

Abstract
We designed 39 new 2-phenylindole derivatives as potential anticancer agents bearing the 3,4,5-trimethox-yphenyl moiety with a sulfur, ketone, or methylene bridging group at position 3 of the indole and with halogen or methoxy substituent(s) at positions 4–7. Compounds 33 and 44 strongly inhibited the growth of the P-glycoprotein-overexpressing multi-drug-resistant cell lines NCI/ADR-RES and Messa/Dx5. At 10 nM, 33 and 44 stimulated the cytotoxic activity of NK cells. At 20–50 nM, 33 and 44 arrested >80% of HeLa cells in the G2/M phase of the cell cycle, with stable arrest of mitotic progression. Cell cycle arrest was followed by cell death. Indoles 33, 44, and 81 showed strong inhibition of the SAG-induced Hedgehog signaling activation in NIH3T3 Shh-Light II cells with IC50 values of 19, 72, and 38 nM, respectively. Compounds of this class potently inhibited tubulin polymerization and cancer cell growth, including stimulation of natural killer cell cytotoxic activity and repression of Hedgehog-dependent cancer.
INTRODUCTION
Microtubules (MTs) are cylindrical structures mainly composed of α,β-tubulin heterodimers. MT assembly is a highly dynamic process because of continuous transitions between polymerization and depolymerization. MTs are involved in a number of essential cellular functions, such as maintenance of cell shape, cell motility, intracellular transport, and cell division. Interfering with the MT dynamic equilibrium, by either inhibiting tubulin polymerization or blocking MT disassembly, prevents proper MT function and ultimately leads to cell death. Interfering with these cellular processes has resulted in a productive strategy for the development of efficient anticancer agents.1–4
Colchicine (1),5,6 combretastatin A-4 (CSA4, 2)7 (Chart 1), and the Catharanthus alkaloids vincristine (VCR) and vinblastine (VBL) inhibit MT assembly by preventing tubulin polymerization, and this leads to cell death. In contrast, taxoids and epothilones bind at a luminal site on the β-subunit8,9 following entry into the MT through pores in its wall10 that are shaped by various tubulin subunits on the MT surface. Some evidence indicates a transient binding of MT-stabilizing agents at a specific pore site. Paclitaxel (PTX) stimulates MT polymerization and stabilization at high concentrations, whereas lower concentrations of PTX inhibit MT dynamics with little effect on the proportion of tubulin in the polymer.11
Chart 1.

General Structures of Compounds 1–45
The development of MT-targeting drugs with different mechanisms of action12 has achieved substantial progress. However, drug resistance, toxicity, and unwanted side effects still remain unsolved problems. Therefore, the quest for new MT inhibitors as components of improved anticancer treatments remains mandatory.13
Arylthioindole (ATI) derivatives are potent inhibitors of tubulin polymerization that bind to the colchicine site on β-tubulin. A number of ATIs proved to be more potent than 1, 2, VBL, and PTX and thus are potential new anticancer agents.14,15
ATI derivatives bearing an aromatic ring at position 2 of the indole (A region) inhibit tubulin polymerization and cancer cell growth. These compounds hamper mitotic progression, thus causing cells to undergo apoptosis (i.e., ATI 3: tubulin assembly, IC50 = 3.3 μM; MCF-7, IC50 = 52 nM)14,15 (Chart 1). In contrast, chemical modification of positions 4–7 of the indole (D region) were not exhaustively explored, although a few ATI derivatives bearing an ethoxycarbonyl functionality at position 2 and a halogen atom or a methoxy group at position 5 proved to be potent tubulin assembly inhibitors (i.e., ATI 4: tubulin assembly, IC50 = 2.0 μM; MCF-7, IC50 = 13 nM).16 It should also be noted that major differences in structure–activity relationships (SARs) were found in comparing ATIs with the 3-aroylindoles reported by Hsieh and co-workers.17 These compounds are characterized by the presence of a methoxy group at position 6 of the indole.
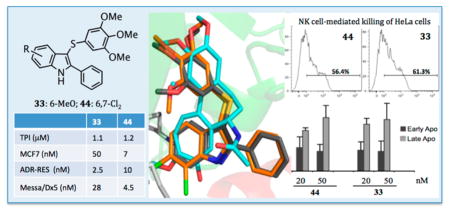
Docking studies of compounds 1 and 3 into the colchicine site of tubulin showed a good superimposition of the trimethoxyphenyl (TMP) moieties and of the tropolone ring C with the phenyl core of the indole14,15 (Figure 1). These findings suggested that appropriate substituents at positions 4–7 of the indole could resemble the methoxy and carbonyl groups at positions 10 and 9, respectively, of the tropolone ring. Docking simulations with PLANTS,18 using our methodology described previously,19 revealed binding poses consistent with the previous ATI series: (i) the TMP group formed polar interactions with βCys241, βMet259, and βLeu255; (ii) the indole NH established a H-bond with αThr179; (iii) the phenyl ring at position 2 of the indole set up hydrophobic interactions with the βLys254 and βLeu248 side chains. In comparison with 3, the chlorine atom(s) of 10, 18, 28, 37, and 44 fitted into a new hydrophobic pocket formed by the βLys353, βAsn258, βMet259, and αVal181 residue side chains (Figure 1S, Supporting Information). The 7-fluoro atom of 40, the most potent tubulin polymerization inhibitor within the series, behaved as a H-bond acceptor with αVal181 (Figure 2S, Supporting Information). The methoxy group of 33 mimicked the corresponding group at position 10 of 1, resembling its interaction with the εN of βLys353.
Figure 1.

Proposed binding modes of 1 (cyan), 3 (gray), and 44 (orange). Tubulin is represented as a cartoon for the α-subunit (red) and β-subunit (green). Residues forming interactions with the ATI D region are depicted in white.
These observations prompted us to undertake SAR investigations at positions 4–7 of the indole (Chart 1 and Table 1). We planned the synthesis of 2-phenyl-1H-indole derivatives, keeping the 2-phenyl group fixed at position 2, because of the better metabolic profile compared to that of indole derivatives bearing an ester function at this position.15
Table 1.
Inhibition of Tubulin Polymerization, Binding of Colchicine to Tubulin, and Growth of MCF-7 Human Breast Carcinoma Cells by Compounds 6–45 and References 1–5
| |||||
|---|---|---|---|---|---|
| compd | R | X | tubulin assembly IC50 ± SDa (μM) | MCF-7 IC50 ± SDb,c (nM) | colchicine binding inhibitionc,d (% ± SD) |
| 6 | 4-Br | S | 2.4 ± 0.1 | 400 ± 70 | 33 ± 2 |
| 7 | 4-Br | C=O | >20 (partial activity)e | 430 ± 100 | ndf |
| 8 | 4-Cl | S | 1.6 ± 0.07 | 290 ± 50 | 48 ± 5 |
| 9 | 4-Cl | C=O | 3.6 ± 0.2 | 260 ± 50 | 38 ± 3 |
| 10 | 4-Cl | CH2 | 17 ± 0.2 | >5000 | nd |
| 11 | 4-F | S | 1.7 ± 0.07 | 80 ± 30 | 80 ± 0.4 |
| 12 | 4-F | C=O | 1.6 ± 0.08 | 65 ± 7 | 63 ± 1 |
| 13 | 4-MeO | S | >20 (partial activity)e | 1400 ± 300 | nd |
| 14 | 4-MeO | C=O | >20 (no activity)g | >5000 | nd |
| 15 | 5-Br | S | 1.3 ± 0.1 | 320 ± 100 | 39 ± 3 |
| 16 | 5-Br | C=O | >40 | 530 ± 100 | nd |
| 17 | 5-Br | CH2 | 4.0 ± 0.5 | 700 ± 300 | 31 ± 2 |
| 18 | 5-Cl | S | 1.5 ± 0.2 | 280 ± 70 | 49 ± 5 |
| 19 | 5-Cl | C=O | >20 | 330 ± 100 | nd |
| 20 | 5-Cl | CH2 | 4.4 ± 0.8 | 310 ± 10 | 38 ± 3 |
| 21 | 5-F | S | 1.5 ± 0.1 | 300 ± 0 | 61 ± 4 |
| 22 | 5-F | C=O | 13 ± 0.4 | 240 ± 90 | nd |
| 23 | 5-OMe | S | 2.1 ± 0.01 | 200 ± 80 | 58 ± 0.5 |
| 24 | 5-OMe | C=O | 8.3 ± 1 | 170 ± 60 | nd |
| 25 | 5-OMe | CH2 | 3.5 ± 0.4 | 100 ± 0 | 41 ± 0.9 |
| 26 | 6-Br | S | 1.9 ± 0.2 | 9.0 ± 2 | 87 ± 0.6 |
| 27 | 6-Br | C=O | 1.3 ± 0.2 | 30 ± 2 | 86 ± 0.4 |
| 28 | 6-Cl | S | 1.2 ± 0.2 | 20 ± 10 | 88 ± 3 |
| 29 | 6-Cl | C=O | 1.4 ± 0.06 | 35 ± 7 | 71 ± 5 |
| 30 | 6-Cl | CH2 | 1.6 ± 0.01 | 55 ± 20 | 72 ± 6 |
| 31 | 6-F | S | 2.4 ± 0.2 | 80 ± 20 | 74 ± 2 |
| 32 | 6-F | C=O | 10 ± 0.4 | 600 ± 0 | nd |
| 33 | 6-MeO | S | 1.1 ± 0.1 | 1.3 ± 0.6 | 96 ± 1 |
| 34 | 6-MeO | C=O | 1.2 ± 0.06 | 30 ± 10 | 95 ± 0.4 |
| 35 | 7-Br | S | 1.6 ± 0.2 | 25 ± 5 | 82 ± 1 |
| 36 | 7-Br | C=O | 1.7 ± 0.1 | 4.0 ± 0.5 | 72 ± 0.3 |
| 37 | 7-Cl | S | 1.3 ± 0.2 | 50 ± 10 | 82 ± 0.3 |
| 38 | 7-Cl | C=O | 1.8 ± 0.05 | 9.0 ± 1 | 69 ± 0.5 |
| 39 | 7-Cl | CH2 | 2.4 ± 0.2 | 200 ± 0 | 50 ± 4 |
| 40 | 7-F | S | 1.0 ± 0.1 | 20 ± 9 | 92 ± 0.3 |
| 41 | 7-F | C=O | 1.7 ± 0.1 | 30 ± 0 | 67 ± 3 |
| 42 | 7-OMe | S | 1.2 ± 0.01 | 19 ± 10 | 87 ± 2 |
| 43 | 7-OMe | C=O | 1.8 ± 0.1 | 200 ± 0 | 62 ± 3 |
| 44 | 6,7-Cl2 | S | 1.2 ± 0.1 | 7.0 ± 3 | 92 ± 2 |
| 45 | 6,7-Cl2 | C=O | 1.5 ± 0.03 | 15 ± 5 | 93 ± 0.7 |
| 1 | 3.2 ± 0.4 | 5.0 ± 1 | nd | ||
| 2 | 1.0 ± 0.1 | 13 ± 3 | 98 ± 0.6 | ||
| 3h | 3.3 ± 0.1 | 52 ± 7 | nd | ||
| 4i | 2.0 ± 0.2 | 13 ± 3 | 93 ± 0.8 | ||
| 5i | 2.5 ± 0.3 | 42 ± 10 | 76 ± 0.2 | ||
Inhibition of tubulin polymerization. Tubulin was at 10 μM in the assembly assay.
Inhibition of growth of MCF-7 human breast carcinoma cells.
Compounds that inhibited tubulin assembly with IC50 ≤ 5 μM were tested in the cellular and colchicine binding assays.
Inhibition of [3H]colchicine binding. Tubulin was at 1 μM. Both [3H]colchicine and inhibitor were at 5 μM.
Partial inhibition at 20 μM.
No data.
Little or no activity at 20 μM.
Reference 15.
Reference 16.
CHEMISTRY
2-Phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indoles 6, 8, 11, 13, 15, 18, 21, 23, 25, 28, 31, 33, 35, 37, 40, 42, and 44 were prepared by reacting 2-phenylindoles 46–62 with bis(3,4,5-trimethoxyphenyl) disulfide16 in the presence of sodium hydride at 130 °C (120 W) for 2 min according to our previously reported venting-while-heating microwave-assisted procedure20 (Scheme 1). Microwave-assisted Friedel–Crafts reaction of the indoles 46–62 with 3,4,5-trimethoxybenzoyl chloride in the presence of AlCl3 in 1,2-dichloroethane at 110 °C (150 W) for 2 min furnished the corresponding methanones 7, 9, 12, 14, 16, 19, 22, 24, 27, 29, 32, 34, 36, 38, 41, 43, and 45.21 Methylene derivatives 10, 17, 20, 25, 30, and 39 were prepared by reducing the appropriate ketones with a borane–tetrahydrofuran complex in acetonitrile/methanol at 50 °C for 1 h22 (10, 20, 25, 30, and 39) or with triethylsilane/trifluoroacetic acid in 1,2-dichloroethane at 250 °C (250 W) for 20 min23 (17).
Scheme 1. Synthesis of Compounds 6–45a.

aReagents and reaction conditions: (a) bis(3,4,5-trimethoxyphenyl) disulfide, anhydrous DMF, closed vessel, 130 °C, 120 W, 2 min, venting-while-heating, 20–77%; (b) 3,4,5-trimethoxybenzoyl chloride, anhydrous AlCl3, 1,2-dichloroethane, closed vessel, 110 °C, 150 W, 2 min, 20–78%; (c) (10, 20, 25, 30, and 39) borane–tetrahydrofuran complex, acetonitrile/methanol, 50 °C, 1 h, 10–41%; (d) (17) triethylsilane, trifluoroacetic acid, 1,2-dichloroethane, 250 °C, 250 W, 20 min, 15%.
Reaction of the appropriate 1-methyl-2-nitrobenzene with benzaldehyde in the presence of sodium ethoxide in anhydrous dimethyl sulfoxide at 25 °C for 12 h furnished the alcohols 63–65. After oxidation of 63–65 to ketones 66–68 with pyridium chlorochromate in anhydrous dichloromethane at 25 °C for 1.5 h, the nitro group was reduced to amino with tin(II) chloride dihydrate/1 N HCl in acetic acid at reflux temperature for 12 h; the intermediate aminoketone underwent intra-molecular cyclization to give the corresponding 2-phenylindoles 46, 54, and 56 (Scheme 2a).24 2-Phenylindoles 47–49, 55, 57, and 61 were prepared by reaction of the N-(2-tolyl)benzamides 69–74 with tert-butyllithium in anhydrous tetrahydrofuran at –40 °C.25 Amides 69–74 were obtained by treatment of an appropriate o-toluidine with benzoyl chloride in the presence of triethylamine in anhydrous tetrahydrofuran at reflux temperature for 2 h (Scheme 2b). Polyphosphoric acid-mediated cyclization at 110 °C for 1 h of phenylhydrazones 75–80, prepared from the appropriate phenylhydrazine hydrochloride and acetophenone in the presence of sodium acetate in ethanol at 80 °C (250 W) for 5 min,26 gave the corresponding 2-phenylindoles 50–52, 58, 60, and 6227 (Scheme 2c).
Scheme 2. Synthesis of Intermediates 46–62a.

aReagents and reaction conditions: (a) benzaldehyde, sodium ethoxide, anhydrous DMSO, 25 °C, 12 h, 40–44%; (b) pyridinium chlorochromate, anhydrous dichloromethane, 25 °C, 1.5 h, 16–40%; (c) tin(II) chloride dihydrate/1 N HCl, acetic acid, reflux temperature, 12 h, 19–24%; (d) benzoyl chloride, triethylamine, anhydrous THF, reflux temperature, 2 h, 49–88%; (e) (i) tert-butyllithium, anhydrous THF, −40 °C, Ar stream; (ii) 1 h, 0 °C; (iii) 25 °C, 12 h, 18–52%; (f) acetophenone, sodium acetate, ethanol, open vessel, 80 °C, 250 W, 5 min, cooling-while-heating, 25–95%; (g) polyphosphoric acid, 110 °C, 1 h, 25–82%.
