

Abstract
Hepatocyte-enriched nuclear factor (HNF)6 and CUX2 are GH and STAT5-regulated homeobox transcription factors. CUX2 shows female-specific expression and contributes to liver sex differences by repressing many male-biased genes and inducing many female-biased genes, whereas HNF6 is expressed at similar levels in male and female liver. In cell-based transfection studies, CUX2 inhibited HNF6 transcriptional regulation of the sex-specific gene promoters CYP2C11 and CYP2C12, blocking HNF6 repression of CYP2C11 and HNF6 activation of CYP2C12. These inhibitory actions of CUX2 can be explained by competition for HNF6 DNA binding, as demonstrated by in vitro EMSA analysis and validated in vivo by global analysis of the HNF6 cistrome. Approximately 40 000 HNF6-binding sites were identified in mouse liver chromatin, including several thousand sites showing significant sex differences in HNF6 binding. These sex-biased HNF6-binding sites showed strong enrichment for correspondingly sex-biased DNase hypersensitive sites and for proximity to genes showing local sex-biased chromatin marks and a corresponding sex-biased expression. Further, approximately 90% of the genome-wide binding sites for CUX2 were also bound by HNF6. These HNF6/CUX2 common binding sites were enriched for genomic regions more accessible in male than in female mouse liver chromatin and showed strongest enrichment for male-biased genes, suggesting CUX2 displacement of HNF6 as a mechanism to explain the observed CUX2 repression of male-biased genes in female liver. HNF6 binding was sex independent at a majority of its binding sites, and HNF6 peaks were frequently associated with cobinding by multiple other liver transcription factors, consistent with HNF6 playing a global regulatory role in both male and female liver.
The liver is characterized by widespread sex differences in gene expression (1–5) controlled by a complex network of GH-regulated transcription factors, including STAT5, BCL6, CUX2, and several hepatocyte-enriched nuclear factors (HNFs) (6–10). GH regulates liver STAT5 activity in a sex-dependent manner, with STAT5 activated intermittently in males and in a persistent manner in females in direct response to the sex-dependent patterns of pituitary GH secretion (8, 11). Stat5b deficiency leads to global loss of sex-biased expression in male mouse liver but affects many fewer sex-biased genes in female liver (3). Liver-specific deletion of HNF4α also leads to a major loss of sex differences in the liver, indicating that STAT5b and HNF4α are both required for maintenance of sex differences in the liver (12, 13).
STAT5 and HNF4α interact with each other and with a network of other liver-enriched transcription factors, including HNF6/Onecut (14, 15). In primary rat hepatocytes, GH-activated STAT5 and HNF4α both bind to and transactivate the promoter of HNF6, a OC divergent homeodomain (HD) gene (16). Other studies show, however, that Hnf6 expression is increased in livers of male mice deficient in HNF4α, indicating negative regulation of Hnf6 by HNF4α (17). HNF4α positively regulates the expression of the variant HD-containing protein Hnf1α, which negatively regulates Hnf4α as well as itself (18, 19). HNF6 can transactivate the genes encoding HNF4α and HNF3β (FOXA2), and can negatively regulate HNF3α (FOXA1) by inhibition of TGFβ signaling (20). Coordinated action between HNF6, HNF4α, and the coactivator PGC-1α regulates glucose-6-phosphatase during liver development (21). Further, STAT5 can enhance HNF4α activation of the ApoCIII promoter, whereas HNF4α can inhibit GH-stimulated STAT5 activation of the β-casein and Ntcp promoters (22). HNF3β is subject to positive auto-regulation and, like HNF6, is negatively regulated by HNF4α in male mouse liver. HNF3β displays cross talk with STAT5b and can inhibit STAT5b activation of a reporter gene by blocking STAT5b tyrosine phosphorylation (23). Conversely, STAT5b can inhibit HNF3β activation of the male-specific CYP2C11 promoter as well as the synergistic activation of the female-specific CYP2C12 promoter by HNF3β and HNF6 (23, 24).
STAT5b regulation of sex-specific gene expression is modulated by cross talk with the male-biased repressor BCL6 (7), which competes with STAT5 for binding at a subset of STAT5-binding sites (7, 25, 26) and preferentially binds to female-biased genes in male liver (8, 27). Recent studies have identified a complementary regulatory network in female liver, involving CUX2 (28), a highly female-specific repressor that is positively regulated by the more continuous plasma GH pattern in female liver, and is negatively regulated by HNF4α and STAT5b in male mouse liver (29). CUX2, which belongs to the same HD superfamily of transcription factors as HNF6 and HNF1α, binds to, and negatively regulates a subset of male-biased genes in female liver (9). CUX2 can also positively regulate a subset of female-biased genes, however, in a majority of cases this regulation is not associated with direct CUX2 binding and apparently proceeds via an indirect mechanism (9). CUX2-binding sites identified in female mouse liver are enriched at male-biased DNase I hypersensitive sites (DHSs), ie, genomic regions where chromatin is more accessible in male liver than in female liver (30). CUX2-binding sites also show significant enrichment for sites where STAT5 binding is greater in male than female liver (9), and which are linked to male-biased liver gene expression (8). Thus, CUX2 binding may repress male-specific gene expression in female liver by alteration of chromatin accessibility and/or by inhibition of STAT5 activation of male-biased genes.
CUX2 binding in mouse liver chromatin is described by a motif (9) most similar to the motifs for HNF6 (31), CUX1/CDP (32), and another HD protein, PBX1 (33). These 3 factors and several other HD-containing proteins are regulated by CUX2, as indicated by their responsiveness to CUX2 knockdown in female liver and/or CUX2 overexpression in male liver (9), suggesting a complex interplay between CUX2 and other HD transcription factors. In this study, we investigate the cross talk between CUX2 and HNF6 in mouse liver using cell-based transfections and electrophoretic mobility shift assay (EMSA) analysis, and by analysis of the liver HNF6 cistrome identified by chromatin immunoprecipitation (ChIP)-sequence (seq). Our findings show that a large majority of CUX2-binding sites in female mouse liver are also binding sites for HNF6, and that CUX2 can inhibit HNF6-dependent transcription, both transcriptional activation and transcriptional inhibition, by a mechanism that involves binding site competition. Unlike CUX2, which is expressed in and binds exclusively to chromatin in female mouse liver, HNF6 binding was observed in both male and female liver, with distinct subsets of HNF6-binding sites occupied in each sex, enabling this liver-enriched transcription factor to contribute both to the sex-dependent and the sex-independent expression of many genes, in part by an apparently cooperative binding to chromatin in association with multiple other liver-expressed transcription factors.
Materials and Methods
Transient transfection
293T cells grown in DMEM containing 10% fetal bovine serum were seeded in triplicate wells of 48-well tissue culture plates (∼75% confluency; ∼90 000 cells/well) and transfected 24 hours later using polyethylenimine (Polysciences, Inc). All transfections were carried out using 100 ng of reporter plasmid, 25–100 ng of transcription factor expression plasmid, and salmon sperm (control) DNA added to give a total of 275-ng DNA. Plasmid amounts used were based on initial titration studies carried out to determine the amounts of each plasmid that give a saturating response and hence the plasmid amounts to include in each cotransfection experiment (see examples in Supplemental Figure 1 Renilla luciferase reporter plasmid pRL-TK (25 ng; Promega) was included as an internal standard for transfection efficiency. Firefly and Renilla luciferase activities were determined using the Dual-Luciferase reporter assay system (Promega) and a Monolight 2010 luminometer (Analytical Luminescence Laboratory) using extracts prepared approximately 16 hours after initiation of transfection. Firefly luciferase values were normalized based on Renilla luciferase activities. Expression plasmids for mouse CUX1 and CUX2 (Dr A. Nepveu, McGill University, Montreal, Canada), HNF6, and OC-2 (Dr F. Lemaigre and Dr G. Rousseau, de Duve Institute, Universite Catholique de Louvain, Brussels, Belgium), HNF1α (Dr F. Gonzalez, National Cancer Institute, National Institutes of Health, Bethesda, MD), and HNF4α (Dr F. Sladek, University of California, Riverside, CA) were obtained from the indicated sources. The HNF6 reporter plasmid 6x-HNF6-Luc was provided by Dr F. Lemaigre and Dr G. Rousseau. Luciferase reporter plasmids pGL3-basic/Luc-1.8kb_CYP2C11promoter (23) and pGL3-basic/Luc-1.95kb_CYP2C12 promoter (24) were described previously.
Electrophoretic mobility shift assays
Cytoplasmic and nuclear extracts were prepared from approximately 900 000 293T cells per 10-cm dish transfected with 16 μg of CUX1, CUX2, HNF6, OC-2, or a control (empty) expression plasmid using the NucBuster protein extraction kit (Novagen). Where indicated, cytoplasmic extract was prepared in passive lysis buffer (Promega). EMSA samples contained 30 μg of cytoplasmic or nuclear extract, 2-μL EMSA buffer (20% glycerol, 5mM MgCl2, 2.5mM EDTA, 2.5mM dithiothreitol, 250mM NaCl, and 50mM Tris Cl; pH 7.5), 2-μg poly(deoxyinosinic-deoxycytidylic)acid (Sigma-Aldrich Corp), 3-μg bovine serum albumin and (where indicated) unlabeled oligonucleotide probe in a total of 14 μL. EMSA gels were run as described (9) using a CUX EMSA probe (9, 28) and other probes listed in figure 2 below. The antisense strand of each probe was end-labeled with 32P using T4 polynucleotide kinase, annealed to the sense strand, and then purified on a BioSpin30 Chromatography Column (Bio-Rad Laboratories, Inc).
Chromatin isolation, sonication and ChIP
All animal protocols were approved by the Boston University Institutional Animal Care and Use Committee. Young adult (7–8 wk) male and female CD-1 mice (crl:CD1 mice; Charles River Laboratories International, Inc) were euthanized, liver tissue was homogenized, and nuclei isolated, cross-linked and sonicated as described (9). Briefly, the nuclear pellet from each liver was cross-linked for 9 minutes at 30°C in 2.5-mL cross-linking buffer containing 1% formaldehyde, followed by glycine addition, isolation of the cross-linked nuclei, resuspension in 2.4 mL of RIPA buffer (50mM Tris-HCl [pH 8.1], 150mM NaCl, 1% IGEPAL CA-630 detergent, and 0.5% sodium deoxycholate) containing 0.5% sodium dodecyl sulfate and Complete Protease Inhibitor Cocktail. Samples were sonicated in 12 × 75-mm borosilicate glass tubes using a Branson Sonifier 250D instrument with a tapered microtip at 35% power output for 15 seconds on and 45 seconds off, for a total on time of 6 minutes (24 cycles). A 15-μL aliquot of the sonicated chromatin was incubated with 0.5 μL of 5M NaCl at 65°C for 6 hours to reverse the cross-links, followed by incubation at 37°C for 30 minutes with 0.5 μL of RNase A (10 mg/mL), then addition of 1 μL (20 μg) of proteinase K and incubation for 2 hours at 56°C. A majority of the DNA fragments ranged from 100–400 bp, as shown on a 1% agarose gel. The remaining sonicated chromatin was snap frozen in liquid nitrogen and stored at −80°C.
