

Abstract
Steroid hormone-regulated differentiation of uterine stromal cells, known as decidualization, is essential for embryo implantation. The role of the estrogen receptor-α (ESR1) during this differentiation process is unclear. Development of conditional Esr1-null mice showed that deletion of this gene in both epithelial and stromal compartments of the uterus leads to a complete blockade of decidualization, indicating a critical role of ESR1 during this process. To further elucidate the cell type-specific function of ESR1 in the uterus, we created WEd/d mice in which Esr1 is ablated in uterine luminal and glandular epithelia but is retained in the stroma. Uteri of WEd/d mice failed to undergo decidualization, indicating that epithelial ESR1 contributes to stromal differentiation via a paracrine mechanism. We noted markedly reduced production of the leukemia inhibitory factor (LIF) in WEd/d uteri. Supplementation with LIF restored decidualization in WEd/d mice. Our study indicated that LIF acts synergistically with progesterone to induce the expression of Indian hedgehog (IHH) in uterine epithelium and its receptor patched homolog 1 in the stroma. IHH then induces the expression of chicken ovalbumin upstream promoter-transcription factor II, a transcription factor that promotes stromal differentiation. To address the mechanism by which LIF induces IHH expression, we used mice lacking uterine epithelial signal transducer and activator of transcription 3, a well-known mediator of LIF signaling. Our study revealed that LIF-mediated induction of IHH occurs without the activation of epithelial signal transducer and activator of transcription 3 but uses an alternate pathway involving the activation of the ERK1/2 kinase. Collectively our results provide unique insights into the paracrine mechanisms by which ESR1 directs epithelial-stromal dialogue during pregnancy establishment.
Steroid hormones 17β-estradiol (E) and progesterone (P) play crucial roles during early pregnancy. These hormones function via their cognate receptors to orchestrate waves of cell proliferation, differentiation, and remodeling in the uterine tissue of the pubertal animal to prepare it for embryo implantation and establishment of pregnancy (1–4). In mice and humans, one of the prerequisites for successful implantation is the differentiation of endometrial stromal cells into a unique secretory tissue, known as the decidua, which controls embryonic growth and survival. This remarkable transformation event, known as decidualization, comprises morphogenetic and biochemical changes driven by the E and P receptors (5–7). Although the cellular events that regulate various phases of decidualization are well described, the steroid-regulated molecular pathways that underlie this differentiation process are not fully understood.
The development of mutant mouse models lacking the receptors for P and E has established the necessity of steroid signaling for implantation. Mice lacking the progesterone receptor (PGR) displayed a refractory uterus that fails to respond to an experimentally induced deciduogenic stimulus, establishing that the PGR plays a critical role in regulating the decidualization process (8, 9). In contrast, mice lacking the estrogen receptor-α (ESR1), developed by Lubahn et al and tested by others (10–12), although impaired in epithelial receptivity for blastocyst attachment, exhibited a partial decidual response when subjected to artificial decidual stimulation. This finding raised questions regarding the essentiality of ESR1 in the control of stromal differentiation. Later it was reported that the ESR1-null mice developed by Lubahn et al still expressed a truncated ESR1 that retained DNA binding, hormone binding, and partial transcription regulatory function, raising the possibility that this residual receptor activity mediated the decidual response observed in these mutant mice (13, 14). Thus, the true role of ESR1 in the regulation of decidualization remained unresolved.
In this study, we used two distinct conditional knockout mouse models to assess the role of ESR1 in the regulation of endometrial stromal differentiation. In the first model, the Esr1 gene was deleted from both epithelial and stromal cells of the uterus. Our results revealed that these mice failed to execute experimentally induced decidualization. In the second model, Esr1 was specifically ablated in the uterine luminal and glandular epithelia but was retained in the stroma. Interestingly, this epithelium-specific deletion also resulted in a complete loss of decidualization, indicating that epithelial ESR1 controls critical aspects of endometrial stromal differentiation via a paracrine mechanism. Further studies demonstrated that epithelial ESR1 directs the glandular production of the leukemia inhibitory factor (LIF), a well-known regulator of implantation (15). LIF, in turn, acts on the luminal epithelium by regulating the downstream paracrine factor Indian hedgehog (IHH), which then acts on the stromal cells to induce their differentiation. Our findings therefore suggest that the ESR1 orchestrates a tripartite molecular communication between the glandular epithelium, luminal epithelium and stroma by initiating a sequential cascade of paracrine signals that play key roles in regulating both epithelial and stromal functionality. Dysregulated uterine ESR1 signaling during pregnancy therefore impairs the epithelial-stromal dialog and contributes to endometrial dysfunction and early pregnancy loss.
Materials and Methods
Mice, hormone treatments, and tissue collection
All experiments involving mice were conducted in accordance with National Institutes of Health standards for the use and care of mice. The animal protocols were approved by the University of Illinois Institutional Animal Care and Use Committee. Mice carrying the floxed ESR1 allele (Esr1fl/fl) were developed by the laboratory of Dr Pierre Chambon (Institut de Genetique et de Biologie Moleculaire et Cellulaire, Strasbourg, France) and were provided to us by Dr Jay Ko (University of Illinois, Urbana-Champaign) (13). These mice were mated with PR-Cre or Wnt7a-Cre mice to generate Esr1d/d or WEd/d mice, respectively. The PR-Cre mice were provided by Drs Francesco J. DeMayo and John P. Lydon (Baylor College of Medicine, Houston, TX) (16). The Wnt7a-Cre mice were provided by Dr Richard Behringer (University of Texas M. D. Anderson Cancer Center, Houston, TX). The Stat3f/f mice, provided by Dr Shizuo Akira (Osaka University, Osaka, Japan) and Dr Hua Yu (City of Hope, Duarte, CA), were crossed with Wnt7a-Cre mice to generate SWd/d mice. For breeding studies, Esr1fl/fl or Esr1d/d or WEd/d female mice were housed with wild-type C57BL/6 male mice (Charles Rivers). The presence of a vaginal plug after mating was designated as day 1 of pregnancy.
Decidualization was experimentally induced in nonpregnant mice as described previously (17). Briefly, mice were first ovariectomized. Two weeks after the ovariectomy, mice were injected with 100 ng of E in 0.1 mL of sesame oil for 3 consecutive days. This was followed by daily injections of 1 mg of P and 10 ng of E for 3 consecutive days. Decidualization was then initiated in one horn by injection of 50 μL oil. The other horn was left unstimulated. The mice were treated with E+P additionally after the stimulation and then killed to collect the uterine tissue at 20 hours after the stimulus (for cell proliferation studies) and 72 hours after the stimulus (for differentiation studies). For the rescue of decidualization, 100 μL of vehicle (PBS) or LIF (1 μg in 100 μL PBS) was added ip every 2 hours for 8 hours before the stimulus. Uteri were collected at 72 hours after the stimulus. Frozen uterine tissue collected at 72 hours after the stimulus was used to assess alkaline phosphatase activity, which was measured as described previously (18).
For gene and protein expression analyses during pregnancy, uteri were collected at different days of pregnancy for RNA isolation and immunohistochemistry (IHC). One horn of the uterus was fixed in 10% formalin prior to IHC, and the other horn was flash frozen in liquid N2 prior to RNA isolation.
Intraluminal administration of the MAPK kinase (MEK) inhibitor
This procedure was performed as described previously (19). The MEK inhibitor PD184352, was initially dissolved in dimethylsulfoxide (DMSO) and then diluted in Hanks' balanced salt solution. Ten microliters of inhibitor (50 μM) was injected into one horn intraluminally 12 hours before the stimulus, whereas the other horn served as vehicle control. LIF was administered as described above and uterine horns were collected 20 hours after the stimulus. Similarly, 10 μL of inhibitor (50 μM) was injected into one horn intraluminally on day 2 of pregnancy, whereas the other horn served as vehicle control. Uterine horns were collected on day 3 of pregnancy.
