

Abstract
Glycodiversification of natural products is an effective strategy for small molecule drug development. Recently, improved methods for chemo-enzymatic synthesis of glycosyl donors has spurred the characterization of natural product glycosyltransferases (GTs), revealing that the substrate specificity of many naturally occurring GTs as too stringent for use in glycodiversification. Protein engineering of natural product GTs has emerged as an attractive approach to overcome this limitation. This review highlights recent progress in the engineering/evolution of enzymes relevant to natural product glycodiversification with a particular focus upon GTs.
Introduction
Glycosyltransferases (GTs) are a large class of enzyme responsible for decorating natural products with an incredible variety of sugar moieties, including d/l-configured, amino-, deoxy-, and methoxy sugars [1], typically from nucleotide diphosphate (NDP) donors. Many therapeutically relevant natural products are glycosylated (Figure 1) and the sugar residues attached to such natural products by GTs are typically indispensable for biological activity [2,3]. Furthermore, the exact identity and pattern of glycosyl moieties can influence pharmacology/pharmacokinetics, invoke biological specificity at the molecular/tissue/organism level, and even define the precise mechanism of action [4,5]. This fact, coupled with the importance of natural products in drug development [6–9], has spurred the development of both chemical and enzymatic methods for glycosylating natural products.
Figure 1.
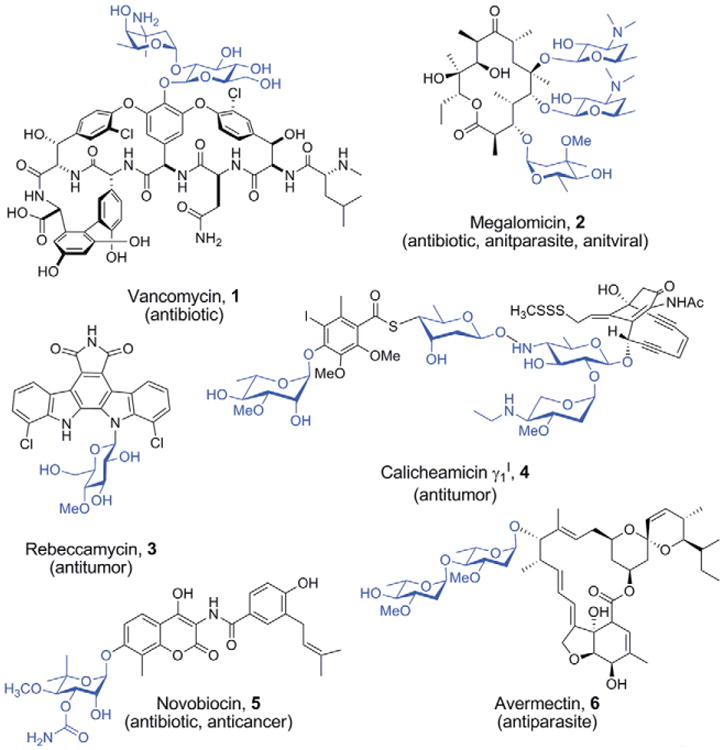
Natural product glycosides and their therapeutic properties. Sugar moieties are highlighted blue.
While the sheer architectural complexity of natural products (e.g. 4, Figure 1) often renders traditional chemical glycosylation strategies impractical for glycodiversification, emerging chemical glycosylation strategies for small molecules such as O'Doherty's de novo glycosylation strategy [10–12] and the neoglycorandomization strategy developed by Thorson and coworkers are rapidly expanding the role of glycosylation in small molecule therapeutic development [13,14•,15]. Alternatively, enzyme-dependent approaches have enabled both in vivo (e.g. pathway engineering) and in vitro (e.g. chemoenzymatic glycorandomization, Figure 2) strategies [16•,17]. Critically, both in vivo and in vitro approaches depend on the inherent GT promiscuity toward the glycosyl-donor and acceptor substrate and, in the case of in vivo approaches, are further restricted by stringency of the NDP-donor and/or aglycon biosynthetic enzymes. While the promiscuity of several wild-type GTs, such as those involved in the biosynthesis of 1 and 5 (Figure 1) is sufficient for efficient glycorandomization [18,19,20••,21,22•,23], the stringency of many GTs restrict their synthetic utility [24,25]. Thus, a major objective is to expand GT promiscuity toward the creation of ‘universal’ catalysts capable of accommodating a broad range of acceptors and sugars. This review will highlight recent applications of enzyme engineering in the context of enzyme-catalyzed sugar nucleotide synthesis and GT-catalyzed small molecule glycosylation.
Figure 2.
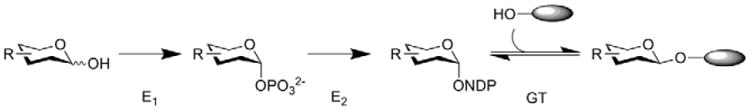
Chemo-enzymatic glycorandomization. E1 represents a promiscuous anomeric sugar kinase, E2 represents a promiscuous sugar-1-phosphate nucleotidyltransferase, GT represents a promiscuous glycosyltransferase and the gray oval represents a complex natural product scaffold.
