical staining with primary antibodies such as Ca2+/calmodulin-dependent protein kinases II (rabbit anti-human Cam kinase II, Abcam) and βIII-tubulin (mouse anti-human βIII-tubulin, Abcam).
Result
Characterization of primary cultured Stem Cells from Apical Papilla (hSCAPs)
Papilla tissues were procured from pre-molar and third molar tooth of patients aged from 6 to 24 which information of the used teeth is noticed in Table 1, and the minced papilla tissues were placed to be adhered on culture flasks. The outgrowth of cells from papilla tissues was detectable in one or two weeks of tissue attachment (Figure 1 IIA). During the expansion of primary isolated cells from apical papilla, two distinguished cell populations were detected under different culture conditions. When the cells were expanded in α-MEM supplemented with 10% FBS, most cells showed fibroblastic morphology, and small population showing spherical shape was found (Figure 1 IIB). Such spherical shaped cell population was largely found during the expansion in the presence of EGF, FGF2 and LIF (Figure 1 IIE). This morphological difference of cell populations showed different molecular expression, and their stem cell characteristic was evaluated by immunocytochemical staining with stem cell markers. As shown in Figure 1 IIC and D, most of fibroblastic cells derived from papilla tissues showed strongly positive reactions against anti-Stro-1and CD44, and, in contrast, spherical shaped cells did not show positive expression of CD44 (Figure 1 IIF), but showed relatively stronger expressions of nestin and CD133 (Figure 1 IIG and H).
Table 1.
Information of premolar and third molar teeth used in this study
| Sample # | Sex | Age | Teeth | Sample # | Sex | Age | Teeth |
|---|---|---|---|---|---|---|---|
| 1 | Male | 6 | Pre-molar | 11 | Male | 19 | Third-molar |
| 2 | Male | 11 | Pre-molar | 12 | Female | 23 | Third-molar |
| 3 | Male | 6 | Pre-molar | 13 | Female | 24 | Third-molar |
| 4 | Male | 9 | Pre-molar | 14 | Female | 12 | Pre-molar |
| 5 | Male | 10 | Pre-molar | 15 | Female | 17 | Third-molar |
| 6 | Male | 12 | Pre-molar | 16 | Female | 12 | Pre-molar |
| 7 | Male | 11 | Pre-molar | 17 | Female | 13 | Pre-molar |
| 8 | Male | 14 | Pre-molar | 18 | Female | 11 | Pre-molar |
| 9 | Male | 10 | Pre-molar | 19 | Female | 6 | Pre-molar |
| 10 | Male | 14 | Pre-molar | 20 | Female | 7 | Pre-molar |
Mycoplasma detection and elimination of primary cultured hSCAPs
Mycoplasma contamination of primary cultured hSCAPs was evaluated by PCR analysis and by mycoplasma-specific staining analysis. PCR analysis was done using BioMycoX mycoplasma PCR detection kit which primers is designed to react specifically with the highly conserved coding region in the mycoplasma genome. All expanded hSCAPs which derived from patients aged from 6 to 24 were tested, and, as shown in Figure 2 I, gel running with PCR products showed two bands in all tested samples: one band with approximate size of 700 bp was an internal DNA band to confirm the well PCR reaction, and the other bands which positioned around 250–300 bp indicated mycoplasma contamination. Even though sample number 11 showed relatively week band intensity in comparison with other samples, PCR analysis showed most clear evidence of mycoplasma contamination of all tested hSCAPs.
Accompanied with the analysis of mycoplasma contamination in gene level, mycoplasma contamination was also evaluated by mycoplasma-specific staining analysis using MycoFluor™ mycoplasma detection kit. As shown in Figure 2 II, four samples (patient number 4, 8, 15, and 16) of hSCAPs were selected randomly for staining, and all tested hSCAPs cultures showed positive staining of mycoplasma colony in live cells. Relatively small sized blue fluorescent spots indicate stained mycoplasma colonies within cytosol of hSCAPs, and nucleus within dead cells showed larger fluorescent spots. In addition, mycoplasma-positive staining was frequently detectable around nucleus.
