

Abstract
The pain response to urinary tract infection (UTI) is largely uncharacterized, but the symptomatic response to UTI contrasts with the lack of pain response among individuals with asymptomatic bacteriuria (ASB). Quantifying pelvic pain in a murine UTI model, uropathogenic E. coli (UPEC) induce transient pelvic pain, whereas an ASB E. coli isolate causes no pain, thus recapitulating the spectrum of clinical responses to intravesical E. coli. These differential pain responses are not correlated with bladder colonization or inflammation but instead are intrinsic to E. coli lipopolysaccharide (LPS) and dependent upon the LPS receptor TLR4. Epidemiologic data suggest a link between interstitial cystitis (IC) and a history of UTI, so it was evaluated whether repetitive UPEC instillation would result in chronic pain via central sensitization. While repeated infection with wild type UPEC result in only transient episodes of acute pain, a UPEC mutant lacking O-antigen causes chronic, post-UTI pelvic pain. Similarly, a K-12 E. coli strain lacking O-antigen induces chronic pain that persisted long after bacterial clearance, and expressing O-antigen nullified the pain phenotype. Spinal cords isolated from mice with post-UTI chronic pain exhibited deficits in short term depression consistent with central sensitization. Deleting O-antigen gene complex from a UPEC strain and subsequent heterologous expression of O-antigen gene clusters demonstrates that a single bacterial isolate can exhibit pain phenotypes ranging from a null phenotype, an acute pain phenotype, to a chronic pain phenotype. Post-UTI chronic pain is also associated with voiding dysfunction and anxious/depressive behavior. These effects are also mediated by TRPV1 at the level of pain establishment and CCR2 at the level of pain maintenance. Together, these findings demonstrate that transient infection with E. coli may result in chronic visceral pain with the hallmarks of neuropathic pain. This pattern of behaviors mimics the spectrum of IC symptoms, thus supporting the possibility of an infectious etiology of IC.
Keywords: infection, pain, O-antigen, lipopolysaccharide, centralization
INTRODUCTION
Urinary tract infection (UTI) is the leading cause of acute pelvic pain, occurring in 20% of women each year and resulting in indirect and direct health-care expenditures exceeding $2 billion annually in the US (1). Although it is not clear if acute and chronic pelvic pain share etiology, epidemiologic evidence for interstitial cystitis/bladder pain syndrome (IC) supports past UTI as a risk factor (2).
Uropathogenic E. coli (UPEC) are the most frequent cause of adult UTI, accounting for 80% of all cases, and the majority of UPEC isolates express type 1 pili on their surface that promote adherence by binding to mannosylated host glycoproteins (3, 4). Many of the key events in UTI pathogenesis associated with UPEC infection have been reviewed in detail elsewhere (5). UPEC interactions with urothelium lead to local cytokine/chemokine production (IL-6/IL-8) that, in turn, promotes neutrophil influx. Simultaneously, UPEC promote urothelial apoptosis and subsequent urothelial injury, where the FimH subunit of type 1 pili acts as a tethered toxin (6, 7). An intracellular lifecycle has been identified for UPEC within the urothelium (8). The apoptotic response to UPEC is thus a double-edged sword: it facilitates purging of adherent and invasive UPEC interacting with superficial urothelial cells but also provides access to underlying cells in which UPEC establishes quiescent intracellular reservoirs thought to underlie recurrent infection. Stimulation of bladder sensory afferent fibers, causing the sensation of pain, might correlate with local bacteria-induced inflammation, but the mechanisms of afferent activation during UTI are unknown. Pelvic pain associated with acute UTI is typically transient (9). Therefore, dogma suggests that infection should cause pain, and resolution of infection should lead to resolution of pain.
It is increasingly apparent that both microbial virulence factors and host innate immune response affect the broad range of inflammation and disease that specific bacterial pathogens may produce (10). An example at one end of the spectrum is asymptomatic bacteriuria (ASB) (11). The prototypical ASB E. coli strain 83972 has been characterized at the molecular level, in vitro, in murine studies, and in clinical studies (12–17). 83972 lacks type-1 pili and does not lead to urothelial apoptosis. In contrast to symptomatic UTI associated with UPEC, 83972 can reside in the bladder at high concentrations and for extended periods without producing symptoms (12, 18), raising the possibility that FimH is a key virulence factor mediating the symptomatic response to UTI.
Beyond the intrinsic importance of understanding microbial pathogenesis, determining why alternate pain states develop during and after UTI may reveal novel therapeutic targets. Thus, identifying the molecular mechanisms and physiologic pathways mediating differential UTI pain responses is vital.
MURINE MODEL OF UTI-ASSOCIATED PELVIC PAIN
Murine cystitis models are used widely in mechanistic studies of pelvic pain, but such studies often rely on substrates that lack obvious clinical relevance (i.e., cyclophosphamide, zymosan). Studies using transurethral instillation with clinical E. coli isolates closely approximate clinical infection, in particular mimicking the inflammatory responses of patients, with the caveat that the initial inoculum likely does not recapitulate the early infection kinetics of ascending infection in patients (19, 20). We recently employed a murine UTI model to investigate mechanisms of UTI-associated pelvic pain initiated by NU14, an archetypal human UPEC strain of the B2 group isolated from the urine of a cystitis patient (21). To do so, we instilled 108 CFU bacteria via transurethral catheterization of female C57BL/6J mice and then characterized bacterial loads, inflammation, and pain over time.
Pain is a subjective sensation for which surrogate and behavioral markers must be measured in animal models. Numerous methods of pain testing are available (22). Visceral pain, such as emanating from the bladder, may be manifested as cutaneous hypersensitivity that is quantifiable by mechanical allodynia (23). For these studies, mechanical allodynia was quantified as responses to the non-painful stimulus of graded von Frey filaments (24). Individual fibers of variable force were applied to the mouse pelvic region or hindpaw. Positive behavioral responses, including sharp retraction of the abdomen, immediate licking or scratching of the stimulated area, or jumping were recorded, and the frequency of responses was compared across control and experimental groups. Testing with von Frey filaments allows for differentiation between isolated pelvic pain found in our model, and global pain (pelvis and hindpaw) that is found in other models (e.g., (24)). The test can be repeatedly performed over extended periods of time and requires no surgical or pharmacological intervention.
