

Abstract
Cytochrome P450 21A2 is a key player in steroid 21-hydroxylation and converts progesterone to 11-deoxycorticosterone and 17α-hydroxy progesterone to 11-deoxycortisol. More than 100 mutations in P450 21A2 have been established in patients thus far; these account for the vast majority of occurrences of congenital adrenal hyperplasia (CAH), which is among the most common heritable metabolic diseases in humans. CAH phenotypes range from the most severe, salt-wasting (SW), to the simple virilizing (SV), and nonclassical (NC) CAH forms. We recently determined the crystal structure of human P450 21A2 in complex with progesterone. To gain more insight into the structural and stability changes underlying the phenotypes of individual mutations, we analyzed 24 SW, SV, and NC mutants in the context of the crystal structure of the human enzyme. Our analysis reveals clear differences in the localization of SW, SV, and NC mutations, with many of the first type mapping to the active site and near the heme and/or substrate and mostly resulting in complete loss of enzyme activity. Conversely, NC mutations are often found near the periphery and close to the surface of the protein, and mutant enzymes retain partial activity. The main conclusion from the mutation-structure-activity study is that the severity of the CAH clinical manifestations can be directly correlated with the degree of mutation-induced damage in terms of protein fold stability and active site changes in the structural model. Thus, the NC phenotype is typically associated with mutations that have a compensatory effect, ie, H-bonding replacing hydrophobic interactions and vice versa.
Cytochrome P450 (P450 or CYP) 21A2 is the major steroid 21-hydroxylase, is primarily expressed in the adrenal cortex, and catalyzes conversion of progesterone and 17α-hydroxy (OH) progesterone to 11-deoxycorticosterone and 11-deoxycortisol, respectively (1–3). Mutations in P450 21A2 are responsible for more than 95% of cases of congenital adrenal hyperplasia (CAH), an autosomal recessive disorder that is among the most common heritable metabolic diseases in humans (1, 2, 4), with an overall incidence worldwide of about 1 in 15 000 births (5, 6). More than 100 different amino acid variants have been catalogued in the clinic to date, and phenoytpes include classical and nonclassical (NC) CAH forms (1, 2, 7). The first form is characterized by a complete or almost complete loss of enzymatic activity, leading to salt-wasting (SW) and simple virilizing (SV) CAH, respectively. The SW type is the most severe form with patients exhibiting impaired cortisol synthesis and aldosterone deficits, whereby treatment during the neonatal period is required to prevent life-threatening salt loss and hypotonic shock. By contrast, SV CAH patients show no defects in aldosterone biosynthesis and are able to maintain sodium homeostasis. However, other symptoms, eg, ambiguous genitalia in female neonates as well as growth acceleration in childhood, are reminiscent of the SW phenotype. In the NC form, mutant P450 21A2 enzymes retain between about 20% and 60% of the 21-hydroxylation activity, resulting in a milder form of CAH that does not cause a cortisol deficiency but is associated with signs of postpubertal androgen excess (2, 7). Interestingly, in most cases the CAH genotype and phenotype appear to be correlated (8).
The region on chromosome 6 that the 21-hydroxylase gene (CYP21A2) maps to is characterized by mutations and variations in gene size and copy number. The bimodular haplotype in chromosome band 6p21.3 found in more than 70% of all Caucasians includes the nonfunctional or pseudogene of P450 21A2 (CYP21A1P). Crossovers, sequence exchanges, and apparent gene conversions between the functional gene and the corresponding pseudogene (that is 98% identical to the former) contribute to the frequent variations, with only about 5% of disease-causing CYP21A2 alleles harboring mutations whose origin cannot be traced to the pseudogene. The number of these more rare, spontaneous mutations continues to expand (eg, Ref. 9). An updated list of P450 21A2 mutations (irrespective of origin, along with the clinical phenotypes and, where known, in vitro activities of mutant enzymes in regard to the conversions of progesterone and 17α-OH progesterone) can be found at http://www.cypalleles.ki.se/.
Using a 3-dimensional (3D) model of the human P450 21A2 structure derived from our crystal structure of the bovine P450 21A2 (10), which exhibits 80% sequence identity with the human enzyme, New and coworkers carried out a comprehensive correlation of structure and clinical CAH phenotypes (7). They concluded from this analysis that there is good agreement between the extent and type of structural disruption caused by the CYP21A2 mutations and the clinical CAH phenotype and the severity of the disorder. Thus, mutations that alter heme binding or the substrate binding pocket or interfere with the anchoring of the enzyme within the membrane result in the SW phenotype and typically destroy enzyme activity. Similarly, the most severe CAH phenotype can also be caused by mutations that lead to significant destabilization of the P450 fold. The authors concluded that this includes approximately 35% of the amino acids surrounding the heme and ligand pocket (7). Conversely, mutations that go along with a near-loss of enzyme activity and are located near the transmembrane domain or in conserved hydrophobic patches are associated with the less severe SV disease type. Lastly, mutations that affect nonconserved hydrophobic patches, salt bridges, H-bonds, interactions with partner proteins or that can be compensated by other residues or accommodated sterically, track with the milder NC form of CAH. A potential shortcoming of any correlative analysis is that the activity of P450 21A2 mutants has only been assessed at the level of recombinantly produced protein in a few cases (11–16) or is unknown. Another point to be made is that correlation of clinical cases with biochemical and structural results is complicated by the fact that there are 2 gene copies; in almost all cases, the individuals are heterozygotes, and a detailed description of the properties of one mutant protein must be considered in the context of the other allele of the patient.