Isolation of uterine epithelial and stromal cells
Isolation of uterine cells was performed as previously described (20). Briefly, uteri were removed at 0 hours, 20 hours, or 72 hours after the stimulus. With the aid of a dissecting microscope, each uterine horn was cut transversely into two equal segments. The tissues were then incubated in 10 mL of a solution of 0.5% bovine pancreatic trypsin (Cooper Biomedical) in Ca- and Mg-free PBS (pH 7.4). Tissues were first incubated at 4°C for 1 hour and then at 37°C for 40 minutes. After incubation, the uterine tissue was squeezed with forceps dissociating the epithelium from the uterine pieces. The epithelial fragments in the supernatant were collected with a pipette; this procedure was repeated three or four times. The epithelial fragments were collected by centrifugation at 100 × g for 5 minutes. The resulting cell pellet was subjected to RNA isolation protocol. The remaining uterine tissue was incubated with 0.5 g/L collagenase at 37°C for 40 minutes. After incubation, the tube was shaken vigorously, and the supernatant was passed through a 70-μm mesh to remove tissue debris. Stromal cells were collected by centrifugation at 1700 × g for 5 minutes. For primary stromal cell culture, cells were grown in DMEM/F-12 medium containing 5% charcoal-stripped fetal bovine serum. To induce in vitro decidualization, the cells were treated with or without a hormone cocktail containing 1 nM E and 1 mM P for 24–72 hours and has been described previously (18).
Real-time PCR analysis
Uterine tissue was either homogenized or uterine epithelium was isolated and total RNA was extracted by using a TRIZOL reagent (Sigma-Aldrich), according to the manufacturer's protocol. cDNA was prepared by standard protocols. The cDNA was amplified to quantify gene expression by real-time PCR, using gene-specific primers and SYBR Green (Applied Biosystems). The expression level of Rplp0 (36B4) or cytokeratin 18 (Ck18) was used as the internal control. For each treatment, the mean cycle threshold (Ct) and SD were calculated from individual Ct values obtained from three replicates of a sample. The normalized δCt in each sample was calculated as mean Ct of target gene subtracted by the mean Ct of internal control gene. The δδCt was then calculated as the difference between the δCt values of the control and treatment sample. The fold change of gene expression in each sample relative to a control was computed as 2-δΔa]Ct. The mean fold induction and SEs were calculated from two or more independent experiments.
IHC, image capture, and quantification of immunostaining
Formalin-fixed uterine pieces were processed for paraffin embedding. Cross-sections (5 μm thickness) were mounted onto microscope slides (Fisher Scientific). For immunostaining, uterine sections were deparaffinized in xylene (three times for 5 min each), rehydrated through a graded series of treatment with ethanol (100%, 95%, 85%, and 70% for 5 min each), and rinsed in tap water. For all samples, antigen retrieval was performed by boiling the sections in 0.01 M sodium citrate buffer (pH 6.0) for 20 minutes, followed by incubation at room temperature for 30 minutes. A 5% solution of normal donkey serum (Jackson ImmunoResearch) in PBS was used as a blocking buffer. Sections were incubated with the following primary antibodies diluted in blocking solution (0.25% BSA, 0.3% Triton X-100, sterile PBS) overnight at 4°C: Ki67 (BD Pharmingen), phosphorylated signal transducer and activator of transcription 3 (STAT3)-TY 705 (Abcam), phosphorylated (p) ERK1/2 (Santa Cruz Biotechnology), ERK1/2 (Santa Cruz Biotechnology), pShp2 (Abcam), and ESR1 (Novus Biologicals). The sections were washed and incubated with either biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories Inc) secondary antibodies for 60 minutes. This was followed by incubation with streptavidin-conjugated horseradish peroxidase (Histostain kit; Zymed Laboratories Inc) for 45 minutes. Sections were stained with 3-amino-9-ethyl carbazole solution (Zymed Laboratories Inc) and counterstained with Mayer's hematoxylin (Sigma). Stained sections were mounted using diazabicyclo (2, 2, 2) octane, Tris-HCl (pH 8.5), and sterile double-distilled water. Negative controls included incubation with donkey serum and omission of the primary antibody for all samples.
The images of IHC staining were captured by using a Leica DM2500 light microscope fitted with a Qimaging Retiga 2000R camera (Qimaging). For quantification of the IHC data, five to six individual ×20 images from each sample were captured. All images were set to have an identical lower threshold. Background was subtracted from each image, and three to four fields in each image were measured to obtain average intensity. Average intensity was plotted as the percentage of highest pixel intensity for each image. Images were processed using Adobe Photoshop version 8. The SD was determined for each averaged total. An ANOVA single-factor analysis was conducted on the grouped means to determine statistical significance at a significance of a value of P < .01.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5. Normally distributed data were analyzed by a one-way ANOVA with a Tukey posttest, a two-way ANOVA with Bonferroni correction, or a Student's t test as appropriate to the number of comparisons to be made. A value of P < .05 was considered significant.
Results
Transgenic mice expressing Cre under the control of the Pgr promoter were used previously to efficiently ablate floxed genes in the uterus (17, 19). To create a conditional knockout of the Esr1 gene in the uterus, we crossed Pgr-Cre mice with those harboring floxed Esr1 gene (Esr1fl/fl) (13). Cre-mediated excision of floxed exon 3 of the Esr1 gene, which contains the DNA binding region, created Esr1d/d mice in which the Esr1 gene is deleted in all uterine cells expressing PGR. The absence of the ESR1 protein in the luminal epithelium, glandular epithelium, and stroma of Esr1d/d uteri was confirmed by IHC (Figure 1A, a and b).
Figure 1.

Ablation of Esr1 in the uterus epithelium leads to a defect in stromal cell decidualization. A, Uteri were collected from day 4 pregnant Esr1fl/fl and Esr1d/d mice. Uterine sections were subjected to IHC using an anti-ESR1 antibody targeting the C terminus of ESR1. Esr1d/d mice displayed efficient deletion of ESR1 (b) when compared to Esr1fl/fl control mice (a). B, Esr1fl/fl and Esr1d/d mice were subjected to experimentally induced decidualization. Uteri were collected 72 hours after administration of oil stimulus. Esr1d/d uteri failed to give a decidual response. C, Ratio of wet weight gains of right and left horns. **, P < .01. D, ALP activity, indicated by purple color, was detected in the decidualized stroma of Esr1fl/fl uteri (a) but not in the stroma of Esr1d/d uteri (b).
To investigate the function of ESR1 during decidualization, Esr1fl/fl and Esr1d/d mice were subjected to experimentally induced decidualization. These mice were ovariectomized, treated sequentially with E and P, and subjected to a mechanical stimulation as described previously (17). This artificial stimulus mimics the embryonic signal during implantation and initiates the decidualization program. Examination of the gross anatomy of the stimulated and unstimulated uterine horns of Esr1fl/fl and Esr1d/d mice indicated that, as expected, the uterine horns of Esr1fl/fl mice exhibited a robust decidual response within 72 hours after receiving the artificial stimulation (Figure 1B). In contrast, the Esr1-deficient uteri under identical conditions failed to show any decidualization (Figure 1B). When the decidual response was assessed by measuring uterine wet weight gain, the Esr1-deficient uteri exhibited a markedly reduced weight gain relative to that seen in Esr1fl/fl uteri (Figure 1C).