MECHANISMS OF UTI-ASSOCIATED PAIN
Acute UTI Pain is Dissociated From Inflammation and Apoptosis
Our initial studies determined that NU14 cystitis produces acute, self-limited pelvic pain (25). In contrast to our earlier observations that herpes-induced pelvic pain is dependent upon mast cells (26), NU14 pain is unchanged in mast cell-deficient KitW-sh mice, suggesting that UTI pain is independent of mast cells. ASB strain 83972 produced no pelvic pain when tested parallel with NU14. Bladder colonization was greater in NU14 compared to ASB 83972, but colonization did not correlate with pain at early or late times following inoculation. Urinary accumulation of neutrophil myeloperoxidase, a marker of bladder inflammation, was similar between both strains and also did not correlate with pain. Finally, gross pathology was not different among bladders of mice infected with NU14 and 83972 (27). Taken together, these findings suggest that cystitis pain is independent of bacterial loads and inflammation.
Type-1 pili, and the pilus adhesin FimH, are associated with urothelial apoptotic lesions after cystitis (28). Given that type-1 piliation is a major difference between NU14 and 83972, we next evaluated the role of type 1 pili in the UTI pain response by employing the FimH-deficient strain NU14-1 and 83972 harboring a plasmid encoding type 1 pili. Both NU14 and NU14-1 induced significantly increased allodynia, whereas 83972 failed to induce allodynia, even when expressing type 1 pili (25). Thus, the disparate pain phenotypes of NU14 and 83972 are unaltered by modulating FimH expression, suggesting that UTI pain is independent of type 1 pili and the urothelial damage associated with UTI.
Acute UTI Pain Is Mediated by Lipopolysaccharide
Lipopolysaccharide (LPS), or endotoxin, is a glycolipid that is a major component of the outer leaflet of outer membrane of Gram-negative bacteria (29). Prior studies showed that LPS is a major mediator of bladder inflammatory response through its interactions with TLR4 (30–33). Although pain was not associated with inflammation, we tested the hypothesis that LPS mediates the differential pain phenotypes of NU14 and 83972 (25). LPS was purified from NU14 and 83972 and instilled into the mice as described above. Although the pain kinetics induced by LPS instillation were more rapid, likely due to instillation of concentrated free ligand, the pain responses to NU14 and 83972 LPS recapitulated the responses to intact bacteria. Despite the clear role of LPS in mediating the differential pain phenotypes of E. coli, NU14 and 83972 LPS induced similar levels of urinary myeloperoxidase. Nonetheless, the pain induced by NU14 LPS instillation was abrogated in TLR4-deficent mice. Together, these findings bolster the conclusion that the UTI pain response is independent of inflammation (Figure 1). And while the role for TLR4-mediated inflammatory responses is well established in diverse systems, these findings identify a novel role for TLR4 as a mediator of infection-associated pain responses independent of inflammation (25).
Figure 1.
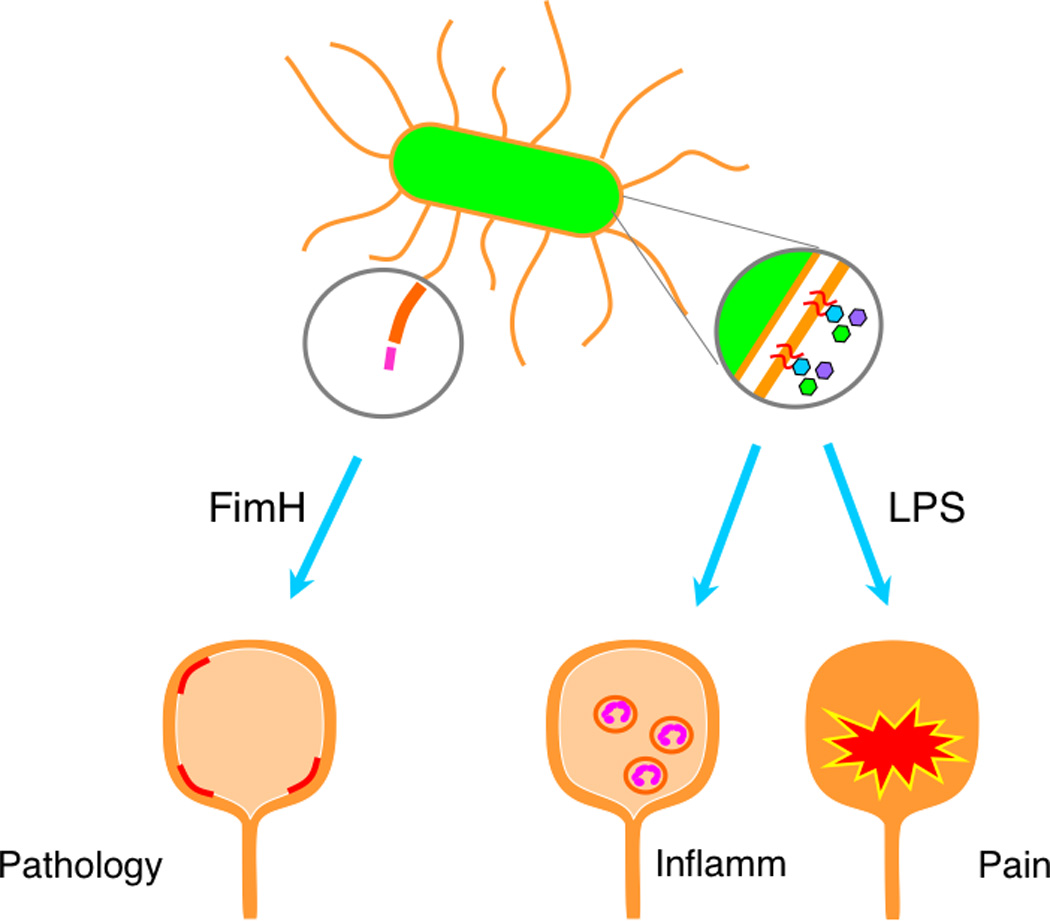
LPS mediates UTI pain independent of inflammation. FimH mediates UPEC adherence and induces urothelial apoptosis but is dispensable for pain. LPS drives both inflammation and pain, but these activities are separable.
