

Sources of the acid mantle
Although the acid mantle initially was assumed to result from exogenous sources, such as sebum-derived free fatty acids (FFA), several endogenous acidifying mechanisms instead account for up to 2 pH units (a 200-fold increase in protons) of the overall pH of the stratum corneum (SC) (Table 1). The sodium–protein antiporter, type 1 (NHE1), which selectively acidifies extracellular domains at the stratum granulosum (SG)/SC interface (1), accounts for ≈¼ unit of the bulk pH of the SC (1). This critically important site is where sphingomyelin and glucosylceramides are processed into ceramides by two acidic pH-dependent enzymes, acidic sphingomyelinase (aSMase) and β-glucocerebrosidase (β-GlcCer’ase), thereby generating the permeability barrier.
Table 1.
Impact of endogenous acidifying mechanisms on SC acidification and epidermal functions
| Acidifying mechanisms | Change in bulk pH | Localization | Known impact on epidermal functions | Changes during postnatal development | Changes in disease | |
|---|---|---|---|---|---|---|
| PL→FFA (sPLA2G2F) | ≈1 unit | ? | ↑Barrier, ↑Cohesion | ↓Neonatal | ? | |
| NHE1 | ≈1/4 unit | SG-SC interface | ↑Barrier | ↓Aged | ? | |
| FLG→PCA (t-UCA) | ≈1/2 unit | ? | ↑Barrier | ? | ? | |
| Melanin persistence/extrusion | Transient | SG-SC interface | ↑Barrier, ↑Cohesion | ? | ? | |
|
|
? | ? | ↑Cohesion | ? | ? |
Chol, cholesterol; CSO4, cholesterol sulphate; FFA, free fatty acid; FLG, filaggrin; NHE1, Na+/H+ antiporter 1; PCA, polycarboxylic acid; PL, phospholipids; SC, stratum corneum; SG, stratum granulosum; SO4, sulphate; t-UCA, trans-urocanic acid.
Secretory phospholipase (sPLA2) releases FFA from the sn.2 position of glycerophospholipids (PL). While one subset releases arachidonic acid that is subsequently converted to eicosanoids, other sPLA2s release FFAs that are required for permeability barrier homoeostasis (2), accounting for ≈1 unit of SC pH. Of the various sPLA2 isoforms, only PLA2G2F is regulated and required for permeability barrier homoeostasis (3). Notably, Pla2g2f ko mice display an elevated SC pH and abnormal permeability barrier homoeostasis (3).
Deimination of amino acid constituents of filaggrin (FLG) generates abundant polycarboxylic acids (PCA) that contribute to SC pH, demonstrated by the ≈½ pH unit increase in pH that occurs in FLG-deficient (noninflammatory) ichthyosis vulgaris (4). Within the FLG→PCA mechanism, the generation of trans-urocanic acid (tUCA) from histidine, which comprises about 10% of FLG, could contribute to pH (S1). However, histidase-deficient (Peruvian) mice do not display a defect in SC acidity (S2). Finally, the pH of darkly pigmented human epidermis is ≈½ pH unit lower (50-fold more acidic) than the SC of lightly pigmented skin (5), due in part to the persistence and extrusion of melanin granules at the SG-SC interface (S3) (Figure S1). The acidic pH of darkly pigmented skin dictates the superior function in such individuals, because acidification of lightly pigmented human skin ‘resets’ functions to darkly pigmented levels (5). But it is likely that one additional mechanism contributes to SC acidification. Cholesterol sulphate (CSO4) is the most abundant and widely distributed of sulphated sterols, and a critical regulator of epidermal differentiation (S4). The CSO4 content of the epidermis climbs to ≈5% of lipid mass in the SG (S5), due to enhanced expression of the gene that encodes SULT2B1b, the enzyme that sulfonates cholesterol. The hydrolytic enzyme, steroid sulfatase (SSase), then hydrolyses CSO4 until its levels decline to ≈1% in the outer SC. In X-linked ichthyosis (XLI), CSO4 levels increase to ≥10% of lipid mass, and the SC in XLI is more acidic than normal (S6), consistent with the low pKa (3.1) of CSO4 (S4). While CSO4 ionization would generate H2SO4in situ, the ongoing hydrolysis of CSO4 to across normal SC could also form H2SO4 in aqueous compartments, perhaps contributing to the selective reduction of pH that persists within extracellular domains at all levels of SC (1).