ChIP was carried out using the above-prepared sonicated cross-linked chromatin samples as described (9). Briefly, 45 μL of washed Protein A Dynal magnetic beads (Invitrogen) were incubated overnight at 4°C on a rocking shaker with 0.5-mL blocking solution containing 8-μg HNF6 antibody, sc-13 050 (epitope corresponds to amino acids 11–110 of human HNF6, which is 98% identical in mouse; Santa Cruz Biotechnology, Inc), followed by incubation overnight at 4°C with 150 μg of sonicated chromatin diluted 5-fold with 1× RIPA buffer containing Complete Protease Inhibitor Tablet but no SDS. Samples were washed then incubated in a 65°C water bath for 30 minutes with 120 μL of elution buffer (50mM Tris-HCl [pH 8.1], 10mM EDTA, and 1% SDS) with periodic vortex mixing. The eluted ChIP samples were purified using a QIAquick Gel Extraction kit (QIAGEN) and analyzed by quantitative polymerase chain reaction (qPCR) in comparison with input DNA controls (9) using primers listed in Supplemental Table 1.
High throughput sequencing
HNF6 ChIP-seq data was obtained from 4 independent young adult male mouse livers (biological replicates) and 4 independent young adult female mouse livers (biological replicates). DNA isolated by ChIP was prepared for 35–40 nt, single end read sequencing on an Illumina GAII instrument carried out at the BioMicro Center at MIT, as described (9). Sequenced reads were mapped to the mouse genome (mm9) using Bowtie, v0.12.7. Raw and mapped sequence reads are available as Gene Expression Omnibus (GEO) series GSE60014. Mapped reads from individual male and female livers were combined for each sex to give one combined adult male and one combined adult female HNF6 ChIP-seq read set. Each read set was analyzed using MACS 2.0 software using default parameters (34) to identify HNF6 peaks (genomic regions showing significant HNF6 binding) at MACS -log10(q value) more than 25 in male liver, and separately in female liver. Liver chromatin immunoprecipitated with rabbit IgG antibody served as a negative control for MACS analysis. MAnorm analysis (35) was used to normalize the HNF6 ChIP-seq data for male and female liver and to identify sets of HNF6 peaks that showed a more than 2-fold enrichment in normalized peak intensity (normalized read counts) in male liver compared with female liver, or vice versa (M value > 1) after merging peaks overlapping between male and female liver samples. This analysis resulted in a genome-wide listing of 39 612 HNF6-binding sites, including 4258 male-enriched binding sites and 2428 female-enriched binding sites, as detailed in Supplemental Table 2.
HNF6 peak set and gene target enrichment analysis
HNF6 peaks (HNF6-binding sites) were examined for their overlap with the sets of 72 862 liver DHS and their sex-biased subsets (30), 15 094 liver STAT5-binding sites and their sex-biased subsets (8), and 1471 female liver CUX2-binding sites (9). Enrichment scores (ESs) for the overlap were calculated in comparison with those of background sets comprised of all HNF6 peaks not in the HNF6 peak set being examined. Significance was assessed by 2-tail Fisher's exact test, with ES > 1.5 combined with P < .002 deemed significant. Gene targets of HNF6-binding sites were defined as all genes whose gene body extends to within 10 kb of an HNF6 ChIP-seq peak. Where indicated, HNF6 peaks were mapped to genes using GREAT (36) based on the basal plus extension association rule, extending 10 kb upstream of genes, 2 kb downstream, and up to 50 kb distally. Gene targets of sets of male-enriched, female-enriched, and sex-independent HNF6-binding sites identified by MAnorm (35) were examined for enrichment for clusters of gene ontology and other annotation terms using DAVID (37). DAVID analysis of targets of sex-independent HNF6 peaks was carried out using 3000 gene targets of sex-independent HNF6 peaks having the lowest MACS 2.0 q value (ie, the strongest HNF6 peaks). Enrichment for HNF6 target genes that show sex-biased expression in mouse liver, as determined by RNA-seq (27), were calculated for all liver-expressed male and female biased genes, and for 6 subsets of male-biased genes (M1 to M6) and 6 subsets of female-biased genes (F1 to F6) classified by their proximal sex-specific chromatin mark patterns, as defined earlier (27) and specified in Supplemental Table 2. Significance was assessed by 2-tail Fisher's exact test in comparison with a background set of gene targets of sex-independent + sex-opposite HNF6-binding sites.
Motif discovery
De novo motif discovery using CisGenome (38) was performed using HNF6 peak summit regions (51-bp window centered on the MACS 2.0 peak summit), with the full sets of sex-enriched HNF6 peaks and the top approximately 3500 sex-independent HNF6 peaks (lowest MACS 2.0 q values) analyzed separately. Motifs were also discovered for the subsets of HNF6 peaks that overlap, or do not overlap, CUX2-binding sites (9). STAMP, a Unweighted Pair Group Method with Arithmetic Mean (UPGMA) distance-based-method (39), was used to quantify information content differences between motifs. Motif enrichment in the HNF6 peak summit regions (+/−200 bp of peak summit at false discovery rate < 1E-4) was determined using CENTDIST (40) for the full set of vertebrate motifs from the TRANSFAC database (41) supplemented by a set of 97 motif families defined previously (42) and by the sets of de novo discovered motifs describing HNF6-binding sites identified as described above. Summit regions for the full sets of male- and female-enriched HNF6 peaks, the set of 3000 top sex-independent HNF6 peaks that do not overlap a CUX2-binding site, and the set of 1228 sex-independent HNF6/CUX2 common peaks were each analyzed by CENTDIST separately. For the sex-independent HNF6 peak sets, the summit of the stronger peak in male or female liver was used for the analysis. CENTDIST results were analyzed to identify the set of top scoring motifs (score > 12) that occur in ≥ 10% of the peak summit regions (+/−200 bp) in at least 1 of the HNF6 peak sets examined, based on a motif scan performed by CENTDIST, after eliminating duplicate or redundant motifs (UPGMA ≤ 0.001).
Cobinding with other liver transcription factors
Peak summit regions of the 39 612 HNF6 peaks were analyzed for proximity to ChIP-seq-binding sites identified in adult male mouse liver for 6 other liver-expressed transcription factors: HNF1A (ArrayExpress E-MTAB-941), HNF3B/FOXA2 (GEO series GSE32244), HNF4A (GEO series GSE22078), CEBPB (GEO series GSE42321), STAT5 (GEO series GSE31578), and glucocorticoid receptor (GR)/NR3C1 (GEO series GSE45674). Sequenced reads from these studies were used to call binding sites using MACS in analyses reported previously (8) or carried out by Gracia Bonilla and Tisha Melia of this laboratory. Peak lists are provided in Supplemental Table 3. A liver factor was defined as being cobound with HNF6 if its ChIP-seq peak center was within 200 nt of the HNF6 peak summit identified by MACS2. For each HNF6 peak set examined (including the subsets of HNF6 peaks whose peak summits overlapped with, or that did not overlap with a DHS region), we calculated the expected distribution of the number of liver transcription factor cobinding events. Thus, for each of the 6 factors, the percentage p of HNF6 peaks cobound represents a probability of factor cobinding with HNF6, and 1-p represents the probability that a factor is not cobound with HNF6. Using these probabilities, expected cobinding frequencies were calculated for each of 7 bins, representing 0 to 6 cobinding events (ie, ranging from only HNF6 bound to HNF6 cobound with all 6 factors). For example, there are 15 possible combinations of 4 cobound factors in the set of 6 factors. For each of these 15 combinations, probabilities p and 1-p for bound and unbound are assigned for each of the 6 factors. The product of these 6 probabilities represents the probability of observing 4 cofactors bound for a given transcription factor combination. The sum of these 15 products, one for each possible combination, represents the total probability of having 4 factors cobound. The expected and the observed cobinding frequencies were then compared using a χ2 test, and P < 1.0E-6 was used to identify distributions significantly different from the expected. HNF6 peaks were considered to contain an HNF6 motif, or a cohesin-non-CTCF site (CNC site) (43), if such a motif (or CNC site) was found within 200 nt of the HNF6 peak summit.
Results
CUX2 inhibition of HNF6 activity on CYP2C11 and CYP2C12 promoters
CYP2C12, a prototypical female-specific gene expressed in rat liver, is developmentally induced in female liver after 4 weeks of age, in parallel to the induction of CUX2, whereas CYP2C11, a prototypical male-specific rat gene, is induced in male liver after 4 weeks of age, in parallel to the loss of CUX2 expression (29). We used transient transfection to investigate whether CUX2 alters CYP promoter activity, either alone or in combination with other liver transcription factors that were previously shown to regulate these promoters (23, 24). The CYP2C11 promoter is activated by HNF1α (23), but not by HNF6, which strongly repressed the HNF1α-dependent activity (Figure 1A). CUX2 alone had no effect on the CYP2C11 promoter; however, it increased its activation by HNF1α and, most notably, it fully reversed the repression by HNF6 (Figure 1A). A similar reversal of the inhibitory actions of HNF6 were seen with CUX1, which displays 48% amino acid sequence similarity to CUX2 (Supplemental Figure 1A).
Figure 1.