RESULTS AND DISCUSSION
Inhibition of Tubulin Polymerization, the Binding of Colchicine to Tubulin, and MCF-7 Breast Cancer Cell Growth
We synthesized compounds 6–45, including ATI derivatives and some corresponding ketone and methylene compounds, to obtain SAR information on the substituent(s) introduced at positions 4–7 of the indole nucleus. The activities of compounds 6–45 as inhibitors of tubulin polymerization in vitro, the growth of MCF-7 human breast cancer cells, and the binding of [3H]colchicine to tubulin are shown in Table 1. The majority of these new derivatives (23 compounds: 8, 11, 12, 15, 18, 21, 26–30, 33–38, and 40–45) inhibited tubulin polymerization with IC50 values in the 1.0–2.0 μM range, as compared with colchicine (1) (IC50 = 3.2 μM) and CSA4 (2) (IC50 = 1.0 μM).
Among the group of compounds 6–14, bearing the substituent at position 4 of the indole, the 4-chloro derivative 8 inhibited tubulin assembly with an IC50 of 1.6 μM and was 2.2-fold more active than the corresponding ketone 9 (IC50 = 3.6 μM). The thio/keto 4-fluoro derivatives 11 (IC50 = 1.7 μM) and 12 (IC50 = 1.6 μM) were almost equipotent as tubulin assembly inhibitors and showed the greatest inhibition of MCF-7 cell growth (11, IC50 = 80 nM; 12, IC50 = 65 nM). Neither the bromine atom nor the methoxy group was found among the most active compounds with position 4 substituents in both the thio and ketone series. In terms of effects on MCF-7 cell growth, there is no evident major difference between the thio and keto series with substituents at position 4, although the data are limited. Compounds 15–25 were characterized by substituents at position 5 of the indole nucleus. As tubulin polymerization inhibitors, the arylthioindoles 15 (IC50 = 1.3 μM), 18 (IC50 = 1.5 μM), 21 (IC50 = 1.5 μM), and 23 (IC50 = 2.1 μM) were all significantly more potent than the corresponding ketones 16, 19, 22, and 24 and slightly more active than the methylene compounds 17, 20, and 25. The tubulin inhibitory activity of these compounds seemed to be weakly affected by the nature of the substituent. In contrast to the corresponding ester derivatives, all these compounds were weak inhibitors of MCF-7 cell growth with IC50 ≥ 100 nM (compare 4 (IC50 = 13 nM) with 23, and 5 (IC50 = 42 nM) with 18). Furthermore, within the limitations of the data, there are no major differences in the effects on MCF-7 cell growth dependent on the bridging group (thio, ketone, or methylene).
Compounds 26–34, bearing the substituent at position 6, inhibited tubulin polymerization with IC50 < 2.5 μM, with the exception of the 6-fluoro derivative 32 (IC50 = 10 μM). As inhibitors of MCF-7 cell growth, ketones were less potent than the corresponding thio derivatives. Among the thio derivatives, the 6-bromo (26, IC50 = 9.0 nM) and 6-methoxy (33, IC50 = 1.3 nM) derivatives were the most potent cell growth inhibitors. Compounds 35–43, with the substituent at position 7 of the indole, were all potent inhibitors of tubulin assembly, with IC50 < 2.5 μM. With the exception of 39 and 43, these compounds potently inhibited the growth of the MCF-7 cells, with compounds 36 (IC50 = 4 nM) and 38 (IC50 = 9 nM) being the most active. In contrast to the findings with the 6-substituted indoles, in the 7-substituted series, the bromo and chloro ketones tended to be more potent than the corresponding thio counterparts as inhibitors of MCF-7 cell growth (compare 26 and 27 with 35 and 36, and 28 and 29 with 37 and 38). Finally, we tried to join the features of 28 and 29 with those of 37 and 38 to further enhance activity. This effort yielded the 6,7-dichloroindole derivatives 44 and 45, both of which indeed were highly potent inhibitors of tubulin polymerization (IC50 = 1.2 and 1.5 μM, respectively) and MCF-7 cell growth (IC50 = 7 and 15 nM, respectively). A SAR summary of tubulin polymerization inhibition (TPI) and inhibition of MCF-7 cell growth of ATI derivatives 6–45 is depicted in Figure 2.
Figure 2.
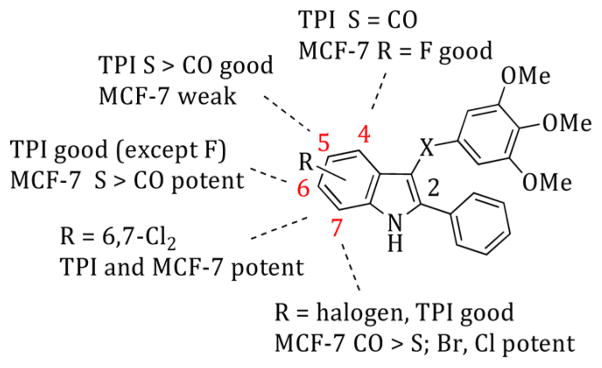
SAR summary of tubulin polymerization inhibition (TPI) and inhibition of MCF-7 cell growth of ATI derivatives 6–45.
The results obtained from the docking simulations described above provided us with a general binding mode that was able to justify the biological activity of the compounds. However, there were some exceptions that could not be fully rationalized (7, 13, 14, and 16), and we did not see any correlation between docking scores and experimental data. This has prompted us to further investigate the binding mode of ATIs with another set of docking simulations using a more recent tubulin structure (PDB code 4O2A),29 crystallized at higher resolution than the structure used previously (2.50 Å vs 3.58 Å, respectively).29 Furthermore, we have performed the docking calculations with two other software programs that each rely on a different search algorithm: Glide30 and Autodock.31
The docking results obtained from these simulations were entirely consistent with the one obtained using PLANTS. Indeed, we observed a virtually identical binding pose for the ATIs, regardless of the tubulin structure or the algorithm used in the simulation (Figure 3S, Supporting Information). It should be noted that we have not observed any correlation between the docking score and the experimental data in any of the calculations (Table 1S, Supporting Information). This observation is not entirely surprising considering this correlation is rarely observed.28
We analyzed the data from Table 1 comparing the microtuble assembly inhibitory concentrations (μM) with the percent inhibition of colchicine binding since these data provide an indirect measure of the affinity of the compounds for the colchicine site (Figure 3). The inhibition of tubulin polymerization was in good agreement with inhibition of colchicine binding. The compounds that inhibited tubulin assembly with IC50 values in the 1.0–1.5 μM range inhibited colchicine binding by 39–96% (mean value of 79%), those that inhibited assembly with IC50 values in the 1.6–2.0 μM range inhibited colchicine binding by 48–82% (mean value of 70%), and assembly inhibitors with IC50 values in the 2.1–3.0 μM range inhibited colchicine binding by 33–74% (mean value of 53%) (CSA4: assembly, IC50 = 1.0 μM; colchicine binding inhibition, 98%). We analyzed the MCF-7 cell growth inhibition (nM) of ATIs 6–45 with the corresponding inhibitory concentration (μM) on tubulin assembly (Figure 4SA, Supporting Information) and with the percent inhibition on the binding of [3H]colchicine to tubulin (Figure 4SB). In these plots we observed good correlation of the antiproliferative data with the biochemical data, particularly with the ligand binding data (inhibition of colchicine binding).
Figure 3.

Correlation between inhibition of tubulin assembly (IC50 values, μM) and inhibition of colchicine binding (%). Data of ATI derivatives 6–45 are shown as open circles. The black circle represents CSA4 as the reference compound.
Cell Growth Inhibition
ATIs 33 and 44 were assayed as growth inhibitors of a panel of cancer cell lines, including MDA-MB-468, MDA-MB-436, MDA-MB-231, A-549, MV4–11, NB4, and NCI-H1975, using PTX as the reference drug. As a growth inhibitor of MDA-MB-468, MDA-MB-436, and MDA-MB-231 breast cancer cells, using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, ATI 33 at 72 h showed IC50 values of 37, 62, and 39 nM, respectively, with the corresponding values for 44 being 33, 75, and 47 nM, respectively (Table 2). Compounds 33 and 44 induced a dose-and time-dependent growth inhibition of each treated cell line (Figures 5S–10S, Supporting Information). Compounds 33 and 44 inhibited the human acute myelocytic leukemia (AML) cell lines MV4–11 and NB4, showing at 48 h IC50 values of 2.5 and 10.5 nM, and 4 and 10 nM, respectively. As growth inhibitors of the A-549 and NCI-H1975 human lung adenocarcinoma cancer cells bearing the KRAS mutation and resistance to EGFR inhibitors, respectively, ATIs 33 and 44 yielded at 72 h IC50 values in the nanomolar concentration range.
Table 2.
Growth Inhibition of the MDA-MB-468, MDA-MB-436, MDA-MB-231, A-549, MV4-11, NB4, and NCI-H1975 Cell Lines by Compounds 33 and 44 and Reference PTX
| compd | IC50 ± SD (nM)
|
||||||
|---|---|---|---|---|---|---|---|
| MDA-MB-468a | MDA-MB-436a | MDA-MB-23a | MV4–11b (AML_M9) | NB4b (AML_M3) | A-549a | NCI-H1975a | |
| 33 | 37 ± 0.5 | 62 ± 1 | 39 ± 1.2 | 2.5 ± 2.1 | 4 ± 1 | 28 ± 6 | 195 ± 158 |
| 44 | 33 ± 0.3 | 75 ± 1.1 | 47 ± 0.1 | 10.5 ± 0.7 | 10 ± 4 | 120 ± 10 | 305 ± 122 |
| PTX | 5 ± 1 | 8 ± 1.5 | 7 ± 2 | ndc | 2.3 ± 0.3 | 7 ± 2 | 2.5 ± 5 |
Incubation time was 72 h.
Incubation time was 48 h.
No data.
Multi-Drug-Resistant (MDR) Cell Lines
Compounds 33 and 44 were evaluated as inhibitors of the ovarian carcinoma cell line OVCAR-8 and its cognate P-glycoprotein (Pgp)-over-expressing line NCI/ADR-RES and of the human uterine sarcoma cell line Messa and its cognate MDR line Messa/Dx5, using 1, 2, vinorelbine (VRB), VBL, and PTX as reference compounds (Table 3). The reference agents 1, VRB, VBL, and PTX weakly inhibited these MDR cell lines. Compounds 33 and 44 and the reference 2 were potent inhibitors of the MDR cell lines. ATI 33 (IC50 = 2.5 ± 1 nM) was comparable to 2 (IC50 = 1.8 ± 1 nM) as an inhibitor of the NCI/ADR-RES cell line, and 44 (IC50 = 4.5 ± 1 nM) was comparable to 2 (IC50 = 2.6 ± 1 nM) in the Messa/Dx5 cells.
Table 3.
Inhibition of Growth of the OVCAR-8 and NCI/ADR-RES, and Messa and Messa/Dx Cell Line Pairs by Compounds 33 and 44 and Reference Compounds 1, 2, VRB, VBL, and PTXa
| compd | IC50 ± SD (nM)
|
|||
|---|---|---|---|---|
| OVCAR-8 | NCI/ADR-RES | Messab | Messa/Dx5b | |
| 33 | 4.3 ± 1 | 2.5 ± 1 | 20.7 ± 1.7 | 28.0 ± 1.0 |
| 44 | 14 ± 2 | 10 ± 4 | 3.5 ± 0.9 | 4.5 ± 1.9 |
| 1 | 30 ± 1 | 420 ± 100 | 11 ± 6 | 329 ± 166 |
| 2 | 2.8 ± 1 | 1.8 ± 1 | 2.7 ± 2 | 2.6 ± 1 |
| VRB | 300 ± 0 | 5000 ± 1000 | ndc | nd |
| VBL | 15 ± 7 | 200 ± 0 | 3 ± 2 | 144 ± 61 |
| PTX | 3.7 ± 1 | 6000 ± 500 | 4 ± 1 | 1764 ± 477 |
Inhibition of growth of the indicated cell lines.
Incubation time was 72 h.
No data.
Arrest of Mitotic Progression and Concomitant Cell Death Induction
To assess whether the growth-suppressive effect of 33 and 44 reflected their antimitotic activity, we assessed their ability to induce mitotic arrest. We previously found that treatment with 20 nM VBL effectively arrested the cell cycle of HeLa cells in mitosis; at lower concentrations VBL did not fully prevent mitotic progression, such that cells assemble defective mitotic spindles, “slip” through the mitotic checkpoint, and progress toward aberrant chromosome segregation.14 We examined HeLa cell cultures treated with 20, 50, and 100 nM 33 or 44 in DMSO, 20 nM VBL, or DMSO vehicle. Treatments were carried out for 24 h, allowing all cells to enter mitosis during the treatment. Cell cultures were then harvested, and their cell cycle profile was analyzed by flow cytometry after incubation with propidium iodide (PI) to reveal their genomic DNA content (linear scale). ATIs 33 and 44 at 20 nM induced >80% of HeLa cells to arrest with a G2/M genomic content, similar to VBL; very few cells progressed past the G2/M phase (Figure 4A). Thus, both 33 and 44 effectively arrested mitotic progression and prevented mitotic slippage, and hence the generation of hyper-diploid or polyploid cells. To assess whether G2/M-arrested cells underwent cell death over time, PI-stained cell samples were analyzed by plotting their DNA content on a logarithmic scale to resolve the sub-G1 region, in which hypodiploid cells represent the terminal products of cell death. ATIs 33 and 44 at 20 nM induced both cell death and concomitant G2/M phase arrest and were comparable to VBL; these effects were even more substantial at 50 nM (Figure 4B). The induction of cell death by 33 seemed to be superior to 44 at all tested concentrations. Biparametric analysis after simultaneous incubation of non-permeabilized cells with annexin V, which reacts with phosphatidylserine residues on the outer cell membrane during early apoptosis, and PI, to which viable cells are not spontaneously permeable, discriminates early and late stages of apoptotic cell death (Figure 4C) from necrotic cells, which are permeable to PI but do not react with annexin V. We found that 33 and 44 induced cell death with apoptotic-like phenotypic features with comparable effectiveness and early-to-late kinetics as observed with VBL.
Figure 4.

(A) Cell cycle profiles of HeLa cell cultures exposed to 33, 44, or VBL for 24 h at the indicated concentrations. The percentage of cells with 2C (G1 phase, black bars), 4C (G2/M phases, white bars), or between 2C and 4C (S phase, dashed bars) genomic DNA content is shown. Mean values were calculated from three independent experiments. (B) Flow cytometry analysis of PI-stained cells with sub-G1 DNA content, representing terminal cell death, after 24 h. Mean values were calculated from four independent experiments. (C) Distribution of cells simultaneously processed for annexin V reactivity and PI incorporation, representing early (reactive to annexin V, not permeable to PI) and late (reactive to both annexin V and PI) stages of the cell death process in cultures treated for 24 h. Mean values were calculated from three independent experiments.
ATIs 33 and 44 Strongly Inhibit Mitotic MT Assembly
To correlate cell cycle arrest and cell death induction with inhibition of MT dynamics in HeLa cells, we employed immunofluorescence (IF) to examine the effects of ATIs on tubulin and MTs. HeLa cells were treated with 20 or 50 nM 33 or 44 for 24 h, the same conditions that induced cell cycle arrest (Figure 4), and then processed for tubulin IF and examined under fluorescence microscopy. We found that at both concentrations 33 or 44 arrested cells in mitosis with a prometaphase-like appearance. Consistent with the absence of hyperdiploid cells in flow cytometry analysis, both ATIs at 20 nM or higher effectively prevented mitotic slippage. At 20 nM, the ATIs inhibited MT polymerization and left tubulin foci and/or small asters of short MT fragments (an example is shown in Figure 5, right, middle panel). The proportion of mitotic cells displaying this inhibited MT phenotype was very similar to that observed with VBL (Figure 5, left). Raising the ATI concentrations to 50 nM yielded a phenotype with no recognizable mitotic MT remnants, but only tubulin aggregates that formed an unstructured meshwork throughout the mitotic cells (Figure 5, right, top panel). In summary, the newly synthesized ATIs strongly affect cellular MT polymerization, resulting in effective inhibition of formation of the mitotic apparatus, particularly when used at 50 nM, and this phenotype was associated with a durable mitotic arrest and concomitant induction of cell death.