After the detection of mycoplasma contamination via PCR and mycoplasma-specific staining analysis, Mycoplasma was eliminated from the contaminated hSCAPs using BioMycoX® Mycoplasma Elimination Kit. After mycoplasma elimination of randomly selected hSCAPs sample, the presence of mycoplasma was evaluated by mycoplasma-specific staining PCR analysis. As shown in Figure 2 III, the mycoplasma-eliminated hSCAPs culture did not show any bands at approximate size of 250–300 bp in PCR analysis, which indicates well elimination of contaminated mycoplasma.
Proliferation activity of mycoplasma-contaminated and -eliminated hSCAPs
Four samples (patient number 4, 8, 15, and 16) were selected randomly to evaluate the effect of mycoplasma contamination on cell proliferation, and the proliferation activity of mycoplasma-contaminated and -eliminated hSCAPs was analyzed by BrdU incorporation assay. As shown in Figure 2 IV, although the extent of differences in proliferation activities between mycoplasma-contaminated hSCAPs and mycoplasma-eliminated hSCAPs was varied in the selected samples, mycoplasma-eliminated hSCAPs showed generally much higher proliferation activities than mycoplasma-contaminated hSCAPs throughout whole culture period. Some hSCAPs showed two fold increase in proliferation activity at 5 days of cultures after mycoplasma elimination, which demonstrates the negative effect of mycoplasma contamination on cell proliferation activity.
Osteogenic and neural differentiation of mycoplasma-eliminated hSCAPs
With mycoplasma-eliminated hSCAPs, 2D and 3D osteogenic and neural differentiation were induced under certain osteogenic and neural differentiation conditions.
For 2D osteogenic differentiation of mycoplasma-eliminated hSCAPs, ostegenic differentiation of hSCAPs was induced in the presence of osteogenic supplements such as ascorbic acid, dexamethasone and β-glycerophosphate, and, as shown in Figure 3 IA-C, culture time-dependent increase of phenotypic ALP expression during osteogenic differentiation. After 20 days of osteogenic differentiation, mineralized nodules were formed in osteogenic culture of hSCAPs throughout whole culture area, which proved by strong positive staining with alizarin red-S (Figure 3 ID, F and G), and such mineralized nodules were found to have high content of calcium and phosphate, which proved by high fluorescence in alizarin red-S staining under fluorescent microscope (Figure 3 IE). In addition to 2D osteogenic differentiation of mycoplasma-eliminated hSCAPs, 3D osteogenic differentiation of hSCAPs were induced by dynamic osteogenic culture of alginate encapsulated hSCAPs in a rotating HARV bioreactor (Figure 3 II). 3D highly mineralized nodules formed by differentiated hSCAPs within alginate hydrogel were found at 20 days of dynamic osteogenic culture (Figure 3 IID and E)
Figure 3.
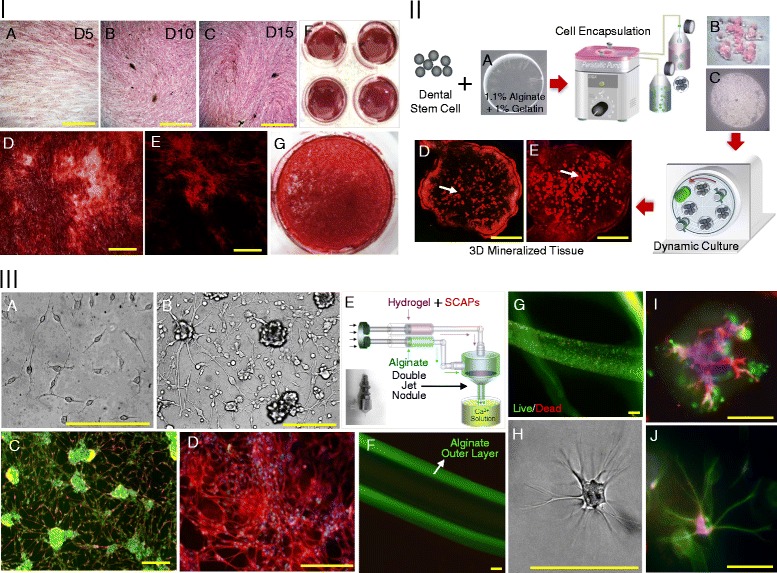