Post-UTI Chronic Pain Is Modulated by O-antigen
Classically, chronic pain is thought to result from a repetitive, noxious stimulus that trigger molecular changes in the CNS and drive a central sensitization of ascending pain-processing pathways (34). Because of the association between IC and UTI history, we hypothesized that repetitive infection of mice with UPEC might trigger chronic pelvic pain, but we observed no such induction of a chronic response following repeated infection with NU14 (27). However, since we had previously identified an LPS mutant of NU14 that triggered altered inflammatory responses in low-inoculum models of early infection (35), we also evaluated this mutant for pain responses following serial infections.
LPS is a modular glycolipid consisting of three major constituents: acylated lipid A, a core oligosaccharide, and O-antigen, a complex carbohydrate structure (29). While lipid A is considered the chief mediator of inflammation by virtue of its interactions with TLR4 (36), diversity of E. coli serotypes is determined largely by O-antigen structure, where more than 180 serotypes are known (29). Surprisingly, despite the extensive diversity that suggests important biologic functions for O-antigen, there is limited understanding of the role for O-antigen in bacterial pathogenesis (37). In a genetic screen for transposon mutants defective for NU14 modulation of inflammatory responses, we previously identified waaL, a disruption of the gene encoding O-antigen ligase (35).
Whereas serial infections with wild NU14 induced only transient allodynia in response to an initial infection and subsequent infections, the waaL mutant induced no allodynia in response to initial infection. However, upon subsequent infections, waaL triggered pronounced pain responses, exceeding NU14 acute pain in magnitude and persisting at weeks despite rapid clearance of bacteria from the bladder (27). The persistence of pain in response to the O-antigen mutant was not merely due to persistent colonization because waaL is highly attenuated in murine UTI and was cleared rapidly (27, 35). The chronic pain produced by the waaL mutant was unaltered upon serial infection of T and B cell-deficient Rag1/2 knockout mice, thereby excluding the possibility that adaptive immune responses mediated the sensitized pain response triggered by waaL.
In order to explore whether UPEC-specific virulence factors act in concert with LPS O-antigen to define E. coli pain phenotypes, we tested a K-12 E. coli strain, SΦ874 (27). SΦ874 strain lacks the entire wz* encoding O-antigen biosynthesis (38, 39). A single infection with SΦ874 produced chronic allodynia lasting more than 4 weeks, yet the chronic pain phenotype of SΦ874 was abrogated by O-antigen expression. Bacterial clearance from the bladder was rapid after infection by SΦ874 with or without O-antigen. These data support the possibility that chronic allodynia may occur subsequent to a single, transient bacterial infection and that O-antigen determines pain phenotype. To confirm this possibility, a wz* deletion mutant of UPEC strain NU14 was generated, resulting in a chronic pain phenotype that could be restored to an acute phenotype by complementation with an NU14 wz* fosmid or entirely abrogated by complementation with an 83972 fosmid. These data suggest that a single UPEC strain may exhibit a spectrum of pain phenotypes, depending upon O-antigen expression (27).
Finally, a combination of TLR4-deficient mice and bone marrow chimeras revealed that post-UTI chronic pain was dependent upon TLR4, but the requirement for TLR4 was independent of hematopoietic lineages, and inflammation was not correlated with pain chronicity (27). Collectively, these findings reveal a novel pain phenotype for some E. coli strains, including a post-UTI chronic pain phenotype that is modulated by O-antigen. Like acute UTI pain, post-UTI chronic pain is mediated by TLR4 but independent of inflammation.
Murine Post-UTI Chronic Pain Models Recapitulate Aspects of Human Disease
Animal studies are unable to purely replicate subjective human reporting of pain and emotional experience. However, measurement of allodynia as a pain phenotype is classic, reproducible, and has an important physiologic basis. Other experimental methods can measure visceral hypersensitivity and relative states of anxiety that are both frequently associated with chronic pain in humans (40, 41).
We evaluated visceral hypersensitivity by testing visceromotor response (VMR) to bladder distension (42). Modified from a previously described protocol (43), we filled the mouse bladder with saline by transurethral catheter at discrete pressures and measured electromyographic (EMG) activity from the external oblique muscles using acutely implanted electrodes. Bladder pressures were computer-controlled with continuous feedback from pressure transducers ensuring stable, reproducible distension. The quantifiable EMG activity was compared to baseline occurring just prior to bladder distension. Tests of mice with chronic allodynia after SΦ874 infection demonstrated hypersensitivity to bladder distension compared to control (J. Rosen, in preparation). Consistent with increased EMG activity in response to bladder filling, dysuria was also in mice with post-UTI chronic pain induced by waaL (Rudick and Klumpp, unpublished observations). Dysuria was observed as void-induced grooming behavior that was increased only transiently during acute NU14 infection but persisted at least 14 days in waaL-infected mice. These findings suggest that post-UTI chronic pain induced by E. coli is also associated with voiding dysfunction, a hallmark of IC.
IC is often associated with co-morbid conditions, and depression has been previously reported among IC patients (reviewed in (44)). Novelty suppressed feeding (NSF) is a rodent correlate to anxious/depressive behavior that is sensitive to chronic anti-depressant therapy (45). Briefly, NSF exploits competing inherent desires of a mouse for food against its fear of open, brightly lit areas. Mice are fasted for 24-hours, then placed in the corner of an open field that contains a single food-pellet in the center. The latency to begin eating the pellet increases if mice are anxious or depressed, approximating anorexia or anhedonia in human depression. Mice with chronic allodynia after SΦ874 infection demonstrated increased latency to eat (J. Rosen, in preparation). Thus, as in human chronic pain, the mice exhibited cognitive change correlating to increased anxiety or depression.