Recently, we expressed human P450 21A2 in Escherichia coli, purified and crystallized the enzyme, and determined its crystal structure in complex with progesterone at 2.64-Å resolution (Figure 1) (17). We also carried out a detailed kinetic analysis that provided evidence that human P450 21A2 is more catalytically efficient than the bovine enzyme and appears not to be limited by product dissociation but by the rate of C-H bond breaking (17). Comparison of the human and bovine crystal structures revealed significant conformational deviations between the two with a root mean square deviation for main chain atoms amounting to 4.7 Å. The availability of the structure of human P450 21A2 and the fact that its structure deviates considerably from the structure of the bovine enzyme (10) and the humanized model based on the latter (7) prompted us to revisit the correlation between structure, CAH phenotype, and P450 21A2 enzymatic activity.
Figure 1.
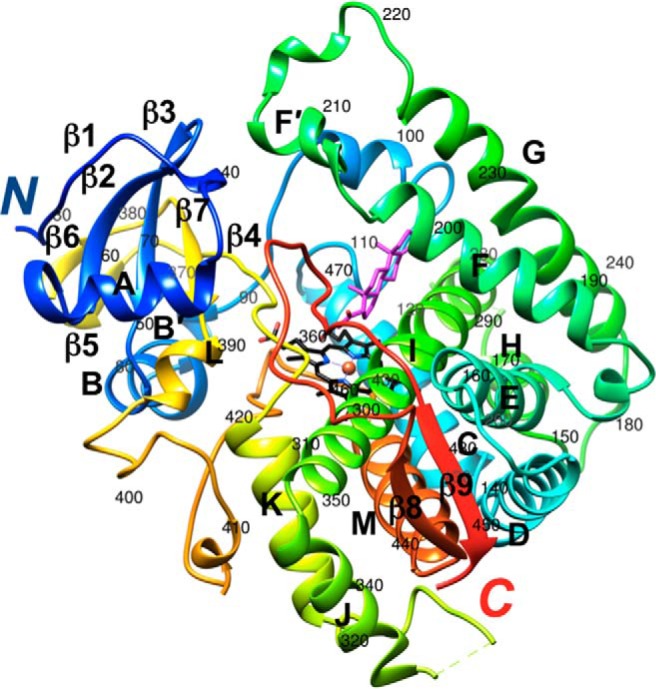
Crystal structure of human P450 21A2 in complex with progesterone (PDB ID code 4Y8W) (17). The protein main chain is shown in ribbon mode and rainbow coloring, from N terminus (blue) to C terminus (red). Carbon, nitrogen, and oxygen atoms of the heme moiety are colored in black, blue, and red, respectively, and Fe3+ is shown as an orange sphere. Carbon and oxygen atoms of progesterone are colored in magenta and red, respectively. Secondary structure elements are labeled and the residue ranges for α-helices A to M and β-strands 1 to 9 are as follows (every 10th residue in the chain is labeled): β1 32–36, A 45–56, β2 59–64, β3 67–72, B 75–84, B́ 86–89, C 115–131, D 136–153, E 161–178, F 188–202, F́ 205–212, G 224–246, H 257–264, I 279–310, J 312–322, K 344–356, β4 364–368, β5 372–374, β6 377–379, β7 384–387, L 389–394, M 432–447, β8 450–454, and β9 479–483.
Materials and Methods
Structural data
Coordinates and crystallographic structure factors for the complex between human P450 21A2 and progesterone are deposited in the Protein Data Bank (http://www.rcsb.org) with the ID code 4Y8W. The asymmetric unit of this crystal contains 3 complexes (A to C), and we used coordinates of complex A for the analysis of side chain interactions of mutant P450 proteins. The sequence of crystallized human P450 21A2 enzyme is that of the human 21-hydroxylase B gene with GenBank number M26856.1 (GI: 180963), based on the work by Rodrigues et al (18). Owing to the polymorphic nature of the 21A2 gene, the numbering of amino acids in the protein encoded in M26856.1 differs by +1 for most residues from that encoded in the human 21-hydroxylase gene with GenBank number M12792.1 (GI: 187895), based on the work by Higashi et al (19). The different numberings arise as a consequence of a single deletion in the N-terminal region of the hP450 21A2 (M12792.1 gene) with the sequence M1L2L3L4G5L6L7L8L9P10… relative to hP450 21A2 (M26856.1 gene), with the sequence M1L2L3L4G5L6L7L8L9L10P11… (bold face letters highlight the different number of leucine residues in the two proteins). Information on P450 21A2 mutants and their phenotypes and activities was taken from the Human Cytochrome P450 (CYP) Allele Nomenclature Database (http://www.cypalleles.ki.se/). Note that the amino acid numbering of P450 21A2 proteins in the database refers to products of the (mutated) genes with GenBank number M12792.1 (19). To avoid confusion, we have used a single numbering system for P450 21A2 amino acids in the present contribution. Thus, residue numbers in the text and illustrations refer to proteins encoded by the human 21-hydroxylase gene with GenBank number M26856.1 (wild-type [wt] protein) or the corresponding genes with mutations and are therefore consistent with the numbering used in the publication reporting the crystal structure of human P450 21A2 (17).