Figure 5.

Mitotic phenotypes in HeLa cell cultures exposed to ATIs 33 and 44 for 24 h and processed for tubulin IF (in red). The bar graph on the left indicates the statistical distribution of the cytological phenotypes depicted under the indicated conditions. The frequency of scored phenotypes is shown as the percentage of all counted mitotic figures (330–400 counted mitotic figures per condition). Gray bars represent the frequency of mitotic cells with severe inhibition of tubulin polymerization, with no recognizable MTs and unstructured tubulin aggregates (exemplified in the top IF panel). Purple bars represent the frequency of mitotic cells with inhibited MT polymerization, yielding short MT stretches (middle panel). Black bars represent the frequency of normal mitoses with polymerized MT arrays forming a bipolar spindle (bottom panel).
Inhibition of PC-3, RD, and HepG2 Cancer Cell Growth
We determined the effects of ATIs 33 and 44 as growth inhibitors of the human prostate cancer PC-3, rhabdomyosarcoma RD, and human liver hepatocellular carcinoma HepG2 cell lines, with VBL and PTX as reference compounds (Table 4). ATIs 33 and 44 strongly inhibited these cell lines at nanomolar concentrations and were superior to the references VBL and PTX. Compound 33 was generally more effective than 44 as an inhibitor of the growth of these three cell lines.
Table 4.
Growth Inhibition of PC-3, RD, and HepG2 Cell Lines by Compounds 33 and 41 and References VBL and PTX
| compd | IC50a ± SD (nM)
|
||
|---|---|---|---|
| PC3 | RD | HepG2 | |
| 33 | 0.3 ± 0.06 | 0.2 ± 0.04 | 0.1 ± 0.02 |
| 44 | 19 ± 1 | 16 ± 1.3 | 62 ± 2 |
| VBL | 766 ± 1000 | 53 ± 2.5 | 81 ± 2.4 |
| PTX | 4900 ± 1.3 | >10000 | 2600 ± 1.5 |
Incubation time was 48 h.
PC-3, RD, and HEPG2 cell cultures were treated for 24 h with increasing concentrations of ATIs 33 and 44 and of PTX as the reference drug at 500, 1000, and 2000 nM. After treatment, cells and vehicle controls (0.1% DMSO) were incubated with PI to analyze their DNA content in flow cytometry assays. Both ATIs arrested cell cycle progression in all three cell lines at low concentrations. ATIs 33 and 44 induced an accumulation in the G2/M phase in PC-3 and RD cells, as did PTX, whereas in HepG2 cells these compounds caused a stronger effect on cell cycle progression as compared with the reference drug (Figure 11S, Supporting Information).
After a 24 h exposure and subsequent incubation in drug-free medium for 24 h, the PC-3, RD, and HepG2 cell lines treated with 33 or 44 showed a strong accumulation of cells in G2/M (Figure 6). Moreover, 33 and 44 induced an irreversible cell cycle arrest at concentrations of 500, 1000, and 2000 nM in the PC-3 and RD cells.
Figure 6.

Cell cycle analysis of PC-3 (A), RD (B), and HepG2 (C) cells treated with 0.1% DMSO or 500, 1000, or 2000 nM 33, 44, or PTX for 24 h, followed by a 24 h recovery in drug-free medium. Histograms represent the percentage of cells with G0/G1, S, and G2/M DNA content expressed as mean values ± SD of three independent experiments.
A correlation between cell cycle arrest and cell death was investigated by exposing the cells to a 500, 1000, or 2000 nM concentration of 33, 44, or PTX, followed by incubation with fluorescently conjugated annexin V and PI (Figure 7). All the cell lines treated with 33 or 44 at 500 nM showed higher rates of cell death than the same populations treated with 500 nM PTX. A dose–response trend in cell death was observed in the RD cell line after a 48 h exposure with either 44 or 33 (Figure 7B) and in the PC3 and HepG2 cell lines following treatment with PTX (Figure 7A,C).
Figure 7.

Cell death cytometric analysis of PC-3 (A), RD (B), and HepG2 (C) cells treated with 0.1% DMSO or 500, 1000, or 2000 nM 33, 44, or PTX for 48 h. Flow cytometric profiles of cell populations following treatment with DMSO or 2000 nM 33 or 44 are at the top of each panel (annexin V–FITC staining on the x axis and PI on the y axis). Histograms represent the percentage of cells in early apoptosis (annexin V–FITC staining) and late apoptosis (annexin V–FITC and PI staining) expressed as mean values ± SD calculated from three independent experiments.
Inhibition of T98G and U343MG Cancer Cell Growth
Malignant gliomas develop from gradual accumulation of multiple genetic alterations, resulting in either activation of oncogenes or inactivation of tumor suppressor genes.32 Human glioblastoma multiforme T98G and U343MG cells show typical hallmarks of glioblastoma multiforme tumors in patients. We evaluated the ability of compounds 33 and 44 to inhibit the growth of T98G and U343MG cancer cells, which show different genetic profiles for the expression of key cell survival proteins, such as p53, MDM2, EGFR, RB, cyclin D, and MMPs.33 Treatment of T98G and U343MG cells with increasing concentrations of 33 or 44 for 24, 48, or 72 h significantly inhibited cell growth in a dose- and time-dependent manner (Figures 12S and 13S, Supporting Information). The IC50 values were calculated taking into account the relative doubling time (CDT),34,35 after 48 h for the T98G cells and after 72 h for the U343MG cells. As a cell growth inhibitor, compound 33 yielded IC50 values of 15.2 ± 1.6 nM in T98G cells and 0.5 ± 0.05 nM in U343 cells; for 44, IC50 values of 16.3 ± 1.5 nM nM in T98G cells and 0.6 ± 0.05 nM nM in U343 cells were obtained.
Expression of MICA and MICB Ligands in HeLa Cells, Resulting in Enhanced Natural Killer (NK) Cell Degranulation
In previous studies,36 treatment of HeLa and HepG2 tumor cell lines with sodium butyrate, a potent repressor of histone deacetylases that causes spindle abnormalities and mitotic arrest, resulted in up-regulation of the expression of NK cell receptor-activating ligands MICA and MICB at both the mRNA and protein levels and in enhanced susceptibility of both cell lines to NK lysis. We evaluated the expression of NKG2D and DNAM-1 ligands in HeLa cells after treatment with ATI 33, 37, or 44, in particular whether the compounds could modulate their expression. We first characterized HeLa cell growth inhibition by 33, 37, or 44, at a sublethal concentration after a 48 h treatment (MTT assay). HeLa cells were more sensitive to 33 and 44 (IC50 = 10 nM) than to 37 (IC50 = 76 nM). After a 48 h treatment with 10 nM ATI, flow cytometric biparametric analysis of HeLa cells by annexin V/PI staining showed only a weak increase of early apoptotic cells compared to control cultures (Figure 14S, Supporting Information).
NK cell receptor-activating ligand analysis by combined IF and flow cytometry revealed a different modulation of NKG2D and DNAM-1 ligands in ATI-treated HeLa cells after a 48 h treatment with sublethal doses. ATIs 33, 37, and 44 behaved as strong enhancers of MICA, ULBP3, and PVR expression, while treatment with the compounds had weaker effects on MICB, ULBP1, and ULBP2 ligand expression (Figure 8) and no effect on the expression of the Nec-2 ligand (data not shown). Interestingly, (1-(3-aminophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone, a potent tubulin polymerization inhibitor belonging to the ARAP class,37 was unable to induce the NKG2D and DNAM-1 ligands (data not shown). The expression of the ligand surface on treated HeLa cells was not accompanied by a corresponding increase in mRNA levels, as indicated by real-time PCR data (Figure 15S, Supporting Information).
Figure 8.
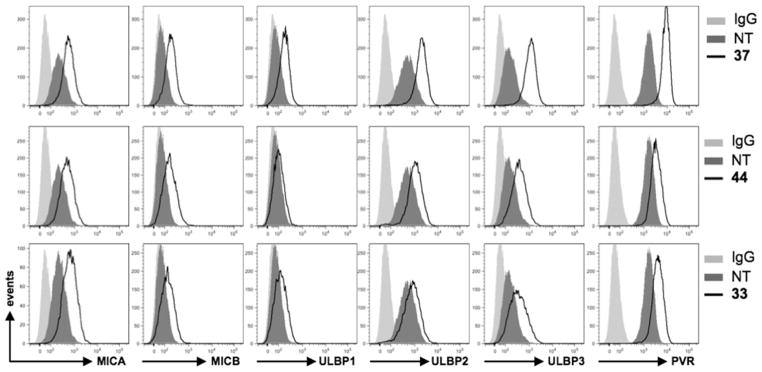
ATIs 33, 37, and 44 up-regulate DNAM-1 and NKG2D ligands in HeLa cells. NKG2D and DNAM-1 ligand surface expression was analyzed by flow cytometry after a 48 h treatment with the indicated ATI compound. Data are representative of one out of three independent experiments.
On the basis of these findings, we evaluated whether ATIs 33 and 44 would increase NK cell degranulation toward HeLa cells. The expression of the lysosomal marker CD107a, which correlates with NK cell cytotoxicity,38 was evaluated by IF and flow cytometry analysis by gating on NK cells upon their interaction with treated or untreated HeLa cells, used as targets. The up-regulation of NKG2D and DNAM-1 ligands was verified before the degranulation assay (data not shown). The expression of CD107a on NK cells contacting treated HeLa target cells indicated that those cells were more susceptible to NK cell lysis (Figure 9).
Figure 9.
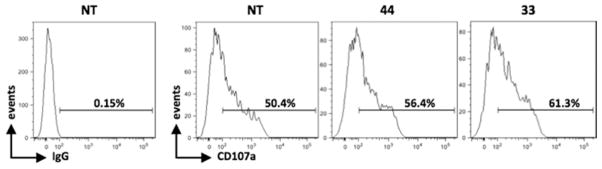
NK cell-mediated killing of HeLa cells increased after a 48 h treatment with ATI 33 or 44. Results are expressed as the percentage of CD107a+ cells after subtraction of the percentage of the control antibody and represent one of two independent experiments. The basal CD107a expression on NK cells was about 2%.
Hedgehog-Inhibiting Activity
The hedgehog (Hh) signaling pathway is deeply involved in tumorigenesis, and inhibitors of the Hh pathway have shown great potential as cancer therapeutics. In recent years, an ever-growing interest in the Hh pathway has led to the development of a variety of small molecules targeting key Hh components, i.e., smoothened (Smo), sonic hedgehog protein (Shh), and Gli1.39 Currently, some antagonists of Smo, the positive signaling transducer in the Hh pathway, are undergoing clinical trials.
We wished to investigate whether ATI compounds, like the structurally related ARAP derivatives,37 could behave as inhibitors of Hh signaling. We selected three highly potent ATI derivatives, 33, 44, and 2-(1H-imidazol-1-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (81),15 and their effects were evaluated by a luciferase assay performed in NIH3T3 Shh-Light II (Shh-LII) cells. In these cells, in which is stably incorporated an Hh-responsive (Gli-RE) reporter, the induction of the pathway occurs following treatment with the Smo agonist SAG. ATIs 33, 44, and 81 showed a strong reduction of luciferase activity in cells treated with SAG in a dose-dependent manner (Figure 10), yielding IC50 values of 19, 72, and 38 nM, respectively. The treatment did not decrease the control Renilla luciferase activity, preventing any cytotoxicity-mediated effects on the inhibition of Hh signaling. To address the ability of these compounds to affect D283 medulloblastoma cell proliferation and survival, we performed a trypan blue count assay. The in vitro treatment with ATIs 33, 44, and 81 at 1 μM impaired cell growth and the percentage of cell death, as shown in Figure 16S, Supporting Information.
Figure 10.
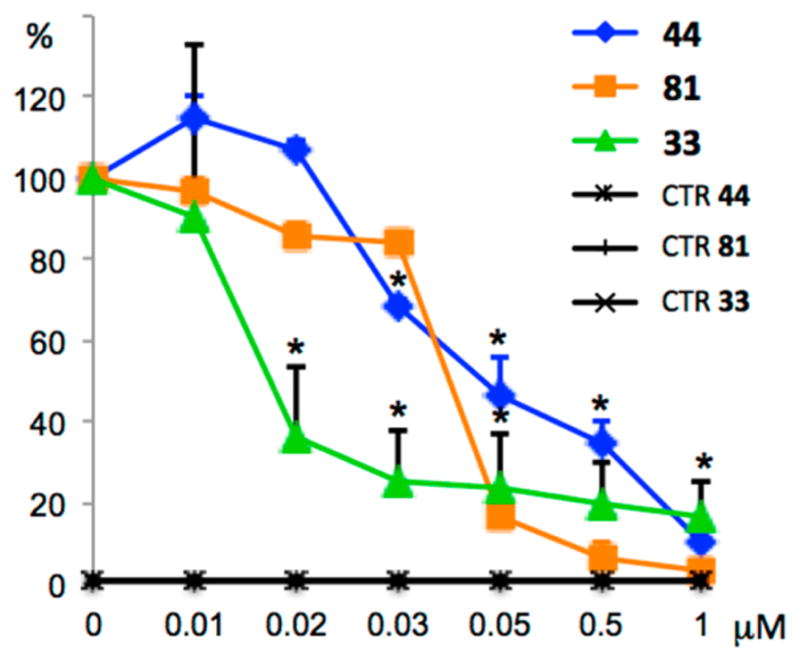
Inhibition of endogenous Hh signaling in Shh-LII cells by 33, 44, and 81. Dose–response curve of the indicated compounds in SAG-treated cells in comparison with untreated Shh-Light II cells as a control (CTR). The treatment time was 48 h, and normalization was against Renilla luciferase. Data are from three independent experiments. Error bars indicate SD. An asterisk indicates P = 0.05 vs CTR.
Hh inhibitors are attracting ever-growing attention because of benefits displayed in the treatment of Hh-dependent cancers, such as medulloblastomas.40 However, the problem of drug resistance to Smo mutations, arising during clinical treatment, has led to a quest for new Hh inhibitors. ATIs 33, 44, and 81 showed potent inhibition of the Hh signaling pathway, suggesting that the ATI class has potential as Hh-dependent anticancer agents.
Metabolic Stability
Compounds 33 and 44 were examined in a microsomal stability assay in comparison with 7-ethoxy-coumarin and propranolol as control compounds, using both human and mouse liver microsomes, to estimate compound stability to phase I oxidative metabolism (Table 5). Compound 44 showed metabolic stability of 25.6 and <3 μL/min/mg of protein in mouse and human liver microsomes, respectively. Compound 33 showed medium metabolic stability with human liver microsomes and low to medium metabolic stability with mouse liver microsomes (relative stabilities are defined in footnote a of Table 5).
Table 5.
Metabolic Stability with Human and Mouse Liver Microsomesa and Aqueous Solubility of Compounds 33 and 44
| compd | clearanceb,c (μL/min/mg of protein)
|
solubilityd at pH 7.4 (μM) | |
|---|---|---|---|
| human liver microsomes | mouse liver microsomes | ||
| 33 | 15.2 ± 8.0 | 43.1 ± 7.9 | 0.99 ± 0.13 |
| 44 | <3 | 25.6 ± 1.7 | 3.18 ± 0.50 |
| 7-ethoxycoumarin | 209 ± 10.2 | 710.8 ± 1.2 | |
| propranolol | 19.1 ± 2.2 | 235.1 ± 24.1 | |
Metabolic stability: <3, good; 3–60, medium; >60, low.
Results are expressed as the mean ± SD, n = 2.
The standard compounds 7-ethoxycoumarin and propranolol showed metabolic stability in agreement with the literature and internal validation data REF.
High-throughput screening solubility assay.
Aqueous Solubility
The solubility in aqueous pH 7.4 buffer of compounds 33 and 44 was measured in a high-throughput screening solubility assay. The solubility of compound 33 was about 1 μM, while, under the same conditions, compound 44 showed a solubility of 3.2 μM (Table 5).