Effect of CUX2 on HNF6-regulated CYP2C11 and CYP2C12 promoter activity. 293T cells were transiently transfected with reporter plasmids for (A) CYP2C11 promoter, (B) CYP2C12 promoter, or (C) a reporter containing 6x-HNF6-binding sites, in the absence (“Empty”) or in the presence of the indicated transcription factor expression plasmids, either individually or in the combinations indicated, to determine their effects of CUX2 on HNF6-regulated reporter activity. The following amount of plasmid DNA was used per well for each transfection. A, Promoter (100 ng), HNF1 (25 ng), HNF6 (100 ng), and CUX2 (100 ng). B, Promoter (100 ng), HNF4α (25 ng), HNF6 (25 ng), and CUX2 (100 ng). C, Reporter (100 ng), HNF6 (25 ng), and CUX2 (100 ng). Data shown are firefly luciferase reporter activities normalized to Renilla luciferase activities based on n = 3 transfections (mean ± SD) with the empty plasmid sample set to 1. Statistical analysis: *, P < .05; **, P < .01; ***, P < .001, for reporter combined with transcription factor(s) vs reporter alone; +, P < .05; ++, P < .01; +++, P < .001, for reporter combined with 2 transcription factors vs reporter combined with 1 transcription factor that activates (ie, HNF1 for A, HNF4 for B, and HNF6 for C); and #, P < .05; ##, P < .01; ###, P < .001 for reporter with 3 transcription factors vs reporter with 2 transcription factors.
In the case of the CYP2C12 promoter, which is moderately activated by HNF4α, a synergistic increase in reporter activity was seen when HNF6 was combined with HNF4α (Figure 1B). CUX2 alone had no effect on CYP2C12 promoter activity; however, CUX2 reversed the stimulatory effects of HNF6 on HNF4α-activated CYP2C12, reducing CYP2C12 activity down to the level seen in the presence of HNF4α alone (Figure 1B). A very similar result was seen with CUX1 (Supplemental Figure 1B). CUX2 inhibition of HNF6 activity was confirmed in the experiment shown in Figure 1C, where CUX2 strongly inhibited HNF6 activation of a reporter gene under control of multiple HNF6-binding sites (6x-HNF6-Luc). A similar result was obtained for CUX1 (Supplemental Figure 1C). Thus, CUX2, as well as CUX1, can inhibit the transcriptional actions of HNF6: both factors reverse the inhibitory activity of HNF6 on CYP2C11 and block the stimulatory activity of HNF6 on both CYP2C12 and the 6x-HNF6-Luc reporter.
Further experiments evidenced the specificity of these effects of HNF6 and CUX2. HNF6 did not inhibit either basal or HNF4-stimulated CYP2C11 activity, demonstrating specificity for HNF6 inhibition of HNF1-stimulated promoter activity (Supplemental Figure 1D). Moreover, HNF6, but not HNF4 or CUX2, was inhibitory to HNF1-stimulated CYP2C11 activity. Further, CUX2 did not further increase HNF4-stimulated CYP2C11 activity (Supplemental Figure 1D); thus, the stimulatory actions of CUX2 are specifically seen in the context of reversing the inhibitory actions of HNF6. Finally, both CUX1 and CUX2 reversed the transcriptional actions of HNF6 in a dose-dependent with all 3 reporters (Supplemental Figure 1, E–G). Other experiments showed that OC-2, which is 67% identical in amino acid sequence to HNF6 (44), acts in a qualitatively similar manner to HNF6 on CYP2C11, CYP2C12, and the 6x-HNF6-Luc reporter; however, neither CUX2 nor CUX1 inhibited the activity of OC-2 (Supplemental Figure 1, A–C, last 2 bars). Accordingly, we focused our further studies on HNF6. Moreover, given the strong female-specific expression of CUX2 in adult mouse liver (29), as well as the absence of CUX1 protein after postnatal day 2 (45), we directed our further investigations to the interactions of HNF6 with CUX2.
EMSA analysis of CUX2 and HNF6-binding site specificities
Next, we considered whether the inhibition of HNF6 activity by CUX2 results from binding site competition. Supporting this hypothesis, EMSA analysis using an HNF6-binding site probe from the CYP2C12 promoter (Figure 2A, HNF6 probe) yielded strong complexes for HNF6, CUX2, and the related factor CUX1 (Figure 3A, lanes 4, 8, and 12). In contrast, a CUX-binding site EMSA probe (Figure 2A, CUX probe) formed a weak EMSA complex with HNF6 as compared with CUX1 and CUX2 (Figure 3B, lane 12 vs lanes 4 and 8). EMSA signals were reduced by excess unlabeled competitor probe (lanes 5, 9, and 13; cf untransfected cell extract [control] in lane 1), indicating their specificity.
Figure 2.

DNA-binding site sequence specificities and motifs for HNF6 and CUX2. A, EMSA probes, with modifications made to the core-binding sequence of the HNF6 and CUX EMSA probes, as indicated. Upper case letters indicate core-binding site, and lower case letters indicate flanking sequence. Modifications to the original probe sequence are marked in red. For all 7 HNF6 EMSA probes, the sequences shown are preceded by 5′-CCA and are followed by GCTTTTGGCAAACTT-3′. The wild-type HNF6 EMSA probe sequence corresponds to the natural HNF6 site found 58 nt upstream of rat CYP2C12. For all 4 CUX EMSA probes, the sequences shown are preceded by 5′-TCGAG and are followed by TTCTTTTC-3′. The probes were used for EMSA analysis using cytoplasmic or nuclear extract from 293T cells transfected with HNF6, CUX2, CUX1, OC-1, or an empty (control) plasmid as shown in Figure 3 and Supplemental Figures 2 and 3. B, DNA-binding motifs are represented by sequence logos, and are based on the Transfac database (motifs numbered 1, 3A, and 3B) or were determined by de novo discovery using the indicated subsets of the present HNF6 ChIP-seq datasets (motifs 4–7) or the published CUX2 dataset (motif 2) (9). Motifs 4A and 4B were discovered using the top approximately 3500 HNF6 peaks that do not overlap with a CUX2-binding site; the major motif, 4A, is based on 1450 HNF6-binding sites, and the minor motif, 4B, is based on 401 sequences present within the full list of approximately 3500 HNF6 peaks. Motifs similar to 4B were also discovered de novo in the analyses that yielded motifs 5, 6, and 7 (data not shown). Motifs 5, 6, and 7 are, respectively, based on 1089, 561, and 466 HNF6-binding site-containing sequences.
Figure 3.
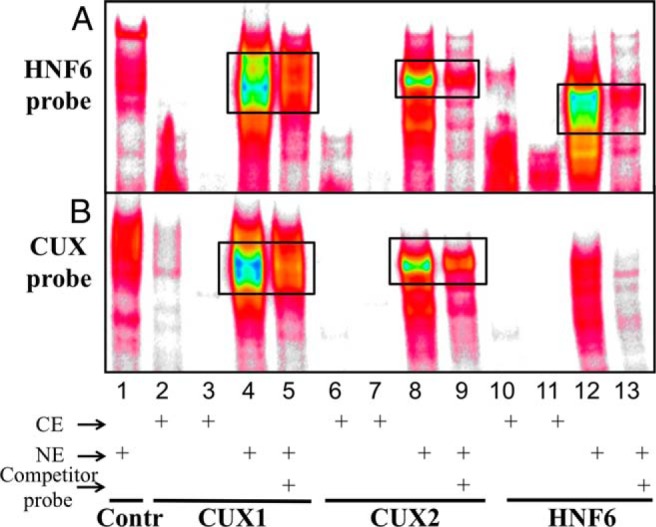
EMSA analysis of CUX1, CUX2, and HNF6 DNA-binding activity. Cytoplasmic extract (CE) and nuclear extract (NE) from 293T cells transfected with CUX1, CUX2, or HNF6 expression plasmid, or from cells transfected with an empty, control plasmid (Contr) were prepared. Cell extracts (30 μg per lane) were analyzed by EMSA using a double-stranded DNA probe containing an HNF6-binding site from the CYP2C12 promoter (top) or a probe containing a CUX1/CUX2-binding site (bottom), as shown in Figure 2A. A 10-fold molar excess of unlabeled (competitor) probe was included where indicated. Black boxes indicate relevant EMSA complexes. CE and NE were isolated using NucBuster (Materials and Methods), except for lanes 2, 6, and 10, where CE was prepared using passive lysis buffer.
CUX1 and CUX2 bind DNA through different combinations of their 3 Cut repeats (CRs) and/or HD (ie, CR1/CR2, CR2/CR3/HD, or CR3/HD) (28). For both CUX1 and CUX2, CR1/CR2 preferentially bind sequences containing 2 nearby CGAT sequences, whereas CR2/CR3/HD and CR3/HD bind with high affinity to an ATCGAT sequence (28). HNF6 can bind DNA via its Cut domain (as seen with the HNF3β/FOXA2 promoter) (46) or using both the Cut domain and the HD (eg, on the TTR promoter) (31). These 2 different modes of HNF6 binding are associated with recruitment of distinct coactivators, CBP and p/CAF, respectively. Moreover, the DNA recognition sequence of the 2 HNF6-binding modes differs by 1 nt: TATTGATTTA for Cut domain binding (46) vs TATTGACTTA for Cut + HD binding (31). Therefore, subtle differences in DNA sequence can lead to significantly dissimilar binding and activation by HNF6 (31). To better understand the differential sequence requirements for CUX and HNF6 binding, we modified the CUX EMSA probe sequence in an effort to increase HNF6 binding. Changing the core CUX motif ATCGAT to ATCAAT (CUX-M1), to make it more HNF6-like, resulted in a large decrease in binding by CUX2 and CUX1, but did not increase binding by HNF6 or OC-2 (Figure 2A; also see Supplemental Figure 2). Further modification of the 5′-flanking sequence from AT to AA (CUX-M2) increased binding by HNF6 but not by the other factors; however, when this modification alone was introduced (CUX-M3), ie, without alteration of the core CUX motif, OC-2 but not HNF6 binding increased (Figure 2A and Supplemental Figure 2).
Next, we attempted to modify the HNF6 probe sequence to either decrease binding by CUX2 while retaining HNF6 binding, or eliminate binding by both CUX and HNF6 family proteins. Changing the core HNF6 motif from AATCAAT to AATCGAT (HNF6-M1) or to TATCGAT (HNF6-M2) had small effects on factor binding. A further modification of the 3′-flanking A to C (HNF6-M5) failed to elicit large changes in binding activity for HNF6, CUX1, or OC-2, but substantially reduced CUX2 binding (Figure 2A and Supplemental Figure 3). However, changing the core sequence from AATCAAT to AGTCAAT (HNF6-M3), which matches the HNF6-binding site on the TTR promoter, nearly abolished binding by all 4 factors, as did the use of a probe with a modified core sequence AATTGAT (HNF6-M4). However, introduction of an additional modification of the 5′-A of the HNF6 core to a T (HNF6-M6) restored binding by all 4 proteins and yielded an additional, higher EMSA complex, which could result from the binding of another protein (Figure 2A and Supplemental Figure 3). The overall similarities of the binding specificities of CUX2 and HNF6, as well as those of CUX1 and OC-2, with CUX-M2 and with all of the HNF6 probes except HNF6-M3 (Figure 2A), highlights their potential to compete with one another for DNA binding in vivo. The extent of competition is expected to be dependent on both the core sequence and the flanking sequence context, as exemplified by the distinct binding activities seen with the CUX-M2 and HNF6 probes, which have the identical core sequence AATCAAT but different flanking sequences.