Side chains of amino acids for which mutation gives rise to the SW, SV, and NC CAH phenotypes are mapped separately in the crystal structure of human P450 21A2 (Figure 2). All structural illustrations were generated with the program UCSF Chimera (20). Activities of P450 21A2 mutant enzymes in the conversions of progesterone to 11-deoxycorticosterone and 17α-OH progesterone to 11-deoxycortisol are depicted in separate panels for the SW, SV, and NC forms in Figure 3. Information on all mutants, including the nucleotide change at the cDNA level, the amino acid change, and activities with the progesterone and 17α-OH progesterone substrates are also provided in tabulated form (Supplemental Tables 1–4 and Supplemental Materials and Methods). Regarding the activities, it is important to keep in mind that most of these were measured using cell-based assays, where mutant P450 21A2 enzymes are transiently expressed and radioactively labeled substrates are used to estimate conversion to the corresponding products (at a fixed time point) by liquid scintillation spectrometry after extraction and separation by thin layer chromatography, in the absence of kinetic considerations (see, eg, Ref. 9).
Figure 2.

Amino acids whose mutation gives rise to the (A) SW, (B) SV, and (C) NC CAH phenotypes mapped in the crystal structure of P450 21A2. Carbon atoms of wt residues are highlighted in blue (SW), green (SV), and purple (NC). The orientation of P450 in the top panels corresponds approximately to that in Figure 1.
Figure 3.
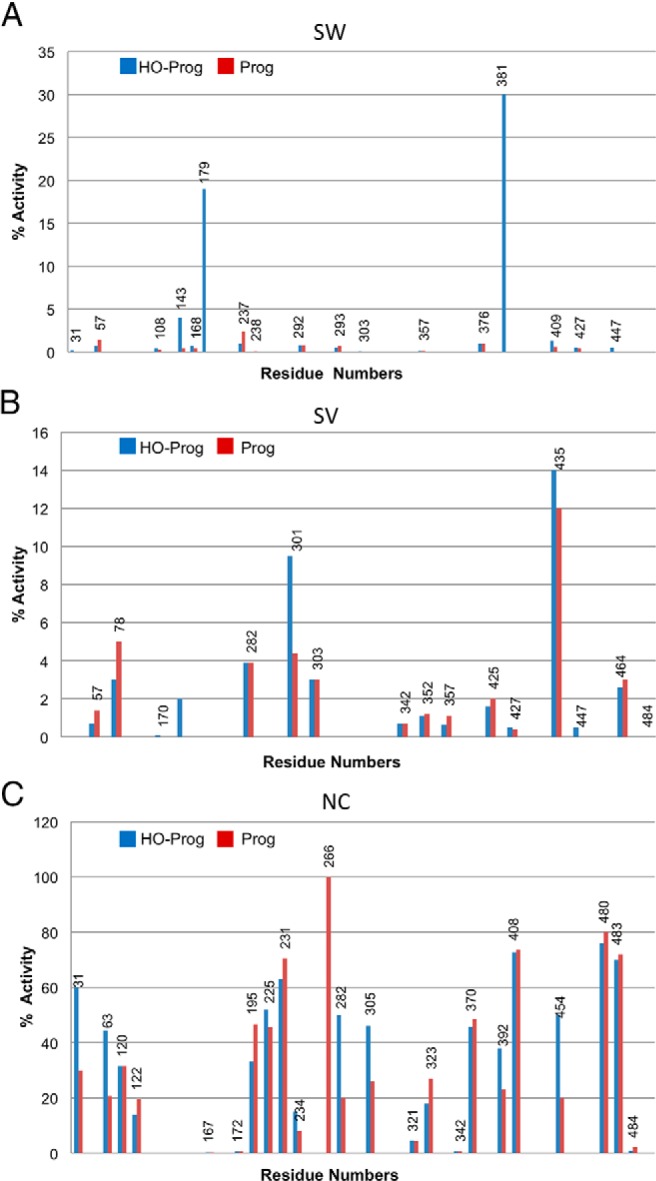
Activities of hP450 21A2 mutant enzymes giving rise to the (A) SW, (B) SV, and (C) NC phenotypes. See also Supplemental Tables 1–3.
Site-directed mutagenesis and protein purification
The human P450 21A2 mutants described for steady-state kinetic analysis were generated using the Quick Change mutagenesis method. The sequences of primers designed for point mutations are listed in Supplemental Table 5. The PET17b plasmid containing the gene encoding human P450 21A2 was used as a template to generate P450 21A2 mutants. cDNAs with the desired mutations were cotransformed with a chaperone pGro12 plasmid into E. coli BL21-Gold DE (3) competent cells. Recombinant mutants of P450 21A2 were overexpressed and then purified using the previously described purification procedures (17).
Catalytic activity assays
The catalytic activity measurements were carried out as described previously (17). The enzyme concentration and incubation time were varied for different P450 21A2 variants, depending on the catalytic activity of the mutants. Optimal conditions for steady-state kinetic assays were determined by time course experiments. The extracted steroid products were separated and analyzed using Ultra Performance Liquid Chromatography as described (17), and catalytic rate constant (kcat) and Michaelis constant (Km) values were estimated using hyperbolic plots in GraphPad Prism software (GraphPad).
