

Abstract
The Hippo pathway plays a major role in organ size control, and its dysregulation contributes to tumorigenesis. The major downstream effectors of the Hippo pathway are the YAP/TAZ transcription co-activators, which are phosphorylated and inhibited by the Hippo pathway kinase LATS1/2. Here, we report a novel mechanism of TAZ regulation by the tight junction protein PARD3. PARD3 promotes the interaction between PP1A and LATS1 to induce LATS1 dephosphorylation and inactivation, therefore leading to dephosphorylation and activation of TAZ. The cytoplasmic, but not the tight junction complex associated, PARD3 is responsible for TAZ regulation. Our study indicates a potential molecular basis for cell growth-promoting function of PARD3 by modulating the Hippo pathway signaling in response to cell contact and cell polarity signals.
Keywords: Hippo pathway, PARD3, TAZ, tight junction
Introduction
The first Hippo pathway component Warts was discovered in Drosophila by genetic mosaic screens for growth regulators 1. The same approach was used to discover several other components of Hippo pathway, including Hippo, Sav, and Mob. Yki, the downstream effector, was identified by yeast two-hybrid screen. The Hippo pathway is highly conserved in mammals, in both pathway components and functions in organ size control and tissue homeostasis 2. Key components of Hippo pathway consist of a kinase cascade of MST1/2 (Hippo homologs) and LATS1/2 (Warts homologs), downstream transcription co-activator YAP/TAZ (Yki homologs), and the transcription factor TEADs (Sd homologs). Forming complex with activated Mob1, LATS1/2 are phosphorylated and activated by MST1/2, the active LATS1/2 phosphorylate and inhibit YAP/TAZ. LATS1/2-dependent phosphorylation retains YAP/TAZ in cytoplasm and promotes YAP/TAZ degradation. Therefore, phosphorylation provides an important mechanism for YAP regulation (inhibition). The dephosphorylated YAP/TAZ are localized in cell nucleus and function as transcription co-activators to induce gene expression. However, YAP/TAZ have no DNA binding domain. Several YAP/TAZ target transcription factors have been identified. Among them, the TEAD family transcription factors are the most important to mediate the growth stimulating function of YAP/TAZ 3.
PARD3 is a PDZ-domain-containing scaffold protein that forms a trimetric complex with PAR6 and atypical protein kinase C (aPKC) to regulate the initial cell polarity cues 4. Localized to the tight junctions, the PAR complex plays a critical role in cell polarity. It is required for neuroblast and epithelial polarization during Drosophila embryogenesis, and regulates various modes of polarization during neuronal development, migration, and tight junction formation in vertebrates 5-7,8,9. PARD3 contains several evolutionarily conserved regions (CRs). The N-terminal domain CR1 is important for apical localization and dimerization of PARD3; the central CR2 consists of three PDZ domains that can interact with proteins with PDZ binding motifs; the CR3 is responsible for the binding and inhibition of aPKC and the C-terminal coil–coil domain 10.
PARD3 has been implicated to act as an invasion and metastasis suppressor. Inhibiting PARD3 causes loss of cell polarity and induces breast tumorigenesis and metastasis 11,12. However, there are other reports showing that PARD3 may function as an oncogene 13. Therefore, PARD3 may have dual functions, either promotion or suppression, in tumorigenesis in a manner dependent on cancer types. In this report, we show that PARD3 activates YAP/TAZ to promote cell growth.