Functions of the acid mantle
In addition to antimicrobial defense, pH regulates at least three additional functions in normal skin (Table 1). First, it is critical for epidermal permeability barrier homoeostasis, because the hydrolysis of sphingomyelin and glucosylceramides into ceramides is dependent upon aSMase and β-GlcCer’ase (S7), which display pH optima of ≈5. Only at the reduced acidity of normal SC can these enzymes generate enough Cer to form lamellar bilayers. Conversely, when the pH of the SC rises with inflammation or under experimental conditions (6), the activities of these enzymes decline in parallel with a deterioration of barrier function.
A second cohort of pH-linked functions includes SC integrity (=resistance to stripping), SC cohesion (=protein removed per stripping) and desquamation. At the low pH of normal SC, corneodesmosomes (CD) are slowly, but progressively degraded by serine proteases (kallikreins, KLK), and then more avidly by aspartate and thiolproteases that exhibit acidic pH optima (5). Conversely, elevations in pH activate KLK, which rapidly degrade CD, accelerating desquamation and compromising SC integrity/cohesion (6) (S8).
A third pH-linked function is pro-inflammatory cytokine activation (S8). Large preformed pools of the 33 kDa precursors of pro-IL-1α and pro-IL-1β are stored in the corneocyte cytosol. As the pH of the SC rises with barrier perturbations and in inflammation, KLK activity increases, releasing the active 17 kDa forms of IL-1α /IL-1β (S9), which in turn initiate divergent, downstream cytokine cascades (S8, S10). While repeated insults lead to inflammation, single perturbations instead unleash beneficial, cytokine-initiated, homoeostatic responses (e.g. accelerated DNA and lipid synthesis) that help to restore barrier homoeostasis (S11).
Developmental variations in pH
Full-term neonatal skin exhibits a near-neutral surface pH, with a permeability barrier which, though sufficient for terrestrial life under basal conditions, recovers more slowly than adult skin from acute perturbations (S12). Moreover, adjustment of SC with acidic buffers normalizes barrier function in neonatal rats (S12), and topical PPARα, PPARβ/δ or LXR activators normalize SC pH and epidermal function by increasing sPLA2 activity (S13). Neonatal skin also displays a well-known propensity to blister, likely due to poor SC cohesion (S12), and impaired antimicrobial defense (both pH-dependent functions). Moderately aged skin (>55 years) also suffers from a flawed permeability barrier and impaired SC integrity, in parallel with an elevated SC pH, due to decreased NHE1 expression (7). Again, acidifying the SC normalizes both permeability barrier function and SC integrity/cohesion in moderately aged mice (7).
Pathophysiology of SC pH – inflammatory dermatoses
The surface pH of inflamed skin inevitably climbs towards neutrality, an increase thought to have dire consequences for the pathogenesis of atopic dermatitis (AD) through increased kallikreins (KLK) activity (S14, S15) (Fig. 1). The article by Lee et al. (8) in a past issue of Experimental Dermatology highlights the importance of the acid mantle for the pathogenesis of AD, and now for the atopic march. Repeated topical applications of the universal hapten, oxazolone, produce an AD-like dermatoses (Ox-AD) in hairless mice in parallel with emergence of an elevated SC pH and a progressive barrier abnormality (9). Lee et al. now show that additional topical and aerosol applications of dust mite antigens further aggravate the Ox-AD dermatoses, ultimately yielding the next temporal component of the atopic diathesis, that is, asthma. These authors show further that maintenance of an acidic pH also not only prevents the emergence of AD, but also the later, asthmatic component of the atopic march. Notably, these studies teach us that AD and the atopic march can develop with repeated insults that compromise barrier function, even against a normal genetic background, but they raise new questions: Would genetically compromised (e.g. filaggrin-deficient) skin demonstrate an even great propensity to develop the atopic march? Would other methods of acidification, such as achieved with topical ingredients like hesperidin (10), prove as effective as the topical cream; and to what extent is the cream alone beneficial? Is asthma already present with topical dust mite antigen applications alone, even prior to aeroallergen exposure? Finally, is the increase in pH in AD due to disease-specific alterations in acidifying mechanisms (Fig. 1), or instead to a broad decline in acidifying mechanisms, perhaps due to downregulation by Th2 cytokines? Nonetheless, it seems highly likely that pH plays a key role in the pathogenesis not only of AD, but likely also in other inflammatory dermatoses.