Results and Discussion
Differences between the experimental and modeled structures of human P450 21A2
Before the crystal structure determination of the complexes between human P450 21A2 and progesterone substrate (17), Haider et al derived a model of the 3D structure of the human enzyme based on the crystal structure of bovine P450 21A2 (7). The sequence identity between the bovine and human enzymes is high (80%), motivating a threading approach combined with energy minimization to build the “humanized model.” However, comparison of the crystal structures of bovine (complex with 17α-OH progesterone) (10) and human P450 21A2 proteins (complex with progesterone) (17) revealed considerable deviations between the two that vary along the chain and are more pronounced in the loop regions. Because the humanized model was used to examine the consequences of mutations for structure and stability and a correlation of the severity of the changes caused by mutation, the experimental model of the 3D structure of human P450 21A2 offers a chance to reevaluate the potential effects of mutations on 21A2 fold and stability. Thus, the availability of the human P450 21A2 crystal structure provides a more reliable model for the structure-based interpretation of CAH phenotypes.
The coordinates of the humanized model (7) are not deposited and we therefore relied on published illustrations of interactions among side chains in the computed structure for comparison with our crystal structure of the P450 21A2 progesterone complex in order to demonstrate similarities and differences between the two for a subset of interactions. Thus, the configurations of some of these salt bridges in the crystal structure and in the humanized model are compared in Figure 4, A–D. With the exception of the interaction shown in Figure 4A (between D408 and R317; crystal structure numbering), the relative orientations of interacting partners in the crystal structure differ significantly from those in the computational model (7). The predicted direct interaction between D112 and R367 is instead water-mediated in the crystal structure, as the 2 side chains are not as tightly spaced as anticipated in the computational model (Figure 4B). The side chain of E141 was modeled in a conformation that would allow formation of an ideal salt bridge with R4445. However, the crystal structure reveals that the carboxylate moiety of E141 interacts with the guanidino group of R334 (Figure 4C). This arginine also contacts E137, whereas R445 is too far removed to interact with either glutamate. The crystal structure confirms that E352, R355, and R409 in the highly conserved EXXR motif are closely spaced and forming interactions, but the comparison with the computational model nevertheless exposes significant differences in the relative orientations of residues at that site (Figure 4D).
Figure 4.
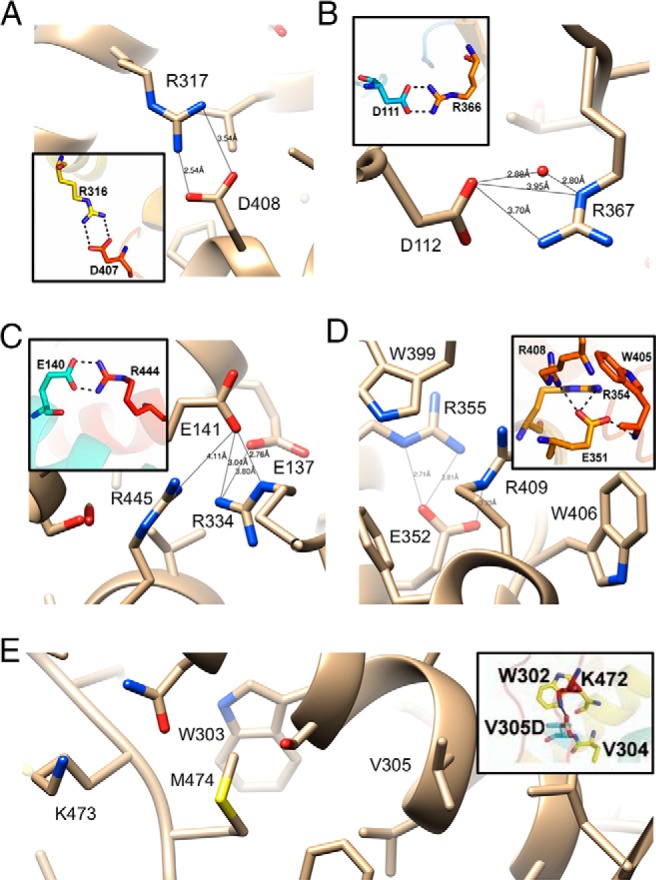
Comparisons between amino acid side chain interactions in the crystal structure of the human P450 21A2 progesterone complex (17) and the humanized model of the enzyme based on the crystal structure of bovine 21A2 (insets) (7). Notice the difference of 1 between residue numbers in the calculated and experimental models. Predicted and actual configurations of salt bridges involving (A) D408, (B) D112, (C) E141, and (D) E352. E, The V305D mutant was assumed to result in salt bridge formation with K473 and increased rigidity (7). However, in the crystal structure of hP450 21A2, K473 and V305 are spaced far apart with M474 inserted between them. Selected contacts in the experimental model are indicated with thin solid lines and distances in Å.
As far as the consequences of mutation on structure are concerned, V305D that results in the SV phenotype (7) offers a good example of the alternative interpretations that one can arrive at by either considering the computational model or the experimental structure of P450 21A2. Thus, the former placed the aspartate moiety in the mutant in H-bonding distance from K473 (Figure 4E). Such an interaction was expected to impart rigidity on the loop region preceding β9 by tying it together with helix I (Figure 1). This view is not supported by the crystal structure, in that K473 is situated at a distance of more than 10 Å from V305 and a direct interaction between aspartate (in the mutant) and lysine is blocked by M474 (Figure 4E).