CONCLUSION
We designed 39 new 2-phenylindole derivatives as potential anticancer agents bearing the 3,4,5-trimethoxyphenyl moiety linked through a sulfur, ketone, or methylene bridging group at position 3 of the indole and with halogen or methoxy substituent(s) at positions 4–7. Twenty-three new derivatives inhibited tubulin polymerization with IC50 values of 1.0–2.0 μM and inhibited colchicine binding with a mean value of >70%; 15 derivatives inhibited the growth of human MCF-7 cells with IC50 ≤ 50 nM.
Compounds 33 and 44, representative members of this series, uniformly inhibited at nanomolar concentration a panel of cancer cells, including MDA-MB-468, MDA-MB-436, MDA MB-231, MV4–11, NB4, A-549, NCI-H1975, T98G, and U343 cells. As inhibitors of PC-3, RD, and HepG2 cancer cell growth, 33 and 44 were superior to the references VBL and PTX. ATI 33 and 44 were comparable to 2 as inhibitors of the MDR NCI/ADR-RES and Messa/Dx5 cell lines. Besides the ability to inhibit tubulin polymerization, for which they were originally designed, ATIs 33 and 44 exhibited an unexpected stimulation of the cytotoxic activity of NK cells. Our findings demonstrated that 33 and 44 at doses of 10 nM, which did not severely affect cell viability, increased NKG2D and DNAM-1 ligand up-regulation of HeLa cells, resulting in an enhanced stimulation of NK cell cytotoxic activity. This novel effect of ATIs, elicited at sublethal doses, along with a stronger expression of NK cell receptor-activating ligand, led to an increased propensity of NK cells to degranulate against tumor cells. At higher concentrations of 20–50 nM, ATIs 33 and 44 induced >80% of HeLa cells to arrest with a G2/M DNA content, an effect similar to that observed with VBL, with very few cells progressing past the 4N G2/M phase. These compounds stably arrested mitotic progression, prevented mitotic slippage and the ensuing formation of aneuploid cells—a hallmark of aggressive cancers—and induced cell death. These findings suggest that the new ATIs 33 and 44 can arrest proliferation of cancer cells with effectiveness comparable or superior to that obtained with VBL. ATIs 44 and 81 showed strong inhibition of the Hh signaling pathway, inhibiting medulloblastoma D283 cells with IC50 values of 72 and 38 nM, respectively.
In summary, these novel ATI compounds show potential to treat cancer via both MT-based and MT-independent pathways. Even at low concentrations where the ATIs do not fully prevent the assembly of the mitotic apparatus, and hence mitotic cell death, these agents effectively up-regulate NK ligands and trigger an alternative cytotoxic response via NK cells. Here we have described the broad potential of the new ATIs in an in-depth characterization of the cell response to increasing drug concentrations. We aim to assess whether specific MT-associated proteins (MAPs) can modulate cell sensitivity to these drugs and favor one pathway over the other. Such MAPs may serve as effective biomarkers to predict the prevalent response to ATI treatment. Compounds 33 and 44 represent novel lead compounds that will prompt further development of the ATI class to obtain new promising anticancer agents with enhanced stimulation of NK cell cytotoxic activity and repression of Hh-dependent cancers. Derivative 44 showed higher metabolic stability than 33 in human and mouse liver microsomes and greater water solubility. The present results highlight the therapeutic potential of the ATI class as anticancer agents and prompt further developmental studies.
EXPERIMENTAL SECTION
General Chemistry Procedures
Microwave-assisted reactions were performed on a CEM Discover SP single-mode reactor. The instrument settings were controlled with PC-running CEM Synergy 1.49 software. Closed vessel experiments were carried out in capped microwave-dedicated vials (10 mL) with a cylindrical stirring bar (length 8 mm, diameter 3 mm). Open vessel experiments were carried out in 100 mL round-bottom flasks equipped with a Dimroth reflux condenser and a cylindrical stirring bar (length 20 mm, diameter 6 mm). Stirring, temperature, irradiation power, maximum pressure (Pmax), PowerMAX (simultaneous cooling-while-heating), ActiVent (simultaneous venting-while-heating), and ramp and hold times were set as indicated. The temperature of the reaction was monitored by an external fiber optic temperature sensor. After completion of the reaction, the mixture was cooled to 25 °C via air-jet cooling. Melting points (mp’s) were determined on a Stuart Scientific SMP1 apparatus and are uncorrected. Infrared (IR) spectra were run on a PerkinElmer SpectrumOne FT-ATR spectrophotometer. Band position and absorption ranges are given in inverse centimeters. Proton nuclear magnetic resonance (1H NMR) spectra were acquired on a Bruker 400 MHz FT spectrometer in the indicated solvent by TopSpin 2.1 software and the files processed by MestreLab Research S.L. MestreReNova 6.2.1–769 software. Chemical shifts are expressed in δ units (ppm) from tetramethylsilane. Multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broadened), br s (broadened singlet), and coupling constants (J) are reported in hertz. Flash chromatography was carried out on an Interchim Spot II Flash system, using Merck SuperVarioFlash D26 cartridges packed with Merck Geduran 60 (0.040–0.063 mm) silica gel. Column chromatography was performed on columns packed with alumina from Merck (70–230 mesh) or silica gel from Macherey-Nagel (70–230 mesh). Aluminum oxide thin-layer chromatography (TLC) cards from Fluka (aluminum oxide-precoated aluminum cards with fluorescent indicator visualizable at 254 nm) and silica gel TLC cards from Macherey-Nagel (silica gel-precoated aluminum cards with fluorescent indicator visualizable at 254 nm) were used for TLC. Developed plates were visualized by a Spectroline ENF 260C/FE UV apparatus. Organic solutions were dried over anhydrous sodium sulfate. Evaporation of solvents was carried out on a Büchi Rotavapor R-210 equipped with a Büchi V-850 vacuum controller and a Büchi V-700 or V-710 vacuum pump. All commercially available reagents were used without further purification. Elemental analyses of the compounds were found within ±0.4% of the theoretical values. The purity of the tested compounds was determined by high-pressure liquid chromatography (HPLC) and was >95%. The HPLC system (Dionex UltiMate 3000, Thermo Fisher Scientific Inc.) was equipped with an SR-3000 solvent rack, an LPG-3400SD quaternary analytical pump, a TCC-3000SD column compartment, a DAD-3000 diode array detectorn and an analytical manual injection valve with a 20 μL loop. Compounds were dissolved in acetonitrile (10 mg/mL). HPLC analysis was performed by using an Acclaim 120 C18 reversed-phase column (5 μm, 4.6 × 250 mm, Thermo Fisher Scientific Inc.) at a temperature of 30 ± 1 °C, an isocratic gradient (acetonitrile:water = 90:10), a flow rate of 1.0 mL/min, and detector signals 254 and 365 nm. Chromatographic data were acquired and processed by Chromeleon 6.80 software (Thermo Fisher Scientific Inc.).
Synthesis of Compounds 6–45
General Procedure for the Preparation of 6, 8, 11, 13, 15, 18, 21, 23, 26, 28, 31, 33, 35, 37, 40, 42, a n d 44. Example : 4 - Bromo - 2 - phenyl - 3 - ((3, 4, 5 -trimethoxyphenyl)thio)-1H-indole (6)
A mixture of 4-bromo-2-phenyl-1H-indole (46) (0.27 g, 1 mmol), bis(3,4,5-trimethoxyphenyl) disulfide (0.44 g, 1.1 mmol), and sodium hydride (0.053 g, 2.2 mmol; 60% in mineral oil) in anhydrous DMF (3 mL) was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). Starting microwave irradiation of 120 W was used, the temperature being ramped from 25 to 130 °C, with rapid stirring and venting (pressure set point 100 psi, times at set point 100, delta pressure 20 psi). Once 130 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min. The mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine and dried. Removal of the solvent gave a residue that was purified by flash chromatography (silica gel, ethyl acetate/n-hexane as eluent) to furnish 6 (0.13 g, 27%). Mp: 202–205 °C (from ethanol). 1H NMR (CDCl3): δ 3.66 (s, 6H), 3.79 (s, 3H), 6.37 (s, 2H), 7.10 (t, J = 7.8 Hz, 1H), 7.38–7.41 (m, 5H), 7.71 (d, J = 6.7 Hz, 2H), 8.72 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3344 cm−1. Anal. (C23H20BrNO3S (470.38)) C, H, Br, N, S.
4-Chloro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (8)
Synthesized as 6, starting from 4-chloro-2-phenyl-1H-indole (47). Yield: 62%. Mp: 178–180 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.57 (s, 3H), 6.26 s, 2H), 7.10–7.11 (m, 1H), 7.18–7.20 (m, 1H), 7.44–7.54 (m, 4H), 7.77–7.80 (m, 2H), 12.51 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3345 cm−1. Anal. (C23H20ClNO3S (425.93)) C, H, Cl, N, S.
4-Fluoro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (11)
Synthesized as 6, starting from 4-fluoro-2-phenyl-1H-indole (48). Yield: 30%. Mp: 168–170 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.53 (s, 6H), 3.56 (s, 3H), 6.28 (s, 2H), 6.80–6.85 (m, 1H), 7.14–7.19 (m, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 7.1 Hz, 1H), 7.51 (t, J = 7.1 Hz, 2H), 7.80–7.82 (m, 2H), 12.30 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3307 cm−1. Anal. (C23H20FNO3S (409.47)) C, H, F, N, S.
4-Methoxy-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (13)
Synthesized as 6, starting from 4-methoxy-2-phenyl-1H-indole (49). Yield: 41%. Mp: 133–136 °C (from toluene). 1H NMR (CDCl3): δ 3.66 (s, 6H), 3.78 (s, 6H), 6.44 (s, 2H), 6.57–6.59 (m, 1H), 7.05 (d, J = 8.2 Hz, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.38–7.48 (m, 3H), 7.73 (d, J = 7.2 Hz, 2H), 8.52 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3261 cm−1. Anal. (C24H23NO4S (421.51)) C, H, N, S.
5-Bromo-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (15)
Synthesized as 6, starting from 5-bromo-2-phenyl-1H-indole (50). Yield: 18%. Mp: 152–155 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.53 (s, 6H), 3.56 (s, 3H), 6.27 (s, 2H), 7.32–7.35 (m, 1H), 7.44–7.58 (m, 5H), 7.86–7.88 (m, 2H), 12.29 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3316 cm−1. Anal. (C23H20BrNO3S (470.38)) C, H, Br, N, S.
5-Chloro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (18)
Synthesized as 6, starting from 5-chloro-2-phenyl-1H-indole (51). Yield: 20%. Mp: 158–160 °C (from ethanol). 1H NMR (CDCl3): δ 3.65 (s, 6H), 3.78 (s, 3H), 6.33 (s, 2H), 7.21–7.24 (m, 1H), 7.36–7.49 (m, 4H), 7.66–7.67 (m, 1H), 7.77–7.80 (m, 2H), 8.69 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3319 cm−1. Anal. (C23H20ClNO3S (425.93)) C, H, Cl, N, S.
5-Fluoro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (21)
Synthesized as 6, starting from 5-fluoro-2-phenyl-1H-indole (52). Yield: 26%. Mp: 158–160 °C (from toluene). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.57 (s, 3H), 6.30 (s, 2H), 7.1 (t, J = 8.0 Hz, 1H), 7.16 (d, J = 9.2 Hz, 1H), 5.45 (t, J = 7.2 Hz, 1H), 7.50–7.55 (m, 3H), 7.88 (d, J = 7.4 Hz, 2H), 12.21 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3231 cm−1. Anal. (C23H20FNO3S (409.47)) C, H, F, N, S.
5-Methoxy-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (23)
Synthesized as 6, starting from 5-methoxy-2-phenyl-1H-indole (53). Yield: 20% as an oil. 1H NMR (CDCl3): δ 3.63 (s, 6H), 3.77 (s, 3H), 3.83 (s, 3H), 6.35 (s, 2H), 6.90–6.93 (m, 1H), 7.10–7.11 (m, 1H), 7.32–7.46 (m, 4H), 7.76–7.77 (m, 2H), 8.58 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3337 cm−1. Anal. (C24H23NO4S (421.51)) C, H, N, S.
6-Bromo-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (26)
Synthesized as 6, starting from 6-bromo-2-phenyl-1H-indole (54). Yield: 22%. Mp: 200–203 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.57 (s, 3H), 3.58 (s, 6H), 6.78 (s, 2H), 7.23–7.27 (m, 3H), 7.35–7.41 (m, 3H), 7.48 (d, J = 8.3 Hz, 1H), 8.09 (s, 1H), 12.40 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3320 cm−1. Anal. (C23H20BrNO3S (470.38)) C, H, Br, N, S.
6-Chloro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (28)
Synthesized as 6, starting from 6-chloro-2-phenyl-1H-indole (55). Yield: 25%. Mp: 207–210 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.57 (s, 3H), 6.29 (s, 2H), 7.14 (dd, J = 1.1 and 7.7 Hz, 1H), 7.43–7.55 (m, 5H), 7.87 (d, J = 7.9 Hz, 2H), 12.23 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3319 cm−1. Anal. (C23H20ClNO3S (425.93)) C, H, Cl, N, S.
6-Fluoro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (31)
Synthesized as 6, starting from 6-fluoro-2-phenyl-1H-indole (56). Yield: 37% as an oil. 1H NMR (CDCl3): δ 3.64 (s, 6H), 3.78 (s, 3H), 6.35 (s, 2H), 6.96 (t, J = 8.8 Hz, 1H), 7.14 (d, J = 9.0 Hz, 1H), 7.41–7.50 (m, 3H), 7.58–7.59 (m, 1H), 7.78 (d, J = 8.0 Hz, 2H), 8.56 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3320 cm−1. Anal. (C23H20FNO3S (409.47)) C, H, F, N, S.
6-Methoxy-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (33)
Synthesized as 6, starting from 6-methoxy-2-phenyl-1H-indole (57). Yield: 24%. Mp: 121–124 °C (from toluene). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.60 (s, 3H), 3.81 (s, 3H), 6.31 (s, 2H), 6.77 (dd, J = 2.2 and 8.6 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H), 7.34–7.42 (m, 2H), 7.50 (t, J = 7.4 Hz, 2H), 7.85 (d, J = 7.2 Hz, 2H), 11.88 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3322 cm−1. Anal. (C24H23NO4S (421.51)) C, H, N, S.
7-Bromo-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (35)
Synthesized as 6, starting from 7-bromo-2-phenyl-1H-indole (58). Yield: 39%. Mp: 142–145 °C (from toluene). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.57 (s, 3H), 6.29 (s, 2H), 7.08 (t, J = 7.7 Hz, 1H), 6.97 (d, J = 7.7 Hz, 1H), 7.43–7.54 (m, 5H), 7.82 (d, J = 7.1 Hz, 1H), 12.11 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3335 cm−1. Anal. (C23H20BrNO3S (470.38)) C, H, Br, N, S.
7-Chloro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (37)
Synthesized as 6, starting from 7-chloro-2-phenyl-1H-indole (59). Yield: 77%. Mp: 62–65 °C (from ethanol). 1H NMR (CDCl3): δ 3.66 (s, 6H), 3.78 (s, 3H), 6.36 (s, 2H), 7.12–7.17 (m, 1H), 7.43–7.57 (m, 4H), 7.58–7.60 (m, 1H), 7.83–7.85 (m, 2H), 8.72 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3259 cm−1. Anal. (C23H20ClNO3S (425.93)) C, H, Cl, N, S.
7-Fluoro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (40)
Synthesized as 6, starting from 7-fluoro-2-phenyl-1H-indole (60). Yield: 32%. Mp: 98–100 °C (from toluene). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.57 (s, 3H), 6.30 (s, 2H), 7.05–7.10 (m, 2H), 7.31 (d, J = 7.1 Hz, 1H), 7.46 (t, J = 7.2 Hz, 1H), 7.53 (t, J = 7.1 Hz, 2H), 7.88 (d, J = 7.2 Hz, 2H), 12.46 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3247 cm−1. Anal. (C23H20FNO3S (409.47)) C, H, F, N, S.
7-Methoxy-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (42)
Synthesized as 6, starting from 7-methoxy-2-phenyl-1H-indole (61). Yield: 57%. Mp: 158–160 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.53 (s, 6H), 3.57 (s, 3H), 3.97 (s, 3H), 6.29 (s, 2H), 6.79–6.81 (m, 1H), 7.04–7.08 (m, 2H), 7.40–7.49 (m, 3H), 7.83–7.86 (m, 2H), 12.09 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3300 cm−1. Anal. (C24H23NO4S (417.45)) C, H, N, S.