Enzymatic synthesis of NDP-donors
The remarkable catalytic efficiency and the keen regio- and stereo-chemical control typically exhibited by enzymes favorably positions chemo-enzymatic strategies for sugar nucleotide synthesis. Sugar-1-phosphates are the common precursor to both enzymatic and chemical NDP-sugar syntheses, and enzyme engineering has been utilized to expand the utility of galactose kinase for sugar-1-phosphate synthesis [26–28]. Sugar-1-phosphates are subsequently converted to sugar nucleotides by nucleotidyltransferases and the nucleotide promiscuity of the uniquely malleable nucleotidyltransferase RmlA [29,30] has also recently been modified via enzyme engineering. Specifically, an examination of the RmlA purine nucleotide specificity revealed significant activity toward a panel of eight natural NTPs with glucose-1-phosphate, although specificity constants were reduced 15 000-fold [31]. Inspection of the RmlA structure suggested that steric bulk provided by the side chain of Gln-83 contributes to pyrimidine bias. Mutation of this residue to aspartic acid shifted specificity toward GTP from dTTP by several orders of magnitude. Similarly, substitution with serine provided a mutant with enhanced activity toward ATP. These ‘generalized’ RmlA variants with broad sugar-1-phosphate and nucleotide promiscuity should prove as useful biocatalysts for the generation of diverse purine-based sugar nucleotide libraries. For example, the RmlA mutant Q83D was recently used to synthesize a library of GDP-donors to probe the specificity of the polyene GTs AmphDI and NysD1 [32]. These protein engineering efforts add to the growing list of ‘substrate engineering’ studies revealing surprising plasticity of the nucleotidyltransferase catalytic machinery [33,34,35•,36,37]. Moreover, a newly disclosed high-throughout assay for nucleotidyltransferase activity should allow further expansion of the RmlA promiscuity via directed evolution [38].
Within the last few years the widely recognized reversibility of reactions catalyzed by GTs has also facilitated the in situ generation of NDP-donors via so-called ‘sugar-exchange’ and ‘aglycon-exchange’ reactions [19,20••,25]. Enzyme engineering efforts may improve reaction rates and/or GT stability to further expand the scope of these reactions. Other approaches to circumvent the strict requirement for NDP-donors, such as the use of analogs bearing simplified esters in place of the ‘activating’ NDP have proved less productive [39], although protein engineering is a likely route to improvements with such unnatural substrates as well.
In vivo approaches to NDP-donors and aglycons via ‘pathway engineering’, embrace gene deletion/insertion approaches [16•,17,40] that often suffer from technical issues associated with genetic manipulation of the producing strain and a significant reduction in production levels of metabolites. Some of these limitations have been overcome by the development of ‘plug and play’ gene cassettes for NDP-donor biosynthesis which provide for their in vivo syntheses [41–43]. Given the ease of transformation and genetic manipulation in Escherichia coli (E. coli), the reconstitution of natural product biosynthetic pathways in E. coli offers an attractive route to in vivo production of natural product glycosides. Recently, Khosla and co-workers reported the use of a simple bioassay screen for macrolide production to improve the biosynthesis of glycosylated macrolide antibiotics in E. coli [44•]. Alternatively, the overexpression of an engineered sugar kinase in E. coli led to the in vivo production of a library of unnatural sugar-1-phosphates as the first step toward in vivo glycorandomization [45].
Engineering natural product GTs
Rational (sequence-guided) design
The CAZY database of GTs (http://www.cazy.org/fam/acc_GT.html) contains ∼24 000 sequences of predicted or known GT sequences. As such, this database provides a wealth of sequence information that can be used to guide GT engineering efforts, as illustrated by the landmark study of Hoffmeister et al. aimed at identifying the residues responsible for donor specificity in the urdamycin GTs [46]. In vivo analysis of chimeras between UrdGT1c and UrdGT1b revealed that a single region conferred donor and acceptor specificity in these two highly related GTs. Specific residues capable of conferring specificity alterations were subsequently identified by mutagenesis of this region in UrdGT1b [47], and while some certain natural activities could only be supported by multiple sets of several amino acid mutations, completely novel activities could be generated by single mutations. In other examples guided by sequence alignments, changing the donor selectivity has proven more complex, such as engineering the donor C4-epimer specificity in a plant GT [48]. Thus, even very subtle specificity alterations are typically dictated by more than one residue. At the same time however, swapping larger sequence elements of plant GTs failed to achieve the desired donor specificity changes [49].
Rational (structure-based) design
Most (if not all) natural product GT's characterized to date belong to the GT-B superfamily of protein fold which consists of two closely associated Rossman-like domains. Within the GT-B fold, the catalytic site is located between the two domains as illustrated by the vancomycin GT GtfD [50], the macrolide GT OleD [51•], and the retaining avilamycin GT AviGT4 [52]. The highly conserved C-terminal domain is responsible for nucleotide binding, while the N-terminal domains show considerable sequence diversity, likely due to the variation in acceptor structures utilized by GT-B GTs. The approximate division of donor and acceptor binding between each domain of the GT-B fold, led this fold to be described as modular, and has served as the basis for the postulated engineering of hybrid GTs with altered substrate specificity by domain swapping [53]. However, the only successful examples citing alteration of GT-B specificity via domain-swapping have used highly homologous GTs, and the true modular nature of GT-B enzymes have yet to be demonstrated. For example, domain shuffling between two related plant flavonoid GTs (90% similarity at the amino acid level) revealed that a 40 amino acid region in the N-terminal domain could improve regios-electivity of glucosylation [54]. Subsequent site-directed mutagenesis illustrated that a single amino acid change was sufficient for this change in function.