Location, structural context, and altered activity of protein mutants
Visualizing the locations of SW, SV, and NC CAH-causing mutations in the 3D structure of P450 21A2 reveals clear differences in the distribution of residues (Figure 2). Thus, SW-causing mutations appear to be quite evenly spread throughout the entire protein, including the N- and C-terminal regions. In addition, a look at the side chains in the illustrations (ie, carbon only vs presence of oxygen or nitrogen) shows that many of the mutations involve hydrophobic amino acids (see also Supplemental Table 1). Mutations causing the SW type are also scattered around the active site and the heme binding pocket, whereas the SV and NC type mutations clearly concern areas that are farther removed from the heart of the P450 enzyme. Mutations causing the more severe SW CAH form are also more common relative to SV (Figure 2, A and B, and Supplemental Tables 1 and 2), whereby there appears to be an absence of the latter in many regions of the P450 fold, eg, in helices F, G, and I as well as in the N- and C-terminal β-sheets. NC-type mutations are more numerous relative to SV and are probably even more common than is suggested by the list shown in Supplemental Table 3, because such mutations may often remain undetected owing to a lack of clear symptoms and the need for clinical intervention. From the illustration in Figure 2C, it is also apparent that many NC mutations map to the protein surface, where they probably do not affect P450 fold or stability, or are seemingly of little consequence for interactions with auxiliary proteins, such as P450 reductase (21) or cytochrome b5 (22–24).
Turning to the consequences of CAH-causing mutations for P450 21A2 activity, an overview of the activities of SW, SV, and NC type mutations (Figure 3) shows a clear trend in that SW mutations that cause the most severe form of the disease generally abolish activity. This is also supported by the kinetic activity data gathered for a selection of P450 21A2 mutants and listed in Table 1. Conversely, SV mutations retain a low level of activity in many cases and NC mutations often only affect activity marginally. These observations, along with the notion that protein structure and activity are tightly linked, raise the possibility that inspection of 3D structural models of P450 21A2 and the visualization of potential changes at the tertiary structural level caused by mutations can lead to a better understanding of the relation between mutation/structure and severity of CAH and that a correlation of the two is meaningful, at least at a qualitative level (7).
Table 1.
Summary of Kinetic Data for P450 21A2 Mutant Proteins With the PROG or 17OHP Substrates
| P450 21A2 | Substrate | kcat (min−1) | Km (μM) | kcat/Km (μM−1 min−1) | Activity (%), wt |
|---|---|---|---|---|---|
| wt | PROG | 170 ± 4 | 0.21 ± 0.03 | 800 ± 120 | (100) |
| 17OHP | 240 ± 5 | 1.5 ± 0.1 | 160 ± 11 | (100) | |
| G65E (SW) | PROG | 3.7 ± 0.2 | 5.5 ± 0.8 | 0.67 ± 0.10 | 0.08 |
| 17OHP | 1.9 ± 0.2 | 13 ± 2 | 0.14 ± 0.03 | 0.08 | |
| T296N (SV/SW) | PROG | 0.46 ± 0.16 | 1.1 ± 0.2 | 0.42 ± 0.16 | 0.05 |
| 17OHP | 2.7 ± 0.3 | 0.46 ± 0.2 | 6.0 ± 2.7 | 3.7 |
PROG, progesterone; 17OHP, 17α-OH progesterone.
For example, a mutation that has dire consequences for P450 21A2 function and activity concerns the switch from proline to glutamine at position 31 (SW type) (Supplemental Table 1). The N-terminal region anchors microsomal P450s such as 21A2 to the cytoplasmic side of the membrane (25), and one can imagine that the P31Q mutation does not just affect conformation but also hydrophobicity and therefore association with the membrane. On the contrary, the P31L mutation exhibits the NC CAH clinical phenotype (Supplemental Table 3). Apparently leucine is better tolerated than glutamine and may actually aid in tying the protein to the membrane. The structural context of a mutation is crucial in terms of the functional consequences and that the SW type is not only caused by drastic amino acid changes, as demonstrated by the A363V mutation. This conservative change causes SW CAH, and the absorption spectrum of the protein is devoid of a ferrous-carbon monoxide complex spectral peak at 450 nm but instead shows a maximum at 420 nm (Lei, L., unpublished data). Inspection of the P450 21A2 crystal structure at that site and replacement of the methyl group with an isopropyl reveals that the larger side chain of valine results in a steric conflict with heme, perhaps causing the latter to shift slightly and resulting in the loss of activity (Supplemental Table 3). Another relatively conservative change concerns residue L301, which is located in helix I (Figure 1), and somewhat farther away from the heme moiety (∼8-Å distance to Cα) than A363. The L301F mutation causes SV CAH, with the activity of the mutant protein reduced to less than 10% of wt P450 21A2 (Supplemental Table 2). L301 juts into a hydrophobic pocket lined by F165, L168, T169, A298, V305, V442, L439, and L443, and accommodating phenylalanine at that position is expected to only require minor adjustments of surrounding residues. However, some of these are in direct contact with the heme, eg, L439, and this may explain the SV phenotype rather than the milder NC form. In the next sections, we discuss in more detail the anticipated structural consequences for 24 selected P450 21A2 mutations found in patients and correlate structural changes with CAH phenotype and altered enzyme activity.