Peripheral and Central Mechanisms in Post-UTI Chronic Pain
Peripheral sensory stimulation and central nervous system sensitization may both contribute to chronic pain (46). Our post-UTI models rely on initial O-antigen mediated, TLR4-dependent event. Bone marrow chimeras excluded the involvement of hematopoietic TLR4, and we did not observe TLR4 co-localized with bladder sensory fibers ((27) and unpublished observations). These findings suggest that the TLR4 which mediates UTI pain resides in the urothelium, but the downstream receptors mediating sensory fiber activation were unclear. We exploited the post-UTI chronic pain of SΦ874 to assess the potential role of two candidate receptors, the transient receptor potential vanilloid type 1 (TRPV1) and C-C chemokine receptor type 2 (CCR2), as mediators of chronic pelvic pain.
TRPV1 is a widely expressed nociceptor activated by a variety of endogenous (i.e., low pH, endocannibanoids) and exogenous stimuli (i.e., heat, capsaicin). TRPV1 is implicated in various chronic pain syndromes and is a mediator of bladder responses (47–51). Previously it was demonstrated that LPS, in a TLR4-dependent manner, increased TRPV1 expression and sensitivity in trigeminal nerve culture (52). We administered the classic TRPV1 antagonist capsazepine concurrent with SΦ874 infection and found that increased allodynia, VMR, and NSF did not develop (J. Rosen, in preparation). However, capsazepine had no effect when administered after SΦ874-associated allodynia and anxiety were established. Dependence on TRPV1 was further supported by failure of SΦ874 to induce allodynia, VMR, or NSF in TRPV1−/− mice. These findings suggest that TRPV1 mediates both the urologic and cognitive changes associated with post-UTI chronic pain. Furthermore, it is likely that TRPV1 mediates these urologic and cognitive changes at the level of initiation but is otherwise dispensable for maintenance of post-UTI manifestations.
Monocyte chemotactic protein 1 (MCP-1) is the cognate ligand for CCR2, and the MCP-1/CCR2 axis has been implicated previously in chronic pain downstream of TRPV1 in mouse DRGs following sciatic nerve injury (53). Although MCP-1 and receptor CCR2 are not normally expressed in dorsal root ganglia (DRG), expression increases after experimental nerve injury, suggesting a role in neuropathic pain (54). We employed transgenic mice where the promoters for CCR2 and MCP-1 drive expression of green and red fluorescent proteins, respectively (55). SΦ874 infection resulted in expression of both CCR2 and MCP-1 at two weeks after infection, and expression was co-localized in DRG cell bodies (J. Rosen, in preparation). Interestingly, while infection with SΦ874 expressing O-antigen (null pain phenotype) induced CCR2, MCP-1 expression was not induced. Furthermore, chronic allodynia was significantly reduced in mice treated with a CCR2 antagonist. These findings are in contrast with those of the role of TRPV1 in post-UTI chronic pain: whereas TRPV1 mediates pain initiation, the MCP-1/CCR2 is required for pain maintenance.
Central nervous system excitability was also hypothesized to play a role in chronic pain after UTI. An isolated preparation of murine sacral spinal cord, the primary location innervated by bladder sensory afferent nerves, was evaluated for spontaneous and induced activity. Initial testing revealed similarly increased spontaneous ventral root action potential firing in spinal cords isolated from mice infected with NU14, waaL, or SΦ874 (27). Correlating with resolution of allodynia, the increased spontaneous activity associated with acute NU14 infection returned to control levels by 14 days after infection, but spontaneous firing rate was not a marker suitable for discriminating between acute and chronic hyperexcitability. To quantify inhibitory control, we measured short-term depression in sacral spinal cords by repeatedly stimulating dorsal roots and measuring ventral responses, responses that typically diminish with successive stimuli as a consequence of normal inhibitory mechanisms (56). Spinal cords of NU14-infected mice exhibited the diminished responses characteristic of inhibitory control that were not significantly different from controls. In contrast, spinal cords of waaL- and SΦ874-infected mice showed significantly less inhibition, and these deficits in inhibited responses were observed at multiple stimulus intensities (27). These findings demonstrate CNS hyperexcitability in models of post-UTI chronic pain consistent with central sensitization. Thus, taken together with findings of TRPV1 and MCP-1/CCR2 involvement, these studies suggest both peripheral and central mechanisms of post-UTI chronic pain (Figure 2).
Figure 2.
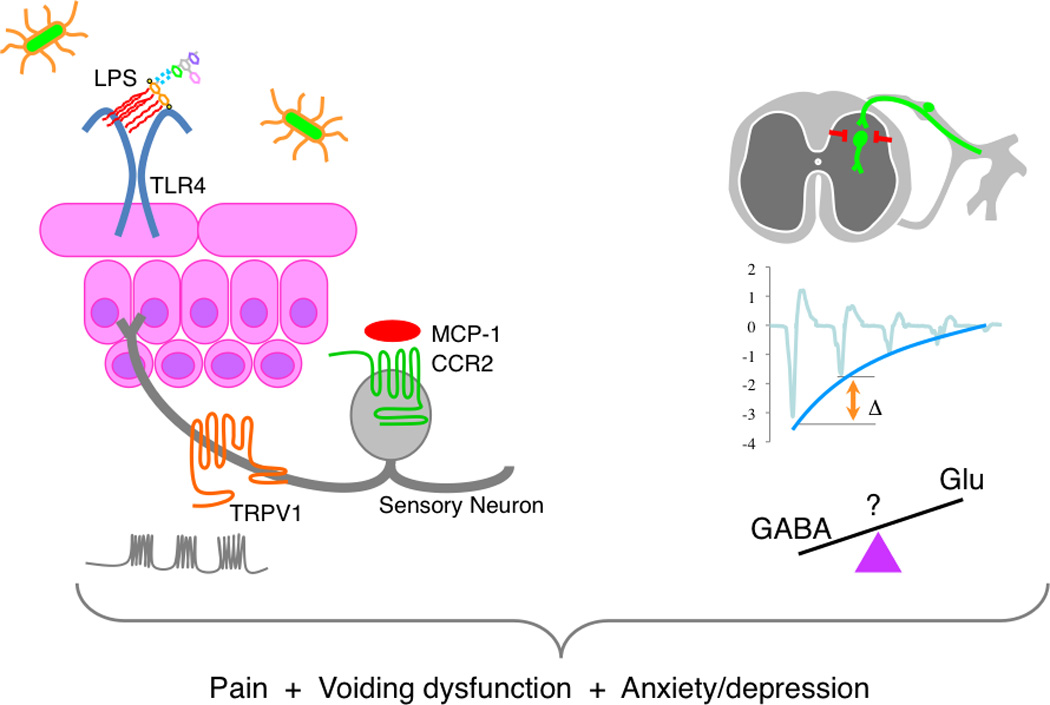
Post-UTI chronic pain is mediated by multiple receptors. E. coli induce pain responses through TLR4, TRPV1, and CCR2 (left panel). Post-UTI chronic pain is centralized and associated with defects in inhibitory control. It remains unclear to what extent defects in inhibition are due to diminished inhibitory circuit function or increased excitatory function (e.g., GABAergic or glutamatergic signaling, respectively).