In a spectacular example of engineering via domain shuffling, the dual N- and O-glucosylation activity (Figure 3a) of a novel plant GT was altered [55••]. Specifically, shuffling between the bifunctional N/O-GT UGT72B1 and the monofunctional N-GT BnUGT revealed that only chimeras containing residues 259-370 from UGT72B1 could support N-glucosylation. Incredibly, mutation at just two positions in BnUGT were sufficient for N-glycosyltransfer, while the reciprocal mutations in UGT72B1 (N312D-Y315F) decreased the kcat/Km ratio toward the amine acceptor by ∼560-fold (Figure 3b). Solution of the crystal structure of UGT72B1, revealed some clues as to the origin of the novel bifunctional N/O-glucosylation activity. It was hypothesized that a non-canonical geometry of the catalytic base His-19 (Figure 3c and compare to Figure 3d), and a nearby Asp-117 (forming a so-called ‘catalytic triad’ with the acceptor nucleophile) could allow nucleophilic attack by the amine acceptor, while not necessarily abstracting a proton as would otherwise be absolutely required for a corresponding hydroxyl-containing acceptor (Figure 3a). Examination of the chimeric GT bound to trichlorophenol or UDP revealed that Tyr-315 may play a role in maintaining His-19 and Asp-117 in their non-canonical positions, while Asn-312 locates to the same niche but does not make direct interactions with these critical residues. Interestingly, the structure of the bifunctional N/O-GT OleD [51•] reveals a catalytic base geometry that resembles the canonical arrangement (Figure 3e) (see also Section ‘directed evolution of glycosyltransferases’ below).
Figure 3.
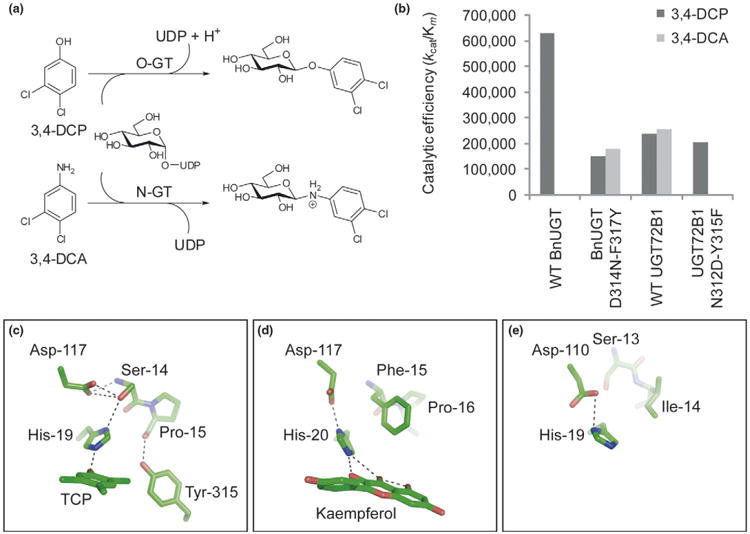
Engineering N/O-glucosylation activities in plant GTs. (a) Glycosylation of hydroxyl containing acceptors necessarily proceeds through proton abstraction whereas N-glycosyltransfer does not. 3,4-DCP, 3,4-dichlorophenol; 3,4-DCA, 3,4-dichloroaniline. (b) Catalytic efficiency of WT and mutant GTs toward N- and O-glucosyltransfer. WT BnUGT does not catalyze N-glucosyltransfer, 314N- F317Y displays considerable activity toward 3,4-DCA. The reciprocal mutations in UGT72B1 shifts the WT dual N/O-glucosyltransfer activity toward a more specific O-GT. (c) Active site of WT UGT72B1 showing the non-canonical interaction of His-19 with Ser-14. (d) Canonical geometry of the catalytic base in VvGT1 and (e) OleD (in this view, the substrate was excluded for clarity).
Alterations of natural product GT specificity by point mutation alone are less prevalent than those achievements guided by domain-shuffling [56]. This contrasts with the relative success of engineering the substrate specificity of the oligosaccharide forming GTs that belong to the GT-A family of protein fold [57,58], which may in part be due to the more recent emergence of natural product GT structures and the less diverse aglycon range utilized by the GT-A family. Nevertheless, several structures of natural product GTs with bound glycosyl donor (or at least modeled donor) have emerged, revealing some details regarding substrate binding and catalysis [59]. Yet, this information remains insufficient to guide even simple engineering exercises such as altering sugar C4 specificity (e.g. converting a glucosyltransferase to a galactosyltransferase) [51,60]. Therefore, continued efforts to obtain new GT-B structures (including different conformational/ligand-bound states of existing solved structures) remain high priority. The recent elucidation of the crystal structure of the C-GT UrdGT2 [61] stands as a notable milestone and may pave the way for engineering the production of novel C-glycosides. Further, structural studies targeted toward multiple GTs from single natural product pathways, which are likely evolutionary related may prove fruitful. For example, the recent disclosure of the crystal structure of CalG3 from the calicheamicin biosynthetic pathway sets the stage to determine the structural features that dictate the differing regioselectivity and promiscuity exhibited by the remaining three GTs involved in calicheamicin biosynthesis [62].
Directed evolution of glycosyltransferases
The lack of high-throughput GT assays has hampered GT directed evolution experiments, which typically require screening at least a few thousand variants. Recently, a high-throughput method was developed for screening oligosaccharide-forming (GT-A) sialyl-transferases (STs). While not directly relevant to classical natural product GTs, this inventive assay was based upon trapping the cell-permeable fluorescent acceptor bodipy-lactose via intracellular ST-catalyzed sialylation (attachment of a charged sugar) and fluorescent activated cell sorting [63••]. The result was the identification of a mutant which displayed a 153-fold better kcat/Km towards the fluorescent acceptor versus the non-fluorescent analog, lactose. The WT enzyme could not support the transfer of CMP-sialic acid to the fluorescently tagged acceptor. Determination of the mutant ST crystal structure revealed the formation a new hydrophobic pocket, to accommodate the fluorescent tag. Although this screen requires charged donors accessible via natural in vivo pathways and non-cytotoxic fluorescent acceptors, it stands as a powerful new tool in GT directed evolution.