Results and Discussion
PARD3 stimulates YAP/TAZ dephosphorylation
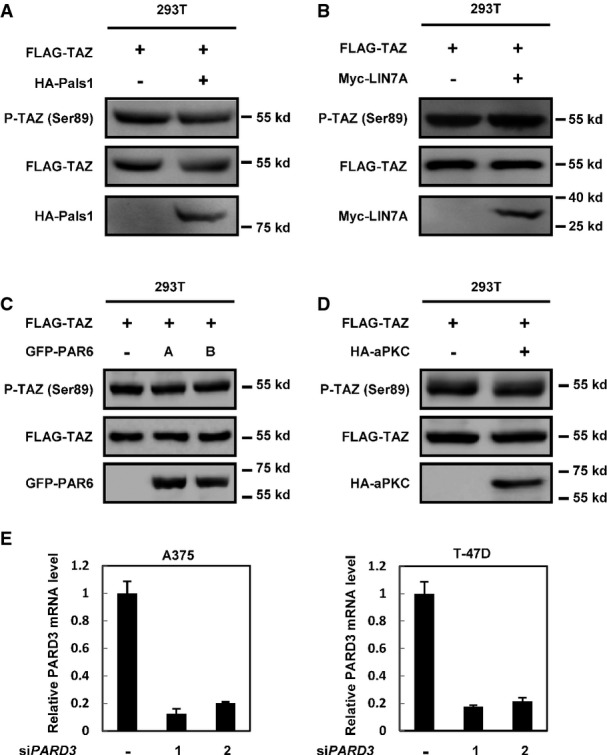
In order to investigate TAZ regulation, we performed TAZ affinity purification and mass spectrometry (MS) and identified multiple putative TAZ-interacting proteins 3. Notably, many polarity proteins and tight junction proteins were identified as TAZ-interacting proteins, results consistent with previous reports 14,15. We then tested whether these putative interacting proteins regulated TAZ. Interestingly, overexpression of PARD3 reduced the phosphorylation of TAZ at Ser89 (Fig1A), which is important for TAZ cytoplasmic localization and inhibition 16, while expression of other tight junction proteins had no significant effect on TAZ phosphorylation (Fig EV1A and B), suggesting that PARD3 has a unique function in TAZ regulation. YAP is a TAZ homolog and similarly regulated by the Hippo pathway. We found that PARD3 expression also reduced YAP2 (a splicing form of YAP) phosphorylation at Ser127 (Fig1B), which is important for YAP cytoplasmic localization. These data suggest that PARD3 activates YAP/TAZ by reducing phosphorylation.
Figure 1.

PARD3 decreases the phosphorylation and induces the activation of YAP/TAZ
- PARD3 dephosphorylates TAZ at Ser89. HEK293T cells were transiently transfected with HA-PARD3 and FLAG-TAZ. Cells were harvested for Western blot analysis.
- PARD3 dephosphorylates YAP2 at Ser127. HEK293T cells were transiently transfected with the HA-PARD3 and FLAG-YAP2 as indicated; the cells were harvested for Western blot analysis.
- Knockdown of PARD3 increases the phosphorylation of TAZ at Ser89 at low but not at high cell density. A375 cells were transfected with two different siRNAs for PARD3 under different cell densities (four times more cells were plated for high density than the low density), and cells were harvested for Western blot analysis.
- Knockdown of PARD3 increases the phosphorylation of YAP at Ser127 at low cell density. T-47D cells were transiently transfected with the indicated siRNAs at different cell densities (4:1), and the cells were harvested for Western blot analysis as indicated.
- Co-expression of PARD3 enhances the activity of the transcriptional co-activator TAZ. Plasmids encoding the indicated proteins or empty PRK7 vector were cotransfected in HEK293T cells together with the TEAD reporter plasmids as indicated in Materials and Methods. The results are average ± SEM of three independent experiments. *P < 0.05, **P < 0.01.
PARD3 dephosphorylates and inactivates YAP/TAZ
- A, B Overexpression of tight junction proteins Pals1 (A) or LIN7A (B) has no effect on the phosphorylation of TAZ on Ser89. HEK293T cells were transiently transfected with HA-Pals1 and Myc-LIN7A in the presence of TAZ as indicated, and the cells were harvested for Western blot analysis.
- C, D The PAR complex components have no effects on TAZ phosphorylation. HA-aPKC and GFP-PAR6A/B were transfected into HEK293T cells, and cell lysates were harvested for Western blot analysis.
- E Verification of PARD3 siRNA knockdown efficiency in A375 and T-47D cells. Samples were detected by real-time PCR. The results are average ± SEM of three independent experiments.