We further analyzed the decidual response of Esr1d/d uteri by monitoring the expression of alkaline phosphatase (ALP), a classical biomarker of decidualization (18). As shown in Figure 1Da, the expression of ALP was prominent in the decidual cells of the control Esr1-intact uteri at 72 hours after the decidual stimulation. In contrast, no expression of this decidual marker was seen in the uteri of Esr1d/d mice at a similar time point, indicating a complete blockade of stromal cell differentiation in these uteri (Figure 1Db). These results established that endometrial ESR1 plays a critical role in decidualization.
Uterine epithelial ESR1 is indispensable for stromal decidualization
Because Esr1 was ablated from both epithelial and stromal cells in Esr1d/d uteri, this model could not inform on the cell type-specific role of this receptor during decidualization. To gain mechanistic insight into the cell-type-specific role of ESR1 and its downstream signaling pathway in the regulation of stromal differentiation, we next performed an epithelium-specific deletion of Esr1 in the uterus. We crossed Esr1fl/fl mice with transgenic mice expressing Cre under the control of a Wnt-7a promoter as described previously (21). This created the WEd/d mice in which the Esr1 gene is deleted exclusively in the uterine glandular and luminal epithelial cells expressing Wnt-7a. The ablation of Esr1 gene in the uterine epithelium of WEd/d mice was confirmed by the greatly reduced levels of Esr1 transcripts in this tissue (Figure 2A). Furthermore, analysis of ESR1 protein by IHC failed to show any expression in the luminal and glandular epithelium of WEd/d uteri, whereas its expression in the stroma remained unaltered (Figure 2B, compare panels a and b).
Figure 2.

Uterine epithelial ESR1 is indispensable for stromal cell decidualization. A, Uterine epithelial RNA was extracted from Esr1fl/fl and WEd/d mice on day 4 of pregnancy (n = 3–4) and analyzed by real-time PCR. Relative levels of Esr1 mRNA expression in uterine epithelium WEd/d mice are compared with those in Esr1fl/fl control mice. Ck18 was used as internal control to normalize gene expression. The data are represented as the mean fold induction ± SEM. ***, P < .001. B, Immunohistochemical localization of ESR1 in the uterus of Esr1fl/fl and WEd/d 18 hours after E treatment. Note the absence of glandular and luminal epithelial expression of ESR1 in WEd/d uteri, whereas stromal expression remains intact. G, glandular epithelium; L, luminal epithelium; S, stroma. C, The right uterine horns (R) of Esr1fl/fl and WEd/d mice were stimulated, whereas the left horns (L) were left unstimulated. When examined after 72 hours, only the right horn of Esr1fl/fl exhibited a robust decidual response after stimulation. The stimulated right horn of WEd/d failed to show any decidual response after 72 hours. D, Ratio of wet weight gains of right and left horns. **, P ≤ .01 (t test, n = 5).
A 6-month breeding study demonstrated that WEd/d mutant mice are infertile (Table 1). Whereas Esr1fl/fl mice exhibited normal litter size and pregnancy rates, the WEd/d females failed to get pregnant when mated with wild-type males. Analysis of the ovarian function of these mice indicated that the reproductive cycle, ovulation, and steroidogenesis occurred normally (data not shown). These results are consistent with those reported previously by the group of Korach and colleagues (22).
Table 1.
Ablation of Uterine Epithelial Esr1 Leads to Infertility
| Genotype | Number | Number of Litters | Number of Litters per Animal (Mean ± SEM) | Number of Pups |
|---|---|---|---|---|
| Esr1fl/fl | 6 | 33 | 5.5 ± 0.4 | 233 |
| WEd/d | 6 | 0 | 0 | 0 |
Results of 6-month breeding study are shown.
Because the circulating levels of E and P were unaltered in the WEd/d mice, we assessed the ability of these mice to undergo stromal differentiation by subjecting them to experimentally induced decidualization as described above. As expected, the decidual response was pronounced in the uterine horns of control Esr1fl/fl mice (Figure 2C, left panel). However, WEd/d uteri failed to show any decidualization under identical conditions (Figure 2C, right panel). Consequently, the WEd/d uteri exhibited a markedly reduced wet weight gain relative to that seen in the Esr1fl/fl uteri (Figure 2D). Taken together, these data indicated that uterine epithelial ESR1 is indispensable for stromal cell decidualization.
Uterine epithelial ESR1 controls stromal proliferation and differentiation
During decidualization, the uterine stromal cells initially undergo proliferation for 24–48 hours followed by differentiation (2, 6, 23–25). To investigate the decidualization defect in mice lacking Esr1 in uterine epithelium, we first monitored the proliferation of steroid hormone-primed stromal cells in response to decidual stimulation. Uteri of Esr1fl/fl mice exhibited widespread immunostaining for Ki67 in the stromal compartment within 20 hours of receiving the decidual stimulus, indicating extensive stromal cell proliferation (Figure 3A, a and b). In contrast, sections from WEd/d uteri exhibited significantly reduced Ki67 staining under identical conditions (Figure 3A, c and d). Quantification of the Ki67 signal revealed a significant reduction in the number of Ki67-positive cells in the stromal compartment of the WEd/d uteri (Figure 3B).
Figure 3.

Uterine stromal cell proliferation during decidualization is impaired in WEd/d mice. A, Cell proliferation was measured by Ki67 immunostaining in uterine sections of Esr1fl/fl (upper panel) and WEd/d (lower panel) mice at 20 hours after decidual stimulation. Magnification (a and c), ×10; (b and d), ×20. B, Percentage of Ki67-positive cells in the stromal compartment of Esr1fl/fl and WEd/d mice 20 hours after decidual stimulation. The data represent the average number of cells from multiple fields of multiple uterine sections. *, P ≤ .05 (t test, n = 4). C, Stromal cells were isolated from uteri 20 hours after decidual stimulation and total RNA was prepared. Real-time RT-PCR was performed to monitor the expression of mRNAs corresponding to the cell cycle genes Ccne1, Ccne2, Ccnd1, Cdk2, Cdk4, and Cdk6. Rplp0 encoding a ribosomal subunit protein was used as internal control to normalize gene expression. The data are represented as the mean fold induction ± SEM. *, P ≤ .05; **, P ≤ .01. ns, not significant. D, ALP activity was detected in the decidualized stroma. a, sections of Esr1fl/fl uteri; b), sections of WEd/d uteri. Purple color indicates ALP activity.
To further investigate the proliferation defect in WEd/d uterine stroma, we analyzed the expression of key cell cycle regulatory factors, including various cyclins and cyclin-dependent kinases (Cdks) (26). Our results revealed a marked down-regulation of cyclin D1 (Ccnd1), cyclin E1 (Ccne1), cyclin E2 (Ccne2), and the cyclin-dependent kinase Cdk2 in the stromal cells of WEd/d uteri at 20 hours after the administration of the decidual stimulus (Figure 3C). These cell cycle-regulatory factors primarily control the G1-S phase transition of the cell cycle. However, not all the factors involved in the G1-S transition exhibited altered the expression in WEd/d uteri. For example, the expression of Cdk4 and Cdk6, which regulate progression through G1, remained unchanged in these uteri. These results are consistent with a cell cycle defect in stromal cells of uteri lacking epithelial ESR1.
We also tested the biochemical markers of stromal differentiation in WEd/d mice. Whereas we observed prominent expression of ALP in the decidual cells of the Esr1fl/fl uteri at 72 hours after decidual stimulation, no expression of this decidual marker was seen in the uteri of WEd/d mice at a similar time point, indicating a complete absence of stromal differentiation in these mice (Figure 3D, compare panels a and b). Collectively our results indicated that uterine epithelial ESR1 is a critical regulator of stromal cell proliferation and differentiation during decidualization.