DISCUSSION
Post-UTI pain is modulated by bacterial O-antigen structure and is separable from well-characterized mechanisms inducing bladder inflammation. These findings call into question any assumption that infection pain is merely a consequence of inflammation, as distinct pain phenotypes are not correlated with inflammation. Indeed, since it has been suggested from classic pain models that inflammation mediates pain (46), our data argue for the importance of continued studies of clinically relevant pain models. Similarly, although central sensitization is classically thought to require a repetitive, noxious stimulus (34), we observe features of central sensitization following a single transient infection with SΦ874, including chronic allodynia and spinal hyperexcitability (27). Further studies are needed to determine if this discrepancy from classic models of chronic pain is unique to processing of bladder afferent signals, E. coli infection, or infectious disease in general.
The relevance of animal models must always be considered carefully, particularly in the context of a syndrome such as IC, where the clinical entity remains an enigma and is generally thought to reflect a symptom complex likely to result from multiple potential etiologies (reviewed in (44)). We find that murine models of post-UTI chronic pain exhibit pelvic allodynia consistent with bladder pain, distension-evoked EMG activity, and dysuria. These models therefore recapitulate key aspects of pelvic pain and voiding dysfunction associated with IC and are consistent with our observations of spinal hyperexcitability. It should not be overlooked that these are all spinal reflex phenomena that alone do not fully capture the complexity of IC. However, like other chronic pain conditions, IC also results in cognitive changes that can be manifested as depression (57). Thus, we find it compelling that mice with post-UTI chronic pain also exhibit increased NSF latency indicative of anxious/depression behavior. This suggests that E. coli-induced pain in mice represent clinically relevant models that are useful for studying both urologic and cognitive dysfunctions associated with chronic pelvic pain.
We find that E. coli cystitis may induce differential pain responses across a spectrum from no pain to acute pain or chronic pain even after a single infection. We also observe that chronic pain may result from a single E. coli infection with SΦ874, whereas waaL requires multiple infections for a pain phenotype, and we find differential induction of CCR2 and MCP-1 by SΦ874, depending upon O-antigen expression. These observations are consistent with a peripheral sensitization mechanism whereby an initial infectious stimulus potentiates bladder nociception pathways, possibly at the level of MCP-1/CCR2, thus facilitating enhanced responses to subsequent insults. Peripheral sensitization has been previously demonstrated in rodents using various noxious chemicals, but E. coli would represent the first example of a clinically relevant agent to produce such sensitization. Taken together, these rodent models suggest at least one mechanism by which UTI may result in chronic pelvic pain, where E. coli trigger CCR2 up-regulation and thus sensitize individuals to a later infection and the potential for centralized nociceptive effects. That all E. coli can produce such chronic pain in all individuals is unlikely. Rather, it is more likely that chronic pelvic pain results from the unfortunate combination of an E. coli strain with a chronic pain phenotype infecting an individual whose is susceptible to developing chronic pain at the genetic/epigenetic level. UTIs are very common, and diverse serotypes and O-antigen phenotypes (smooth versus rough) are associated with UTI (58, 59). So it is possible that such unfortunate combinations of susceptible individuals and infection with chronic pain E. coli may underlie the association of IC with UTI history (Figure 2).
It is likely that defining the mechanisms of UTI-associated pain will inform future clinical approaches to the management of chronic pelvic pain. For example, resiniferatoxin (RTX) is a potent agonist of TRPV1 that can also be used to desensitize afferents (60). RTX was thus employed in clinical trials for chronic pain in IC patients but has yielded variable results (61–63). This may be consistent with our findings that TRPV1 mediates chronic pain establishment but is dispensable for pain maintenance in post-UTI chronic pain. Thus, although RTX may indeed prove effective against IC pain in future studies, our data suggest that patients must be stratified in such trial, perhaps to focus on patients early in disease progression, before any UTI-associated pain becomes refractory to therapies targeting TRPV1. In contrast, we observe that a CCR2 antagonist reduces post-UTI chronic pain. Increased MCP-1 has previously been associated with chronic prostatis/chronic pelvic pain syndrome (CP/CPPS), and an E. coli strain isolated from a CP/CPPS patient induces chronic pelvic pain in mice (64, 65). Many drugs targeting the MCP-1/CCR2 axis are currently in development. By analogy with our murine studies, we speculate that such CCR2 antagonists would be effective against post-UTI chronic pelvic pain in humans, perhaps both in IC and CP/CPPS. Patients with a known history of UTI preceding the onset of chronic pelvic pain would represent an attractive cohort for such studies of CCR2 antagonists for pelvic pain.
CONCLUSION
In summary, the murine UTI model can be used to characterize mechanisms of pelvic pain responses in response to E. coli infection. E. coli strains may exhibit a spectrum of pain phenotypes ranging from a null phenotype to a phenotype that induced chronic pelvic pain following a single, transient infection. The pain phenotype is independent of inflammation but is nonetheless mediated by LPS and the LPS receptor, TLR4, and subsequent afferent responses involving TRPV1 and CCR2. Post-UTI chronic pain in mice can recapitulate key features of urologic and cognitive dysfunction observed in IC patients, and these findings from clinically relevant infection models have significant implications for our understanding of chronic pain mechanisms and therapeutic targets for IC.