In another illustration of the power of directed evolution, the promiscuity of a natural product GT was recently expanded [64••]. This advance was based on the key observation that the oleandomycin GT OleD could glucosylate a range of small aromatic phenolics, including the fluorescent umbelliferone, 7-hydroxy-4-methylcoumarin (Figure 4, 7) [65]. Specifically, the ability of OleD-catalyzed umbelliferone 7-OH glycosylation to mask fluorescence enabled screening a library of OleD variants created by error-prone PCR and resulted in the identification of several variants with improved activity toward 7/UDP-Glc [66]. A triple mutant (P67T/S132F/A242V) created by recombination of functional mutations produced a 60-fold improvement in catalytic efficiency with 7/UDP-Glc. Incredibly, this variant displayed a staggering improvement in promiscuity toward a diverse panel of glycosyl donors, 15 of 22 NDP-donors were substrates for the evolved GT, 12 of these were not detectable substrates for WT OleD. This study [64••], and a comprehensive follow-up study, revealed an unprecedented level of tolerance to acceptor scaffold and nucleophiles (RW Gantt, GJ Williams, and JS Thorson, unpublished data). Specifically, the evolved OleD catalyzed the glucosylation of 71 acceptors out of a 137-member panel. Successful acceptors included diverse drug-like scaffolds such as macrolides (e.g. 8 and 17, Figure 4), flavonoids (e.g. 9), isoflavones (e.g. 10), aminocoumarins (e.g. 11), anthraquinones (e.g. 12), indolocarbozoles (e.g. 13), polyenes (e.g. 14), cardenolides (e.g. 15), steroids (e.g. 16), beta-lactams (e.g. 18), enediynes (e.g. 19 and 20) and alkaloids (21). Putative glycosidic bonds were formed with N-, S-, and O-containing nucleophiles (e.g. 22, 23, and 24, respectively) with predicted pKa's ranging 4–18 (e.g. 25–26). The NMR structures of the OleD 22-, 23-, and 24-glucoside products conclusively confirmed, for the first time, the ability of OleD to catalyze heteroatom glycosidic bond formation. In addition, the variant OleD catalyzed iterative glycosylation in several cases, leading to the exciting possibility of using variant OleDs for synthesis of disaccharide or trisaccharide natural product analogs.
Figure 4.
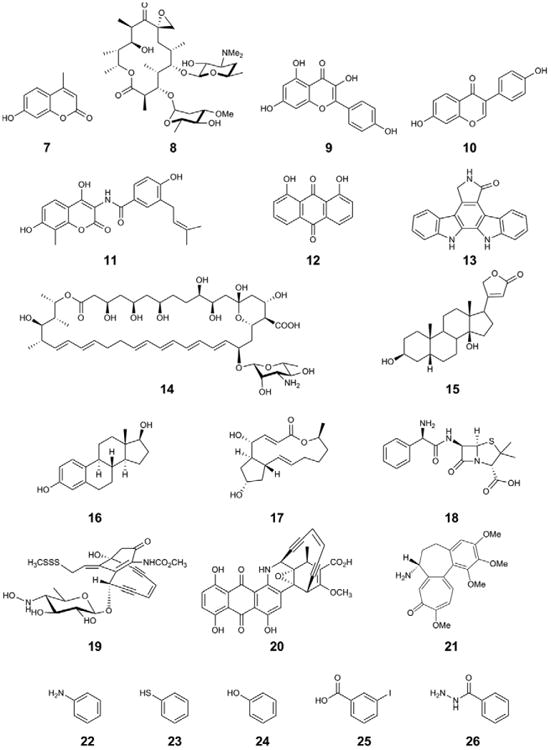
Diversity of acceptor substrates for evolved OleD variants.
While the triple mutant OleD glucosylated 11 acceptors that were not detectable substrates for the WT enzyme (e.g. 16 and 19, Figure 4), and displayed rate improvements toward other non-natural acceptors as high as 180-fold [64••], other enhancements were more modest, such as with the aminocoumarin novobiocic acid (Figure 4, 11) [64••]). Given the particular medicinal importance of glycosylated 11 analogs [67], and the stringency of the natural GT (NovM) responsible for 11-glycosylation [24], a two-phase engineering strategy was employed to further increase activity toward this target scaffold. It was reasoned that while amino acid mutations found in the original high-throughput screen (HTS) improve promiscuity in general, these mutations may not be optimal for a given acceptor. For the first phase, site-directed mutagenesis was used to discover the best combination of mutations found in the original progeny. In the second phase, small libraries created by saturation mutagenesis were screened by HPLC which led to the identification of a mutant with a several-hundred fold improvement in catalytic efficiency toward 11, while maintaining an impressive glycosyl donor promiscuity [68]. Notably, this was accomplished in the absence of a genuine HTS for 11-glucosylation, and suggests a similar strategy to further tailor activity to other non-natural acceptors. Directed evolution has previously produced promiscuous enzymes [69], even when such promiscuity has not been screened for directly. In the case of the evolved OleD variant, promiscuity may have arisen via several scenarios, such as changes to product release or improved NDP-donor binding. Functional mutations occur near the diphosphate-binding site, as well as within a loop that forms part of the acceptor binding site that is highly variable amongst other natural product GTs. These regions may prove to be effective hotspots in future engineering programs. Cumulatively, these studies exemplify the evolved OleD as a truly multifunctional biocatalyst for glycosidic bond formation.
In other recent developments, a method for screening glycosyltransferase saturation libraries was reported based upon the change in color of a pH indicator when a proton is released during the transfer of sugar moiety from the glycosyl donor to the acceptor [70]. It is not yet clear whether the strict assay conditions required to detect small changes in proton concentration are generally suitable for high throughput crude extract screening. In addition, a strategy for attaching the E. coli bacterial cell wall biosynthetic GT MurG, to the surface of phage, along with suitable acceptor substrate, was recently described [71]. The implementation this screen toward MurG directed evolution has yet to be reported.