Characterization of the liver HNF6 cistrome
To ascertain whether CUX2 and HNF6 compete for binding to liver chromatin in vivo, we used ChIP-qPCR to interrogate HNF6 binding at sites known to bind CUX2 in female mouse liver (9). HNF6 binding was seen at CUX2-binding sites near the male-biased genes Cml5 and Fh1, and near the sex-independent genes Mad1l1 and Adhfe1 (Figure 4, A–D) in both male and female liver. Genes that contained no significant CUX2 ChIP-seq reads displayed no significant binding by HNF6 and served as negative controls (Figure 4, E and F).
Figure 4.

ChIP-qPCR analysis of HNF6 binding in adult male and female mouse liver. ChIP samples were prepared from sonicated, cross-linked chromatin samples prepared from 4 individual male and 4 individual female mouse livers using antibody to HNF6 or IgG as a control antibody. The chromatin immunoprecipitated DNA was analyzed by qPCR using primers corresponding to 4 CUX2-binding sites identified by ChIP-seq (9). These sites are nearby the indicated male-biased genes (A and B) or sex-independent genes (C and D). No significant binding was observed at genomic regions that did not contain CUX2-binding sites (E and F). ChIP-qPCR using normal rabbit IgG antibody showed almost no detectable background binding for all 6 genomic regions. Each bar represents an individual liver. Y-axis, qPCR quantification as a percentage of the sonicated chromatin input to the ChIP reaction.
Next, we used ChIP-seq to characterize mouse liver HNF6-binding sites genome-wide and establish the global extent of CUX2 and HNF6-binding site competition. A total of 39 612 peaks of HNF6 binding were identified in male and female mouse liver (Supplemental Table 2). Of these peaks, 43.7% overlapped with the set of 72 862 mouse liver DHSs (merged DHS set) (30), which correspond to open chromatin regions and are highly enriched in gene regulatory regions. The percentage of HNF6-binding sites that overlap with a DHS increased to approximately 75% when the top 10 000 HNF6-binding sites (ranked by MACS score) were considered (Supplemental Figure 4). In contrast, the overlap with DHS is ≥ 90% for other liver-enriched transcription factors, with the exception of CEBPA (67%) and the pioneer factor FOXA2 (68%) (8, 30).
Quantitative comparison of the male and female liver HNF6 ChIP-seq datasets normalized using MAnorm (35) identified 4258 HNF6 peaks showing significantly higher binding activity in male liver and 2428 HNF6 peaks showing significantly higher binding activity in female liver (Supplemental Table 2). These sex-enriched HNF6-binding sites showed strong enrichment for the corresponding sets of sex-specific DHS: male-enriched HNF6 peaks were 6.6-fold enriched for being at a male-specific DHS (P = 3.41E-198), and correspondingly for female-enriched HNF6 peaks being at a female-specific DHS (ES = 10.48, P = 2.11E-64) when compared with background sets of HNF6 peaks not in the HNF6 test peak set (Table 1). Further, when considering the subsets of sex-enriched HNF6 peaks overlapping liver DHS (809 male-enriched HNF6 peaks (19% of total set) and 755 female-enriched HNF6 peaks (31% of total set), the enrichments increased to 45-fold (male-enriched HNF6 peaks; P = 0) and 16.8-fold (female-enriched HNF6 peaks; P = 8.8E-80) (Table 1). Thus, there is a strong association between sex-dependent HNF6 binding and the sex-dependence of chromatin accessibility. Comparison with the set of 15 094 mouse liver-binding sites for STAT5 identified previously (8) showed that the full set of male-enriched HNF6 peaks was significantly associated with male-enriched STAT5-binding sites (ES = 4.12, P = 2.37E-84) and correspondingly for female-enriched HNF6 peaks and female-enriched STAT5-binding sites (ES = 3.45, P = 4.87E-20) (Table 1). These enrichments increased to 11.3-fold (P = 4.03E-128) and 5.08-fold (P = 3.41E-26) when the subsets of sex-specific HNF6 sites at DHS were considered. Figures 5 and 6 show examples of female-enriched and male-enriched HNF6-binding sites (marked in pink and blue, respectively), along with nearby sex-biased genes, DHS, and binding sites for other GH-regulated transcription factors.
Table 1.
Enrichment of HNF6 Peak Sets for Overlap With DHS and With Sex-Enriched Binding Sites for STAT5 in Mouse Liver
| Genomic Region Considered | Male-Enriched HNF6 Peaks (4258) |
Female-Enriched HNF6 Peaks (2428) |
HNF6/CUX2 Common Peaks (1338) |
|||
|---|---|---|---|---|---|---|
| ES | P | ES | P | ES | P | |
| All DHS (72 862) | 0.27 | 9.69E-282 | 0.56 | 3.41E-39 | 6.29 | 3.83E-191 |
| Female-biased DHS (1379) | 0.02 | 3.07E-35 | 10.5 | 2.11E-64 | 1.86 | NS |
| 0.14 | 1.96E-04 | 16.8 | 8.80E-80 | |||
| Male-biased DHS (2800) | 6.59 | 3.41E-198 | 0.06 | 5.82E-12 | 4.28 | 3.22E-16 |
| 45.1 | 0 | 0.03 | 6.94E-25 | |||
| Female-enriched STAT5 sites (1791) | 0.10 | 3.82E-17 | 3.45 | 4.87E-20 | 0.77 | 3.82E-08 |
| Male-enriched STAT5 sites (1765) | 4.12 | 2.37E-84 | 0.17 | 2.63E-17 | 5.96 | 4.45E-67 |
Shown are ES and P values for overlap between the sets of male-enriched, female-enriched, or CUX2-overlapping HNF6 peaks, and the indicated sets of DHS, and STAT5-binding sites. ESs for overlap with male and female-biased DHS were calculated for the full sets of male-enriched and female-enriched HNF6 peaks, and for the subsets comprised of 809 male-enriched HNF6 peaks and 755 female-enriched HNF6 peaks that are at DHS (ES and P values in third and fifth rows, shown in italics). The ES values shown indicate the bias of a given HNF6 peak set for overlap with the indicated set of DHS, or the indicated set of transcription factor-binding sites, compared with a background set comprised of all HNF6 peaks not in the HNF6 peak set being examined. For example, male-enriched HNF6-binding sites are 6.59-fold enriched for overlapping male-biased DHS compared with HNF6 peaks that are not male-enriched, with the enrichment increasing to 45-fold when the set of 809 male-enriched HNF6-binding sites at DHS is considered. Significant enrichments are highlighted in bold. ES values of less than 1 at a P < .002 indicate significant depletion. The numbers of HNF6 peaks in each peak set are shown at the top, and the numbers of DHS, and STAT5 sites are shown in the first column (numbers in parenthesis). NS, not significant (P > 0.002).
Figure 5.

Female-enriched HNF6-binding sites associated with subclass F3 female-biased genes. Shown are examples of subclass F3 genes (27) along with browser tracks indicating the locations of DHS, binding sites for STAT5, BCL6, CUX2, and HNF6. Sex-independent DHS and transcription factor-binding sites are colored gray, female-enriched sites are colored pink, and sites of BCL6 binding in male liver are colored blue. Binding sites for STAT5 and BCL6 are shown as a 600-nt-wide window surrounding the ChIP-seq peak summit. For the DHS track, a greater intensity of pink color indicates a larger sex-difference in chromatin accessibility. Also shown are normalized wig files of HNF6 ChIP-seq reads for male and female liver. A, the Sult3a1 + Rsph4a genomic region is associated with 5 female-enriched and 1 sex-independent HNF6 site; only 3 of these 6 HNF6 sites coincide with a DHS. B, Cyp2b9 is associated with 11 nearby female-enriched and 1 sex-independent HNF6 site, with none found at a DHS. C, Cyp3a44 is associated with 2 female-enriched and 2 sex-independent HNF6 sites, of which 2 are located at DHS regions. One of the female-enriched HNF6 sites downstream of Cyp2b9 and 1 of the sex-independent HNF6 sites upstream of Cyp3a44 overlaps a CUX2-binding site. Data for DHS (30), and binding sites for CUX2 (9), STAT5, and BCL6 (8), determined by ChIP-seq, are based on previous publications.
Figure 6.

Male-enriched HNF6-binding sites associated with male-biased genes. Shown are examples of male-biased genes along with browser tracks indicating the locations of DHS and binding sites for the indicated transcription factors, and wig files for HNF6 ChIP-seq reads for male and female liver, as in Figure 5. Blue indicates male-enriched DHS and male-enriched binding sites for the indicated factors. A, the C6 genomic region is associated with 7 male-enriched HNF6 sites, 5 of which overlap a DHS. B, Cyp4a12a is associated with 3 male-enriched HNF6 sites and 1 sex-independent HNF6 site, with 2 of these 4 sites found at a DHS. C, Hsd3b5 is associated with 4 male-enriched and 5 sex-independent HNF6 sites, 6 of which are located at DHS regions. Locations of binding sites for the other factors are as shown.
Relationship to CUX2-binding sites
More than 90% of the 1471 CUX2-binding sites identified in female mouse liver (9) overlap with a peak of HNF6 binding (Supplemental Table 2), consistent with the close similarity of HNF6 and CUX2 motifs (Figure 2B) and supporting the proposal that CUX2 competes with HNF6 for binding to these sites in female liver. De novo motif discovery using summit regions of the strongest HNF6 peaks that do not overlap a CUX2 peak yielded a motif very similar to the Transfac HNF6 motif (motif 4A vs motif 1) and to the CUX2 motif identified previously (motif 2) (Figure 2B) (9). A subset of sequences in the HNF6 peak set yielded a second, related motif containing a 5′-extension of several adenines (motif 4B) (Figure 2B). The male-enriched and female-enriched HNF6 peak summit regions yielded de novo discovered motifs that were indistinguishable from each other (UPGMA distance < 0.001) and from the top HNF6 motif (motifs 5 and 6 vs motif 4A) (Figure 2B). Thus, sequence differences at the HNF6-binding site do not determine the sex-differences in HNF6 binding. Further, HNF6/CUX2 common peaks and HNF6 peaks not bound by CUX2 yielded indistinguishable motifs (motif 7 vs motif 4A), indicating that sequences differences at the binding site, such as those suggested by the EMSA analyses (Figure 2A), do not explain DNA-binding preferences in mouse liver chromatin for HNF6/CUX2 common binding site vs HNF6 binding alone.