6,7-Dichloro-2-phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (44)
Synthesized as 6, starting from 6,7-dichloro-2-phenyl-1H-indole (62). Yield: 57%. Mp: 158–160 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.55 (s, 6H), 3.57 (s, 3H), 6.29 (s, 2H), 7.32 (d, J = 8.5 Hz, 1H), 7.44–7.55 (m, 4H), 7.83–7.85 (m, 2H), 12.44 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1610, 3268 cm−1. Anal. (C23H19Cl2NO3S (460.37)) C, H, Cl, N, S.
General Procedure for the Preparation of 7, 9, 12, 14, 16, 19, 22, 24, 27, 29, 32, 34, 36, 38, 41, 43, and 45. Example: (4-Bromo-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (7)
A mixture of 46 (0.27 g, 1 mmol), 3,4,5-trimethoxybenzoyl chloride (0.23 g, 1 mmol), and anhydrous aluminum chloride (0.13 g, 1 mmol) in anhydrous 1,2-dichloroethane (2 mL) was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C, with stirring. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min. The reaction mixture was quenched on 1 M HCl/crushed ice and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by flash chromatography (silica gel, ethyl acetate/n-hexane as eluent) to furnish 7 (0.09 g, 20%). Mp: 228–230 °C (from ethanol). 1H NMR (CDCl3): δ 3.74 (s, 6H), 3.88 (s, 3H), 7.13–7.16 (m, 3H), 7.31–7.37 (m, 4H), 7.42–7.46 (m, 3H), 8.58 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1618, 3344 cm−1. Anal. (C24H20BrNO4 (466.32)) C, H, Br, N.
(4-Chloro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (9)
Synthesized as 7, starting from 47. Yield: 35%. Mp: 232–235 °C (from ethanol). 1H NMR (CDCl3): δ 3.72 (s, 6H), 3.94 (s, 3H), 7.17 (s, 2H), 7.19–7.23 (m, 2H), 7.30–7.33 (m, 3H), 7.38 (dd, J = 2.0 and 7.0 Hz, 1H), 7.45–7.48 (m, 2H), 8.73 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1618, 3169 cm−1. Anal. (C24H20ClNO4 (421.87)) C, H, Cl, N.
(4-Fluoro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (12)
Synthesized as 7, starting from 48. Yield: 25%. Mp: 212–215 °C (from toluene). 1H NMR (DMSO-d6): δ 3.62 (s, 6H), 3.64 (s, 3H), 6.84–6.89 (m, 1H), 6.99 (s, 2H), 7.18–7.22 (m, 1H), 7.30–7.35 (m, 4H), 7.42–7.44 (m, 2H), 12.34 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1618, 3205 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, F, N.
(4-Methoxy-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (14)
Synthesized as 7, starting from 49. Yield: 51%. Mp: 144–146 °C (from toluene). 1H NMR (DMSO-d6): δ 3.53 (s, 3H), 3.64 (s, 6H), 3.69 (s, 3H), 6.55–6.57 (m, 1H), 7.02 (s, 2H), 7.09–7.16 (m, 2H), 7.29–7.39 (m, 3H), 7.49–7.51 (m, 2H), 11.95 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1622, 3220 cm−1. Anal. (C25H23NO5 (417.45)) C, H, N.
(5-Bromo-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (16)
Synthesized as 7, starting from 50. Yield: 55%. Mp: 200–203 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.57 (s, 3H), 3.58 (s, 6H), 6.78 (s, 2H), 7.23–7.27 (m, 3H), 7.35–7.41 (m, 3H), 7.48 (d, J = 8.3 Hz, 1H), 8.09 (s, 1H), 12.40 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1610, 3305 cm−1. Anal. (C24H20BrNO4 (466.32)) C, H, Br, N.
(5-Chloro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (19)
Synthesized as 7, starting from 51. Yield: 54%. Mp: 222–225 °C (from ethanol). 1H NMR (CDCl3): δ 3.69 (s, 6H), 3.81 (s, 3H), 6.94 (s, 2H), 7.24–7.30 (m, 4H), 7.33–7.40 (m, 3H), 8.07–8.08 (m, 1H), 8.81 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1610, 3340 cm−1. Anal. (C24H20ClNO4 (421.87)) C, H, Cl, N.
(5-Fluoro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (22)
Synthesized as 7, starting from 52. Yield: 66%. Mp: 198–200 °C (from toluene). 1H NMR (DMSO-d6): δ 3.58 (s, 3H), 3.59 (s, 6H), 6.78 (s, 2H), 7.10–7.15 (m, 1H), 7.24–7.27 (m, 3H), 7.34–7.37 (m, 2H), 7.51 (q, J = 4.6 Hz, 1H), 7.66 (dd, J = 2.5 and 10.1, 1H), 12.30 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1620, 3225 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, F, N.
(5-Methoxy-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (24)
Synthesized as 7 starting from 53. Yield: 78%. Mp: 202–205 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.58 (s, 3H), 3.59 (s, 6H), 3.78 (s, 3H), 6.78 (s, 2H), 6.90 (dd, J = 2.6 and 8.8 Hz, 1H), 7.24–7.25 (m, 3H), 7.32–7.34 (m, 2H), 7.41 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 2.4 Hz, 1H), 12.11 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1652, 3195 cm−1. Anal. (C25H23NO5 (417.45)) C, H, N.
(6-Bromo-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (27)
Synthesized as 7, starting from 54. Yield: 40% as an oil. 1H NMR (CDCl3): δ 3.68 (s, 6H), 3.81 (s, 3H), 6.94 (s, 2H), 7.23–7.27 (m, 3H), 7.33–7.36 (m, 3H), 7.59 (s, 1H), 7.87 (d, J = 8.6 Hz, 1H), 8.84 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1610, 3304 cm−1. Anal. (C24H20BrNO4 (466.32)) C, H, Br, N.
(6-Chloro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (29)
Synthesized as 7, starting from 55. Yield: 67%. Mp: 188–190 °C (from ethanol). 1H NMR (CDCl3): δ 3.63 (s, 6H), 3.79 (s, 3H), 6.93 (s, 2H), 7.20–7.24 (m, 4H), 7.31–7.33 (m, 2H), 7.41–7.43 (m, 1H), 7.91 (d, J = 8.6 Hz, 1H), 8.84 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3319, 1618 cm−1. Anal. (C24H20ClNO4 (421.87)) C, H, Cl, N.
(6-Fluoro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (32)
Synthesized as 7, starting from 56. Yield: 25% as an oil. 1H NMR (CDCl3): δ 3.68 (s, 6H), 3.80 (s, 3H), 6.94 (s, 2H), 7.05 (t, J = 8.9 Hz, 1H), 7.14 (d, J = 9.0 Hz, 1H), 7.26–7.33 (m, 5H), 7.99–8.02 (m, 1H), 8.58 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1621, 3265 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, F, N.
(6-Methoxy-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (34)
Synthesized as 7, starting from 57. Yield: 63%. Mp: 186–188 °C (from toluene). 1H NMR (DMSO-d6): δ 3.59 (s, 9H), 3.83 (s, 3H), 6.80 (s, 2H), 6.85 (dd, J = 2.3 and 8.8 Hz, 1H), 6.97 (d, J = 2.2 Hz, 1H), 7.24–7.26 (m, 3H), 7.32–7.34 (m, 2H), 7.79 (d, J = 8.8 Hz, 1H), 12.01 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1610, 3302 cm−1. Anal. (C25H23NO5 (417.45)) C, H, N.
(7-Bromo-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (36)
Synthesized as 7, starting from 58. Yield: 58%. Mp: 188–190 °C (from ethanol). 1H NMR (CDCl3): δ 3.71 (s, 6H), 3.82 (s, 3H), 6.97 (s, 2H), 7.18 (t, J = 7.8 Hz, 1H), 7.28–7.32 (m, 3H), 7.42–7.44 (m, 2H), 7.48 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H), 8.67 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1618, 3258 cm−1. Anal. (C24H20BrNO4 (466.32)) C, H, Br, N.
(7-Chloro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (38)
Synthesized as 7, starting from 59. Yield: 64%. Mp: 148–150 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.61 (s, 6H), 6.80 (s, 2H), 7.20–7.29 (m, 4H), 7.34 (dd, J = 1.0 and 7.6 Hz, 1H), 7.38–7.41 (m, 2H), 7.88 (dd, J = 1.0 and 8.0 Hz, 1H), 12.41 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1607, 3215 cm−1. Anal. (C24H20ClNO4 (421.87)) C, H, Cl, N.
(7-Fluoro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (41)
Synthesized as 7, starting from 60. Yield: 65%. Mp: 172–175 °C (from toluene). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.61 (s, 6H), 6.81 (s, 2H), 7.07–7.18 (m, 2H), 7.26–7.27 (m, 3H), 7.38–7.40 (m, 2H), 7.72 (d, J = 7.8 Hz, 1H), 12.60 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1615, 3254 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, F, N.
(7-Methoxy-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (43)
Synthesized as 7, starting from 61. Yield: 20%. Mp: 185–188 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.60 (s, 6H), 3.98 (s, 3H), 6.79 (s, 2H), 6.83 (d, J = 7.4 Hz, 1H), 7.11 (t, J = 7.9 Hz, 1H), 7.20–7.23 (m, 3H), 7.35–7.38 (m, 2H), 7.47–7.49 (m,1H), 12.24 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1606, 3270 cm−1. Anal. (C25H23NO5 (417.45)) C, H, N.
(6,7-Dichloro-2-phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (45)
Synthesized as 7, starting from 62. Yield: 23%. Mp: 218–220 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.61 (s, 6H), 6.81 (s, 2H), 7.25–7.32 (m, 3H), 7.39–7.42 (m, 3H), 7.86 (d, J = 8.5 Hz, 1H), 12.58 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1607, 3271 cm−1. Anal. (C24H19Cl2NO4S (456.32)) C, H, Cl, N, S.
General Procedure for the Preparation of 10, 20, 25, 30, and 39. Example: 4-Chloro-2-phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (10)
A borane–tetrahydrofuran complex (1.0 mL, 1.0 M in tetra-hydrofuran) was slowly added to a cold solution of 9 (0.1 g, 0.24 mmol) in anhydrous acetonitrile (1.4 mL) containing anhydrous methanol (0.02 mL) under an Ar stream. The reaction was stirred at 50 °C for 1 h. After cooling, the mixture was carefully diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by flash chromatography (silica gel, ethyl acetate/n-hexane as eluent) to furnish 10 (0.01 g, 10%). Mp: 178–180 °C (from ethanol). 1H NMR (CDCl3): δ 3.74 (s, 6H), 3.84 (s, 3H), 4.45 (s, 2H), 6.44 (s, 2H), 7.10–7.15 (m, 2H), 7.33 (dd, J = 2.2 and 6.8 Hz, 1H), 7.41–7–47 (m, 3H), 7.51–7.53 (m, 2H), 8.29 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3358 cm−1. Anal. (C24H22ClNO3 (407.89)) C, H, Cl, N.
5-Chloro-2-phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (20)
Synthesized as 10, starting from 19. Yield: 41%. Mp: 172–175 °C (from ethanol). 1H NMR (CDCl3): δ 3.76 (s, 6H), 3.82 (s, 3H), 4.16 (s, 2H), 6.41 (s, 2H), 7.15 (dd, J = 2.0 and 8.6 Hz, 1H), 7.31 (d, J = 8.6 Hz, 1H), 7.37–7.39 (m, 1H), 7.42–7.46 (m, 3H), 7.51–7.54 (m, 2H), 8.23 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3340 cm−1. Anal. (C24H22ClNO3 (407.89)) C, H, Cl, N.
5-Methoxy-2-phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (25)
Synthesized as 10, starting from 24. Yield: 26%. Mp: 137–139 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.60 (s, 6H), 3.72 (s, 3H), 4.14 (s, 2H), 6.48 (s, 2H), 6.76 (dd, J = 2.4 and 8.9 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 7.27 (d, J = 8.9 Hz, 1H), 7.35–7.39 (m, 1H), 7.50 (t, J = 7.4 Hz, 2H), 7.61–7.64 (m, 2H), 11.12 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3361 cm−1. Anal. (C25H25NO4 (403.47)) C, H, N.
6-Chloro-2-phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (30)
Synthesized as 10, starting from 29. Yield: 41%. Mp: 187–185 °C (from ethanol). 1H NMR (CDCl3): δ 3.73 (s, 6H), 3.84 (s, 3H), 4.21 (s, 2H), 6.44 (s, 2H), 7.07 (dd, J = 2.0 and 8.4 Hz, 1H), 7.38–7.42 (m, 3H), 7.45–7.49 (m, 2H), 7.53–7.56 (m, 2H), 8.25 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3332 cm−1. Anal. (C24H22ClNO3 (407.89)) C, H, Cl, N.
7-Chloro-2-phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (39)
Synthesized as 10, starting from 38. Yield: 41%. Mp: 156–158 °C (from ethanol). 1H NMR (CDCl3): δ 3.74 (s, 6H), 3.83 (s, 3H), 4.22 (s, 2H), 6.44 (s, 2H), 7.05 (t, J = 7.8 Hz, 1H), 7.22–7–24 (m, 1H), 7.39–7.45 (m, 2H), 7.48–7.51 (m, 2H), 7.59–7.61 (m, 2H), 8.34 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3347 cm−1. Anal. (C24H22ClNO3 (407.89)) C, H, Cl, N.
Preparation of 5-Bromo-2-phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (17)
A mixture of 16 (0.25 g, 0.54 mmol), triethylsilane (0.14 g, 0.19 mL, 1.2 mmol), and trifluoroacetic acid (0.63 g, 0.41 mL, 5.5 mmol) in 1,2-dichloroethane (2.0 mL) was placed into the microwave cavity (closed vessel mode, Pmax =250 psi). A starting microwave irradiation of 250 W was used, the temperature being ramped from 25 to 250 °C, with rapid stirring and cooling. Once 250 °C was reached, taking about 2 min, the reaction mixture was held at this temperature for 20 min. The reaction mixture was diluted with a saturated aqueous solution of NaHCO3 and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by flash chromatography (silica gel, ethyl acetate/n-hexane as eluent) to furnish 17 (0.04 g, 15%) as a colorless oil. 1H NMR (CDCl3): δ 3.73 (s, 6H), 3.83 (s, 3H), 4.17 (s, 2H), 6.42 (s, 2H), 7.28 (s, 2H), 7.37–7.47 (m, 3H), 7.53–7.55 (m, 2H), 7.62 (s, 1H), 8.25 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3338 cm−1. Anal. (C24H22BrNO3 (452.34)) C, H, Br, N.
Synthesis of Compounds 46–62
General Procedure for the Preparation of 46, 54, and 56. Example: 4-Bromo-2-phenyl-1H-indole (46)
A mixture of 66 (0.1 g, 0.4 mmol), tin(II) chloride dihydrate (1.35 g, 6 mmol), and 1 N HCl (0.92 mL) in glacial acetic acid (4.80 mL) was heated at reflux temperature for 12 h. After cooling, the reaction mixture was diluted with a saturated aqueous solution of potassium carbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, chloroform as eluent) to furnish 46 (0.02 g, 19%). Mp: 98–100 °C, lit.41 100–102 °C.
6-Bromo-2-phenyl-1H-indole (54)
Synthesized as 46, starting from 67. Yield: 46%. Mp: 188–190 °C (from ethanol), lit.42 187 °C.
6-Fluoro-2-phenyl-1H-indole (56)
Synthesized as 46, starting from 68. Yield: 24%. Mp: 170–171 °C (from ethanol), lit.43 171–172 °C.
General Procedure for the Preparation of 47–49, 55, 57, and 61. Example: 4-Chloro-2-phenyl-1H-indole (47)
A solution of 69 (2.0 g, 8 mmol) in anhydrous tetrahydrofuran (120 mL) was cooled at −40 °C, and then a solution of tert-butyllithium (9.4 mL, 16 mmol, 7.7 M in pentane) was added dropwise under an Ar stream. The reaction mixture was stirred at 0 °C for 1 h and at 25 °C for 12 h, diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, dichloromethane as eluent) to furnish 47 (0.56 g, 31%). Mp: 73–76 °C (from ethanol), lit.41 73–75 °C.