Structural changes caused by mutations exhibiting the SW phenotype
P31Q
The first residue (from the N terminus) completely visible in the electron density of the P450 21A2 crystal structure is L30. The next residue, P31, is lodged in a hydrophobic cavity constituted by L30, I59, V70, I374, Y377, and I379 on the surface of the protein (Figure 5A). Thus, it changes the direction of the chain together with P32, serves as an anchor for the hydrophobic N-terminal tail that protrudes from the surface at this site, and helps attach the P450 to the membrane. In addition to the above hydrophobic contacts, a H-bond between the keto oxygen of P31 and Nϵ of R61 helps tie down the proline. Mutation to glutamine disrupts this hydrophobic network, induces steric conflicts with I59 and V70, and likely affects the flexibility of the tether between P450 and the membrane.
Figure 5.

Selected mutations causing the SW phenotype. (A) P31Q, (B) G65E, (C) L108R, and (D) V140E. Side chain carbon atoms of mutated amino acids are turquois and selected residues are labeled.
G65E
G65 is embedded in a hydrophobic patch of the N-terminal β-sheet and surrounded by β-strand residues L38, L40, L64, and L66, plus V212 and I213 from helix F' (Figure 5B). Mutation to E abolishes activity (Table 1), inserts a bulky, negatively charged moiety into this hydrophobic environment, results in a steric clash with V212, and alters the interaction between the β-sheet and the loop region between helices F and G on the surface of the protein (Figure 1). Preliminary steady-state kinetic analysis of this variant showed that the effects of the substitution are seen in both the kcat and Km parameters (Table 1), although the exact meaning of these is not yet clear.
L108R
L108 is located in a loop that precedes helix C (Figure 1) and is in direct contact with methyl and propionic acid substituents of heme. It also lies less than 10 Å away from the progesterone substrate and direct hydrophobic neighbors include A285 and L430. Basic amino acids that interact directly with the carboxylate moiety of the propionic acid substituent adjacent to L108 include R92, K121, and R427. Mutation of L108 to R introduces an additional charged residue to the existing trio of positively charged amino acids and would likely upset the balance of interactions that stabilize heme. In particular, the L108R mutation could lead to repulsions with the above lysine and arginine residues that will affect the orientation of heme and may in fact result in a direct clash with heme as well as neighboring amino acids (eg, T124) (Figure 5C).
V140E
V140 is located in helix D, which is part of an α-helical bundle with helices E, I, and M, whereby hydrophobic residues of helix D stabilize coil-coil interactions with helices E and M (Figure 1). V140 is surrounded by hydrophobic residues L143, I172, L434, L437, and V441 (Figure 5D). Replacing valine with glutamate disturbs the hydrophobic core of the α-helical bundle and possibly creates repulsions with E137 and E438, which reside in the core and jut away from the outside of the bundle, respectively. Because one of the edges of heme is wedged between helices I and M (Figure 1), tampering with the coil-coil interface will likely have dire consequences.
For additional examples of SW mutations (T296N, W303R, R355C, and F405S), see Supplemental Materials and Methods and Supplemental Figure 1.
Structural changes caused by mutations exhibiting the SV phenotype
I78T
I78 is situated in helix B and surrounded by hydrophobic residues L72, M82, P387, L389, L420, and C424. Mutation to the more polar threonine is expected to somewhat destabilize the interactions among hydrophobic residues (Figure 6A). However, residue I78 is preceded by T77 and followed by E79, and H393 is situated nearby. A water molecule could potentially enter the site and link the side chain of one of these residues to T78 in the mutant enzyme. In any case, I78 lies at some distance from both heme and substrate and the mutation is not expected to result in a dramatic change of protein stability, although the activity of the I78T mutant was reported to be reduced to less than 10% of that of the wt enzyme (Figure 3B).
Figure 6.

Selected mutations causing the SV phenotype. (A) I78T, (B) C148R, (C) C170R, and (D) V282G.
C148R
C148 is located near the C-terminal end of helix D (Figure 1), with the sulfhydryl group on the surface and at the floor of a shallow depression. The rim of this depression is lined by polar residues Q145, E149, R152, S333, R445, Q448, and R484 (Figure 6B). Introduction of another arginine at that site will likely result in Coulombic repulsions with R145 and R445 that can be relieved by slightly pushing apart helix D and the β8/β9-sheet at the very C-terminal end of the P450 enzyme (Figure 1).
C170R
C170 lies near the C-terminal end of helix E, at a site where it crosses helix F in a perpendicular fashion (Figure 1). Its neighbors on helix E are L167 and C174, and the hole on helix F that fits the C170 methylenesulfhydryl peg is made up of M188, Y191, Y192, and I195. Mutation to arginine is certain to lead to clashes, as the longer and charged side chain cannot be accommodated at the tight crossover of helices (Figure 6C). Thus, the arginine side chain cannot protrude from the space between helices E, F, and I, and relief may require pushing away the N-terminal half of helix F (Figure 1).
V282G
V282 is located near the N-terminal end of helix I, which is connected via a flexible loop region to a short α-helical segment, helix H (Figure 1, middle right, in the background). Residues A266 to N276 of this loop are not visible in the electron density. The very N-terminal section of helix I and all of helix H engage in a coil-coil interaction, whereby V282 forms a hydrophobic patch together with H-helix amino acids M258, M261, L262, and V265 (Figure 6D). Mutating V265 to glycine would weaken the interface at the periphery of the protein and loosen the anchoring of helix I to some extent. Some 16 Å away (G293), helix I passes by the heme, and, despite the relatively minor change from V to G, this mutation reduces the activity to just 4% of that of the wt enzyme (Figure 3B).