HNF6/CUX2 common binding sites showed significant enrichment for overlap with male-biased DHS (ES = 4.28, P = 3.22E-16) and for genomic regions that show male-enriched STAT5 binding (ES = 5.96, P = 4.45E-67) as compared with HNF6 peaks not overlapping CUX2-binding sites (Table 1). Based on these findings, and given that CUX2 binding occurs exclusively in female liver, owing to the absence of CUX2 protein in male liver (29), we anticipated that the set of HNF6/CUX2 common sites found at DHS would show a significant preference for male-enriched HNF6 binding when compared with the set of HNF6 peaks at DHS that are not bound by CUX2; however, no such enrichment was seen (ES = 1.17, P = .27). However, we did find that the set of HNF6/CUX2 common peaks at DHS was significantly depleted of female-enriched HNF6 peaks at DHS (ES = 0.31, P = 4.33E-08), consistent with CUX2 being preferentially bound at these sites in female liver as compared with HNF6. Thus, although CUX2 binding is not enriched at sites of male-enriched HNF6 binding, CUX2 binding is significantly depleted at female-enriched HNF6-binding sites.
HNF6-associated factor comotifs
Transcription factor comotifs associated with HNF6-binding sites were identified using CENTDIST (40). The set of 3000 strongest sex-independent HNF6 peaks (lowest MACS q values) that do not overlap with a CUX2 peak was analyzed, as was each sex-enriched HNF6 peak set, to discover any differences in comotifs between peak sets (Table 2). As expected, the highest scoring motifs described HNF6 itself and several transcription factors with similar binding site specificities (ie, CUX2, CUX1/CLOX, and PBX). In addition, several comotifs were commonly associated with, and were found near, the HNF6 peak summits at frequencies ranging up to approximately 20%. The highest scoring comotifs described the liver-enriched transcription factors/factor families HNF4, HNF1, HNF3/FOX, and C/EBP, which variously interact with each other and with STAT5 to regulate sex-dependent liver gene expression (15). Other HNF6 comotifs with lower scores include those describing members of the CDX, POU, and SRY transcription factor families. Overall, results were similar for all 3 HNF6 peak sets. No significant differences in comotifs were seen for HNF6/CUX2 common peak set as compared with the sex-independent HNF6 peak set (Table 2), indicating that CUX2 binding is not driven by a differential association with comotifs compared with the HNF6-only peak set.
Table 2.
Top-Scoring Motifs in Sets of Sex-Independent, Male-Enriched and Female-Enriched HNF6 Peak Summits
| Sex-Independent HNF6 Peaks Without CUX2 Binding (3000 Top Peaks) |
Male-Enriched HNF6 Peaks (4258 Peaks) |
Female-Enriched HNF6 Peaks (2428 Peaks) |
Sex-Independent HNF6/CUX2 Common Peaks (1228 Peaks) |
|||||
|---|---|---|---|---|---|---|---|---|
| Motif Score | % | Motif Score | % | Motif Score | % | Motif Score | % | |
| HNF6 de novo | 195 | 47.1 | 161 | 38.3 | 105 | 31.0 | 83.5 | 35.0 |
| V$CLOX_01 | 129 | 31.0 | 123 | 30.1 | 73.3 | 23.5 | 70.4 | 29.8 |
| V$PBX1_01 | 79.5 | 24.0 | 82.2 | 29.1 | 44.6 | 20.3 | 40.6 | 27.2 |
| V$HNF4_Q6_01 | 28.2 | 16.5 | 18.3 | 10.0 | 9.5 | 9.5 | 14.1 | 16.3 |
| V$HNF1_C | 26.4 | 20.0 | 25.8 | 20.4 | 15.5 | 17.9 | 19.6 | 23.5 |
| V$HNF3_Q6_01 | 20.9 | 23.3 | 26.2 | 23.1 | 18 | 20.0 | 12.6 | 22.9 |
| V$CEBP_Q2_01 | 13.4 | 9.2 | 19.8 | 9.7 | 13.5 | 8.9 | 13.3 | 11.6 |
| V$CDX_Q5 | 12.4 | 16.2 | 18.4 | 20.6 | 10.8 | 16.9 | 8.41 | 16.8 |
| V$POU3F2_01 | 13.9 | 12.8 | 16.9 | 17.1 | 8.3 | 13.5 | 8.29 | 12.9 |
| V$SRY_02 | 11.8 | 12.3 | 8.8 | 13.7 | 9.1 | 12.8 | 6.11 | 12.9 |
Shown are the motif center distribution scores, which are based on the frequency and velocity graphs of motif distribution (sum of CENTDIST Z0 and Z1 scores [40], respectively), and percent values, which indicate the percent of HNF6 peak summit regions having one or more motif occurrences within a +/−200-bp window at false discovery rate < 1E-04. Data are shown for all motifs with center distribution score of more than 12 and more than 10% motif occurrence for at least 1 of the 4 HNF6 peak sets shown here. Closely related motifs (UPGMA <0.001), such as CUX2, which is redundant with the de novo HNF6 motif (see Figure 2B), are not listed. The motif V$CLOX_01 is representative of CUX1. The higher motif scores and occurrence frequencies of the male-enriched compared with the female-enriched HNF6 peak sets may reflect the weaker HNF6 peak intensities of the female-enriched peak set.
To test the above prediction that HNF6 binding is significantly associated with binding by other liver transcription factors, we examined published male mouse liver ChIP-seq binding data for 6 transcription factors. Factors whose binding sites are within 200 bp of an HNF6 peak summit were considered as cobound with HNF6. Each of the 6 factors (HNF1A, HNF3B/FOXA2, HNF4, CEBPB, STAT5, and GR/NR3C1) showed substantial cobinding with the set of 3000 top-scoring HNF6-binding sites (Figure 7A). Many more cobinding events were seen at the 2381 HNF6 sites within DHS compared with the 619 HNF6 sites not in DHS regions, consistent with the strong preference of these and other transcription factors for binding to open chromatin (30). Factor cobinding frequency distributions (values ranging from 0 to 6 other factors cobound with HNF6) were significantly different than the expected distributions, both for HNF6-binding sites at DHS (Figure 7B) and for HNF6-binding sites not at DHS (Figure 7C). Specifically, HNF6-binding sites at DHS showed >2-fold enrichment for: 1) cobinding with all 6 other factors and 2) cobinding with no other factors (singleton binding event) or cobinding with only one other factor (Figure 7B). Similarly, HNF6 sites not at DHS showed >2-fold enrichment for cobinding with 4, 5 or 6 other factors, and for no other factors being cobound (Figure 7C). A similar overall pattern of enrichment for multifactor cobinding was seen for the subset of male-specific HNF6 sites within DHS, but with an even greater enrichment for singleton HNF6 sites (Supplemental Figure 5A).
Figure 7.

Cobinding frequency distributions for top-scoring 3000 HNF6 peak summit regions. Frequencies of cobinding events for the peak summit regions of 3000 top scoring HNF6 peaks are shown. A, Heat maps of transcription factor cobinding events for 2381 HNF6 peaks at DHS (top) and for 619 HNF6 peaks not at DHS (bottom). Each black bar indicates a cobinding event for the transcription factor marked at bottom. In each heat map, HNF6 peaks are ordered by the number of factors cobound, ranging from 6 factors (top) to 0 factors (bottom). B, Distribution of number of cobinding events for the sets of HNF6 peak summit regions at DHS, with red bars representing the observed frequencies of 0–6 cobinding events, and gray bars representing the expected distribution based on the percentage overlaps with individual cofactors (see Materials and Methods). C, Distribution of number of cobinding events for the sets of HNF6 peak summit regions not at DHS, with blue bars representing the observed frequencies of 0–6 cobinding events, and gray bars representing the expected distribution based on the percentage overlaps with individual cofactors determined experimentally (see Materials and Methods).
Many (39%) of the top 3000 HNF6-binding sites either do not contain an HNF6 motif, or contain a motif below the detection threshold of our motif scan analysis, suggesting HNF6 binds to these sites, at least in part via interactions with other directly bound transcription factors. We did not, however, see a striking differential distribution of factor cobinding events at HNF6-binding sites with vs without an HNF6 motif (Supplemental Figure 5B). We also examined the association between factor cobinding events and presence of a cohesin-non-CTCF site (CNC site), which is involved in chromosome looping and can stabilize cis-regulatory modules occupied by multiple tissue-specific regulators, including HNF6 (43). This stabilization is reported to enable HNF6 and other liver transcription factors to bind chromatin even in the absence of a strong cognate motif (43). However, in our analysis, the subset of 1453 top 3000 HNF6 sites that overlap with liver CNC sites (43) did not show a preference for HNF6 sites without (or with low-scoring) HNF6 motifs: HNF6 motif frequency of 61.7% in the set of 1453 CNC site-overlapping top HNF6 peaks vs 61% in all top 3000 HNF6 peaks. When considering all 39 612 HNF6 peaks, the corresponding motif frequencies were 53.7% for all HNF6 peaks vs 45.7% for the subset of CNC-overlapping HNF6 peaks. Further, the reported positive impact of the presence of a CNC site on the recruitment of multiple cofactors was not apparent in our analysis, where the presence of a CNC site had no obvious impact on the distribution of cofactor binding events at HNF6 sites localized to DHS (Supplemental Figure 5C).
HNF6 target genes and pathways
Analysis of gene targets of HNF6 peaks, defined as genes within 10 kb of an HNF6-binding site, identified gene targets for 68% of the binding sites (Supplemental Table 2). The set of 3000 genes targeted by the top sex-independent HNF6 peaks was significantly associated with diverse biological functions, including cytochrome P450/microsomal drug metabolism, nucleotide binding, steroid metabolic process, zinc finger/steroid hormone receptors, metal ion binding, lipid transport, negative regulation of transcription, pleckstrin homology, and GTPase regulation (Supplemental Table 4A). Gene targets of female-enriched but not male-enriched HNF6 peaks were also significantly associated with cytochrome P450/microsomal drug metabolism, nucleotide binding, metal ion binding, and zinc finger/steroid hormone receptors but were additionally associated with positive regulation of transcription, ubiquitin conjugation, and chromatin organization (Supplemental Table 4B). Significant associations of gene targets of male-enriched HNF6 peaks were different from that of the sex-independent and female-enriched HNF6 target gene groups. These associations included small GTPase binding, metal ion binding, calcium-dependent membrane targeting, phospholipase C, synapse, and cytoskeletal protein binding, in addition to pleckstrin homology and metal ion binding, which were shared with the other 2 gene sets (Supplemental Table 4C). Thus, male-enriched and female-enriched HNF6-binding targets genes are associated with distinct cellular and metabolic functions.