Epithelial ESR1 regulates decidualization via LIF
We next investigated the paracrine mechanism by which epithelial ESR1 promotes stromal decidualization. An examination of the expression of ESR1 at the onset of decidualization revealed a prominent expression of ESR1 in glandular epithelium of Esr1fl/fl uteri at 20 hours after the administration of decidual stimulation (Figure 4Aa). ESR1 is also present in the stroma but barely detectable in the luminal epithelium at this time. In contrast, in WEd/d uteri, ESR1 is absent from both luminal and glandular epithelium but is still expressed in the stroma (Figure 4Ab) at a level comparable with that expressed in the stroma of Esr1fl/fl uteri (Figure 4B). The high expression of glandular ESR1 in Esr1fl/fl uteri and its absence in WEd/d uteri led us to consider the possibility that ESR1 controls the secretion of a glandular factor that promotes stromal decidualization. It is well known that LIF, a glandular E-regulated factor, is critical for implantation (27, 28). We therefore examined the expression of Lif transcripts in Esr1fl/fl and WEd/d animals during decidualization. As shown in Figure 4C, the expression of Lif was drastically reduced in uteri lacking epithelial ESR1, whereas the levels of PGR and its downstream effector HAND2 (19) remained unaltered between Esr1fl/fl and WEd/d uteri (Figure 4, D and E). These results supported our hypothesis that LIF is a downstream mediator of the effects of epithelial ESR1 in the regulation of stromal decidual response.
Figure 4.

Supplementation of LIF rescues stromal cell decidualization in WEd/d mice. A, ESR1 expression in Esr1fl/fl uteri (a) and WEd/d uteri (b) at 20 hours after experimentally induced decidualization. Black arrows indicate glandular expression of ESR1. B, Percentage intensity of stromal expression of ESR1 in Esr1fl/fl and WEd/d uteri at 20 hours after experimentally induced decidualization. Quantification of IHC staining was performed as described in Materials and Methods. ns, not significant (t test, n = 5–7). C, Real-time PCR was performed to monitor the expression of Lif and PgR in the uteri of Esr1fl/fl and WEd/d mice at 20 hours after stimulation. The level of Ck18 transcripts, which encode an epithelium-specific protein, was used as internal control to normalize gene expression. The data are represented as the mean fold induction ± SEM. **, P ≤ .01. ns, not significant. D, PGR and HAND2 expression in Esr1fl/fl uteri (panels a and c) and WEd/d uteri (panels b and d) at 20 hours after experimentally induced decidualization. E, Percentage intensity of uterine expression of PGR and HAND2 in Esr1fl/fl and WEd/d uteri at 20 hours after experimentally induced decidualization. Quantification of IHC staining was performed as described in Materials and Methods. ns, not significant (t test, n = 3–4). F, LIF rescues stromal cell decidualization in WEd/d mice. Esr1fl/fl and WEd/d mice were subjected to experimentally induced decidualization after administration of vehicle (saline) or LIF. Uteri were assessed for gross anatomy (panels a–c) and ALP activity (panels d–f) at 72 hours after decidual stimulation. Panels a and d show Esr1fl/fl mice treated with vehicle; panels b and e show WEd/d mice treated with vehicle; panels c and f show WEd/d mice treated with LIF. The right horn (R) was stimulated, whereas the left horn (L) was left unstimulated. G, Ratio of wet weights of right and left horns of Esr1fl/fl, WEd/d, and WEd/d plus LIF. **, P < .01 (n = 5–7 for each treatment group).
To further assess the role of LIF in mediating the downstream effects of epithelial ESR1 during decidualization, we investigated whether its supplementation rescues the decidualization defect exhibited by WEd/d uteri. Recombinant LIF was administered ip to WEd/d mice, which were then subjected to experimentally induced decidualization. Uteri were dissected 72 hours later, and the decidual response was measured. Remarkably we observed that the supplementation of LIF restored the decidual response in the WEd/d uteri as measured by morphological examination (Figure 4F, compare panels a–c), ALP activity (Figure 4F, compare panels d–f), and wet weight gain (Figure 4G). These results strongly supported our hypothesis that LIF, acting in a paracrine manner, mediates the function of epithelial ESR1 in the regulation of stromal decidualization.
LIF controls IHH expression in luminal epithelium
We next investigated the mechanism by which LIF controls decidualization. The uterine luminal epithelium expresses LIF receptor and is a known target of glandular LIF (29, 30). We therefore considered the possibility that LIF acts on the luminal epithelium to induce the expression of a factor that in turn acts on stromal cells to promote decidualization. It was previously reported that IHH, produced downstream of P action in the luminal epithelium, is a key factor for implantation (31, 32). It was also reported that IHH production is impaired in LIF-null uteri (33). IHH secreted from the luminal epithelium binds to it receptor patched homolog 1 (PTCH1) in the stroma to induce the expression of the transcription factor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII), also known as NR2F2, which promotes stromal decidualization (32, 34). We therefore examined whether treatment of WEd/d mice with LIF, which rescues the decidualization defect in these mice, also triggers the expression of genes involved in the IHH signaling cascade. As shown in Figure 5A, we observed a significant down-regulation of Ihh, Ptch1, and COUP-TFII transcripts in WEd/d uteri compared with Esr1fl/fl uteri, indicating that decidual impairment is associated with reduced IHH signaling. Importantly, administration of LIF to WEd/d uteri, which rescued decidualization defect, was also able to markedly induce the expression of Ihh, Ptch1, and COUP-TFII (Figure 5B). Collectively these results are consistent with the hypothesis that the glandular LIF regulates uterine stromal differentiation by inducing a paracrine mechanism involving the production of IHH in the luminal epithelium and activation of its downstream targets in the stroma.
Figure 5.
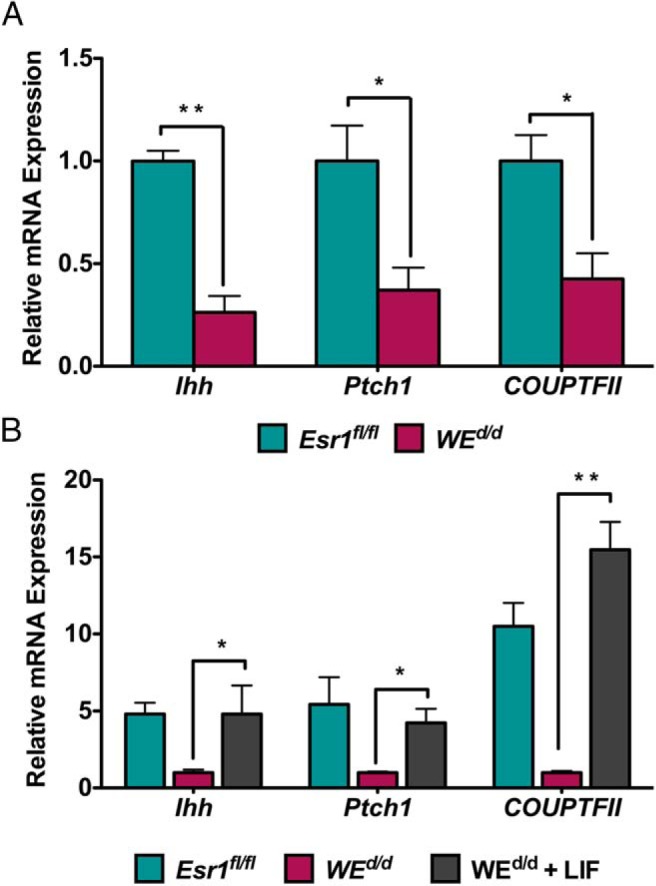
LIF controls the IHH pathway during stromal decidualization. A, Ihh signaling is down-regulated in the uteri of WEd/d mice. Real-time RT-PCR was performed to monitor the expression of Ihh, Ptch1, and COUP-TFII in the uteri of Esr1fl/fl and WEd/d mice at 20 hours after decidual stimulation. The levels of Rplp0 and Ck18 were used as an internal control to normalize gene expression. B, Ihh signaling is restored in WEd/d mice during the rescue of decidualization by LIF. Real-time PCR was performed to monitor the expression of Ihh, Ptch1, and COUP-TFII in the uteri of Esr1fl/fl, WEd/d, and WEd/d plus LIF at 20 hours after decidual stimulation. The levels of Rplp0 and Ck18 were used as an internal control to normalize gene expression. The data are represented as the mean fold induction ± SEM. *, P ≤ .05; **, P ≤ .01.