4-Fluoro-2-phenyl-1H-indole (48)
Synthesized as 47 starting from 70. Yield: 52%. Mp: 60–62 °C (from ethanol), lit.41 62–64 °C.
4-Methoxy-2-phenyl-1H-indole (49)
Synthesized as 47, starting from 71. Yield: 34%. Mp: 100–103 °C (from ethanol), lit.44 103–105 °C.
6-Chloro-2-phenyl-1H-indole (55)
Synthesized as 47, starting from 72. Yield: 18%. Mp: 176–177 °C (from ethanol), lit.45 180–181 °C.
6-Methoxy-2-phenyl-1H-indole (57)
Synthesized as 47, starting from 73. Yield: 31%. Mp: 170–173 °C (from ethanol), lit.46 173–176 °C.
7-Methoxy-2-phenyl-1H-indole (61)
Synthesized as 47, starting from 74. Yield: 28%. Mp: 83–87 °C (from ethanol), lit.47 85–86 °C.
General Procedure for the Preparation of 50–52, 58, 60, and 62. Example: 5-Bromo-2-phenyl-1H-indole (50)
Compound 75 (0.80 g, 2.8 mmol) was added by portions to polyphosphoric acid (8.0 g) preheated at 110 °C. The reaction mixture was stirred at the same temperature for 1 h and then quenched on crushed ice. The solid was filtered and crystallized from ethanol to give 75 (0.65 g, 82%). Mp: 190–192 °C, lit.48 193–196 °C.
5-Chloro-2-phenyl-1H-indole (51)
Synthesized as 50, starting from 76. Yield: 37%. Mp: 198–200 °C (from ethanol), lit.49 203–204 °C.
5-Fluoro-2-phenyl-1H-indole (52)
Synthesized as 50, starting from 77. Yield: 37%. Mp: 180–185 °C (from ethanol), lit.50 181–183 °C.
7-Bromo-2-phenyl-1H-indole (58)
Synthesized as 50, starting from 78. Yield: 14%. Mp: 115–117 °C (from ethanol), lit.51 117–118 °C. 1H NMR (DMSO-d6): δ 6.97 (t, J = 7.7, 1H), 7.02 (s, 1H), 7.31–7.38 (m, 2H), 7.47 (t, J = 7.5 Hz, 2H), 7.56 (d, J = 7.8 Hz, 1H), 7.98–8.00 (m, 2H), 11.36 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3436 cm−1.
7-Fluoro-2-phenyl-1H-indole (60)
Synthesized as 50, starting from 79. Yield: 25%. Mp: 122–125 °C (from ethanol). 1H NMR (DMSO-d6): δ 6.91–7.00 (m, 3H), 7.33–7.38 (m, 2H), 7.47 (t, J = 7.5 Hz, 2H), 7.95 (d, J = 7.4 Hz, 2H), 11.85 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3436 cm−1.
6,7-Dichloro-2-phenyl-1H-indole (62)
Synthesized as 50, starting from 80. Yield: 27%. Mp: 105–108 °C (from ethanol). 1H NMR (DMSO-d6): δ 7.03 (s, 1H), 7.21 (t, J = 8.4 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 7.46–7.55 (m, 3H), 7.99 (d, J = 8.0 Hz, 2H), 11.71 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3248 cm−1.
5-Methoxy-2-phenyl-1H-indole (53) was synthesized according to a literature52 procedure.
7-Chloro-2-phenyl-1H-indole (59) was synthesized according to a literature53 procedure.
Synthesis of Compounds 63–80
General Procedure for the Preparation of 63–65. Example: 2-(2-Bromo-6-nitrophenyl)-1-phenylethanol (63)
To a solution of benzaldehyde (0.12 g, 0.1 mL, 1.1 mmol) and 2-bromo-6-nitrotoluene (0.25 g, 1.2 mmol) in anhydrous DMSO (5 mL) was added a solution of sodium ethoxide in anhydrous ethanol (0.32 mL, 0.43 M). The reaction mixture was stirred at 25 °C for 12 h, carefully diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:3 as eluent) to furnish 63 (0.15 g, 40%) as an oil. 1H NMR (DMSO-d6): δ 3.21–3.24 (m, 2H), 4.64 (d, J = 7.4 Hz, 1H), 5.48 (br s, disappeared on treatment with D2O, 1H), 7.25–7.33 (m, 5H), 7.42 (t, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.2 Hz, 1H) ppm. IR: ν 2961, 3030, 3063, 3325, 3560 cm−1.
2-(4-Bromo-2-nitrophenyl)-1-phenylethanol (64)
Synthesized as 63, starting from 4-bromo-1-methyl-2-nitrobenzene. Yield: 44% as an oil. 1H NMR (DMSO-d6): δ 3.07–3.12 (m, 2H), 4.69–4.73 (m, 1H), 5.43 (d, J = 4.5 Hz, disappeared on treatment with D2O, 1H), 7.21–7.25 (m, 1H), 7.28–7.33 (m, 4H), 7.39 (d, J = 8.3 Hz, 1H), 7.80 (dd, J = 1.7 and 8.2 Hz, 1H), 8.08–8.10 (m, 1H) ppm. IR: ν 2934, 3030, 3065, 3418 cm−1.
2-(4-Fluoro-2-nitrophenyl)-1-phenylethanol (65)
Synthesized as 63, starting from 4-fluoro-1-methyl-2-nitrobenzene. Yield: 44% as an oil. 1H NMR (DMSO-d6): δ 3.12–3.14 (m, 2H), 4.68–4.73 (m, 1H), 5.40 (d, J = 4.4 Hz, disappeared on treatment with D2O, 1H), 7.22–7.33 (m, 5H), 7.46–7.51 (m, 2H), 7.81 (dd, J = 2.3 and 9.0 Hz, 1H) ppm. IR: ν 2932, 3032, 3087, 3404 cm−1.
General Procedure for the Preparation of 66–68. Example: 2-(2-Bromo-6-nitrophenyl)-1-phenylethanone (66)
A solution of 63 (0.1 g, 0.3 mmol) in anhydrous dichloromethane (1.0 mL) was added to a suspension of pyridinium chlorochromate (0.10 g, 0.47 mmol) in the same solvent (2.0 mL). The reaction mixture was stirred at 25 °C for 1.5 h and diluted with water. The layers were separated, and the organic phase was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, chloroform:petroleum ether = 1:1 as eluent) to furnish 66 (0.06 g, 40%). Mp: 113–115 °C (from ethanol). 1H NMR (DMSO-d6): δ 4.89 (s, 2H), 7.52–7.62 (m, 3H), 7.70 (t, J = 7.8 Hz, 1H), 8.05–8.10 (m, 4H) ppm. IR: ν 1680 cm−1.
2-(4-Bromo-2-nitrophenyl)-1-phenylethanone (67)
Synthesized as 66, starting from 64. Yield: 40%. Mp: 114–116 °C (from ethanol). 1H NMR (DMSO-d6): δ 4.87 (s, 2H), 7.53–7.60 (m, 3H), 7.70 (t, J = 7.1 Hz, 1H), 7.96–7.98 (m, 1H), 8.05 (d, J = 7.4 Hz, 2H), 8.28–8.30 (m, 1H) ppm. IR: ν 1686 cm−1.
2-(4-Fluoro-2-nitrophenyl)-1-phenylethanone (68)
Synthesized as 66, starting from 65. Yield: 16%. Mp: 95–98 °C (from ethanol). 1H NMR (DMSO-d6): δ 4.88 (s, 2H), 7.56–7.72 (m, 5H), 8.03–8.06 (m, 3H) ppm. IR: ν 1680 cm−1.
General Procedure for the Preparation of 69–74. Example: N-(3-Chloro-2-methylphenyl)benzamide (69)
A solution of benzoyl chloride (7.93 g, 6.55 mL, 56 mmol) in anhydrous tetrahydrofuran (20 mL) was added dropwise to a solution of 3-chloro-2-methylaniline (6.68 g, 5.64 mL, 47 mmol) and triethylamine (5.70 g, 7.85 mL, 56 mmol) at 0 °C in the same solvent (134 mL). The reaction was heated at reflux for 2 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was triturated with diethyl ether to furnish 69 (5.66 g, 49%). Mp: 168–170 °C, lit.54 170 °C. 1H NMR (CDCl3): δ 2.40 (s, 3H), 7.21 (t, J = 8.0 Hz, 1H), 7.26–7.29 (m, 1H), 7.51–7.55 (m, 2H), 7.58–7.62 (m, 1H), 7.75–7.80 (m, 2H; one proton disappeared after treatment with D2O), 7.90–7.92 (m, 2H) ppm. IR: ν 1647, 3244 cm−1.
N-(3-Fluoro-2-methylphenyl)benzamide (70)
Synthesized as 69, starting from 3-fluoro-2-methylaniline. Yield: 67%. Mp: 150–152 °C (from ethanol), lit.55 157–158 °C. 1H NMR (CDCl3): δ 2.25 (s, 3H), 6.93 (t, J = 9.0 Hz, 1H), 7.20–7.27 (m, 1H), 7.52 (t, J = 7.7 Hz, 2H), 7.59 (t, J = 7.3 Hz, 1H), 7.71–7.75 (m, 2H; one proton disappeared after treatment with D2O), 7.88–7.91 (m, 2H) ppm. IR: ν 1670, 3230 cm−1.
N-(3-Methoxy-2-methylphenyl)benzamide (71)
Synthesized as 69, starting from 3-methoxy-2-methylaniline. Yield: 82%. Mp: 173–175 °C (from ethanol), lit.56 177 °C. 1H NMR (DMSO-d6): δ 2.06 (s, 3H), 3.82 (s, 3H), 6.89 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 7.8 Hz, 1H), 7.19 (t, J = 8.1 Hz, 1H), 7.53 (t, J = 7.6 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.98 (d, J = 7.2 Hz, 2H), 9.89 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1649, 3229 cm−1.
N-(5-Chloro-2-methylphenyl)benzamide (72)
Synthesized as 69, starting from 5-chloro-2-methylaniline. Yield: 58%. Mp: 125–127 °C (from ethanol), lit.57 121–123 °C.
N-(5-Methoxy-2-methylphenyl)benzamide (73)
Synthesized as 69, starting from 5-methoxy-2-methylaniline. Yield: 88%. Mp: 113–115 °C (from ethanol). 1H NMR (DMSO-d6): δ 2.17 (s, 3H), 3.74 (s, 3H), 6.77 (dd, J = 2.6 and 8.4 Hz, 1H), 6.98 (d, J = 2.6 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 7.53 (t, J = 7.6 HZ, 2H), 7.59 (t, J = 7.3 Hz, 1H), 7.98 (d, J = 7.1 Hz, 2H), 9.83 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1650, 3299 cm−1.
N-(2-Methoxy-6-methylphenyl)benzamide (74)
Synthesized as 69, starting from 2-methoxy-6-methylaniline. Yield: 82%. Mp: 127–130 °C (from ethanol). 1H NMR (DMSO-d6): δ 2.50 (s, 3H), 3.74 (s, 3H), 6.87–6.93 (m, 2H), 7.20 (t, J = 8.0 Hz, 1H), 7.49–7.60 (m, 3H), 8.00 (d, J = 7.3 Hz, 2H), 9.57 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 1647, 3360 cm−1.
General Procedure for the Preparation of 75–80. Example: 1-(4-Bromophenyl)-2-(1-phenylethylidene)hydrazine (75)
A mixture of (4-bromophenyl)hydrazine hydrochloride (3.35 g, 15 mmol), aceto-phenone (1.20 g, 1.16 mL, 10 mmol), and sodium acetate (1.23 g, 15 mmol) in ethanol (15 mL) was placed into the microwave cavity (open vessel mode). Microwave irradiation of 250 W was used, the temperature being ramped from 25 to 80 °C. Once 80 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 5 min, with stirring and cooling. The reaction mixture was cooled to 0 °C, filtered, washed with petroleum ether, and dried to give 76 (0.90 g, 30%). Mp: 118–120 °C (from ethanol). 1H NMR (CDCl3): δ 2.25 (s, 3H), 7.08 (d, J = 8.9 Hz, 2H), 7.35–7.41 (m, 6H; one proton disappeared after treatment with D2O), 7.83 (d, J = 8.5 Hz, 2H) ppm. IR: ν 3353 cm−1.
1-(4-Chlorophenyl)-2-(1-phenylethylidene)hydrazine (76)
Synthesized as 75, starting from (4-chlorophenyl)hydrazine hydrochloride. Yield: 25%. Mp: 105–107 °C (from ethanol), lit.58 100–102 °C. 1H NMR (CDCl3): δ 2.25 (s, 3H), 7.12 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H), 7.31–7.35 (m, 2H; one proton disappeared after treatment with D2O), 7.40 (t, J = 7.0 Hz, 2H), 7.79 (d, J = 7.1 Hz, 2H) ppm. IR: ν 3352 cm−1.
1-(4-Fluorophenyl)-2-(1-phenylethylidene)hydrazine (77)
Synthesized as 75, starting from (4-fluorophenyl)hydrazine hydrochloride. Yield: 95%. Mp: 110–112 °C (from ethanol), lit.59 107 °C. 1H NMR (DMSO-d6): δ 2.25 (s, 3H), 7.00–7.10 (m, 2H), 7.22–7.29 (m, 3H), 7.38 (t, J = 7.3 Hz, 2H), 7.78 (d, J = 7.2 Hz, 2H), 9.31 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3380 cm−1.
1-(2-Bromophenyl)-2-(1-phenylethylidene)hydrazine (78)
Synthesized as 75, starting from (2-bromophenyl)hydrazine hydrochloride. Yield: 86%. Mp: >300 °C (from ethanol). Spectral data were consistent with those reported in the literature.60
1-(2-Fluorophenyl)-2-(1-phenylethylidene)hydrazine (79)
Synthesized as 75, starting from (2-fluorophenyl)hydrazine hydrochloride. Yield: 68%. Mp: >300 °C (from ethanol). 1H NMR (DMSO-d6): δ 2.32 (s, 3H), 6.79–6.84 (m, 1H), 7.13–7.17 (m, 2H), 7.34–7.43 (m, 3H), 7.56–7.59 (m, 1H), 7.80–7.83 (m, 2H), 8.64 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3380 cm−1.
1-(2,3-Dichlorophenyl)-2-(1-phenylethylidene)hydrazine (80)
Synthesized as 75, starting from (2,3-dichlorophenyl)hydrazine hydrochloride. Yield: 79%. Mp: 112–115 °C. 1H NMR (DMSO-d6): δ 2.36 (s, 3H), 7.09 (dd, J = 1.4 and 7.9 Hz, 1H), 7.31 (t, J = 8.4, 1H), 7.37–7.45 (m, 3H), 7.60 (dd, J = 1.3 and 8.4 Hz, 1H), 7.84–7.87 (m, 2H), 8.40 (br s, disappeared on treatment with D2O, 1H) ppm. IR: ν 3270 cm−1.
Molecular Modeling
All molecular modeling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 14. The tubulin structure was downloaded from the Protein Data Bank (http://www.rcsb.org/, PDB codes 1SA06 and 4O2A29).6 Ligand structures were built with MOE61 and minimized using the MMFF94x force field until an RMSD gradient of 0.05 kcal mol−1 Å−1 was reached. The docking simulations were performed using PLANTS18 on the 1SA0 crystal structure.