Osteogenic and neural differentiation of mycoplasma-eliminated hSCAPs. I. 2D Osteogenic differentiation of mycoplasma-eliminated hSCAPs: A, B and C. Phenotypic ALPase expression after 5, 10 and 15 days of osteogenic culture, D, F and G. Alizarin red-S stained image of mineralized nodules after 20 days of osteogenic culture under light microscope, E. alizarin red-S stained image of mineralized nodules after 20 days of osteogenic culture under fluorescence microscope, II. 3D Osteogenic differentiation of mycoplasma-eliminated hSCAPs: A. alginate hydrogel, B and C: alginate hydrogel encapsulating hSCAPs, D and E. alizarin red-S stained image of mineralized nodules within alginate hydrogel after 20 days of 3D osteogenic culture under fluorescence microscope, III. 2D and 3D neural differentiation of mycoplasma-eliminated hSCAPs: A and B. microscopic images of hSCAPs in 2D neural differentiation culture. C and D. βIII tubulin (red), Cam kinase II (green) and nuclear staining (DAPI; blue), E. Schematic illustration of hSCAPs encapsulation of hSCAPs for 3D neural differentiation. F. tubular alginate hydrogel, G. live and dead image of encapsulated hSCAPs in tubular alginate hydrogel, H. microscopic images of hSCAPs in 3D neural differentiation culture, I. βIII tubulin (red), Cam kinase II (green) and nuclear staining (DAPI; blue) of differentiated hSCAPs in 3D neural differentiation culture, J. βIII tubulin (green), Cam kinase II (red) and nuclear staining (DAPI; blue) of differentiated hSCAPs in 3D neural differentiation culture, Scale bars indicate 200 μm.
For 2D neural differentiation of mycoplasma-eliminated hSCAPs, hSCAPs were allowed to form neurospheres in the presence of FGF2 and FBS and then hSCAPs-derived neurospheres were plated on laminin-coated culture, 2D neural differentiation was induced by the neural inductive culture in the presence of NGF, EGF, and FGF2 for one week and by the subsequent neural maturation culture in the presence of GDNF and retinoic acid. As shown in Figure 3 IIIA and B, morphological change of hSCAPs to biopolar and multipolar cells was found, and extension of neurite from differentiated hSCAPs was also detected around the plated neurospheres. In addition, the differentiated hSCAPs expressed βIII-tubulin along with the extended neurite and cell bodies and expressed cam kinase II, indicating neural differentiation (Figure 3 IIIC and D). In addition to 2D neural differentiation of mycoplasma-eliminated hSCAPs, neurosphere-derived single cell suspension was mixed with matrigel and encapsulated alginate hydrogel using double jet nodule (Figure 3 IIIE), and 3D neural differentiation was induced under the same culture condition. As shown in Figure 3 IIIF and G, alginate hydrogel well formed the outer layer encapsulating cells, and encapsulated hSCAPs showed well viability within hydrogel, and multipolar cells expressing neural markers such as βIII-tubulin and cam kinase II were found within hydrogel (Figure 3 IIIH-J).