ACKNOWLEDGEMENTS
Original work described here from the Klumpp laboratory was supported by NIH/NIDDK awards DK066112, DK04648, and DK82342 to D.J.K.
REFERENCES
- 1.Foxman B, Barlow R, D'Arcy H, Gillespie B, Sobel JD. Urinary tract infection: self-reported incidence and associated costs. Ann Epidemiol. 2000;10(8):509–515. doi: 10.1016/s1047-2797(00)00072-7. [DOI] [PubMed] [Google Scholar]
- 2.Peters KM, Killinger KA, Ibrahim IA. Childhood symptoms and events in women with interstitial cystitis/painful bladder syndrome. Urology. 2009;73(2):258–262. doi: 10.1016/j.urology.2008.09.014. Epub 2008/11/28. [DOI] [PubMed] [Google Scholar]
- 3.Sauer FG, Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bacterial pili: molecular mechanisms of pathogenesis. Curr Opin Microbiol. 2000;3(1):65–72. doi: 10.1016/s1369-5274(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 4.Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. Bacterial adhesion to target cells enhanced by shear force. Cell. 2002;109(7):913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 5.Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8829–8835. doi: 10.1073/pnas.97.16.8829. Epub 2000/08/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klumpp DJ, Rycyk MT, Chen MC, Thumbikat P, Sengupta S, Schaeffer AJ. Uropathogenic Escherichia coli induces extrinsic and intrinsic cascades to initiate urothelial apoptosis. Infect Immun. 2006;74(9):5106–5113. doi: 10.1128/IAI.00376-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, et al. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282(5393):1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 8.Hunstad DA, Justice SS. Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli. Annual review of microbiology. 2010;64:203–221. doi: 10.1146/annurev.micro.112408.134258. Epub 2010/09/10. [DOI] [PubMed] [Google Scholar]
- 9.Bleidorn J, Gagyor I, Kochen MM, Wegscheider K, Hummers-Pradier E. Symptomatic treatment (ibuprofen) or antibiotics (ciprofloxacin) for uncomplicated urinary tract infection?--results of a randomized controlled pilot trial. BMC medicine. 2010;8:30. doi: 10.1186/1741-7015-8-30. Epub 2010/05/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragnarsdottir B, Svanborg C. Susceptibility to acute pyelonephritis or asymptomatic bacteriuria: host-pathogen interaction in urinary tract infections. Pediatr Nephrol. 2012;27(11):2017–2029. doi: 10.1007/s00467-011-2089-1. Epub 2012/02/14. [DOI] [PubMed] [Google Scholar]
- 11.Lindberg U, Claesson I, Hanson LA, Jodal U. Asymptomatic bacteriuria in schoolgirls. I. Clinical and laboratory findings. Acta paediatrica Scandinavica. 1975;64(3):425–431. doi: 10.1111/j.1651-2227.1975.tb03859.x. Epub 1975/05/01. [DOI] [PubMed] [Google Scholar]
- 12.Andersson P, Engberg I, Lidin-Janson G, Lincoln K, Hull R, Hull S, et al. Persistence of Escherichia coli bacteriuria is not determined by bacterial adherence. Infect Immun. 1991;59(9):2915–2921. doi: 10.1128/iai.59.9.2915-2921.1991. Epub 1991/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez JG, Sunden F, Connolly J, Svanborg C, Wullt B. Genetic control of the variable innate immune response to asymptomatic bacteriuria. PLoS ONE. 2011;6(11):e28289. doi: 10.1371/journal.pone.0028289. Epub 2011/12/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hull RA, Rudy DC, Donovan WH, Wieser IE, Stewart C, Darouiche RO. Virulence properties of Escherichia coli 83972, a prototype strain associated with asymptomatic bacteriuria. Infect Immun. 1999;67(1):429–432. doi: 10.1128/iai.67.1.429-432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klemm P, Roos V, Ulett GC, Svanborg C, Schembri MA. Molecular characterization of the Escherichia coli asymptomatic bacteriuria strain 83972: the taming of a pathogen. Infect Immun. 2006;74(1):781–785. doi: 10.1128/IAI.74.1.781-785.2006. Epub 2005/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roos V, Schembri MA, Ulett GC, Klemm P. Asymptomatic bacteriuria Escherichia coli strain 83972 carries mutations in the foc locus and is unable to express F1C fimbriae. Microbiology (Reading, England) 2006;152(Pt 6):1799–1806. doi: 10.1099/mic.0.28711-0. [DOI] [PubMed] [Google Scholar]
- 17.Vejborg RM, Friis C, Hancock V, Schembri MA, Klemm P. A virulent parent with probiotic progeny: comparative genomics of Escherichia coli strains CFT073, Nissle 1917 and ABU 83972. Mol Genet Genomics. 2010;283(5):469–484. doi: 10.1007/s00438-010-0532-9. Epub 2010/04/01. [DOI] [PubMed] [Google Scholar]
- 18.Darouiche RO, Donovan WH, Del Terzo M, Thornby JI, Rudy DC, Hull RA. Pilot trial of bacterial interference for preventing urinary tract infection. Urology. 2001;58(3):339–344. doi: 10.1016/s0090-4295(01)01271-7. Epub 2001/09/11. [DOI] [PubMed] [Google Scholar]
- 19.Hopkins WJ, Hall JA, Conway BP, Uehling DT. Induction of urinary tract infection by intraurethral inoculation with Escherichia coli: refining the murine model. The Journal of infectious diseases. 1995;171(2):462–465. doi: 10.1093/infdis/171.2.462. [DOI] [PubMed] [Google Scholar]
- 20.Hung CS, Dodson KW, Hultgren SJ. A murine model of urinary tract infection. Nat Protoc. 2009;4(8):1230–1243. doi: 10.1038/nprot.2009.116. Epub 2009/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson JR, Weissman SJ, Stell AL, Trintchina E, Dykhuizen DE, Sokurenko EV. Clonal and pathotypic analysis of archetypal Escherichia coli cystitis isolate NU14. The Journal of infectious diseases. 2001;184(12):1556–1565. doi: 10.1086/323891. [DOI] [PubMed] [Google Scholar]