Conclusions
Glycodiversification of natural products has emerged as an effective approach in the discovery of improved drugs. Chemo-enzymatic strategies making use of anomeric kinases, nucleotidyltransferases and GTs appear attractive due to the high regio- and stereo-control typically exhibited by these enzymes. Yet, the concomitant high substrate specificity of these enzymes also limits their general use. Structure-based and directed evolution approaches have already been used to provide anomeric kinases with expanded promiscuity, and directed evolution of nucleotidyltransferases is likely imminent. Natural product GTs have been traditionally awkward targets for directed evolution due to the lack of suitable screening methods. Novel screening methodologies are now beginning to appear and have already enabled the creation of novel GTs with dramatic changes in substrate specificity and promiscuity. Some of these novel enzymes will prove valuable for altering the glycosylation pattern of natural products and the construction of artificial in vivo glycodiversification pathways. We anticipate that future success will depend on the continued development of novel methods to screen or select for GTs with novel properties. In addition, the continued pursuit of new GT structures from natural product biosynthetic pathways and mutational analysis will further our understanding of the relationship between GT structure and function.
Acknowledgments
This contribution was supported in part by National Institutes of Health Grants CA84374 and U19 CA113297 (JTS). JST is an H.I. Romnes fellow.
References and recommended reading
Papers of particular interest, published within the of the review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Williams GJ, Zhang C, Thorson JS. Natural product glycosyltransferases: Properties and applications. Adv Enzymol Relat Areas Mol Biol. :76. doi: 10.1002/9780470392881.ch2. in press. [DOI] [PubMed] [Google Scholar]
- 2.Weymouth-Wilson AC. The role of carbohydrates in biologically active natural products. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 3.Křen V, Řezanka T. Sweet antibiotics – the role of glycosidic residues in antibiotic and antitumor activity and their randomization. FEMS Microbiol Rev. 2008;32:858–889. doi: 10.1111/j.1574-6976.2008.00124.x. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed A, Peters NR, Fitzgerald MK, Watson JA, Jr, Hoffmann FM, Thorson JS. Colchicine glycorandomization influences cytotoxicity and mechanism of action. J Am Chem Soc. 2006;128:14224–14225. doi: 10.1021/ja064686s. [DOI] [PubMed] [Google Scholar]
- 5.Langenhan JM, Peters NR, Guzei IA, Hoffmann FM, Thorson JS. Enhancing the anticancer properties of cardiac glycosides by neoglycorandomization. Proc Natl Acad Sci U S A. 2005;102:12305–12310. doi: 10.1073/pnas.0503270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 7.Newman DJ. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem. 2008;51:2589–2599. doi: 10.1021/jm0704090. [DOI] [PubMed] [Google Scholar]
- 8.Butler MS, Newman DJ. Mother Nature's gifts to diseases of man: the impact of natural products on anti-infective, anticholestemics and anticancer drug discovery. Prog Drug Res. 2008;65(1):3–44. doi: 10.1007/978-3-7643-8117-2_1. [DOI] [PubMed] [Google Scholar]
- 9.Butler MS. Natural products to drugs: natural product-derived compounds in clinical trials. Nat Product Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 10.Shan M, O'Doherty GA. De novo asymmetric syntheses of SL0101 and its analogues via a palladium-catalyzed glycosylation. Org Lett. 2006;8:5149–5152. doi: 10.1021/ol062076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo H, O'Doherty GA. De novo asymmetric synthesis of the anthrax tetrasaccharide by a palladium-catalyzed glycosylation reaction. Angew Chem Int Ed Engl. 2007;46:5206–5208. doi: 10.1002/anie.200701354. [DOI] [PubMed] [Google Scholar]
- 12.Zhou M, O'Doherty GA. De novo approach to 2-deoxy-β-glycosides: asymmetric syntheses of digoxose and digitoxin. J Org Chem. 2007;72:2485–2493. doi: 10.1021/jo062534+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langenhan JM, Griffith BR, Thorson JS. Neoglycorandomization and chemoenzymatic glycorandomization: two complementary tools for natural product diversification. J Nat Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]
- 14.Griffith BR, Krepel C, Fu X, Blanchard S, Ahmed A, Edmiston CE, Thorson JS. Model for antibiotic optimization via neoglycosylation: Synthesis of liponeoglycopeptides active against VRE. J Am Chem Soc. 2007;129:8150–8155. doi: 10.1021/ja068602r. • This paper describes the installation of all possible N-decanoylglucopyr-anose and N-biphenoylglucopyranose regioisomers onto the vancomycin aglycon. In contrast to prior work, optimal antibacterial activity via lipidation resulted from modification of the glucose C3′ or C4′. This study highlighted the efficiency of neoglycorandomization in small molecule drug discovery. [DOI] [PubMed] [Google Scholar]
- 15.Langenhan JM, Engle JM, Slevin LK, Fay LR, Lucker RW, Smith KR, Endo MM. Modifying the glycosidic linkage in digitoxin analogs provides selective cytotoxins. Bioorg Med Chem Lett. 2008;18:670–673. doi: 10.1016/j.bmcl.2007.11.058. [DOI] [PubMed] [Google Scholar]
- 16.Thibodeaux CJ, Melancon CE, Liu HW. Unusual sugar biosynthesis and natural product glycodiversification. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. • This review expertly summarizes both in vivo and in vitro approaches to natural product glycodiversification. [DOI] [PubMed] [Google Scholar]