Biology
Tubulin Assembly
The reaction mixtures contained 0.8 M monosodium glutamate (pH 6.6 with HCl in a 2 M stock solution), 10μM tubulin, 4% (v/v) DMSO, and varying concentrations of drug. Following a 15 min preincubation at 30 °C, samples were chilled on ice, GTP to 0.4 mM was added, and turbidity development was followed at 350 nm in a temperature-controlled recording spectrophotometer for 20 min at 30 °C. The extent of reaction was measured. Full experimental details were previously reported.62
[3H]Colchicine Binding Assay
The reaction mixtures contained 1.0 μM tubulin, 5.0 μM [3H]colchicine, and 5.0 μM inhibitor and were incubated for 10 min at 37 °C. Complete details were described previously.63
Cell Cultures
Cell lines were obtained from the American Type Culture Collection (ATCC), unless otherwise specified. MCF-7 breast carcinoma, OVCAR-8, and NCI/ADR-RES cells were obtained from the National Cancer Institute drug screening laboratory, and NB4 cells and MV4-11 cells from the Deutsche Sammlung von Mikroorganismen and Zellkulturen. All cell lines, except as indicated, were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 20 mM HEPES, 100 U/mL penicillin, 100 mg/mL streptomycin, and 1% L-glutamine; specific requirements include the addition of sodium pyruvate (1–2% for RD rhabdomyo-sarcoma, HepG2 hepatoma, and the three MDA breast carcinoma cell lines) and glucose (1 g/L for RD and HepG2, 4.5 g/L for PC3 prostate carcinoma). Cell lines were cultured at 37 °C in 5% CO2/95% air in a humidified incubator. Treatments were initiated 24 h after cell seeding using ATI compound diluted in 0.1% DMSO, the indicated reference compound, or 0.1% DMSO vehicle, for 24–72 h as indicated.
Cell Viability Assays
For MCF-7 breast carcinoma and OVCAR-8 and NCI/ADR-RES ovary carcinoma cells, the methodology for evaluation of growth was previously described, except that cells were grown for 96 h for IC50 determinations.64
MV4–11, NB4, A-549, NCI-H1975, Messa, and Messa/Dx5 cells were seeded into 96-well plates (Corning Inc., Costar) at a density of 2 × 103 cells/well in 50 μL of the appropriate medium. For the MDA-MB-468, MDA-MB-436, and MDA-MB-231 breast carcinoma cell lines, the cells were plated in 100 μL of medium in 96-well plates at a density of 3.5 × 103 cells/well for MDA-MB-468, 3 × 103 cells/well for MDA-MB-436, and 2 × 103 cells/well for MDA-MB-436. After 24 h, the cells were treated with the inhibitor (0.39–100 nM) and then evaluated in MTT assays as described.65 Statistical analysis was performed by analysis of variance (ANOVA) with Neumann–Keul’s multiple comparison test or teh Kolmogorov–Smirnov test where appropriate.
For the PC-3, HepG2, and RD cell lines, the cells were seeded in 24-well plates at a density of 95 × 103 cells/100 μL well (PC-3 and RD) or 120 × 103 cells/100 μL well (HepG2). After 24 h, the test compound was added (0.01–25 μM) for 48 h. After removal of the medium, MTT was added (500 μM final concentration in 500 μL/well of phosphate-buffered saline (PBS)) and incubation continued at 37 °C for 2 h in the dark. The formazan crystals were dissolved in 2-propanol containing 0.04 N HCl (200 μL). A550 in the wells was determined using a Multiskan Spectrum Thermo Electron Corp. reader. IC50 values were calculated by nonlinear regression analysis (GraphPad Prism statistics software). Experiments were performed in triplicate. For HeLa cells, 7 × 103 cell aliquots were seeded in a flat-bottom 96-well tissue culture plate and, after 24 h, were exposed to the inhibitor (10–100 nM) for 24 or 48 h. MTT (10 μL, 5 mg/mL) (Sigma-Aldrich) was added to each well, and the cells were further incubated for 3 h at 37 °C. After solubilization of the crystals with 2-propanol/0.04 N HCl, A570 measurements were made with an ELISA reader, and IC50 values were derived from dose–response curves.
For the MV4–11, NB4 (AML), A-549, and NCI-H1975 (lung adenocarcinoma) cell lines, cell growth was measured using CellTiter-Flor (Promega), a nonlytic, single-reagent-addition fluorescence assay that detects the relative number of living cells in samples after experimental manipulation. The CellTiter-Fluor cell viability assay measures the conserved and constitutive protease activity within live cells and, therefore, acts as a marker for cell viability. NB4 and MV4–11 cells in exponential growth were incubated for 48 h with different concentrations of the inhibitors. After 48 h, CellTiter-Fluor reagent was added to the cell culture medium (1:1, v/v) and incubated for at least 90 min at 37 °C. A549 and NCI-H1975 cells were treated with the inhibitor for 72 h, and then CellTiter-Fluor reagent was added to one-fifth of the culture medium volume. Fluorescence was recorded (excitation wavelength, 360 nm; emission wavelength, 535 nm), and the IC50 was calculated using GraphPad software.
For the Messa and Messa/Dx5 (resistant) sarcoma cell lines, the CellTiter-Glo luminescent cell viability assay was used (Promega, Madison, WI). Cells in exponential growth were incubated for 72 h with different concentrations of the inhibitor, and then the same volume of CellTiter-Glo reagent was added. The solution was stirred for 2 min to induce cell lysis. Luminescence was recorded after an additional 10 min. IC50 values were calculated using nonlinear regression analysis (GraphPad Prism statistics software).
For T98G and U343MG cells, growth was measured by a colorimetric MTS conversion assay, as previously reported.15
For D283 medulloblastoma cells, 3 × 105 D283 cells/well were plated in a 24-multiwell dish. After 24 h, ATI derivative 44 or 81 (1 μM) was added to the cells for the indicated time. Viability was evaluated with a trypan blue assay.
Antibodies and Immunostaining
The following unconjugated monoclonal antibodies (mAb’s) were used for immunostaining: anti-MICA (MAB159227), anti-MICB (MAB236511), anti-ULBP1 (MAB170818), anti-ULBP2 (MAB165903), and anti-ULBP3 (MAB166510) from R&D Systems (Minneapolis, MN), anti-PVR (SKII.4) kindly provided by Prof M. Colonna (Washington University, St Louis, MO), anti-Nec-2 (R2.525) from BD Pharmingen (San Diego, CA), and allophycocyanin (APC)-conjugated goat affinity purified F(ab′)2 fragment to mouse IgG (GAM) from Jackson ImmunoResearch Laboratories (West Grove, PA).
Flow Cytometry Analysis
A total of 3 × 105 HeLa cells were seeded in tissue culture dishes. After ATI treatment, cell numbers were counted using a Z1 Coulter particle counter (Beckman Coulter). Cell cycle phase distribution was analyzed in permeabilized cells incubated with PI (Sigma-Aldrich P4170, 0.04 mg/mL). SS and FL-3 parameters were acquired on a linear amplification scale, and FS and Fl2 on a log scale. Cell aggregates were gated out on the biparametric graph FL-3lin/ratio. Apoptosis was determined as the proportion of cells exhibiting a DNA content lower than 2 N after gating out cell debris on the biparametric graph FS/SS using the WinMDI software. Cell death was analyzed in 200 000 cell aliquots in binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) incubated with annexin V–FITC (Immunological Sciences, IK-11120) alone or annexin V–FITC in combination with PI in the absence of permeabilizing agents. Cell samples were analyzed in a Coulter Epics XL cytofluorometer (Beckman Coulter) equipped with EXPO 32 ADC software. At least 10 000 cells per sample were acquired.
IF and Image Analysis
Cells were seeded on sterile polylysine-coated coverslips placed in tissue culture plates. After treatment with ATI or VBL, as indicated, the cells were fixed with 3.7% para-formaldehyde in PBS for 10 min at room temperature and then permeabilized in 0.1% Triton-X100 in PBS for 5 min. Blocking and antibody reactions were carried out in PBS/0.05% Tween 20 containing 3% BSA at room temperature using mouse anti-α-tubulin (1:2000, B-5-1-2, Sigma-Aldrich) followed by FITC-conjugated goat antimouse IgG (Jackson ImmunoResearch Laboratories). Chromosomal DNA was stained with 4,6-diamidino-2-phenylindole (DAPI; 0.1 μg/mL) and mounted in Vectashield (Vector Laboratories). Images were analyzed using a Nikon Eclipse 90i microscope equipped with a Qicam Fast 1394 CCD camera (Qimaging). To resolve MT remnants or unstructured tubulin foci, some of the acquired images were deconvoluted and analyzed using the extended depth of focus on Z-serial optical sections using a Nis-Elements AR 4.2 (Nikon).
IF and Flow Cytometry Ligand Analysis
NKG2D and DNAM-1 ligand surface expression on HeLa cells was analyzed by IF staining using anti-MICA, anti-MICB, anti-ULBP1/2/3, anti-PVR, or anti-Nec2 unconjugated mAb’s, followed by secondary GAM/APC. Samples were analyzed using a FACSCanto II (BD Biosciences, San Jose, CA). Flow cytometric analysis was performed using the FlowJo software, version 8.8.7 (TreeStar, Ashland, OR).
Degranulation Assay
NK cell-mediated cytotoxicity was evaluated using the degranulation lysosomal marker CD107a as previously described.66 As a source of effector cells, we used human peripheral blood mononuclear cells (PBMCs) isolated from healthy donors by Lymphoprep (Nycomed, Oslo, Norway) gradient centrifugation and then cocultured for 10 days with an irradiated (30 Gy) Epstein–Barr virus (EBV)-transformed B-cell line. Cells were grown in RPMI 8866 at 37 °C in a humidified 5% CO2 atmosphere. On day 10, the cell population was routinely more than 90% CD56+CD16+CD3−, as assessed by IF and flow cytometric analysis. After a 48 h treatment with 33 or 44, HeLa cells were incubated with activated NK cells at effector:target (E:T) ratios of 1:1 in a flat-bottom 96-well tissue culture plate in complete medium (DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% FCS). The plates were incubated at 37 °C in a 5% CO2 atmosphere for 2 h. Thereafter, the cells were washed with PBS and incubated with anti-CD107a/APC (or cIgG/APC) for 45 min at 4 °C. The cells were also stained with anti-CD3/FITC and anti-CD56/PE to gate the CD3−CD56+ NK cell population.
Real-Time PCR
MICA, MICB, ULBP1, ULBP2, ULBP3, and PVR mRNA expression was analyzed by real-time PCR. Total RNA from HeLa cells was extracted using Trizol (Invitrogen) after a maximum of 24 h of drug treatment. Total RNA (1 μg) was used for cDNA first-strand synthesis using oligo-dT (Promega, Madison, WI) in a 25 μL reaction volume. To analyze ligand mRNA expression, the cDNA was amplified in triplicate with the following primers: Hs00792952_m1 for MICA, Hs00792952_m1 for MICB, Hs00197846_m1 for PVR, Hs00607609_m1 for ULBP2, Hs00225909_m1 for ULBP3, and Hs99999903_m1 for β-actin, all conjugated with fluorochrome FAM (Applied Biosystems). The level of ligand expression was measured using the threshold cycle value (Ct). The ΔCt was obtained by subtracting the Ct value of the gene of interest (MICA, MICB, or PVR) from the housekeeping gene (β-actin) Ct value. We used ΔCt of the NT sample as the calibrator. The fold change was calculated according to the formula 2−Δ ΔCt, where ΔΔCt was the difference between ΔCt of the sample and that of the calibrator (according to the formula, the value of the calibrator in each run is 1).
Hh-Dependent Luciferase Reporter Assay
The luciferase assay was performed in Shh-Light II (Shh-LII) cells, stably incorporating a Gli-responsive luciferase reporter and the pRL-TK Renilla (normalization control), treated for 48 h with SAG (200 nM) and the studied compounds. Luciferase and Renilla activity were assayed with a dual-luciferase assay system according to the manufacturer’s instructions (Promega, Madison, WI). The results are expressed as luciferase/Renilla ratios and represent the mean ± SD of three experiments, each performed in triplicate.
LC–MS/MS Analytical Method
Samples were analyzed under the following conditions: UFLC (Shimadzu) AC20 coupled with an API 3200 triple-quadrupole (ABSciex); eluents, (phase A) 95% water, 5% acetonitrile + 0.1% HCOOH, (phase B) 5% water, 95% acetonitrile + 0.1% HCOOH; flow rate, 0.3 mL/min; column, Gemini-Nx 5 μm C18 110A (50 × 2.00 mm) at 35 °C; injection volume, 5 μL. LC–MS/MS analyses were carried out using an ESI(+) interface in multiple reaction monitoring mode.
Metabolic Stability
Compounds 33 and 44 were dissolved in DMSO in duplicate at a final concentration of 1 μM and preincubated for 10 min at 37 °C in potassium phosphate buffer (pH 7.4), 3 mM MgCl2, with human or mouse liver microsomes (Xenotech) at a final concentration of 0.5 mg/mL. After the preincubation period, the reactions were started by adding the cofactor mixture (NADP, Glc6P, G6P-DH). Samples were taken at times 0, 10, 20, 30, and 60 min. Acetonitrile was added to stop the reaction and centrifuged. Supernatants were analyzed and quantified by LC–MS/MS. A control sample without cofactors was always added to check the stability of the test compounds in the reaction mixtures. The reference standards were 7-ethoxycoumarin and propranolol. A fixed concentration of verapamil was added in every sample as an internal standard for LC–MS/MS. The percentage of the area of the test compound remaining at the various incubation times was calculated with respect to the area of the compound at time 0 min.
The rate constant, k (min−1), derived for the exponential decay equation (peak area/IS vs time) was used to calculate the rate of intrinsic clearance (CL) of the compound using the following equation:
Aqueous Solubility
The solubilities of compounds 33 and 44 were measured using a high-throughput screening assay format. Samples prepared at the target concentration of 200 μM were placed in a 96-well filter plate and incubated at room temperature for 90 min. The plate was then filtered, and solutions were analyzed by LC–MS/MS. Final concentrations were evaluated by comparing the area under the curve of the MeOH stock solution with those of the test compound solutions.
Supplementary Material
Acknowledgments
This research was supported by grants from PRIN 2010-2011 (2010W7YRLZ_001 and 2010W7YRLZ_001006), PRIN 2012-2013 (2012C5YJSK002), Bando Futuro in Ricerca 2010 (RBFR10ZJQT), Progetti di Ricerca di Università, Sapienza Università di Roma (C26H135FL5 and C26A14TLFT), and the Istituto Pasteur-Fondazione Cenci Bolognetti and Grants AIRC IG 14534 (P.L.) and AIRC IG 14723 (L.D.M.). We are grateful to Giulia Guarguaglini and Italia Anna Asteriti for microscopy analysis. We also thank Professor Claudia Piccoli and Dr. Matteo Landriscina, University of Foggia, Italy, for providing the cell lines. We are grateful to Dr. Enrico Cundari for help with the flow cytometric analysis.
ABBREVIATIONS USED
- MT
microtubule
- CSA4
combretastatin A-4
- VLB
vinblastine
- VCR
vincristine
- VRB
vinorelbine
- PTX
paclitaxel
- ATI
arylthioindole
- TMP
trimethoxyphenyl
- Hh
Hedgehog
- DMSO
dimethyl sulfoxide
- SAR
structure–activity relationship
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MDR
multi-drug-resistant
- P-gp
P-glycoprotein
- NK
natural killer
- SAG
smoothened (Smo) agonist, 3-chloro-N[trans-4-(methylamino)cyclohexyl]-N-[[3-(4-pyridinyl)-phenyl]methyl]benzo[b]thiophene-2-carboxamide
- DAPI
4′,6-diamidino-2-phenylindole
- PBMC
peripheral blood mononuclear cell
- PI
propidium iodide
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
- IF
immunofluorescence
- APC
allophycocyanin
- GAM
mouse IgG
Footnotes
The authors declare no competing financial interest.
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
DEDICATION
In memory of Prof. Alberto Gulino, an outstanding scientist and a dear colleague.
ASSOCIATED CONTENT
Figures showing the proposed binding modes of 10, 18, 28, and 44, those of 33 and 40, and that of 40, correlation of MCF-7 cytotoxicity data with inhibition of tubulin assembly and inhibition of colchicine binding, MDA-MB-468, MDA-MB-436, and MDA-MB-231 cancer cell growth inhibition by compounds 33 and 44, cell cycle analysis of PC-3, RD, and HepG2 cells treated with 33, 44, or PTX for 24 h, cell growth inhibition of 33- or 44-treated T98G and U343 cells, cell growth inhibition of ATI-treated HeLa cells after a 48 h treatment, mRNA abundance by real-time PCR after a 24 h treatment with the indicated ATI at 10 nM, and effects of ATI derivatives 33, 44, and 81 on D283 cell growth, tables listing the correlation docking score vs biological activity and elemental analysis of compounds 6–45, and the simplified molecular-input line-entry system (SMILES) stereochemistries of compounds 6–45. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00310.