Discussion
Recently, postnatal stem cells from dental tissues have been received great attention in the field of tissue engineering and regenerative medicine in terms of relatively easy accessibility, multi-differentiation capacity and possible autologus implantation. Implantation studies showed in vivo bone and neural tissue regeneration capacity of implanted dental tissue-derived stem cell [13,14,20]. Regarding developmental origin, so called neural crest derived ectomesenchyme, of stem cells to participate in tooth morphogenesis, different lineage differentiation, mesenchymal differentiation and neuroectodermal differentiation, might be derived from postnatal stem cells from dental tissues under certain circumstance [1,2]. The residence of postnatal stem cells showing multi-differentiation capacity have been identified within various dental tissues such as dental pulp, periodontal ligament, and papilla tissues [3,6,7], and, among these dental tissues, papilla tissue which generally found in a developing tooth is known to be a pool of stem cells [21]. In our study, different stem cell populations could be generated by the culture under different culture conditions during expansion of stem cells from apical papilla (hSCAPs), and a stem cell population showing fibroblastic spindle shape during the expansion expressed mesenchymal markers such as Stro-1 and CD44 which consistent to the previous report to show the derivation of stem cells with mesenchymal characteristic from dental papilla tissue [3], but, during hSCAPs expansion in the presence of EGF, FGF2 and LIF, distinct cell population with spherical shape appeared and expressed of neural stem cell markers such as nestin and CD133 [22]. Both expressions of mesenchymal and neural stem cell maker of papilla-derived stem cells might be related to their stem cell characteristics which derived from neural crest-derived ectomesenchyme in tooth developmental process [9,23]. However, it might be necessary to investigate more about the characteristics of postnatal stem cells from dental tissues to broaden their therapeutic applications.
In spite of potent usefulness of stem cells from dental tissues in tissue engineering and regenerative medicine, for clinical application and stem cell researches using postnatal stem cells from dental tissues, bacterial contamination should be considered in fact most teeth are usually extracted due to oral disease such as oral cavity which might be frequently related with bacterial infections. Especially, mycoplasma is one of most popular microorganism frequently found in oral cavity [16], and is the smallest prokaryote with below 1 μm in a diameter which small size allows mycoplasma to pass through conventional filters used to prohibit bacterial and fungal contamination, and mycoplasma easily absorb into host animal cells [15]. In that dental tissues can be frequently exposed to bacterial infection, mycoplasma infection of propagated dental tissue-derived cell cultures is probably unavoidable. In our study, most premolar and third molar teeth were extracted from patient for orthopedic surgery to cure oral disease, and papilla tissues was procured from the discarded teeth. After isolation, mycoplasma contamination was evaluated with primary cultured hSCAPs, and most hSCAPs were found to be contaminated by mycoplasma, which were proved by positive staining and mycoplasma-specific DNA expression. To confirm the possible cross-contamination from other cells in our laboratory, adipose-derived stem cell, human dermal fibroblasts and primary cultured periodontal ligament cells was used to test mycoplasma contamination, but there was no sign of mycoplasma contamination (data not shown). Such mycoplasma contamination is known to affect many aspects of cell physiology such as cell proliferation, chromosomal aberration and also immunological reactions [17,18]. For example, the 50% of inhibition in cell proliferation was reported by influencing nutrient consumption of cells during cell culture [17]. In addition, upon in vivo implantation of mycoplasma-contaminated cells, mycoplasma from infected cells might affect host cells participating in immunological reactions such as macrophage activation and inhibition of antigen presentation [18]. In our study, the contaminated mycoplasma in hSCAPs were eliminated, and it was found that mycoplasma contamination affected cell proliferation activity of primary culture hSCAPs. Finally, the mycoplasma-eliminated hSCAPs showed multi-differentiation capacity into osteogenic and neural lineage cells under certain culture conditions, and also 3D osteogenic and neural differentiation could be successfully induced using hydrogel encapsulation technique or dynamic culture system for their application to tissue engineering. Our study might suggest that the experimental process of mycoplasma elimination may be required for primary cell culture and for the application of primary cells to tissue engineering and regenerative medicine, but its time-consuming and expensive process, which takes approximately for 7–10 days, is still a problem to be overcome for its practical application to conventional culture process.
Conclusion
Our study showed that dental papilla-derived stem cells could be isolated from the discard human premolar and third molar teeth after orthodontic therapy and that almost dental papilla-derived stem cell was shown to be infected by mycoplasma, which mycoplasma infection affected cell proliferation activity. Such infected mycoplasma in dental papilla-derived stem cells could be clearly eliminated using the mycoplasma elimination kit, and mycoplasma-eliminated stem cells showed multi-differentiation activity into osteogenic and neural lineage differentiation. Based on scientific knowledge that infected mycoplasma can affect cell proliferation activity, chromosomal aberration and immune response in cell culture and implantation, mycoplasma detection and elimination process should be considered as one of essential steps prior to stem cell research and potent clinical application using postnatal stem cells from dental tissues.