- 17.Salas JA, Mendez C. Engineering the glycosylation of natural products in actinomycetes. Trends Microbiol. 2007;15:219–232. doi: 10.1016/j.tim.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Fu X, Albermann C, Zhang C, Thorson JS. Diversifying vancomycin via chemoenzymatic strategies. Org Lett. 2005;7:1513–1515. doi: 10.1021/ol0501626. [DOI] [PubMed] [Google Scholar]
- 19.Zhang C, Albermann C, Fu X, Thorson JS. The in vitro characterization of the iterative avermectin glycosyltransferase AveBI reveals reaction reversibility and sugar nucleotide flexibility. J Am Chem Soc. 2006;128:16420–16421. doi: 10.1021/ja065950k. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, Thorson JS. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science. 2006;313:1291–1294. doi: 10.1126/science.1130028. •• In this seminal report, the reversibility of four GTs was used to prepare more than 70 differentially glycosylated calicheamicin and vancomycin analogs in ‘sugar-exchange’ and ‘aglycone-exchange’ reactions. [DOI] [PubMed] [Google Scholar]
- 21.Fu X, Albermann C, Jiang J, Liao J, Zhang C, Thorson JS. Antibiotic optimization via in vitro glycorandomization. Nat Biotechnol. 2003;21:1467–1469. doi: 10.1038/nbt909. [DOI] [PubMed] [Google Scholar]
- 22.Minami A, Uchida R, Eguchi T, Kakinuma K. Enzymatic approach to unnatural glycosides with diverse aglycon scaffolds using glycosyltransferase VinC. J Am Chem Soc. 2005;127:6148–6149. doi: 10.1021/ja042848j. • This study reports on the substrate promiscuity of a macrolactam GT VinC. The authors discovered that the enzyme accepted both α- and β-anomers of several dTDP-2-deoxy-d-sugars to produce the β- and α-glycosides, respectively. Subsequently, a set of 22 diverse glycosides was constructed using VinC, and a mechanistic model proposed to explain the finding. [DOI] [PubMed] [Google Scholar]
- 23.Minami A, Eguchi T. Substrate flexibility of vicenisaminyltransferase VinC involved in the biosynthesis of vicenistatin. J Am Chem Soc. 2007;129:5102–5107. doi: 10.1021/ja0685250. [DOI] [PubMed] [Google Scholar]
- 24.Albermann C, Soriano A, Jiang J, Vollmer H, Biggins JB, Barton WA, Lesniak J, Nikolov DB, Thorson JS. Substrate specificity of NovM: implications for novobiocin biosynthesis and glycorandomization. Org Lett. 2003;5:933–936. doi: 10.1021/ol0341086. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C, Fu Q, Albermann C, Li L, Thorson JS. The in vitro characterization of the erythronolide mycarosyltransferase EryBV and its utility in macrolide diversification. ChemBioChem. 2007;8:385–390. doi: 10.1002/cbic.200600509. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmeister D, Yang J, Liu L, Thorson JS. Creation of the first anomeric d/l-sugar kinase by means of directed evolution. Proc Natl Acad Sci U S A. 2003;100:13184–13189. doi: 10.1073/pnas.2235011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Liu L, Thorson JS. Structure-based enhancement of the first anomeric glucokinase. ChemBioChem. 2004;5:992–996. doi: 10.1002/cbic.200400041. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Fu X, Liao J, Liu L, Thorson JS. Structure-based engineering of E. coli galactokinase as a first step toward in vivo glycorandomization. Chem Biol. 2005;12:657–664. doi: 10.1016/j.chembiol.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Barton WA, Lesniak J, Biggins JB, Jeffrey PD, Jiang J, Rajashankar KR, Thorson JS, Nikolov DB. Structure, mechanism and engineering of a nucleotidylyltransferase as a first step toward glycorandomization. Nat Struct Biol. 2001;8:545–551. doi: 10.1038/88618. [DOI] [PubMed] [Google Scholar]
- 30.Barton WA, Biggins JB, Jiang J, Thorson JS, Nikolov DB. Expanding pyrimidine diphosphosugar libraries via structure-based nucleotidylyltransferase engineering. Proc Natl Acad Sci U S A. 2002;99:13397–13402. doi: 10.1073/pnas.192468299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moretti R, Thorson JS. Enhancing the latent nucleotide triphosphate flexibility of the glucose-1-phosphate thymidylyltransferase RmlA. J Biol Chem. 2007;282:16942–16947. doi: 10.1074/jbc.M701951200. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C, Jiang J, Moretti R, Thorson J. The in vitro characterization of polyene glycosyltransferases AmphDI and NysDI. Chembiochem. doi: 10.1002/cbic.200800349. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Timmons SC, Mosher RH, Knowles SA, Jakeman DL. Exploiting nucleotidylyltransferases to prepare sugar nucleotides. Org Lett. 2007;9:857–860. doi: 10.1021/ol0630853. [DOI] [PubMed] [Google Scholar]
- 34.Huestis MP, Aish GA, Hui JP, Soo EC, Jakeman DL. Lipophilic sugar nucleotide synthesis by structure-based design of nucleotidylyltransferase substrates. Org Biomol Chem. 2008;6:477–484. doi: 10.1039/b716955h. [DOI] [PubMed] [Google Scholar]
- 35.Timmons SC, Hui JP, Pearson JL, Peltier P, Daniellou R, Nugier-Chauvin C, Soo EC, Syvitski RT, Ferrieres V, Jakeman DL. Enzyme-catalyzed synthesis of furanosyl nucleotides. Org Lett. 2008;10:161–163. doi: 10.1021/ol7023949. • Bacterial nucleotidyltransferases are usually used for the synthesis of hexopyranosyl nucleotides. This report describes the ability of several bacterial enzymes to couple four hexofuranosyl-1-phosphates with dTDP. This discovery should allow more extensive analysis of GT donor promiscuity. [DOI] [PubMed] [Google Scholar]