- 22.Barrot M. Tests and models of nociception and pain in rodents. Neuroscience. 2012;211:39–50. doi: 10.1016/j.neuroscience.2011.12.041. Epub 2012/01/17. [DOI] [PubMed] [Google Scholar]
- 23.Laird JM, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain. 2001;92(3):335–342. doi: 10.1016/S0304-3959(01)00275-5. [DOI] [PubMed] [Google Scholar]
- 24.Guerios SD, Wang ZY, Boldon K, Bushman W, Bjorling DE. Lidocaine prevents referred hyperalgesia associated with cystitis. Neurourology and urodynamics. 2009;28(5):455–460. doi: 10.1002/nau.20670. Epub 2009/03/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudick CN, Billips BK, Pavlov VI, Yaggie RE, Schaeffer AJ, Klumpp DJ. Host-pathogen interactions mediating pain of urinary tract infection. The Journal of infectious diseases. 2010;201(8):1240–1249. doi: 10.1086/651275. Epub 2010/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudick CN, Bryce PJ, Guichelaar LA, Berry RE, Klumpp DJ. Mast cell-derived histamine mediates cystitis pain. PLoS ONE. 2008;3(5):e2096. doi: 10.1371/journal.pone.0002096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudick CN, Jiang M, Yaggie RE, Pavlov VI, Done J, Heckman CJ, et al. O-antigen modulates infection-induced pain States. PLoS ONE. 2012;7(8):e41273. doi: 10.1371/journal.pone.0041273. Epub 2012/08/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thumbikat P, Berry RE, Zhou G, Billips BK, Yaggie RE, Zaichuk T, et al. Bacteria-induced uroplakin signaling mediates bladder response to infection. PLoS Pathog. 2009;5(5):e1000415. doi: 10.1371/journal.ppat.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annual review of biochemistry. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backhed F, Soderhall M, Ekman P, Normark S, Richter-Dahlfors A. Induction of innate immune responses by Escherichia coli and purified lipopolysaccharide correlate with organ- and cell-specific expression of Toll-like receptors within the human urinary tract. Cell Microbiol. 2001;3(3):153–158. doi: 10.1046/j.1462-5822.2001.00101.x. [DOI] [PubMed] [Google Scholar]
- 31.Jerde TJ, Bjorling DE, Steinberg H, Warner T, Saban R. Determination of mouse bladder inflammatory response to E. coli lipopolysaccharide. Urol Res. 2000;28(4):269–273. doi: 10.1007/s002400000114. Epub 2000/09/30. [DOI] [PubMed] [Google Scholar]
- 32.Samuelsson P, Hang L, Wullt B, Irjala H, Svanborg C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect Immun. 2004;72(6):3179–3186. doi: 10.1128/IAI.72.6.3179-3186.2004. Epub 2004/05/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song J, Duncan MJ, Li G, Chan C, Grady R, Stapleton A, et al. A novel TLR4-mediated signaling pathway leading to IL-6 responses in human bladder epithelial cells. PLoS Pathog. 2007;3(4):e60. doi: 10.1371/journal.ppat.0030060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 35.Billips BK, Schaeffer AJ, Klumpp DJ. Molecular basis of uropathogenic Escherichia coli evasion of the innate immune response in the bladder. Infect Immun. 2008;76(9):3891–3900. doi: 10.1128/IAI.00069-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Plainvert C, Bidet P, Peigne C, Barbe V, Medigue C, Denamur E, et al. A new O-antigen gene cluster has a key role in the virulence of the Escherichia coli meningitis clone O45:K1:H7. Journal of bacteriology. 2007;189(23):8528–8536. doi: 10.1128/JB.01013-07. Epub 2007/10/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Batchelor RA, Haraguchi GE, Hull RA, Hull SI. Regulation by a novel protein of the bimodal distribution of lipopolysaccharide in the outer membrane of Escherichia coli. Journal of bacteriology. 1991;173(18):5699–5704. doi: 10.1128/jb.173.18.5699-5704.1991. Epub 1991/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuhard J, Thomassen E. Altered deoxyribonucleotide pools in P2 eductants of Escherichia coli K-12 due to deletion of the dcd gene. Journal of bacteriology. 1976;126(2):999–1001. doi: 10.1128/jb.126.2.999-1001.1976. Epub 1976/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nickel JC, Tripp DA, Pontari M, Moldwin R, Mayer R, Carr LK, et al. Interstitial cystitis/painful bladder syndrome and associated medical conditions with an emphasis on irritable bowel syndrome, fibromyalgia and chronic fatigue syndrome. J Urol. 2010;184(4):1358–1363. doi: 10.1016/j.juro.2010.06.005. Epub 2010/08/20. [DOI] [PubMed] [Google Scholar]
- 41.Feng B, La JH, Schwartz ES, Gebhart GF. Irritable bowel syndrome: methods, mechanisms, and pathophysiology. Neural and neuro-immune mechanisms of visceral hypersensitivity in irritable bowel syndrome. American journal of physiology Gastrointestinal and liver physiology. 2012;302(10):G1085–G1098. doi: 10.1152/ajpgi.00542.2011. Epub 2012/03/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai HH, Qiu CS, Crock LW, Morales ME, Ness TJ, Gereau RWt. Activation of spinal extracellular signal-regulated kinases (ERK) 1/2 is associated with the development of visceral hyperalgesia of the bladder. Pain. 2011;152(9):2117–2124. doi: 10.1016/j.pain.2011.05.017. Epub 2011/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christianson JA, Gebhart GF. Assessment of colon sensitivity by luminal distension in mice. Nat Protoc. 2007;2(10):2624–2631. doi: 10.1038/nprot.2007.392. Epub 2007/10/20. [DOI] [PubMed] [Google Scholar]