Figure 1.
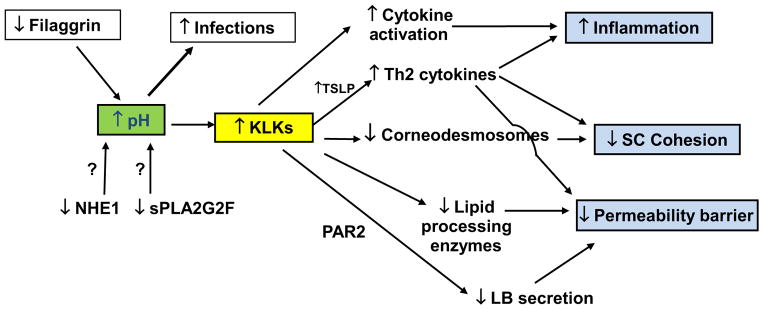
Inherited abnormalities in acidifying mechanisms activate kallikreins (KLKs), compromising multiple functions in inflammatory dermatoses.
Supplementary Material
Figure S1. Pigment granule persistence, with accelerated extrusion after barrier disruption in pigmented hairless (Skh2/J) mice.
Data S1–S15. Supplemental References
Footnotes
Conflict of interest
The author has declared no conflicting interests.
Supporting Information
Additional supporting data may be found in the supplementary information of this article.
References
- 1.Behne MJ, Meyer JW, Hanson KM, et al. J Biol Chem. 2002;277:47399–47406. doi: 10.1074/jbc.M204759200. [DOI] [PubMed] [Google Scholar]
- 2.Man MQ, Jain M, Feingold KR, et al. J Invest Dermatol. 1996;106:57–63. doi: 10.1111/1523-1747.ep12327246. [DOI] [PubMed] [Google Scholar]
- 3.Ilic D, Bollinger JM, Gelb M, et al. Biochim Biophys Acta. 1841;2014:416–421. doi: 10.1016/j.bbalip.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruber R, Elias PM, Crumrine D, et al. Am J Pathol. 2011;178:2252–2263. doi: 10.1016/j.ajpath.2011.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunathilake R, Schurer NY, Shoo BA, et al. J Invest Dermatol. 2009;129:1719–1729. doi: 10.1038/jid.2008.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mauro T, Holleran WM, Grayson S, et al. Arch Dermatol Res. 1998;290:215–222. doi: 10.1007/s004030050293. [DOI] [PubMed] [Google Scholar]
- 7.Choi EH, Man MQ, Xu P, et al. J Invest Dermatol. 2007;127:2847–2856. doi: 10.1038/sj.jid.5700913. [DOI] [PubMed] [Google Scholar]
- 8.Lee HJ, Yoon NY, Lee NR, et al. Exp Dermatol. 2014;23:736–741. doi: 10.1111/exd.12525. [DOI] [PubMed] [Google Scholar]
- 9.Man MQ, Hatano Y, Lee SH, et al. J Invest Dermatol. 2008;128:79–86. doi: 10.1038/sj.jid.5701011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Man G, Mauro TM, Kim PL, et al. Exp Dermatol. 2014;23:645–651. doi: 10.1111/exd.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pigment granule persistence, with accelerated extrusion after barrier disruption in pigmented hairless (Skh2/J) mice.
Data S1–S15. Supplemental References