References
- 1.Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 3.Teicher BA. Newer cytotoxic agents: attacking cancer broadly. Clin Cancer Res. 2008;14:1610–1617. doi: 10.1158/1078-0432.CCR-07-2249. [DOI] [PubMed] [Google Scholar]
- 4.Honore S, Pasquier E, Braguer D. Understanding microtubule dynamics for improved cancer therapy. Cell Mol Life Sci. 2005;62:3039–3056. doi: 10.1007/s00018-005-5330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]
- 6.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 7.Lin MC, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding to colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 8.Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 9.Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha, beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 10.Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, Lopez JA, Andreu JM, Hamel E, Diaz JF. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat Chem Biol. 2007;3:117–125. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- 11.Beckers T, Mahboobi S. Natural, semisynthetic and synthetic microtubule inhibitors for cancer therapy. Drugs Future. 2003;28:767–785. [Google Scholar]
- 12.Mani S, Macapinlac MJ, Goel S, Verdier-Pinard D, Fojo T, Rothenberg M, Colevas D. The clinical development of new mitotic inhibitors that stabilize the microtubule. Anti-Cancer Drugs. 2004;15:553–558. doi: 10.1097/01.cad.0000131681.21637.b2. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updates. 2007;10:162–181. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 14.La Regina G, Bai R, Rensen W, Coluccia A, Piscitelli F, Gatti V, Bolognesi A, Lavecchia A, Granata I, Porta A, Maresca B, Soriani A, Iannitto ML, Mariani M, Santoni A, Brancale A, Ferlini C, Dondio G, Varasi M, Mercurio C, Hamel E, Lavia P, Novellino E, Silvestri R. Design and synthesis of 2-heterocyclyl-3-arylthio-1H-indoles as potent tubulin polymerization and cell growth inhibitors with improved metabolic stability. J Med Chem. 2011;54:8394–8406. doi: 10.1021/jm2012886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La Regina G, Bai R, Rensen WM, Di Cesare E, Coluccia A, Piscitelli F, Famiglini V, Reggio A, Nalli M, Pelliccia S, Da Pozzo E, Costa B, Granata I, Porta A, Maresca B, Soriani A, Iannitto ML, Santoni A, Li J, Cona MM, Chen F, Ni Y, Brancale A, Dondio G, Vultaggio S, Varasi M, Mercurio C, Martini C, Hamel E, Lavia P, Novellino E, Silvestri R. Towards highly potent cancer agents by modulating the C-2 group of the arylthioindole class of tubulin polymerization inhibitors. J Med Chem. 2013;56:123–149. doi: 10.1021/jm3013097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, Wilcox E, Hamel E, Artico M, Silvestri R. Arylthioindoles, potent inhibitors of tubulin polymerization. J Med Chem. 2004;47:6120–6123. doi: 10.1021/jm049360d. [DOI] [PubMed] [Google Scholar]
- 17.Liou JP, Chang YL, Kuo FM, Chang CW, Tseng HY, Wang CC, Yang YN, Chang JY, Lee SJ, Hsieh HP. Concise synthesis and structure-activity relationships of combretastatin A-4 analogues, 1-aroylindoles, and 3-aroylindoles as novel classes of potent antitubulin agents. J Med Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- 18.Korb O, Stutzle T, Exner TE. PLANTS: Application of ant colony optimization to structure-based drug design. In: Dorigo M, Gambardella LM, Birattari M, Martinoli A, Poli R, Stutzle T, editors. Ant Colony Optimization and Swarm Intelligence, Proceedings of the 5th International Workshop, ANTS. Springer; Berlin: 2006. pp. 247–258. Lecture Notes in Computer Science, Series 4150. [Google Scholar]
- 19.Coluccia A, Sabbadin D, Brancale A. Molecular modeling studies on arylthioindoles, as potent inhibitors of tubulin polymerization. Eur J Med Chem. 2011;46:3519–3525. doi: 10.1016/j.ejmech.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 20.La Regina G, Gatti V, Famiglini V, Piscitelli F, Silvestri R. Venting-while-heating microwave-assisted synthesis of 3-arylthioindoles. ACS Comb Sci. 2012;14:258–262. doi: 10.1021/co200165j. [DOI] [PubMed] [Google Scholar]
- 21.La Regina G, Sarkar T, Bai R, Edler MC, Saletti R, Coluccia A, Piscitelli F, Minelli L, Gatti V, Mazzoccoli C, Palermo V, Mazzoni C, Falcone C, Scovassi AI, Giansanti V, Campiglia P, Porta A, Maresca B, Hamel E, Brancale A, Novellino E, Silvestri R. New arylthioindoles and related bioisosteres at the sulfur bridging group. 4. Synthesis, tubulin polymerization, cell growth inhibition, and molecular modeling studies. J Med Chem. 2009;52:7512–7527. doi: 10.1021/jm900016t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapleo CB, Butler RCM, England DC, Myers PL, Roach AG, Smith CFC, Stillings MR, Tulloch IF. Heteroaromatic analogs of the α2-adrenoreceptor partial agonist clonidine. J Med Chem. 1989;32:1627–1630. doi: 10.1021/jm00127a037. [DOI] [PubMed] [Google Scholar]
- 23.Erker T. Chemistry of thieno-annelated O,N- and S,N-containing heterocyclic compounds. Part 14. Syntheses of thieno[2,3-b][1,4]-thiazepine derivatives with calcium antagonistic activity. Sci Pharm. 1996;64:345–352. [Google Scholar]
- 24.Kermack WO, Perkin WH, Jr, Harmine Robinson R, harmaline V. Synthesis of norharman. J Chem Soc, Trans. 1921;119:1602–1642. [Google Scholar]
- 25.Houlihan WJ, Parrino VA, Uike Y. Lithiation of N-(2-alkylphenyl)alkanamides and related compounds. A modified Madelung indole synthesis. J Org Chem. 1981;46:4511–4515. [Google Scholar]
- 26.La Regina G, Gatti V, Piscitelli F, Silvestri R. Open vessel and cooling while heating microwave-assisted synthesis of pyridinyl N-aryl hydrazones. ACS Comb Sci. 2011;13:2–6. doi: 10.1021/co100015b. [DOI] [PubMed] [Google Scholar]
- 27.Robinson B. The Fischer Indole Synthesis. J. Wiley and Sons; Chichester, U.K: 1982. [Google Scholar]
- 28.Warren LW, Andrews CW, Capelli AM, Clarke B, La Londe J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 29.Prota AE, Danel F, Bachmann F, Bargsten K, Buey RM, Pohlmann J, Reinelt S, Lane H, Steinmetz MO. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J Mol Biol. 2014;426:1848–1860. doi: 10.1016/j.jmb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 30.(a) Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]; (b) Small-Molecule Drug Discovery Suite 2015–2: Glide, version 6.7. Schrödinger, LLC; New York: 2015. [Google Scholar]
- 31.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cavenee WK. Accumulation of genetic defects during astrocytoma progression. Cancer. 1992;70:1788–1793. doi: 10.1002/1097-0142(19920915)70:4+<1788::aid-cncr2820701621>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 33.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simonart T, Andrei G, Parent D, Van Vooren JP, De Clercq E, Snoeck R. In vitro sensitivity of Kaposi’s sarcoma cells to various chemotherapeutic agents including acyclic nucleoside phosphonates. Antiviral Chem Chemother. 1999;10:129–134. doi: 10.1177/095632029901000304. [DOI] [PubMed] [Google Scholar]
- 35.Ryu HH, Jung S, Jung TY, Moon KS, Kim IY, Jeong YI, Jin SG, Pei J, Wen M, Jang WY. Role of metallothionein 1E in the migration and invasion of human glioma cell lines. Int J Oncol. 2012;41:1305–1313. doi: 10.3892/ijo.2012.1570. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C, Wang Y, Zhou Z, Zhang J, Tian Z. Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases their susceptibility to NK lysis. Cancer Immunol Immunother. 2009;58:1275–1285. doi: 10.1007/s00262-008-0645-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.La Regina G, Bai R, Coluccia A, Famiglini V, Pelliccia S, Passacantilli S, Mazzoccoli C, Ruggieri V, Sisinni L, Bolognesi A, Rensen WM, Miele A, Nalli M, Alfonsi R, Di Marcotullio L, Gulino A, Brancale A, Novellino E, Dondio G, Vultaggio S, Varasi M, Mercurio C, Hamel E, Lavia P, Silvestri R. New pyrrole derivatives with potent tubulin polymerization inhibiting activity as anticancer agents including Hedgehog-dependent cancer. J Med Chem. 2014;57:6531–6552. doi: 10.1021/jm500561a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. 2005;202:1001–1012. doi: 10.1084/jem.20051143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19:1410–1422. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- 40.MacDonald TJ, Aguilera D, Castellino RC. The rationale for targeted therapies in medulloblastoma. Neuro Oncol. 2014;16:9–20. doi: 10.1093/neuonc/not147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guilarte V, Castroviejo MP, Garcia-Garcia P, Fernandez-Rodriguez MA, Sanz R. Approaches to the synthesis of 2,3-dihaloanilines. Useful precursors of 4-functionalized-1H-indoles. J Org Chem. 2011;76:3416–3437. doi: 10.1021/jo200406f. [DOI] [PubMed] [Google Scholar]
- 42.Valois-Escamilla I, Alvarez-Hernandez A, Rangel-Ramos LF, Suarez-Castillo OR, Ayala-Mata F, Zepeda-Vallejo G. Synthesis of 6-bromo-2-arylindoles using 2-iodobenzoic acid as precursor. Tetrahedron Lett. 2011;52:3726–3728. [Google Scholar]
- 43.Wang H, Li Y, Jiang L, Zhang R, Jin K, Zhao D, Duan C. Ready synthesis of free N-H 2-arylindoles via the copper-catalyzed amination of 2-bromo-arylacetylenes with aqueous ammonia and sequential intramolecular cyclization. Org Biomol Chem. 2011;9:4983–4986. doi: 10.1039/c1ob05549f. [DOI] [PubMed] [Google Scholar]
- 44.Sanz R, Castroviejo MP, Guilarte V, Perez A, Fananas FJ. Regioselective Synthesis of 4- and 7-alkoxyindoles from 2,3-dihalophenols: application to the preparation of indole inhibitors of phospholipase A2. J Org Chem. 2007;72:5113–5118. doi: 10.1021/jo070643y. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki N, Yasaki S, Yasuhara A, Sakamoto T. Convenient indole synthesis from 2-iodoanilines and terminal alkynes by the sequential Sonogashira reaction and the cyclization reaction promoted by tetrabutylammonium fluoride (TBAF) Chem Pharm Bull. 2003;51:1170–1173. doi: 10.1248/cpb.51.1170. [DOI] [PubMed] [Google Scholar]
- 46.Shen M, Leslie BE, Driver TG. Dirhodium(II)-catalyzed intramolecular C-H amination of aryl azides. Angew Chem, Int Ed. 2008;47:5056–5059. doi: 10.1002/anie.200800689. [DOI] [PubMed] [Google Scholar]
- 47.(a) Sridharan V, Perumal S, Avendano C, Menendez JC. Microwave-assisted, solvent-free Bischler indole synthesis. Synlett. 2006;1:91–95. [Google Scholar]; (b) Candito DA, Lautens M. Exploiting the chemistry of strained rings: synthesis of indoles via domino reaction of aryl iodides with 2H-azirines. Org Lett. 2010;12:3312–3315. doi: 10.1021/ol100975b. [DOI] [PubMed] [Google Scholar]
- 48.Deprez NR, Kalyani D, Krause A, Sanford MS. Room temperature palladium-catalyzed 2-arylation of indoles. J Am Chem Soc. 2006;128:4972–4973. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]
- 49.Davies IW, Smitrovich JH, Sidler R, Qu C, Gresham V, Bazaral C. A highly active catalyst for the reductive cyclization of ortho-nitrostyrenes under mild conditions. Tetrahedron. 2005;61:6425–6437. [Google Scholar]
- 50.Maizuru N, Inami T, Kurahashi T, Matsubara S. Nickel-catalyzed cycloaddition of anthranilic acid derivatives to alkynes. Org Lett. 2011;13:1206–1209. doi: 10.1021/ol200090g. [DOI] [PubMed] [Google Scholar]
- 51.Carlin RB, Larson GW. The Fischer indole synthesis. IV. Halogen interchange during zinc halide induced Fischer reactions of acetophenone 2,6-dihalophenylhydrazones. J Am Chem Soc. 1957;79:934–941. [Google Scholar]
- 52.Kaczor AA, Kronbach C, Unverferth K, Pihlaja K, Wiinamaki K, Sinkkonen J, Kijkowska-Murak U, Wrobel T, Stachal T, Matosiuk D. Novel non-competitive antagonists of kainate GluK1/GluK2 receptors. Lett Drug Des Discovery. 2012;9:891–898. [Google Scholar]
- 53.Da Settimo F, Simorini F, Taliani S, La Motta C, Marini AM, Salerno S, Bellandi M, Novellino E, Greco G, Cosimelli B, Da Pozzo E, Costa B, Simola N, Morelli M, Martini C. Anxiolytic-like effects of N,N-dialkyl-2-phenylindol-3-ylglyoxylamides by modulation of translocator protein promoting neurosteroid biosynthesis. J Med Chem. 2008;51:5798–5806. doi: 10.1021/jm8003224. [DOI] [PubMed] [Google Scholar]
- 54.Wibaut JP. Quantitative researches on the nitration of chlorotoluenes. Recl Trav Chim Pays-Bas Belg. 1913;32:244–320. [Google Scholar]
- 55.Barben IK, Suschitzky H. Heterocyclic fluorine compounds. IV. Monofluoroindazoles. J Chem Soc. 1960:672–676. [Google Scholar]
- 56.Ruggli P, Leonhardt W, Isatogens V. Hydroxyhydroquinol derivatives of the isatogen series. Helv Chim Acta. 1924;7:689–702. [Google Scholar]
- 57.Katritzky AR, Hayden AE, Kirichenko K, Pelphrey P, Ji Y. A Novel route to imidoylbenzotriazoles and their application for the synthesis of enaminones. J Org Chem. 2004;69:5108–5111. doi: 10.1021/jo0496594. [DOI] [PubMed] [Google Scholar]
- 58.Ghiglieri-Bertez C, Coquelet C, Alazet A, Bonne C. Dual inhibitors of the cyclooxygenase and lipoxygenase pathways: synthesis and activity of hydrazone derivatives. Eur J Med Chem. 1987;22:147–152. [Google Scholar]
- 59.Suschitzky H. Fluorine-substituted phenylhydrazines. J Chem Soc. 1953:3326–3327. [Google Scholar]
- 60.Woelfle H, Kopacka H, Wurst K, Preishuber-Pfluegl P, Bildstein B. On the way to biodegradable poly(hydroxyl butyrate) from propylene oxide and carbon monoxide via β-butyrolactone: Multisite catalysis with newly designed chiral indole-imino chromium(III) complexes. J Organomet Chem. 2009;694:2493–2512. [Google Scholar]
- 61.Molecular Operating Environment (MOE 2007.09) Chemical Computing Group, Inc; Montreal, Quebec, Canada: [Google Scholar]
- 62.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 63.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;35:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 64.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A, Gray-Goodrich M, Campbell H, Mayo J, Boyd M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor-cell lines. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 65.Caraglia M, Marra M, Leonetti C, Meo G, D’Alessandro AM, Baldi A, Santini D, Tonini G, Bertieri R, Zupi G, Budillon A, Abbruzzese A. R115777 (Zarnestra)/Zoledronic acid (Zometa) cooperation on inhibition of prostate cancer proliferation is paralleled by Erk/Akt inactivation and reduced Bcl-2 and Bad phosphorylation. J Cell Physiol. 2007;211:533–543. doi: 10.1002/jcp.20960. [DOI] [PubMed] [Google Scholar]
- 66.Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, Peruzzi G, Ricciardi MR, Petrucci MT, Cippitelli M, Santoni A. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol. 2014;193:950–960. doi: 10.4049/jimmunol.1400271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.