The association of sex-biased HNF6-binding sites with nearby (within 10 kb) sex-biased genes was examined by enrichment analysis. Female-biased HNF6-binding sites showed 3.05-fold enrichment for mapping to one or more female-biased genes (P = 1.75E-39) and 2-fold depletion for mapping to male-biased genes (P = 8.48E-08) as compared with nonfemale-enriched HNF6-binding sites. In contrast, male-biased HNF6-binding sites did not show enrichment for mapping to male-biased genes (ES = 1.07, P = 4.1E-01), but were 3.57-fold depleted for mapping to a female-biased gene (ES = 0.28; P = 1.21E-25), as compared with nonfemale-enriched HNF6-binding sites (Table 3). When we examined the enrichment of sex-biased HNF6-binding sites at individual subclasses of sex-biased genes grouped based on their sex-dependent local chromatin states (27), we observed an even more striking enrichment of female-enriched HNF6 sites mapping to genes in female gene subclass F2 (ES = 4.46, P = 1.35E-14) as well as subclass F3 (ES = 17.2, P = 2.55E-27) (Table 3 and Figure 5), which has the highest frequency of nearby, sex-biased chromatin marks (27). Further, male-enriched HNF6 sites now showed significant enrichment for a corresponding subclass of male-biased genes with proximal male-biased chromatin marks (subclass M3; ES = 2.86, P = 1.5E-03). The enrichment for male subclass M3 increased to ES = 5.83, P = 2.13E-04 when only the male-enriched HNF6-binding sites within DHS regions were considered. Even more striking enrichments of male-enriched HNF6-binding sites for the male-biased M3 gene subclass were observed when HNF6-binding sites were mapped to genes using GREAT's basal plus extension association rule (36): ES = 4.76, P = 3.42E-06 when all male-enriched HNF6-binding sites were considered; and ES = 14.35, P = 2.61E-09 when male-enriched HNF6-binding sites mapping to DHS were considered. These findings were confirmed when we considered the number of sex-biased gene targets of sex-enriched HNF6-binding sites. Thus, female subclass F3 genes were 6.26-fold enriched for being targets of female-enriched HNF6-binding sites as compared with a background set of gene targets of nonfemale-enriched HNF6-binding sites, and male subclass M3 genes were 3.76-fold enriched for being targets of male-enriched HNF6-binding sites as compared with the gene targets of nonmale-enriched HNF6-binding sites (Supplemental Table 5).
Table 3.
Enrichment of HNF6 Peak Sets for Sets of Sex-Biased Gene Targets
| Gene Set | Female-Enriched HNF6 Peaks |
Male-Enriched HNF6 Peaks |
HNF6/CUX2 Common Peaks |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of Peaks | ES | P | Number of Peaks | ES | P | Number of Peaks | ES | P | |
| All female-biased genes (477) | 224 | 3.05 | 1.75E-39 | 47 | 0.28 | 1.21E-25 | 75 | 1.63 | 1.66E-04 |
| subset F1 (284) | 105 | 2.2 | 9.66E-12 | 31 | 0.3 | 1.24E-15 | 40 | 1.39 | NS |
| subset F2 (126) | 46 | 4.46 | 1.35E-14 | 10 | 0.4 | 1.92E-03 | 15 | 2.2 | NS |
| subset F3 (21) | 38 | 17.2 | 2.55E-27 | 2 | 0.23 | 2.28E-02 | 9 | 4.05 | 7.63E-04 |
| All male-biased genes (423) | 51 | 0.5 | 8.46E-08 | 181 | 1.07 | NS | 93 | 1.83 | 4.00E-07 |
| subset M1 (267) | 29 | 0.43 | 4.51E-07 | 113 | 1 | NS | 51 | 1.50 | NS |
| subset M3 (12) | 3 | 0.87 | NS | 14 (17)a | 2.86 (4.76) | 1.5E-03 (3.42E-06) | 5 | 2.96 | NS |
| subset M4 (17) | 1 | 0.11 | NS | 10 | 0.67 | NS | 19 | 4.85 | 1.11E-07 |
The ES and associated P values shown represent the bias for the members of each sex-enriched HNF6 peak set to map to sex-biased gene targets in gene subclass F1-F6 and M1-M6 compared with a background set of HNF6 peaks not in the sex-enriched HNF6 peak set. For example, female-enriched HNF6 peaks map to subclass F3 gene targets at a 17.2-fold greater frequency than male-enriched + sex-independent HNF6 peaks. Corresponding male-enriched HNF6 peaks are depleted of the indicated classes of female-biased gene targets (ES < 1 at P < .002). The ES for male-enriched HNF6 peaks mapping to male-biased gene set M3 increases from 2.86 to 4.76 when the HNF6 peaks are mapped to genes using the basal plus extension peak-gene association rule of GREAT (see Materials and Methods). Significant enrichments are highlighted in bold. Gene subclasses not showing enrichments at P < .002 for at least 1 of the 3 HNF6 peak sets are not shown. NS, not significant (P > .002).
Twelve of the 17 male-enriched HNF6-binding sites mapping to the M3 subset of male-biased genes by GREAT analysis are in DHS regions (ES = 14.35, P = 2.61E-09, as compared with nonmale-enriched HNF6-binding sites in DHS regions mapping to M3 genes). At 9 of the 17 HNF6-binding sites mapping to M3 genes, HNF6 is bound in the presence of either 0 (7 HNF6 sites) or 1 (2 HNF6 sites) of the 6 other liver transcription factors that often cobind with HNF6 (see Figure 7), with a significant enrichment for M3 genes (ES = 4.85, P = 7.97E-04, as compared with nonmale-enriched HNF6-binding sites cobound with either 0 or 1 other liver factors).
Examination of gene targets of HNF6/CUX2 common peaks revealed their significant enrichment for both male-specific and female-specific genes, with stronger enrichments of the HNF6/CUX2 common peaks in gene subclasses F3 (ES = 4.05, P = 7.63E-04) and M4 (ES = 4.85, P = 1.11E-07) (Table 3), both of which have local, sex-biased chromatin marks (27). Very similar results were obtained when the enrichments were calculated based on the number of gene targets (rather than the number of peaks mapping to gene targets) in each gene class. Thus, gene targets of HNF6/CUX2 common peaks were most highly enriched in sex-biased gene subclasses F1 (ES = 1.95, P = 9.55E-15), F3 (ES = 6.45, P = 7.07E-05), M1 (ES = 2.4, P = 3.33E-06), and M4 (ES = 8.99, P = 9.74E-07) (Supplemental Table 5), consistent with the finding that CUX2 both represses male-biased genes and induces female-biased genes (9).
Discussion
The regulation of sex-biased gene expression in mouse liver involves the action of GH via multiple, interacting transcription factors, including STAT5 and BCL6, whose overlapping binding sites enable BCL6 to preferentially repress a subset of GH and STAT5-regulated female-biased genes in male mouse liver (8). A complementary mechanism has been identified in female liver, whereby CUX2, a highly female-specific transcription factor, represses a substantial subset of male-biased genes. This repression is associated with CUX2 binding to chromatin at sites that are associated with male-enriched binding of STAT5 and are more accessible in male than in female liver (9). Here, we investigate the cross talk of CUX2 with HNF6, a homeobox-containing protein with a DNA-binding specificity similar to CUX2.
CUX2 repression of male-biased genes in female mouse liver is associated with direct CUX2 binding at proximal genomic sites in female mouse liver chromatin (9); however, the mechanism whereby CUX2 effects repression of male-biased genes is not known. CUX1, a related transcription factor, represses gene expression by a combination of binding site competition and active repression (47). Here, we show that CUX2 can modulate transcriptional activity by counteracting both the stimulatory and the inhibitory effects of HNF6, as seen in transfection studies using 2 model sex-specific gene promoters. Thus, CUX2 reversed the stimulatory effects of HNF6 on HNF4α-activated CYP2C12, and it blocked the inhibitory effects of HNF6 on HNF1α-activated CYP2C11. These responses likely result from direct competition between CUX2 and HNF6 for binding to HNF6/CUX shared motifs that regulate these CYP promoters. This was indicated by EMSA analysis of individual binding sites in vitro and confirmed in vivo in mouse liver chromatin, both for individual genes (ChIP-qPCR) and globally (ChIP-seq). Of note, the effects of CUX2 seen in the transfection studies – up-regulation of the male-specific CYP2C11 promoter and down-regulation of the female-specific CYP2C12 promoter, are opposite to the common pattern of CUX2 effects seen in global analysis in mouse liver, ie, down-regulation of male-specific genes and up-regulation of female-specific genes (9). This difference could reflect the absence of other, associated transcription factors in the transfection studies, and/or a requirement for distal gene regulatory elements and native chromatin structure to recapitulate the global regulatory patterns manifested in liver in vivo.
The binding site motif identified for CUX2 (9) is most similar to the motifs describing binding sites used by HNF6, CUX1/CDP (Figure 2B), and another HD protein, PBX1. EMSA analysis confirmed that CUX2 and HNF6 (as well as CUX1 and the related factor OC-2) bind similar DNA sequences; however, each factor also displayed unique DNA-binding site specificity. Nevertheless, even small differences in sequence of the DNA-binding site may lead to diverse outcomes, as HNF6 can bind DNA either with or without its HD and CUX2 can bind DNA through various combinations of its 3 Cut regions and the HD (28, 31). Isoforms of HNF6 that differ in their DNA binding specificities are also known (48). HNF6 interacts with the coactivator p/CAF when bound to the TTR promoter, which involves HNF6's HD and Cut domain, whereas HNF6 interacts with the coactivator CBP when bound to the HNF3β/FOXA2 promoter, which involves only the HNF6 Cut domain (31). The recognition sequences for these 2 DNA binding activities of HNF6 differ by only one base pair (31). HNF6-stimulated transcription may also be modulated by interactions with coactivators, as indicated by the increased stability of HNF6 protein, and by its ability to stimulate Glut2 promoter activity, when acetylated by CBP (49). Although specific DNA sequences that discriminate between HNF6 and CUX2 DNA binding in vitro could be found (Figure 2A), de novo motif discovery did not reveal motif differences between HNF6-binding sites that do vs do not overlap with binding sites for CUX2 in mouse liver in vivo (motif 4A vs motif 7) (Figure 2B). Thus, liver binding specificities for HNF6 binding vs HNF6/CUX2 common binding cannot be explained by the underlying DNA motifs.