LIF induces IHH via a STAT3-independent pathway
Previous studies have shown that glandular LIF acts on the LIF receptors present in the luminal epithelium to activate the STAT3, a transcription factor that regulates the expression of genes involved in epithelial junctional complex remodeling (21, 29). LIF is also reported to act via a STAT3-independent mechanism involving the ERK pathway to exert its effects on certain target cells (35–37). To examine whether the Ihh induction by LIF in steroid-primed uterus occurs in a STAT3-dependent fashion, we used SWd/d mice in which the Stat3 gene has been conditionally deleted from the luminal and glandular epithelial cells of the uterus, thus eliminating LIF signaling via the Janus kinase (JAK)/STAT3 pathway (21). When we administered P and LIF to ovariectomized mice and monitored Ihh expression, we observed that Ihh was modestly induced in uterine epithelial cells within 6 hours of P injection (Figure 6A). We also observed a similar modest induction of Ihh upon LIF administration. However, upon coadministration of P and LIF, there was a dramatic induction of Ihh, indicating a synergistic effect of P and LIF on Ihh expression (Figure 6A). Surprisingly, our results showed that treatment with LIF and P could still support the synergistic induction of Ihh in the uterine luminal epithelium of SWd/d mice (Figure 6B), indicating that LIF-mediated induction of this morphogen occurs via a STAT3-independent mechanism.
Figure 6.
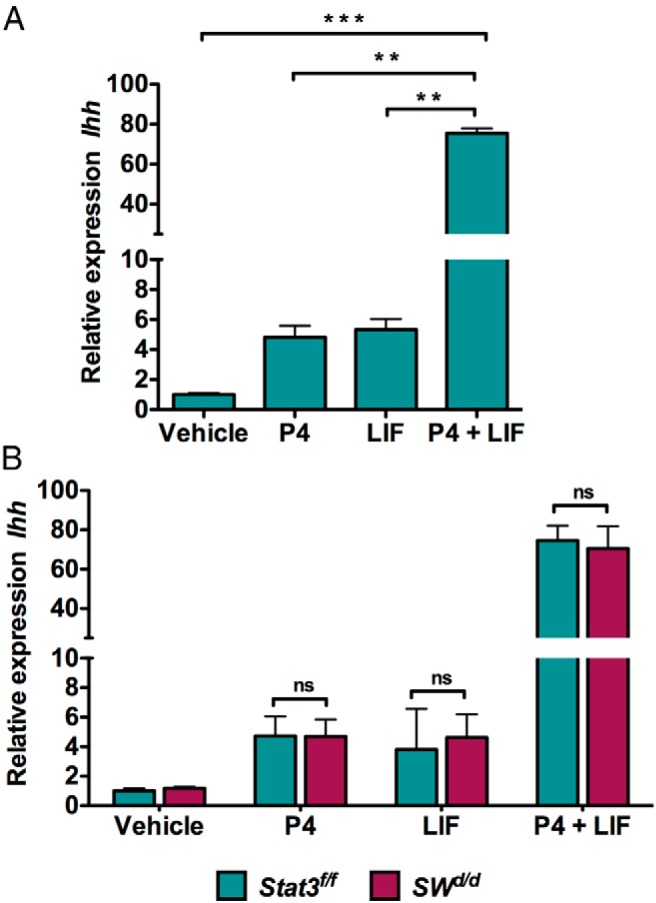
LIF and P act synergistically to induce Ihh expression in uterine epithelium. A, Expression level of Ihh mRNA in the LE of wild-type overectomized mice injected with vehicle (oil), P (1 mg), LIF (1 μg, three injections at 2 h intervals), or P along with LIF. LE was isolated at 6 hours after injection. The data are represented as the mean fold induction ± SEM. **, P ≤ .01; ***, P ≤ .001. ns, not significant. B, Expression level of Ihh mRNA in the LE of Stat3fl/fl and SWd/d ovariectomized mice injected with oil, P (1 mg), LIF (1 μg, three injections at 2 h intervals), or P along with LIF. LE was isolated at 6 hours after injection. The data are represented as the mean fold induction ± SEM. ns, not significant; P4, progesterone.
LIF induces IHH via an ERK1/2-dependent pathway
We next examined whether the LIF-mediated induction of Ihh involves the ERK pathway. For this purpose, we compared the expression levels of phosphorylated-ERK1/2 (p-ERK1/2) in the uteri of animals treated with LIF alone, P alone, and LIF plus P. An increase in the level of pERK1/2 was observed in uterine epithelium upon treatment with LIF alone (Figure 7A, compare panels a and b). Treatment with P alone also activated ERK1/2 signaling pathway in the uterine epithelium but to a relatively lesser extent than with LIF alone (Figure 7A, compare panels a–c). The levels of pERK1/2 were maximally induced in response to combined actions of P and LIF (Figure 7Ad). Similarly, phospho-small heterodimer partner 2 (SHP2), an adaptor molecule in the ERK1/2 signaling pathway that acts as tyrosine phosphatase (38), was also activated in the same manner as pERK1/2 (Figure 7B, panels a–d). Taken together, these results indicated that LIF induces Ihh in P-primed uterine luminal epithelium via activation of ERK1/2 pathway. Consistent with these results, we observed that the intraluminal administration of PD184352, an inhibitor of MEK and ERK1/2 phosphorylation (39), to uteri led to a marked decrease in pERK1/2 levels (Figure 7C, compare panels a and b). Administration of the inhibitor of ERK1/2 phosphorylation also resulted in a concomitant suppression of Ihh expression, whereas the levels of Pgr transcripts remained unaltered in response to this treatment (Figure 7D).
Figure 7.

LIF regulates Ihh expression via ERK1/2 activation. A, Immunohistochemical localization of pERK1/2 in wild-type overectomized mice uteri injected with oil (a), LIF (1 μg, three injections at 2 hour intervals) (b), P (1 mg) (c), or P along with LIF for 6 hours (d). B, Immunohistochemical localization of pSHP2 of wild-type overectomized mice uteri injected with oil (a), LIF (1 μg, three injections at 2 h intervals) (b), P (1 mg) (c),or P along with LIF for 6 hours (d). C, Down-regulation of ERK1/2 in the uterus inhibits Ihh induction by LIF. Immunohistochemical localization of pERK1/2 in wild-type overectomized mice uteri pretreated with DMSO and P along with LIF in saline (a) or PD184352 and P along with LIF (b) for 6 hours. Note the reduced expression of pERK1/2 in the luminal epithelium of uteri treated with PD184352. D, Real-time PCR was performed to monitor the expression of Ihh in the uterus of wild-type mice treated with DMSO and (P along with LIF in saline) or PD184352 and (P along with LIF in saline). Ck18 was used as internal control to normalize gene expression. The data are represented as the mean fold induction ± SEM. **, P ≤ .01. ns, not significant. E, Uterine sections from day 1 to day 4 (panels a–d) of pregnancy were subjected to immunohistochemical analysis using an anti-pERK1/2 antibody. F, Real-time PCR was performed to monitor the expression of Ihh and Pgr in the uterine horns of wild-type day 3 pregnant mice treated with either DMSO or PD184352. Ck18 was used as internal control to normalize gene expression. The data are represented as the mean fold induction ± SEM. *, P ≤ .05. ns, not significant.