- 36.Mizanur RM, Zea CJ, Pohl NL, Ko KS, Zea CJ, Pohl NL. Unusually broad substrate tolerance of a heat-stable archaeal sugar nucleotidyltransferase for the synthesis of sugar nucleotides. J Am Chem Soc. 2004;126:15993–15998. doi: 10.1021/ja046070d. [DOI] [PubMed] [Google Scholar]
- 37.Ko KS, Zea CJ, Pohl NL, Mizanur RM, Zea CJ, Pohl NL. Strategies for the chemoenzymatic synthesis of deoxysugar nucleotides: substrate binding versus catalysis. J Org Chem. 2005;70:1919–1921. doi: 10.1021/jo048424p. [DOI] [PubMed] [Google Scholar]
- 38.Moretti R, Thorson JS. A comparison of sugar indicators enables a universal high-throughput sugar-1-phosphate nucleotidyltransferase assay. Anal Biochem. 2008;377:251–258. doi: 10.1016/j.ab.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khaled A, Piotrowska O, Dominiak K, Auge C. Exploring specificity of glycosyltransferases: synthesis of new sugar nucleotide related molecules as putative donor substrates. Carbohydr Res. 2008;343:167–178. doi: 10.1016/j.carres.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Blanchard S, Thorson JS. Enzymatic tools for engineering natural product glycosylation. Curr Opin Chem Biol. 2006;10:263–271. doi: 10.1016/j.cbpa.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez L, Aguirrezabalaga I, Allende N, Brana AF, Mendez C, Salas JA. Engineering deoxysugar biosynthetic pathways from antibiotic-producing microorganisms. A tool to produce novel glycosylated bioactive compounds. Chem Biol. 2002;9:721–729. doi: 10.1016/s1074-5521(02)00154-0. [DOI] [PubMed] [Google Scholar]
- 42.Lombo F, Gibson M, Greenwell L, Brana AF, Rohr J, Salas JA, Mendez C. Engineering biosynthetic pathways for deoxysugars: branched-chain sugar pathways and derivatives from the antitumor tetracenomycin. Chem Biol. 2004;11:1709–1718. doi: 10.1016/j.chembiol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Perez M, Lombo F, Baig I, Brana AF, Rohr J, Salas JA, Mendez C. Combinatorial biosynthesis of antitumor deoxysugar pathways in Streptomyces griseus: Reconstitution of “unnatural natural gene clusters” for the biosynthesis of four 2,6-D-dideoxyhexoses. Appl Environ Microbiol. 2006;72:6644–6652. doi: 10.1128/AEM.01266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee HY, Khosla C. Bioassay-Guided evolution of glycosylated macrolide antibiotics in Escherichia coli. PLoS Biology. 2007;5:243–250. doi: 10.1371/journal.pbio.0050045. • This article describes the construction and evolutionary-optimized macrolide glycosylation pathway in E. coli and sets the stage for high-level fermentation of novel macrolide glycosides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hui JP, Yang J, Thorson JS, Soo EC. Selective detection of sugar phosphates by capillary electrophoresis/mass spectrometry and its application to an engineered E. coli host. ChemBioChem. 2007;8:1180–1188. doi: 10.1002/cbic.200700116. [DOI] [PubMed] [Google Scholar]
- 46.Hoffmeister D, Ichinose K, Bechthold A. Two sequence elements of glycosyltransferases involved in urdamycin biosynthesis are responsible for substrate specificity and enzymatic activity. Chem Biol. 2001;8:557–567. doi: 10.1016/s1074-5521(01)00039-4. [DOI] [PubMed] [Google Scholar]
- 47.Hoffmeister D, Wilkinson B, Foster G, Sidebottom PJ, Ichinose K, Bechthold A. Engineered urdamycin glycosyltransferases are broadened and altered in substrate specificity. Chem Biol. 2002;9:287–295. doi: 10.1016/s1074-5521(02)00114-x. [DOI] [PubMed] [Google Scholar]
- 48.Kubo A, Arai Y, Nagashima S, Yoshikawa T. Alteration of sugar donor specificities of plant glycosyltransferases by a single point mutation. Arch Biochem Biophys. 2004;429:198–203. doi: 10.1016/j.abb.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 49.Kohara A, Nakajima C, Yoshida S, Muranaka T. Characterization and engineering of glycosyltransferases responsible for steroid saponin biosynthesis in Solanaceous plants. Phytochemistry. 2007;68:478–486. doi: 10.1016/j.phytochem.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 50.Mulichak AM, Lu W, Losey HC, Walsh CT, Garavito RM. Crystal structure of vancosaminyltransferase GtfD from the vancomycin biosynthetic pathway: interactions with acceptor and nucleotide ligands. Biochemistry. 2004;43:5170–5180. doi: 10.1021/bi036130c. [DOI] [PubMed] [Google Scholar]
- 51.Bolam DN, Roberts S, Proctor MR, Turkenburg JP, Dodson EJ, Martinez-Fleites C, Yang M, Davis BG, Davies GJ, Gilbert HJ. • The crystal structure of two macrolide glycosyltransferases provides a blueprint for host cell antibiotic immunity. Proc Natl Acad Sci U S A. 2007;104:5336–5341. doi: 10.1073/pnas.0607897104. The Streptomyces antibioticus GTs OleI and OleD are involved in host cell immunity from endogenous and exogenous agents. The crystal structures of these enzymes in conjunction with active-site mutational analysis provided insight into the mechanism of action and substrate specificity. The failure however to engineer UDP-Glc/UDP-Gal activity or to alter acceptor macrolide specificity by loop grafting and mutagenesis highlights that directed evolution methods are likely required. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez-Fleites C, Proctor M, Roberts S, Bolam DN, Gilbert HJ, Davies GJ. Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem Biol. 2006;13:1143–1152. doi: 10.1016/j.chembiol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 53.Hu Y, Walker S. Remarkable structural similarities between diverse glycosyltransferases. Chem Biol. 2002;9:1287–1296. doi: 10.1016/s1074-5521(02)00295-8. [DOI] [PubMed] [Google Scholar]