The close similarity between HNF6 and CUX2 DNA-binding site motifs, and their ability to bind common sequences in both EMSA and global ChIP-seq analyses, indicate that these 2 factors bind to common sites, and provide strong support for the proposal that the observed inhibitory action of CUX2 on HNF6 transcriptional activity involves binding site competition. Indeed, 91% of the CUX2 peaks identified in female mouse liver (9) overlapped with an HNF6 peak identified by ChIP-seq. The total number of HNF6/CUX2 common binding sites is likely greater than the 1338 sites identified here, given that the number of liver CUX2-binding sites identified previously (1471 sites) is limited by the low titer of the available CUX2-specific antibody, as discussed (9). CUX2 binding to chromatin is seen in female but not male liver, where CUX2 expression is nearly undetectable at the protein and RNA levels (29); consequently, binding site competition between CUX2 and HNF6 is restricted to female liver (Figure 8). Although we thus anticipated that the set of HNF6/CUX2 common peaks would preferentially show a male-enriched pattern of HNF6 binding, this was not seen. The set of HNF6/CUX2 common peaks was, however, significantly depleted of female-enriched HNF6-binding sites, supporting the conclusion that CUX2 binding to the HNF6/CUX2 common sites in female liver competes out HNF6 binding, which decreases the likelihood of female-enriched HNF6 binding at such sites. We also found that HNF6/CUX2 common sites are significantly enriched for chromatin regions more open in male than in female liver (male-biased DHS) (30), which helps explain the depletion of female-enriched HNF6 binding at these sites (Figure 8).
Figure 8.
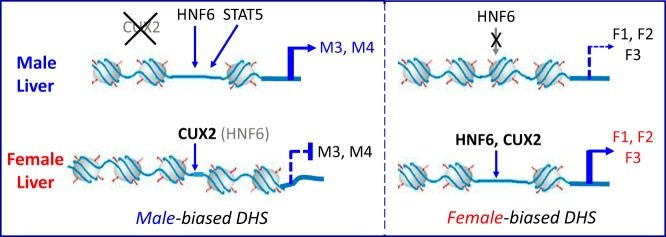
Model for regulation of sex-biased genes by HNF6 in adult male and female mouse liver. Regulation of male-biased genes (left): HNF6 binds to male-enriched binding sites within male-biased DHS regions nearby male-biased genes, in particular class M3 genes (Table 3), leading to transactivation. This transactivation is often associated with male-biased STAT5 binding (Figure 6). CUX2 is exclusively expressed in female liver, where it competes with HNF6 for binding at a subset of HNF6-binding sites based on their common sequence motifs (Figure 2), leading to loss of HNF6-dependent transactivation and repression of male-biased gene expression, in particular class M4 genes (Table 3). Regulation of female-biased genes (right): female-biased HNF6 binding occurs within female-biased DHS regions nearby female-biased gene classes F1, F2, and F3 leading to transactivation. CUX2 binding is seen at a subset of these female-biased HNF6-binding sites (in particular, nearby F3 genes; see Figure 5 and Table 3) and is associated with female-biased gene activation. This activating effect of CUX2 on a subset of female-biased genes contrasts with the suppressive effects of CUX2 on male-biased genes (9). A subset of the sex-enriched HNF6-binding sites identified were not associated with sex-biased DHS (not illustrated). Transfection studies (Figure 1) show that CUX2 can also modulate sex-biased gene transcription by counteracting transcriptional inhibitory effects of HNF6 (not illustrated).
HNF6 is an important global activator of hepatic gene expression (50), and the displacement of HNF6 by CUX2 at a subset of HNF6-binding sites indicated by our findings could be a mechanism by which CUX2 directly and selectively represses gene expression, in particular, male-biased gene expression in female liver (9), where CUX2 is specifically expressed (29). Our finding that HNF6/CUX2 common binding sites are enriched at male-biased DHS, and also at male-enriched STAT5-binding sites, both of which are strongly associated with male-biased gene expression (8, 30), suggests that CUX2 binding counters positive interactions of HNF6 with GH pulse-activated STAT5 in regulating male-biased gene expression. HNF6 but not CUX1 is likely to be involved in this cooperative, positive regulation of male-biased genes, as CUX1 protein levels are strongly down-regulated in mouse liver beginning at embryonic day 18.5 (E18.5), and is nearly extinguished by the action of liver microRNA-122, via its binding site in the 3′-untranslated region of CUX1 mRNA (45), before male-biased gene expression emerges at adulthood. Supporting this model, HNF6/CUX2 common binding sites showed significant enrichment for proximity to male-biased genes, with the strongest enrichment seen at gene targets belonging to male-biased gene subclass M4 (Table 3 and Supplemental Table 5), which is one of two male gene subclasses showing enrichment for proximal male-biased chromatin marks (27). However, we also observed significant enrichment of HNF6/CUX2 common binding sites at female-biased genes, most notably members of female gene subclass F3, which is characterized by local chromatin marks and includes several highly female-biased genes whose expression is apparently regulated by CUX2 via a direct binding mechanism (9), such as Cyp2b9 and Cyp3a44 (Figures 5 and 8).
HNF6 binding to liver chromatin occurred at a similar level in male and female mice at the vast majority (≥80%) of HNF6-binding sites, however, sex-biased HNF6 binding was seen at a few thousand sites. Female-enriched HNF6-binding sites and their target genes were significantly enriched in several classes of female-biased gene targets, most notably genes comprising female subclass F3 (Figure 5), which has the highest frequency of local female-enriched activating chromatin marks and local male-enriched repressive chromatin marks (27). Male-enriched HNF6-binding sites were correspondingly enriched at male subclass M3 gene targets, which are characterized by proximal male-biased chromatin marks (Figure 8) (27). These significant associations with sex-biased genes having proximal sex-biased chromatin marks raise the possibility that the occurrence of such chromatin marks may be a better predictor of sex-biased HNF6 peak-gene target interactions than linear proximity alone. The sets of male-enriched and female-enriched HNF6-binding sites also showed strong enrichment for localization at open chromatin regions (DHS) showing the same sex-bias. Thus, the sex-dependence of HNF6 binding is strongly associated with, and may be directly determined by the sex-dependence of chromatin accessibility, as was also seen for the sex- and plasma GH pattern-dependent binding of STAT5 to mouse liver chromatin (8). Nevertheless, and in contrast to liver STAT5-binding sites, a substantial fraction of the sex-enriched HNF6-binding sites identified here were not associated with sex-specific DHS, and could not be attributed to differences in HNF6 motif between male-biased and female-biased binding sites. Further studies are required to fully understand the factors and mechanisms that determine sex differences in HNF6 binding and the potential role of sex-differential chromatin states in the establishment and maintenance of these sex differences in factor binding.
HNF6 regulation of target genes can be modulated by cross talk with other transcription factors, including cofactors recruited to nearby genomic sequences. Motif analysis of HNF6 peak regions showed high scores for motifs associated with HNF1, HNF4α, HNF3/FOX, and C/EBP family members (Table 2), consistent with earlier reports that these liver-enriched transcription factors work in a combinatorial manner to regulate hepatic gene expression (51, 52), including sex-dependent gene expression (15). Comparison with published male mouse liver ChIP-seq datasets for these 4 liver transcription factors, as well as datasets for STAT5 and GR, revealed a significantly greater than expected frequency of multiple cobinding events than expected by chance, suggesting cooperativity in liver factor binding, in particular for HNF6 sites found in DHS regions (Figure 7). At the same time, HNF6-only binding events (singleton binding), primarily in non-DHS genomic regions, were also more frequent than expected. HNF6 and HNF3β/FOXA2 can synergistically activate the female-specific CYP2C12 promoter, with HNF6 potentially acting as a coactivator and recruiting p300/CBP proteins (23, 53). HNF6 has also been shown to synergize with HNF1α and C/EBPα, and with HNF4α (54), consistent with the motif coassociations seen here. Other HNF6 cofactor motifs identified here include members of the CDX, POU, and SRY families. Cooperation between HNF6 and CDX in regulating postnatal intestinal development has been reported (55), and hepatoblasts stimulated by GH can differentiate into biliary epithelial cells (cholangiocytes) that coexpress HNF6 and the SRY family factor SOX9 (56). Further studies are required to elucidate the interactions of HNF6 with these cofactors and to define roles they might play in modulating HNF6-dependent regulatory events linked to sex-specific gene expression in the liver.
Acknowledgments
We thank Gracia Bonilla and Tisha Melia for MACS analysis of GEO datasets of liver transcription factor ChIP-seq peak sets.
This work was supported in part by the National Institutes of Health Grant DK33765 (to D.J.W.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP
- chromatin immunoprecipitation
- DHS
- DNase I hypersensitive site
- EMSA
- electrophoretic mobility shift assay
- ES
- enrichment score
- GEO
- Gene Expression Omnibus
- GR
- glucocorticoid receptor
- HD
- homeodomain
- HNF
- hepatocyte-enriched nuclear factor
- qPCR
- quantitative PCR
- seq
- sequence
- UPGMA
- Unweighted Pair Group Method with Arithmetic Mean.