To further investigate the regulation of Ihh expression by ERK1/2 in pregnant uterus, we determined the expression profiles of pERK1/2 during days 1–4 of pregnancy. We found that the levels of pERK1/2 reached a maximal level in the uterine luminal epithelium on days 2–3 of pregnancy (Figure 7E), which overlaps with the previously reported initial phase of LIF expression in the uterus during days 1–3 of pregnancy (27, 40). Interestingly, by day 4 of pregnancy, when a transient surge of LIF acts via JAK-STAT3 pathway, pERK1/2 expression in the luminal epithelium declined to undetectable levels (Figure 7E, panels a–d). The expression pattern of pERK1/2 during days 1–3 of gestation also agrees well with the previously reported spatiotemporal expression of Ihh transcripts, which are elevated on days 2–3 of pregnancy but decline sharply on day 4 (31). We next tested whether the blockade of ERK1/2 phosphorylation prevents the induction of Ihh on days 2–3 of pregnancy. Indeed, upon administration of PD184352 to uterine horns on day 2 of pregnancy, we observed that Ihh levels are strongly suppressed on day 3 of gestation in comparison with untreated control uterine horns, whereas the Pgr levels remain unaffected by this treatment (Figure 7F). Collectively these data are consistent with our hypothesis that pERK1/2, acting downstream of the first phase of LIF production in the uterus during days 1–3 of pregnancy, regulates IHH expression in the luminal epithelium during days 2–3, which in turn controls stromal decidualization via PTCH1 and COUP-TFII.
Discussion
Recent studies have revealed that a paracrine dialog between uterine epithelial and stromal cells, guided by the steroid hormone receptors present in these cells, is critical for acquisition of epithelial receptivity as well as functional competence of the stroma during pregnancy establishment (7). A dynamic pattern of expression of the ESR1 protein is seen in both epithelial and stromal compartments of the uterus during the reproductive cycle and pregnancy (41). Earlier studies, using a knockout mouse model that retained a truncated ESR1 in the uterus, concluded that the decidualization process is independent of ESR1 (10–12). In this paper, we showed that if Esr1 is conditionally deleted from both epithelial and stromal compartments of the uterus, using Pgr-Cre, the resulting Esr1d/d mice display a complete loss of decidual response (Figure 1). To our knowledge, this is the first demonstration that uterine ESR1 plays an essential role in the regulation of decidualization. The Esr1d/d model, however, could not inform on the cell type-specific role of ESR1 in the uterus because the receptor was ablated from both epithelial and stromal cells. A clear understanding of the functions of ESR1 in each cell type during the reproductive cycle and pregnancy can be achieved only by developing mouse models harboring the uterine cell type-specific deletion of Esr1.
To achieve this goal, we used Wnt7a-Cre mice to create WEd/d mice harboring a conditional deletion of Esr1 in uterine luminal and glandular epithelium. These mice, which were previously reported by the group of Korach and colleagues (22), provided novel information regarding the functional role of epithelial ESR1. Until recently, it was believed that the epithelial ESR1 mediates E-induced growth of the luminal epithelium during the reproductive cycle. This long-held concept proved to be incorrect. Conditional knockout of Esr1 in uterine epithelium demonstrated unequivocally that the E-induced mitogenic activity in uterine epithelium is not controlled by the ESR1 (22). This surprising finding raised questions regarding the true biological role of epithelial ESR1. In this study, we address this issue by analyzing the reproductive phenotype of WEd/d mice. Consistent with the findings of the group of Korach and colleagues, we observed that the E-induced epithelial cell proliferation remained unaltered in WEd/d mice (data not shown). Most importantly, our study revealed that these mice display an impaired decidualization response, thereby supporting a critical role of epithelial ESR1 in the regulation of stromal differentiation.
Impaired decidualization in WEd/d mice pointed to a paracrine regulation of stromal differentiation directed by epithelial ESR1. When we investigated the expression pattern of ESR1 in the uteri of Esr1f/f mice during early stages of decidualization, we could barely detect its expression in the luminal epithelium but observed strong expression in the glands. In WEd/d uteri, the glandular expression of ESR1 was absent (Figure 4A), raising the possibility that the decidualization defect in these mice could be due to the lack of secretion of an ESR1-regulated paracrine factor from the glands. An obvious candidate for such a role is LIF, which was previously shown to be an E-regulated factor produced in the uterine glands (28, 30). Indeed, our studies demonstrated that treatment of WEd/d mice with LIF rescues the decidualization defect, establishing LIF as the downstream paracrine mediator of epithelial ESR1 that critically controls stromal differentiation.
Although previous studies have shown that LIF plays a critical role in implantation and decidualization (27–30), the molecular mechanism by which LIF controls decidualization of stromal cells is poorly understood. We report here that coadministration of LIF and P leads to a synergistic stimulation of IHH expression in the luminal epithelium during early pregnancy. This result is consistent with a previous report that IHH expression in luminal epithelium is greatly reduced in pseudopregnant uteri of LIF-null mice (33). The group of Demayo and colleagues (31) had previously shown that IHH produced by the luminal epithelium acts on its receptor PTCH1 on the stromal cells to induce COUP-TFII, an essential factor for decidualization. Taken together, these findings provide a plausible mechanistic pathway linking glandular production of LIF to its paracrine action in the luminal epithelium to induce IHH, which then acts on the stroma to promote decidualization. A recent report indicated that LIF can also act directly on human endometrial stromal cells to enhance the expression of the decidualization marker prolactin (42). When we treated primary cultures of mouse endometrial stromal cells isolated from pregnant uteri on day 4 of gestation with LIF in the presence of P, LIF failed to exhibit any additive effect on the expression of well-known decidualization markers, such as ALP, prolactin-related gene Prl8a2, and COUP-TFII (data not shown). These results are not in agreement with the concept that LIF acts directly on the stromal cells to induce decidualization. However, we cannot rule out the possibility that under in vivo conditions LIF may have additional effects on decidualization via LIF receptors reported to be present in the subluminal stromal cells (43).
Previous studies showed that LIF exhibits a biphasic pattern of expression in the preimplantation uterus (27, 39, 40). During the first phase, glandular LIF production is high at proestrus/estrus near the time of ovulation in response to the preovulatory surge of E and continues on days 1 and 2 of pregnancy. The LIF level then declines on day 3. The second phase involves its rise again on day 4 concomitant with the transient surge of nidatory E (27, 40). During the entire preimplantation phase spanning days 1–4 of pregnancy, the LIF receptor is constitutively expressed in uterine luminal epithelium, consistent with the view that this tissue is the primary target of LIF action during this preparatory period (43). Comparison of the uterine expression profiles of LIF and LIF receptor with that of IHH during days 1–3 of pregnancy indicated that the first phase of LIF expression and signaling temporally overlaps with the induction of IHH, which peaks during days 2–3 of pregnancy (31). Interestingly, the expression of IHH drops to very low levels on day 4 (31) when the second surge of glandular LIF expression occurs. Based on these results, we postulate that it is the first phase of LIF expression that is responsible for IHH expression in the preimplantation uterus (Figure 8). IHH then acts on the stromal cells via the PTCH1 receptor to set in motion a cascade of pathways that prepare the uterus to fully respond to the decidual stimulation provided by the attachment of the embryo to the receptive uterus at midnight on day 4. We propose that the second peak of LIF expression, which occurs immediately prior to implantation, plays an important role in inducing signaling pathways that modulate uterine luminal epithelial junctional complexes, thereby facilitating embryo attachment (21, 43).