- 44.Hanno P. Bladder Pain Syndrome (Interstitial Cystitis) and Related Disorders. In: Wein A, Kavoussi L, Novick A, Partin A, Peters C, editors. Campbell-Walsh Urology. 10th ed. Philadelphia: Elsevier; 2012. pp. 357–401. [Google Scholar]
- 45.Bodnoff SR, Suranyi-Cadotte B, Quirion R, Meaney MJ. A comparison of the effects of diazepam versus several typical and atypical anti-depressant drugs in an animal model of anxiety. Psychopharmacology. 1989;97(2):277–279. doi: 10.1007/BF00442264. Epub 1989/01/01. [DOI] [PubMed] [Google Scholar]
- 46.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139(2):267–284. doi: 10.1016/j.cell.2009.09.028. Epub 2009/10/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, et al. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nature neuroscience. 2002;5(9):856–860. doi: 10.1038/nn902. Epub 2002/08/06. [DOI] [PubMed] [Google Scholar]
- 48.Li M, Sun Y, Simard JM, Chai TC. Increased transient receptor potential vanilloid type 1 (TRPV1) signaling in idiopathic overactive bladder urothelial cells. Neurourology and urodynamics. 2011;30(4):606–611. doi: 10.1002/nau.21045. Epub 2011/02/26. [DOI] [PubMed] [Google Scholar]
- 49.Skryma R, Prevarskaya N, Gkika D, Shuba Y. From urgency to frequency: facts and controversies of TRPs in the lower urinary tract. Nature reviews Urology. 2011;8(11):617–630. doi: 10.1038/nrurol.2011.142. Epub 2011/10/06. [DOI] [PubMed] [Google Scholar]
- 50.Wang ZY, Wang P, Merriam FV, Bjorling DE. Lack of TRPV1 inhibits cystitis-induced increased mechanical sensitivity in mice. Pain. 2008;139(1):158–167. doi: 10.1016/j.pain.2008.03.020. Epub 2008/05/01. [DOI] [PubMed] [Google Scholar]
- 51.Yu W, Hill WG, Apodaca G, Zeidel ML. Expression and distribution of transient receptor potential (TRP) channels in bladder epithelium. Am J Physiol Renal Physiol. 2011;300(1):F49–F59. doi: 10.1152/ajprenal.00349.2010. Epub 2010/10/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. Journal of dental research. 2011;90(6):759–764. doi: 10.1177/0022034511400225. Epub 2011/03/12. [DOI] [PubMed] [Google Scholar]
- 53.Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104(1):254–263. doi: 10.1111/j.1471-4159.2007.04969.x. Epub 2007/10/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. Journal of neurophysiology. 2006;96(5):2189–2199. doi: 10.1152/jn.00222.2006. Epub 2006/06/16. [DOI] [PubMed] [Google Scholar]
- 55.White FA, Feldman P, Miller RJ. Chemokine signaling and the management of neuropathic pain. Mol Interv. 2009;9(4):188–195. doi: 10.1124/mi.9.4.7. Epub 2009/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annual review of physiology. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. Epub 2002/02/05. [DOI] [PubMed] [Google Scholar]
- 57.Nickel JC, Tripp DA, Pontari M, Moldwin R, Mayer R, Carr LK, et al. Psychosocial phenotyping in women with interstitial cystitis/painful bladder syndrome: a case control study. J Urol. 2010;183(1):167–172. doi: 10.1016/j.juro.2009.08.133. Epub 2009/11/17. [DOI] [PubMed] [Google Scholar]
- 58.Hultgren SJ, Schwan WR, Schaeffer AJ, Duncan JL. Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect Immun. 1986;54(3):613–620. doi: 10.1128/iai.54.3.613-620.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stenutz R, Weintraub A, Widmalm G. The structures of Escherichia coli O-polysaccharide antigens. FEMS microbiology reviews. 2006;30(3):382–403. doi: 10.1111/j.1574-6976.2006.00016.x. [DOI] [PubMed] [Google Scholar]
- 60.Okun A, DeFelice M, Eyde N, Ren J, Mercado R, King T, et al. Transient inflammation-induced ongoing pain is driven by TRPV1 sensitive afferents. Molecular pain. 2011;7:4. doi: 10.1186/1744-8069-7-4. Epub 2011/01/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Apostolidis A, Gonzales GE, Fowler CJ. Effect of intravesical Resiniferatoxin (RTX) on lower urinary tract symptoms, urodynamic parameters, and quality of life of patients with urodynamic increased bladder sensation. European urology. 2006;50(6):1299–1305. doi: 10.1016/j.eururo.2006.04.006. Epub 2006/05/16. [DOI] [PubMed] [Google Scholar]
- 62.Lazzeri M, Spinelli M, Beneforti P, Malaguti S, Giardiello G, Turini D. Intravesical infusion of resiniferatoxin by a temporary in situ drug delivery system to treat interstitial cystitis: a pilot study. European urology. 2004;45(1):98–102. doi: 10.1016/s0302-2838(03)00418-4. Epub 2003/12/12. [DOI] [PubMed] [Google Scholar]
- 63.Payne CK, Mosbaugh PG, Forrest JB, Evans RJ, Whitmore KE, Antoci JP, et al. Intravesical resiniferatoxin for the treatment of interstitial cystitis: a randomized, double-blind, placebo controlled trial. J Urol. 2005;173(5):1590–1594. doi: 10.1097/01.ju.0000154631.92150.ef. Epub 2005/04/12. [DOI] [PubMed] [Google Scholar]
- 64.Desireddi NV, Campbell PL, Stern JA, Sobkoviak R, Chuai S, Shahrara S, et al. Monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha as possible biomarkers for the chronic pelvic pain syndrome. J Urol. 2008;179(5):1857–1861. doi: 10.1016/j.juro.2008.01.028. discussion 61-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudick CN, Berry RE, Johnson JR, Johnston B, Klumpp DJ, Schaeffer AJ, et al. Uropathogenic Escherichia coli induces chronic pelvic pain. Infect Immun. 2011;79(2):628–635. doi: 10.1128/IAI.00910-10. Epub 2010/11/17. [DOI] [PMC free article] [PubMed] [Google Scholar]