References
- 1. Rinn JL, Rozowsky JS, Laurenzi IJ, et al. Major molecular differences between mammalian sexes are involved in drug metabolism and renal function. Dev Cell. 2004;6(6):791–800. [DOI] [PubMed] [Google Scholar]
- 2. Ahluwalia A, Clodfelter KH, Waxman DJ. Sexual dimorphism of rat liver gene expression: regulatory role of growth hormone revealed by deoxyribonucleic acid microarray analysis. Mol Endocrinol. 2004;18(3):747–760. [DOI] [PubMed] [Google Scholar]
- 3. Clodfelter KH, Holloway MG, Hodor P, Park SH, Ray WJ, Waxman DJ. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol. 2006;20(6):1333–1351. [DOI] [PubMed] [Google Scholar]
- 4. Kwekel JC, Desai VG, Moland CL, Branham WS, Fuscoe JC. Age and sex dependent changes in liver gene expression during the life cycle of the rat. BMC Genomics. 2010;11:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Klein K, Sugathan A, et al. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS One. 2011;6(8):e23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol. 2009;76(2):215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meyer RD, Laz EV, Su T, Waxman DJ. Male-specific hepatic Bcl6: growth hormone-induced block of transcription elongation in females and binding to target genes inversely coordinated with STAT5. Mol Endocrinol. 2009;23(11):1914–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y, Laz EV, Waxman DJ. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol. 2012;32(4):880–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conforto TL, Zhang Y, Sherman J, Waxman DJ. Impact of CUX2 on the female mouse liver transcriptome: activation of female-biased genes and repression of male-biased genes. Mol Cell Biol. 2012;32(22):4611–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J, Wan Y, Na S, Liu X, et al. Sex-dependent regulation of hepatic CYP3A by growth hormone: roles of HNF6, C/EBPα, and RXRα. Biochem Pharmacol. 2015;93(1):92–103. [DOI] [PubMed] [Google Scholar]
- 11. Waxman DJ, Ram PA, Park SH, Choi HK. Intermittent plasma growth hormone triggers tyrosine phosphorylation and nuclear translocation of a liver-expressed, Stat 5-related DNA binding protein. Proposed role as an intracellular regulator of male-specific liver gene transcription. J Biol Chem. 1995;270(22):13262–13270. [DOI] [PubMed] [Google Scholar]
- 12. Holloway MG, Miles GD, Dombkowski AA, Waxman DJ. Liver-specific hepatocyte nuclear factor-4α deficiency: greater impact on gene expression in male than in female mouse liver. Mol Endocrinol. 2008;22(5):1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holloway MG, Laz EV, Waxman DJ. Codependence of growth hormone-responsive, sexually dimorphic hepatic gene expression on signal transducer and activator of transcription 5b and hepatic nuclear factor 4α. Mol Endocrinol. 2006;20(3):647–660. [DOI] [PubMed] [Google Scholar]
- 14. Nagaki M, Moriwaki H. Transcription factor HNF and hepatocyte differentiation. Hepatol Res. 2008;38(10):961–969. [DOI] [PubMed] [Google Scholar]
- 15. Wiwi CA, Waxman DJ. Role of hepatocyte nuclear factors in growth hormone-regulated, sexually dimorphic expression of liver cytochromes P450. Growth Factors. 2004;22(2):79–88. [DOI] [PubMed] [Google Scholar]
- 16. Lahuna O, Rastegar M, Maiter D, Thissen JP, Lemaigre FP, Rousseau GG. Involvement of STAT5 (signal transducer and activator of transcription 5) and HNF-4 (hepatocyte nuclear factor 4) in the transcriptional control of the hnf6 gene by growth hormone. Mol Endocrinol. 2000;14(2):285–294. [DOI] [PubMed] [Google Scholar]
- 17. Wiwi CA, Gupte M, Waxman DJ. Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4α-deficient mice. Mol Endocrinol. 2004;18(8):1975–1987. [DOI] [PubMed] [Google Scholar]
- 18. Bailly A, Torres-Padilla ME, Tinel AP, Weiss MC. An enhancer element 6 kb upstream of the mouse HNF4α1 promoter is activated by glucocorticoids and liver-enriched transcription factors. Nucleic Acids Res. 2001;29(17):3495–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayashi Y, Wang W, Ninomiya T, Nagano H, Ohta K, Itoh H. Liver enriched transcription factors and differentiation of hepatocellular carcinoma. Mol Pathol. 1999;52(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Plumb-Rudewiez N, Clotman F, Strick-Marchand H, et al. Transcription factor HNF-6/OC-1 inhibits the stimulation of the HNF-3α/Foxa1 gene by TGF-β in mouse liver. Hepatology. 2004;40(6):1266–1274. [DOI] [PubMed] [Google Scholar]
- 21. Beaudry JB, Pierreux CE, Hayhurst GP, et al. Threshold levels of hepatocyte nuclear factor 6 (HNF-6) acting in synergy with HNF-4 and PGC-1α are required for time-specific gene expression during liver development. Mol Cell Biol. 2006;26(16):6037–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park SH, Wiwi CA, Waxman DJ. Signalling cross-talk between hepatocyte nuclear factor 4α and growth-hormone-activated STAT5b. Biochem J. 2006;397(1):159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park SH, Waxman DJ. Inhibitory cross-talk between STAT5b and liver nuclear factor HNF3β: impact on the regulation of growth hormone pulse-stimulated, male-specific liver cytochrome P-450 gene expression. J Biol Chem. 2001;276(46):43031–43039. [DOI] [PubMed] [Google Scholar]
- 24. Delesque-Touchard N, Park SH, Waxman DJ. Synergistic action of hepatocyte nuclear factors 3 and 6 on CYP2C12 gene expression and suppression by growth hormone-activated STAT5b. Proposed model for female specific expression of CYP2C12 in adult rat liver. J Biol Chem. 2000;275(44):34173–34182. [DOI] [PubMed] [Google Scholar]
- 25. Chen Y, Lin G, Huo JS, et al. Computational and functional analysis of growth hormone (GH)-regulated genes identifies the transcriptional repressor B-cell lymphoma 6 (Bc16) as a participant in GH-regulated transcription. Endocrinology. 2009;150(8):3645–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chia DJ, Rotwein P. Defining the epigenetic actions of growth hormone: acute chromatin changes accompany GH-activated gene transcription. Mol Endocrinol. 2010;24(10):2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol. 2013;33(18):3594–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gingras H, Cases O, Krasilnikova M, Bérubé G, Nepveu A. Biochemical characterization of the mammalian Cux2 protein. Gene. 2005;344:273–285. [DOI] [PubMed] [Google Scholar]
- 29. Laz EV, Holloway MG, Chen CS, Waxman DJ. Characterization of three growth hormone-responsive transcription factors preferentially expressed in adult female liver. Endocrinology. 2007;148(7):3327–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ling G, Sugathan A, Mazor T, Fraenkel E, Waxman DJ. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol Cell Biol. 2010;30(23):5531–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iyaguchi D, Yao M, Watanabe N, Nishihira J, Tanaka I. DNA recognition mechanism of the ONECUT homeodomain of transcription factor HNF-6. Structure. 2007;15(1):75–83. [DOI] [PubMed] [Google Scholar]
- 32. Vadnais C, Awan AA, Harada R, et al. Long-range transcriptional regulation by the p110 CUX1 homeodomain protein on the ENCODE array. BMC Genomics. 2013;14:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thiaville MM, Stoeck A, Chen L, et al. Identification of PBX1 target genes in cancer cells by global mapping of PBX1 binding sites. PLoS One. 2012;7(5):e36054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012;13(3):R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McLean CY, Bristor D, Hiller M, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 38. Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26(11):1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mahony S, Benos PV. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35(Web Server issue):W253–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Z, Chang CW, Goh WL, Sung WK, Cheung E. CENTDIST: discovery of co-associated factors by motif distribution. Nucleic Acids Res. 2011;39(Web Server issue):W391–W399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matys V, Kel-Margoulis OV, Fricke E, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34(Database issue):D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Macisaac KD, Gordon DB, Nekludova L, et al. A hypothesis-based approach for identifying the binding specificity of regulatory proteins from chromatin immunoprecipitation data. Bioinformatics. 2006;22(4):423–429. [DOI] [PubMed] [Google Scholar]
- 43. Faure AJ, Schmidt D, Watts S, et al. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 2012;22(11):2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jacquemin P, Pierreux CE, Fierens S, van Eyll JM, Lemaigre FP, Rousseau GG. Cloning and embryonic expression pattern of the mouse Onecut transcription factor OC-2. Gene Expr Patterns. 2003;3(5):639–644. [DOI] [PubMed] [Google Scholar]
- 45. Xu H, He JH, Xiao ZD, et al. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52(4):1431–1442. [DOI] [PubMed] [Google Scholar]
- 46. Lannoy VJ, Rodolosse A, Pierreux CE, Rousseau GG, Lemaigre FP. Transcriptional stimulation by hepatocyte nuclear factor-6. Target-specific recruitment of either CREB-binding protein (CBP) or p300/CBP-associated factor (p/CAF). J Biol Chem. 2000;275(29):22098–22103. [DOI] [PubMed] [Google Scholar]
- 47. Sansregret L, Nepveu A. The multiple roles of CUX1: insights from mouse models and cell-based assays. Gene. 2008;412(1–2):84–94. [DOI] [PubMed] [Google Scholar]
- 48. Lannoy VJ, Bürglin TR, Rousseau GG, Lemaigre FP. Isoforms of hepatocyte nuclear factor-6 differ in DNA-binding properties, contain a bifunctional homeodomain, and define the new ONECUT class of homeodomain proteins. J Biol Chem. 1998;273(22):13552–13562. [DOI] [PubMed] [Google Scholar]
- 49. Rausa FM, 3rd, Hughes DE, Costa RH. Stability of the hepatocyte nuclear factor 6 transcription factor requires acetylation by the CREB-binding protein coactivator. J Biol Chem. 2004;279(41):43070–43076. [DOI] [PubMed] [Google Scholar]
- 50. Wang K, Holterman AX. Pathophysiologic role of hepatocyte nuclear factor 6. Cell Signal. 2012;24(1):9–16. [DOI] [PubMed] [Google Scholar]
- 51. Odom DT, Zizlsperger N, Gordon DB, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303(5662):1378–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev. 2006;20(16):2293–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rausa FM, Tan Y, Costa RH. Association between hepatocyte nuclear factor 6 (HNF-6) and FoxA2 DNA binding domains stimulates FoxA2 transcriptional activity but inhibits HNF-6 DNA binding. Mol Cell Biol. 2003;23(2):437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshida Y, Hughes DE, Rausa FM, 3rd, et al. C/EBPα and HNF6 protein complex formation stimulates HNF6-dependent transcription by CBP coactivator recruitment in HepG2 cells. Hepatology. 2006;43(2):276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maier EA, Dusing MR, Wiginton DA. Temporal regulation of enhancer function in intestinal epithelium: a role for Onecut factors. J Biol Chem. 2006;281(43):32263–32271. [DOI] [PubMed] [Google Scholar]
- 56. Dianat N, Dubois-Pot-Schneider H, Steichen C, et al. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology. 2014;60(2):700–714. [DOI] [PMC free article] [PubMed] [Google Scholar]