- 54.Cartwright AM, Lim EK, Kleanthous C, Bowles DJ. A kinetic analysis of regiospecific glucosylation by two glycosyltransferases of Arabidopsis thaliana: domain swapping to introduce new activities. J Biol Chem. 2008;283:15724–15731. doi: 10.1074/jbc.M801983200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brazier-Hicks M, Offen WA, Gershater MC, Revett TJ, Lim EK, Bowles DJ, Davies GJ, Edwards R. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc Natl Acad Sci U S A. 2007;104:20238–20243. doi: 10.1073/pnas.0706421104. • In vitro assay of a library of plant GTs identified a bifunctional N/O-GT. Subsequent crystal structure determination and successful protein engineering was used to provide unique insight into the dual N/O-glucosylation activity of the GT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He XZ, Wang X, Dixon RA. Mutational analysis of the Medicago glycosyltransferase UGT71G1 reveals residues that control regioselectivity for (iso)flavonoid glycosylation. J Biol Chem. 2006;281:34441–34447. doi: 10.1074/jbc.M605767200. [DOI] [PubMed] [Google Scholar]
- 57.Ramakrishnan B, Boeggeman E, Qasba PK. Applications of glycosyltransferases in the site-specific conjugation of biomolecules and the development of a targeted drug delivery system and contrast agents for MRI. Expert Opin Drug Deliv. 2008;5:149–153. doi: 10.1517/17425247.5.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qasba PK, Boeggeman E, Ramakrishnan B. Site-specific linking of biomolecules via glycan residues using glycosyltransferases. Biotechnol Prog. 2008;24:520–526. doi: 10.1021/bp0704034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 60.Offen W, Martinez-Fleites C, Yang M, Kiat-Lim E, Davis BG, Tarling CA, Ford CM, Bowles DJ, Davies GJ. Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 2006;25:1396–1405. doi: 10.1038/sj.emboj.7600970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mittler M, Bechthold A, Schulz GE. Structure and action of the C–C bond-forming glycosyltransferase UrdGT2 involved in the biosynthesis of the antibiotic urdamycin. J Mol Biol. 2007;372:67–76. doi: 10.1016/j.jmb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Zhang C, Bitto E, Goff R, Singh S, Bingman C, Griffith B, Albermann C, Phillips G, Jr, Thorson J. Biochemical and structural insights of the early glycosylation steps in calicheamicin biosynthesis. Chem Biol. 2008;15:842–853. doi: 10.1016/j.chembiol.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aharoni A, Thieme K, Chiu CP, Buchini S, Lairson LL, Chen H, Strynadka NC, Wakarchuk WW, Withers SG. High-throughput screening methodology for the directed evolution of glycosyltransferases. Nat Methods. 2006;3:609–614. doi: 10.1038/nmeth899. • A novel truly high-throughout assay for GT activity was described. A library of 106–107 sialyltransferase mutants was rapidly selected and a variant obtained with a several-hundred fold increase in sialylation activity. [DOI] [PubMed] [Google Scholar]
- 64.Williams GJ, Zhang C, Thorson JS. Expanding the promiscuity of a natural-product glycosyltransferase by directed evolution. Nat Chem Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. • A simple fluorescence based screen employing a surrogate acceptor substrate was used to evolve for the first time a natural product GT. Promiscuity toward donor and acceptor was significantly broadened. This study provides a foundation for the creation of variant GTs with novel activities for use in drug discovery. [DOI] [PubMed] [Google Scholar]
- 65.Yang M, Proctor MR, Bolam DN, Errey JC, Field RA, Gilbert HJ, Davis BG. Probing the breadth of macrolide glycosyltransferases: in vitro remodeling of a polyketide antibiotic creates active bacterial uptake and enhances potency. J Am Chem Soc. 2005;127:9336–9337. doi: 10.1021/ja051482n. [DOI] [PubMed] [Google Scholar]
- 66.Williams GJ, Thorson JS. A high-throughput fluorescence-based glycosyltransferase screen and its application in directed evolution. Nat Protocols. 2008;3:357–362. doi: 10.1038/nprot.2007.538. [DOI] [PubMed] [Google Scholar]
- 67.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BS. Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. J Org Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 68.Williams GJ, Goff RD, Zhang C, Thorson JS. Optimizing glycosyltransferase specificity via “hot spot” saturation mutagenesis presents a catalyst for novobiocin glycorandomization. Chem Biol. 2008;15:393–401. doi: 10.1016/j.chembiol.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carr R, Alexeeva M, Enright A, Eve TS, Dawson MJ, Turner NJ. Directed evolution of an amine oxidase possessing both broad substrate specificity and high enantioselectivity. Angew Chem Int Ed Engl. 2003;42:4807–4810. doi: 10.1002/anie.200352100. [DOI] [PubMed] [Google Scholar]
- 70.Persson M, Palcic MM. A high-throughput pH indicator assay for screening glycosyltransferase saturation mutagenesis libraries. Anal Biochem. 2008;378:1–7. doi: 10.1016/j.ab.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 71.Love KR, Swoboda JG, Noren CJ, Walker S. Enabling glycosyltransferase evolution: a facile substrate-attachment strategy for phage-display enzyme evolution. Chembiochem. 2006;7:753–756. doi: 10.1002/cbic.200600018. [DOI] [PubMed] [Google Scholar]