Acknowledgement
This research was supported by grants NRF-2013R1A1A2009706, 2012R1A2A2A02010181 and 2012R1A5A2051387 from the Basic Research Program through the National Research Foundation of Korea (NRF) funded by the Korea government (MEST) Republic of Korea, and by National Agenda Project (NAP: NAP-09-04) from the National Research Council of Science and Technology.
Abbreviations
- SCAPs
Stem cells from apical papilla
- PBS
Phosphate buffered saline
- FBS
Fibroblast growth factor
- P/S
Penicillin/Streptomycin
- DAPI
4′,6-diamidino-2-phenylindole
- PCR
Polymerase chain reaction
- ALP
Alkaline phosphatase
- EGF
Epithelial growth factor
- FGF
Fibroblast growth factor
- LIF
Leukemia inhibitory factor
- GDNF
Glial cell-derived neurotrophic factor
- HARV
High aspect ratio vessel
- NGF
Nerve growth factor
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BC carried out stem cell characterization, mycoplasma detection and elimination, and experiment about neural differentiation of stem cells, and drafted the manuscript. SY carried out experiment about osteogenic differentiation of stem cells. YD provided tooth for this study and carried out stem cell extraction. SC provided tooth for this study and carried out stem cell extraction. DW helped to draft the manuscript. YS participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Byung-Chul Kim, Email: xikian@naver.com.
So Yeon Kim, Email: goodany00@naver.com.
Yong-Dae Kwon, Email: kwony@khu.ac.kr.
Sung Chul Choe, Email: pedochoi@khu.ac.kr.
Dong-Wook Han, Email: nanohan@pusan.ac.kr.
Yu-Shik Hwang, Email: yshwang@khu.ac.kr.
References
- 1.Chai Y, Jiang X, Ito Y, Bringas P, Han J, Rowitch DH, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–9. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- 2.Miletich I, Sharpe PT. Neural crest contribution to mammalian tooth formation. Birth Defects Res (Part C) 2004;72:200–12. doi: 10.1002/bdrc.20012. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda E, Hirose M, Kotobuki N, Shimaoka H, Tadokoro M, Maeda M, et al. Osteogenic differentiation of human dental papilla mesenchymal cells. Biochem Biophy Res Comm. 2006;342:1257–62. doi: 10.1016/j.bbrc.2006.02.101. [DOI] [PubMed] [Google Scholar]
- 4.Sakai VT, Zhang Z, Dong Z, Neiva KG, Machado MAAM, Shi S, et al. SHED differentiate into functional odontoblasts and endothelium. J Dent Res. 2010;89:791–6. doi: 10.1177/0022034510368647. [DOI] [PubMed] [Google Scholar]
- 5.Zhang W, Walboomers XF, Shi S, Fan M, Jansen JA. Multilineage differentiation potential of stem cells derived from human dental pulp after cryopreservation. Tissue Eng. 2006;12:2813–23. doi: 10.1089/ten.2006.12.2813. [DOI] [PubMed] [Google Scholar]
- 6.Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci. 2000;97:13625–30. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo BM, Miura M, Gronthos S, Bartold PM, Batouli S, Brahim J, et al. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet. 2004;364:149–55. doi: 10.1016/S0140-6736(04)16627-0. [DOI] [PubMed] [Google Scholar]