For additional examples of SV mutations (L309F, R342P, L364W, and G425S), see the Supplemental Materials and Methods and Supplemental Figure 2.
Structural changes caused by mutations exhibiting the NC phenotype
H120R
H120 is located in helix C and pairs with H281 from helix I near the surface of the protein (Figure 1). Mutation to arginine does not disrupt the interaction between the histidine side chains. Thus, Nϵ of the guanidino moiety can maintain the contact to H281, but the longer arginine side chain brings NH1 and NH2 into vicinity of the keto oygens of residues P106, D107, and L108 that are located in the loop region preceding helix C (the average N(H)… O distance is 3.44 Å) (Figure 7A). It appears that arginine can be accommodated at this site with relatively minor adjustments, consistent with the activity of the H120R mutant that amounts to approximately 32% of the activity of wt P450 21A2 enzyme.
Figure 7.

Selected mutations causing the NC phenotype. (A) H120R, (B) K121Q, (C) I172N, and (D) I195N.
K121Q
Carboxylate groups of heme propionic acid substituents are contacted by protonated side chains and, for one of them, the interacting residues include R92, K121, and R427, with formation of 4 H-bonds (Figure 7B). The vicinity of K121 to heme should render its mutation potentially detrimental to P450 21A2 activity. However, the K121Q mutant enzyme retains between 14% (17α-OH progesterone) and 20% (progesterone) of the activity of the wt enzyme (Figure 3C). Modeling reveals that although the mutation alters the electrostatic character of the residue at position 121, the glutamine side chain is able to maintain a H-bond to the heme carboxylate.
I172N
I172 sits in helix E and is surrounded by residues of mixed polarity that are contributed by helices D (V139, E140, and L143), E (L168), and M (L434 and E438). Although mutation significantly alters activity (reduction to <1%) (Figure 3C), most likely as a result of weakened hydrophobic interactions, the amino group of the side chain of N172 is in H-bonding distance from the carboxylate moiety of E438 (Figure 7C). H-bond formation could partly offset the change in stability caused by the loss of hydrophobic interactions.
I195N
I195 is located in helix F and 1 and 2 helical turns away from V198 and W202, respectively, that are directly contacting the substrate at the ceiling of the active site (Figure 1). Its environment is of mixed polarity as in the case of the above I172, and neighboring residues include L290, I291, and T294 from helix I as well as R170 from helix E. Upon mutation, the side chain amino group of N195 lies in H-bonding distance from Nϵ of R170 and Oγ of T294 (Figure 7D). A further H-bond can be established between the main chain amine of I291 and the side chain oxygen of N195. Taken together, this swapping of hydrophobic and electrostatic interactions appears to be of little consequence for enzyme activity (33% and 47% of wt for 17α-OH progesterone and progesterone, respectively) (Figure 3C).
For additional examples of NC mutations (R234G, S302Y, N388K, and P454S), see the Supplemental Materials and Methods and Supplemental Figure 3.
Conclusions
Visualization of P450 21A2 mutations with the SW, SV, and NC phenotypes of CAH mapped onto the crystal structure of the human enzyme reveals some clear trends as far as the severity of the disease and the locations of mutations underlying the clinical manifestations are concerned. The same applies to changes in P450 21A2 enzyme activity as a result of mutations. Those generating the SW form are scattered throughout the enzyme and are more common than those generating the SV form. The former are in many cases associated with the core of the enzyme that includes conserved hydrophobic regions as well as motifs stabilizing the fold via H-bonds or salt bridges. Although most SW mutations involve amino acids near heme and substrate, there are also some that are more removed from the active site. These elicit indirect changes that affect nearest neighbors lining the active site, or perhaps exerting their negative effects allosterically. SW mutations mostly result in a sharp downturn in activity and commonly destroy most of the activity.
By comparison, SV mutations appear to be more rare and asymmetrically distributed along the protein chain (eg, they are absent in helices D, F, and G). Compared with the SW type, they are more typically located nearer to peripheral regions or the surface, although they abrogate enzyme activity in many cases. The hypothesis that the severity of the CAH phenotype is correlated with the degree of structural perturbation caused by an amino acid change is borne out the NC mutations at the other end of the disease spectrum. Many of the amino acids affected are far removed from the active site and in quite a few cases are located near the surface or just below. However, there are a few examples of SV or NC mutations that are located close to the heme/substrate or directly interact with one of them. Thus, L364W (SV) is very likely to affect both heme and substrate directly, and K121Q (NC) and R234G/K (NC) concern amino acids in the active site that contact heme and substrate, respectively. The last 2 NC type mutations, one replacing arginine with lysine and the other with glycine, offer insight into the role of the H-bond established by R234 to the O3 keto oxygen at the ceiling of the active site. Although lysine is expected to be a good mimic of arginine, the mutation to glycine removes the H-bond to the substrate. One may conclude from this observation and the NC phenotype displayed by both mutations that this H-bond does little to orient the substrate or enhance enzyme activity.