Figure 8.
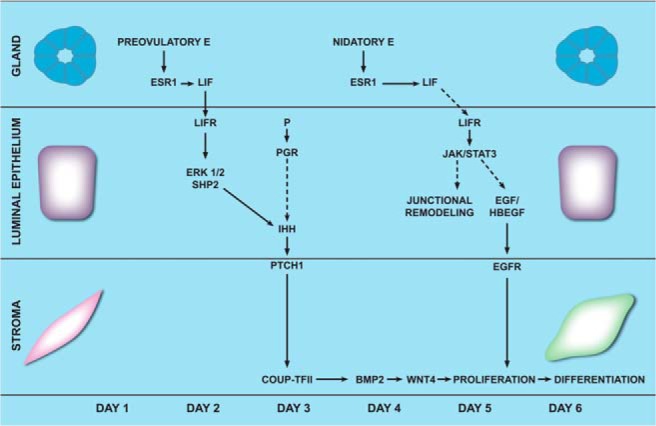
Schematic model of the paracrine networks acting downstream of epithelial ESR1 in the mouse uterus during implantation. In the preimplantation uterus, E contributed by the preovulatory surge at the estrous stage acts through its receptor, ESR1, to induce LIF production in the glands during days 1–3 of gestation. LIF, secreted from the glands, acts via the LIF receptors on the luminal epithelium to activate ERK signaling pathway in a synergistic fashion by acting in concert with P, which acts through its receptor PGR. The JAK-STAT3 pathway is not activated by LIF in the luminal epithelial cells at this time. Activated ERK1/2 then induces the expression of Ihh in the luminal epithelium. IHH secreted from the luminal epithelium activates the IHH-PTCH1-COUPTFII signaling cascade in the uterine stromal cells. This chain of signaling events promotes stromal proliferation and differentiation, which are key events during implantation. BMP2, bone morphogenetic protein 2; LIFR, LIF receptor. EGF, Epidermal growth factor; HBEGF, Heparin-binding EGF-like growth factor.
Signaling by LIF is initiated when it binds to the LIF receptors on the target cell (15, 29, 30). The LIF receptor is known to signal through distinct downstream pathways: JAK-STAT3 or Ras/ERK or phosphatidylinositol 3-kinase/AKT (29, 35, 37). Previous studies have shown that a transient surge of LIF on day 4 of gestation induces embryo attachment by activating the JAK-STAT3 pathway (21, 29). Surprisingly, the induction of Ihh in response to LIF signaling remained unaffected in uteri lacking epithelial STAT3, suggesting that a pathway other than the JAK-STAT3 mediates LIF-induced expression of IHH. We noted that pERK1/2, the active form of ERK1/2, as well as pSHP2, an adaptor molecule in the ERK1/2 pathway, are present in the luminal epithelium on days 2–3 of gestation, and they exhibit a similar temporal expression pattern as that reported for Ihh (Figure 7E). Furthermore, intraluminal administration of the MEK inhibitor PD184352, which prevents activation of ERK1/2, resulted in a marked suppression of Ihh expression, indicating that activation of ERK1/2 is critical for IHH induction in response to LIF signaling (Figure 7F). Collectively these findings are consistent with the concept that the first phase of LIF expression, which spans the preovulatory period and days 1–2 of gestation, activates the ERK1/2 pathway in the luminal epithelium to induce IHH expression in the preimplantation uterus, which then acts on the stromal cells to promote decidualization. Additionally, the second surge of LIF on day 4 activates JAK-STAT3 pathway in the luminal epithelial cells and regulates a distinct set of genes that promote epithelial remodeling, uterine receptivity, and embryo attachment (15, 21).
In summary, this paper reports that uterine ESR1 plays an essential role in decidualization. It also describes a paracrine mechanism by which uterine epithelial ESR1 controls stromal differentiation (Figure 8). An initial step in this mechanism is the induction of LIF in the endometrial glands in response to the preovulatory surge of E. Our study clarifies that LIF acts in a STAT3-independent but ERK-dependent manner to induce IHH expression in the uterus during early pregnancy. The knowledge gained from this study further enriches our understanding of the molecular pathways that interconnect important paracrine factors, such as LIF and IHH, which operate downstream of steroid hormone signaling to critically regulate implantation and decidualization during early pregnancy. Although the present study did not address the role of stromal ESR1 in decidualization, it is likely that it also plays a role during this process. Accumulating evidence suggests that stromal ESR1 regulates the production of growth factors that regulate epithelial proliferation in response to the preovulatory surge of E (7, 19). Future studies in our laboratory, using an in vitro system in which the primary cultures of endometrial stromal cells undergo steroid hormone-induced differentiation, will examine whether stromal ESR1 functions in a cell-autonomous manner to control decidualization.
Acknowledgments
We thank Jason Neff for the artwork, Karen Doty for the histology, and Elizabeth Hunt for the help with phenotypic analyses.
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health through Cooperative Agreement U54 HD055787 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (to I.C.B. and M.K.B.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ALP
- alkaline phosphatase
- Cdk
- cyclin-dependent kinase
- COUP-TFII
- chicken bumin upstream promoter-transcription factor II
- Ct
- cycle threshold
- DMSO
- dimethylsulfoxide
- E
- 17β-estradiol
- ESR1
- estrogen receptor-α
- HAND2
- Heart- and neural crest derivatives-expressed protein 2
- IHC
- immunohistochemistry
- IHH
- Indian hedgehog
- JAK
- Janus kinase
- LIF
- leukemia inhibitory factor
- MEK
- MAPK kinase
- p
- phosphorylated
- P
- progesterone
- PGR
- P receptor
- PTCH1
- receptor patched homolog 1
- SHP2
- Tyrosine phosphatase
- STAT3
- signal transducer and activator of transcription 3.