- 8.Morsczeck C, Gotz W, Schierholz J, Zeilhofer F, Kuhn U, Mohl C, et al. Isolation of precursor cells (PCs) from human dental follicle of wisdom teeth. Matrix Biol. 2005;24:155–65. doi: 10.1016/j.matbio.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Kim BC, Bae HJ, Kwon IK, Lee EJ, Park JH, Khademhosseini A, et al. Osteoblastic/cementoblastic and neural differentiation of dental stem cells and their applications to tissue engineering and regenerative medicine. Tissue Eng Part B. 2012;18(3):235–44. doi: 10.1089/ten.teb.2011.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothová M, Feng J, Sharpe PT, Peterková R, Tucker AS. Contribution of mesoderm to the developing dental papilla. Inter J Dev Biol. 2011;55(1):59–64. doi: 10.1387/ijdb.103083mr. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda E, Morita R, Nakao K, Ishida K, Nakamura T, Takano-Yamamoto T, et al. Fully functional bioengineered tooth replacement as an organ replacement therapy. Proc Natl Acad Sci. 2009;106:13475–80. doi: 10.1073/pnas.0902944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laino G, d’Aquino R, Graziano A, Lanza V, Carinci F, Naro F, et al. A new population of human adult dental pulp stem cells: a useful source of living autologous fibrous bone tissue (LAB) J Bone Miner Res. 2005;20:1394–402. doi: 10.1359/JBMR.050325. [DOI] [PubMed] [Google Scholar]
- 13.Papaccio G, Graziano A, d’Aquino R, Graziano MF, Pirozzi G, Menditti D, et al. Long-term cryopreservation of dental pulp stem cells (SBP-DPSCs) and their differentiated osteoblasts: a cell source for tissue repair. J Cell Phy. 2006;208:319–25. doi: 10.1002/jcp.20667. [DOI] [PubMed] [Google Scholar]
- 14.Arthur A, Rychkov G, Shi S, Koblar SA, Gronthos S. Adult human dental pulp stem cells differentiate toward functionally active neurons under appropriate environmental cues. Stem Cells. 2008;26:17871795. doi: 10.1634/stemcells.2007-0979. [DOI] [PubMed] [Google Scholar]
- 15.Kraus DC, Taylor-Robinson D. Mycoplasmas which infect humans. Mycoplasmas. In: Maniloff J, editor. Molecular biology and pathogenesis. Washington DC: American Society for Microbiology Press; 1992. pp. 417–44. [Google Scholar]
- 16.Nicolson GL, Nasralla MY, Nicolson NL. The pathogenesis and treatment of mycoplasmal infections. Antimicro Infect Dis News. 1999;17(11):81–7. doi: 10.1016/S1069-417X(00)88885-8. [DOI] [Google Scholar]
- 17.Mcgarrity GJ, Vanaman V, Sarama J. Cytogenetic effects of mycoplasmal infection of cell cultures: A review. In Vitro. 1984;20(1):1–18. doi: 10.1007/BF02633326. [DOI] [PubMed] [Google Scholar]
- 18.Mühlradt PF, Meyer H, Jansen R. Identification of S-(2,3-Dihydroxypropyl)cystein in a macrophage-activating lipopeptide from Mycoplasma fermentans. Biochemistry. 1996;35:7781–6. doi: 10.1021/bi9602831. [DOI] [PubMed] [Google Scholar]
- 19.Hwang YS, Cho J, Chan KLA, Heng J, Boccacini AR, Kazarian SG, et al. The use of murine embryonic stem cells, alginate encapsulation, and rotary microgravity bioreactor in bone tissue engineering. Biomaterials. 2009;30:499–507. doi: 10.1016/j.biomaterials.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 20.Arthur A, Shi S, Zannettino AC, Fujii N, Gronthos S, Koblar SA. Implanted adult human dental pulp stem cells induce endogenous axon guidance. Stem Cells. 2009;27:2229–37. doi: 10.1002/stem.138. [DOI] [PubMed] [Google Scholar]
- 21.Sonoyama W, Liu Y, Yamaza T, Tuan RS, Wang S, Shi S, et al. Characterization of the apical papilla and its residing stem cells from human immature permanent teeth: a pilot study. J Endodontics. 2008;34:166–71. doi: 10.1016/j.joen.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wislet-Gendebien S, Hans G, Leprince P, Rigo JM, Moonen G, Rogister B. Plasticity of cultured mesenchymal stem cells: switch from nestin-positive to excitable neuron-like phenotype. Stem Cells. 2005;23:392–402. doi: 10.1634/stemcells.2004-0149. [DOI] [PubMed] [Google Scholar]
- 23.Tucker A, Sharpem P. The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev. 2004;5:499–508. doi: 10.1038/nrg1380. [DOI] [PubMed] [Google Scholar]