The crystal structure of the human cytochrome P450 21A2 enzyme provides a useful model for interpreting the structure and stability changes triggered by CAH-causing mutations and supersedes earlier structures of the bovine enzyme and a humanized model based on it (7). As mentioned earlier, biochemical and structural data on a variant protein must be considered in the context of a second allele in correlating our predictions with clinical phenotypes. However, detailed analysis of mutant proteins is an established method of approaching the metabolic basis of inherited disease. With the experimental structure of the human enzyme now in hand and clear evidence, as demonstrated here, that this model allows a better understanding of the consequences of SW, SV, and NC CAH mutations for P450 21A2 folding and stability, it is now important to expand these insights using experimental data. Thus, crystal structures of selected P450 21A2 mutant enzymes, in combination with kinetic investigations, will discern the actual causes of the negative effects on structure and activity and to some degree dynamics. Such studies are currently underway in our laboratories.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01 GM103937 (to M.E. and F.P.G.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CAH
- congenital adrenal hyperplasia
- CYP
- cytochrome P450 enzyme
- 3D
- 3 dimensional
- NC
- nonclassical
- OH
- hydroxyl
- P450
- cytochrome P450 enzyme
- SV
- simple virilizing
- SW
- salt wasting
- wt
- wild type.
References
- 1. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–291. [DOI] [PubMed] [Google Scholar]
- 2. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, ed. Cytochrome P450: Structure, Mechanism, and Biochemistry. 4th ed New York, NY: Springer; 2015:523–785. [Google Scholar]
- 4. Higashi Y, Hiromasa T, Tanae A, et al. Effects of individual mutations in the P-450(C21) pseudogene on the P-450(C21) activity and their distribution in the patient genomes of congenital steroid 21-hydroxylase deficiency. J Biochem. 1991;109:638–644. [DOI] [PubMed] [Google Scholar]
- 5. New MI, Wilson RC. Steroid disorders in children: congenital adrenal hyperplasia and apparent mineralocorticoid excess. Proc Natl Acad Sci USA. 1999;96:12790–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Therrell BL, Jr, Berenbaum SA, Manter-Kapanke V, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–590. [DOI] [PubMed] [Google Scholar]
- 7. Haider S, Islam B, D'Atri V, et al. Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc Natl Acad Sci USA. 2013;110:2605–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilson RC, Nimkarn S, Dumic M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Gent Metab. 2007;90:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Robins T, Bellanne-Chantelot C, Barbaro M, Cabrol S, Wedell A, Lajic S. Characterization of novel missense mutations in CYP21 causing congenital adrenal hyperplasia. J Mol Med. 2007;85:247–255. [DOI] [PubMed] [Google Scholar]
- 10. Zhao B, Lei L, Kagawa N, et al. Three-dimensional structure of steroid 21-hydroxylase (cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J Biol Chem. 2012;287:10613–10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tusie-Luna MT, Traktman P, White PC. (1990) Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265:20916–20922. [PubMed] [Google Scholar]
- 12. Tusie-Luna MT, Speiser PW, Dumic M, New MI, White PC. A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol Endocrinol. 1991;5:685–692. [DOI] [PubMed] [Google Scholar]
- 13. Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172—Asn or Arg356—Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265:3549–3552. [PubMed] [Google Scholar]
- 14. Robins T, Barbaro M, Lajic S, Wedell A. Not all amino acid substitutions of the common cluster E6 mutation in CYP21 cause congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2005;90:2148–2153. [DOI] [PubMed] [Google Scholar]
- 15. Ohlsson G, Müller J, Skakkebaek NE, Schwartz M. Steroid 21-hydroxylase deficiency: mutational spectrum in Denmark, three novel mutations, and in vitro expression analysis. Hum Mutat. 1999;13:482–486. [DOI] [PubMed] [Google Scholar]
- 16. Lajic S, Clauin S, Robins T, et al. Novel mutations in CYP21 detected in individuals with hyperandrogenism. J Clin Endocrinol Metab. 2002;87:2824–2829. [DOI] [PubMed] [Google Scholar]
- 17. Pallan PS, Wang C, Lei L, et al. Human cytochrome P450 21A2, the major steroid 21-hydroxylase. Structure of the enzyme-progesterone substrate complex and rate-limiting C-H bond cleavage. J Biol Chem. 2015;290:13128–13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodrigues NR, Dunham I, Yu CY, Carroll MC, Porter RR, Campbell RD. Molecular characterization of the HLA-linked steroid 21-hydroxylase B gene from an individual with congenital adrenal hyperplasia. EMBO J. 1987;6:1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Natl Acad Sci USA. 1986;83:2841–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 21. Xia C, Panda SP, Marohnic CC, Martásek P, Masters BS, Kim JJ. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc Natl Acad Sci USA. 2011;108:13486–13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ahuja S, Jahr N, Im SC, et al. A model of the membrane-bound cytochrome b5-cytochrome P450 complex from NMR and mutagenesis data. J Biol Chem. 2013;288:22080–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Estrada DF, Laurence JS, Scott EE. Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J Biol Chem. 2013;288:17008–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Estrada DF, Skinner AL, Laurence JS, Scott EE. Human cytochrome P450 17A1 conformational selection. Modulation by ligand and cytochrome b5. J Biol Chem. 2014;289:14310–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson EF, Stout CD. Structural diversity of eukaryotic membrane cytochrome P450s. J Biol Chem. 2013;288:17082–17090. [DOI] [PMC free article] [PubMed] [Google Scholar]