References
- 1. Finn CA, Martin L. The control of implantation. J Reprod Fertil. 1974;39(1):195–206. [DOI] [PubMed] [Google Scholar]
- 2. Dey SK, Lim H, Das SK, et al. Molecular cues to implantation. Endocr Rev. 2004;25(3):341–373. [DOI] [PubMed] [Google Scholar]
- 3. Bazer FW, Spencer TE, Johnson GA, Burghardt RC. Uterine receptivity to implantation of blastocysts in mammals. Front Biosci. 2011;3:745–767. [DOI] [PubMed] [Google Scholar]
- 4. Cha J, Sun X, Dey SK. Mechanisms of implantation: strategies for successful pregnancy. Nat Med. 2012;18(12):1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vasquez YM, DeMayo FJ. Role of nuclear receptors in blastocyst implantation. Semin Cell Dev Biol. 2013;24(10–12):724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramathal CY, Bagchi IC, Taylor RN, Bagchi MK. Endometrial decidualization: of mice and men. Semin Reprod Med. 2010;28(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pawar S, Hantak AM, Bagchi IC, Bagchi MK. Minireview: steroid-regulated paracrine mechanisms controlling implantation. Mol Endocrinol. 2014;28(9):1408–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lydon JP, DeMayo FJ, Funk CR, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. [DOI] [PubMed] [Google Scholar]
- 9. Franco HL, Jeong JW, Tsai SY, Lydon JP, DeMayo FJ. In vivo analysis of progesterone receptor action in the uterus during embryo implantation. Semin Cell Dev Biol. 2008;19(2):178–186. [DOI] [PubMed] [Google Scholar]
- 10. Curtis Hewitt S, Goulding EH, Eddy EM, Korach KS. Studies using the estrogen receptor alpha knockout uterus demonstrate that implantation but not decidualization-associated signaling is estrogen dependent. Biol Reprod. 2002;67(4):1268–1277. [DOI] [PubMed] [Google Scholar]
- 11. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA. 1993;90(23):11162–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paria BC, Huet-Hudson YM, Dey SK. Blastocyst's state of activity determines the “window” of implantation in the receptive mouse uterus. Proc Natl Acad Sci USA. 1993;90(21):10159–10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development. 2000;127(19):4277–4291. [DOI] [PubMed] [Google Scholar]
- 14. Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER α gene. FASEB J. 2010;24(12):4660–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosario GX, Hondo E, Jeong JW, et al. The LIF-mediated molecular signature regulating murine embryo implantation. Biol Reprod. 2014;91(3):66. [DOI] [PubMed] [Google Scholar]
- 16. Soyal SM, Mukherjee A, Lee KY, et al. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis. 2005;41(2):58–66. [DOI] [PubMed] [Google Scholar]
- 17. Laws MJ, Taylor RN, Sidell N, et al. Gap junction communication between uterine stromal cells plays a critical role in pregnancy-associated neovascularization and embryo survival. Development. 2008;135(15):2659–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finn CA, Hinchliffe JR. Reaction of the mouse uterus during implantation and deciduoma formation as demonstrated by changes in the distribution of alkaline phosphatase. J Reprod Fertil. 1964;8:331–338. [DOI] [PubMed] [Google Scholar]
- 19. Li Q, Kannan A, DeMayo FJ, et al. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science. 2011;331(6019):912–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Q, Kannan A, Wang W, et al. Bone morphogenetic protein 2 functions via a conserved signaling pathway involving Wnt4 to regulate uterine decidualization in the mouse and the human. J Biol Chem. 2007;282(43):31725–31732. [DOI] [PubMed] [Google Scholar]
- 21. Pawar S, Starosvetsky E, Orvis GD, Behringer RR, Bagchi IC, Bagchi MK. STAT3 regulates uterine epithelial remodeling and epithelial-stromal crosstalk during implantation. Mol Endocrinol. 2013;27(12):1996–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci USA. 2010;107(45):19272–19277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das RM, Martin L. Uterine DNA synthesis and cell proliferation during early decidualization induced by oil in mice. J Reprod Fertil. 1978;53(1):125–128. [DOI] [PubMed] [Google Scholar]
- 24. Mantena SR, Kannan A, Cheon YP, et al. C/EBPβ is a critical mediator of steroid hormone-regulated cell proliferation and differentiation in the uterine epithelium and stroma. Proc Natl Acad Sci USA. 2006;103(6):1870–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang W, Taylor RN, Bagchi IC, Bagchi MK. Regulation of human endometrial stromal proliferation and differentiation by C/EBPβ involves cyclin E-cdk2 and STAT3. Mol Endocrinol. 2012;26(12):2016–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brooks G. Cyclins, cyclin-dependent kinases, and cyclin-dependent kinase inhibitors: detection methods and activity measurements. Methods in Mol Biol. 2005;296:291–298. [DOI] [PubMed] [Google Scholar]
- 27. Stewart CL, Kaspar P, Brunet LJ, et al. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 1992;359(6390):76–79. [DOI] [PubMed] [Google Scholar]
- 28. Chen JR, Cheng JG, Shatzer T, Sewell L, Hernandez L, Stewart CL. Leukemia inhibitory factor can substitute for nidatory estrogen and is essential to inducing a receptive uterus for implantation but is not essential for subsequent embryogenesis. Endocrinology. 2000;141(12):4365–4372. [DOI] [PubMed] [Google Scholar]
- 29. Cheng JG, Chen JR, Hernandez L, Alvord WG, Stewart CL. Dual control of LIF expression and LIF receptor function regulate Stat3 activation at the onset of uterine receptivity and embryo implantation. Proc Natl Acad Sci USA. 2001;98(15):8680–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vogiagis D, Salamonsen LA. Review: the role of leukaemia inhibitory factor in the establishment of pregnancy. J Endocrinol. 1999;160(2):181–190. [DOI] [PubMed] [Google Scholar]
- 31. Takamoto N, Zhao B, Tsai SY, DeMayo FJ. Identification of Indian hedgehog as a progesterone-responsive gene in the murine uterus. Mol Endocrinol. 2002;16(10):2338–2348. [DOI] [PubMed] [Google Scholar]
- 32. Lee K, Jeong J, Kwak I, et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat Genet. 2006;38(10):1204–1209. [DOI] [PubMed] [Google Scholar]
- 33. Wakitani S, Hondo E, Phichitraslip T, Stewart CL, Kiso Y. Upregulation of Indian hedgehog gene in the uterine epithelium by leukemia inhibitory factor during mouse implantation. J Reprod Dev. 2008;54(2):113–116. [DOI] [PubMed] [Google Scholar]
- 34. Kurihara I, Lee DK, Petit FG, et al. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Gene. 2007;3(6):e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gough NM. Molecular genetics of leukemia inhibitory factor (LIF) and its receptor. Growth Factors. 1992;7(3):175–179. [DOI] [PubMed] [Google Scholar]
- 36. Auernhammer CJ, Melmed S. Leukemia-inhibitory factor-neuroimmune modulator of endocrine function. Endocr Rev. 2000;21(3):313–345. [DOI] [PubMed] [Google Scholar]
- 37. Krasilnikov M, Ivanov VN, Dong J, Ronai Z. ERK and PI3K negatively regulate STAT-transcriptional activities in human melanoma cells: implications towards sensitization to apoptosis. Oncogene. 2003;22(26):4092–4101. [DOI] [PubMed] [Google Scholar]
- 38. Montiel M, Quesada J, Jimenez E. Activation of calcium-dependent kinases and epidermal growth factor receptor regulate muscarinic acetylcholine receptor-mediated MAPK/ERK activation in thyroid epithelial cells. Cell Signal. 2007;19(10):2138–2146. [DOI] [PubMed] [Google Scholar]
- 39. Allen LF, Sebolt-Leopold J, Meyer MB. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin Oncol. 2003;30(5 suppl 16):105–116. [DOI] [PubMed] [Google Scholar]
- 40. Shen MM, Leder P. Leukemia inhibitory factor is expressed by the preimplantation uterus and selectively blocks primitive ectoderm formation in vitro. Proc Natl Acad Sci USA. 1992;89(17):8240–8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tan J, Paria BC, Dey SK, Das SK. Differential uterine expression of estrogen and progesterone receptors correlates with uterine preparation for implantation and decidualization in the mouse. Endocrinology. 1999;140(11):5310–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shuya LL, Menkhorst EM, Yap J, Li P, Lane N, Dimitriadis E. Leukemia inhibitory factor enhances endometrial stromal cell decidualization in humans and mice. PloS One. 2011;6(9):e25288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ni H, Ding NZ, Harper MJ, Yang ZM. Expression of leukemia inhibitory factor receptor and gp130 in mouse uterus during early pregnancy. Mol Reprod Dev. 2002;63(2):143–150. [DOI] [PubMed] [Google Scholar]