PARD3 forms a polarity complex with PAR6 and the atypical protein kinase C (aPKC). Both PAR6 and aPKC play indispensable roles in the regulation of various cell polarization events 17. We examined the role of PAR6 and aPKC in the regulation of TAZ and observed that expression of either PAR6 or aPKC had no effect on the phosphorylation of TAZ Ser89 (FigEV1C and D). Next, we knocked down PARD3 using two different short hairpin RNAs in the A375 melanotic melanoma cell line and the T-47D breast cancer cell line. The knockdown efficiency was confirmed by real-time PCR (FigEV1E). At low cell density, PARD3 knockdown increased endogenous TAZ phosphorylation at Ser89, while a similar PARD3 knockdown had no effect on TAZ phosphorylation when A375 cells were cultured at high density (Fig1C). Similar observations were made with endogenous YAP phosphorylation in response to PARD3 knockdown in T-47D cell line (Fig1D). These data support a role of endogenous PARD3 in YAP/TAZ regulation. Moreover, our data suggest a cell density-dependent effect of PARD3 in regulating YAP/TAZ phosphorylation.
Next, we examined the functional consequences of PARD3 on YAP/TAZ activity using a well-established GAL4-TEAD luciferase reporter, which is directly regulated by YAP/TAZ activation status 16. As expected, co-expression of PARD3 with TAZ augmented the GAL4-TEAD luciferase reporter activity in comparison with TAZ expression alone (Fig1E). Together, the above observations indicate a physiological role of PARD3 in stimulating TAZ activity.
PARD3 promotes TAZ nuclear localization
Phosphorylation of TAZ at Ser89 creates a binding site for 14-3-3, which binds to and retains TAZ in cytoplasm. The above phosphorylation study suggests that PARD3 should inhibit the interaction between TAZ and 14-3-3. We performed co-immunoprecipitation (Co-IP) experiments with TAZ antibody and detected that overexpression of PARD3 inhibited the interaction between endogenous TAZ and 14-3-3 (Fig2A). Consistently, cell fractionation experiments showed that TAZ protein levels were reduced in the cytoplasm and enriched in the nucleus in the presence of PARD3 (Fig2B). Moreover, when PARD3 was knocked down at low cell density, more TAZ protein was detected in the cytoplasmic fraction (Fig2C). The immuno-fluorescence experiments further confirmed that knockdown of PARD3 shifted TAZ to the cytoplasm (Fig2D). These results are consistent with the phosphorylation data that PARD3 inhibits TAZ S89 phosphorylation. Phosphorylation of TAZ at residues Ser314 is required for binding of β-TrCP, a subunit of the SCF E3 ubiquitin ligase, and promotes TAZ ubiquitination and degradation 18. We had also found that PARD3 expression reduced the interaction between TAZ and β-TrCP (Fig2E). In addition, PARD3 enhanced the binding of TAZ and TEAD4 transcriptional factor (Fig2F). Collectively, these data indicate that PARD3 promotes TAZ nuclear localization and association with TEAD4.
Figure 2.

PARD3 promotes TAZ translocation into the nucleus
- PARD3 decreases TAZ binding to 14-3-3. The indicated constructs were transfected into HEK293T cells. Immunoprecipitation with TAZ antibody was performed as indicated.
- PARD3 overexpression increases TAZ nuclear localization. Cell fractionation experiments were performed in PARD3-overexpressing T-47D cells as indicated for Western blot analysis.
- Left: PARD3 knockdown decreases TAZ/YAP nuclear localization at low cell density. Cell fractionation experiments were performed in PARD3-knockdown T-47D cells as indicated. Right: Validation of siRNAs directed against PARD3. T-47D cells were transfected with the indicated siRNA. Forty-eight hours after transfection, the remaining expression level of PARD3 was determined using real-time PCR.
- PARD3 knockdown promotes TAZ to shift to the cytoplasm. T-47D cells were stained with TAZ antibody, and DAPI stains cell nuclei.
- PARD3 decreases the interaction of endogenous TAZ with β-TrCP. 293T cells were transiently transfected with HA-PARD3. The samples were analyzed by Western blot after immunoprecipitation with TAZ antibody.
- PARD3 increases the interaction between endogenous TAZ and TEAD4. 293T cells were transiently transfected with HA-PARD3. The samples were analyzed by Western blot after immunoprecipitation with TAZ antibody.
The aPKC binding region of PARD3 inhibits LATS1 by promoting its association with PP1A and dephosphorylation
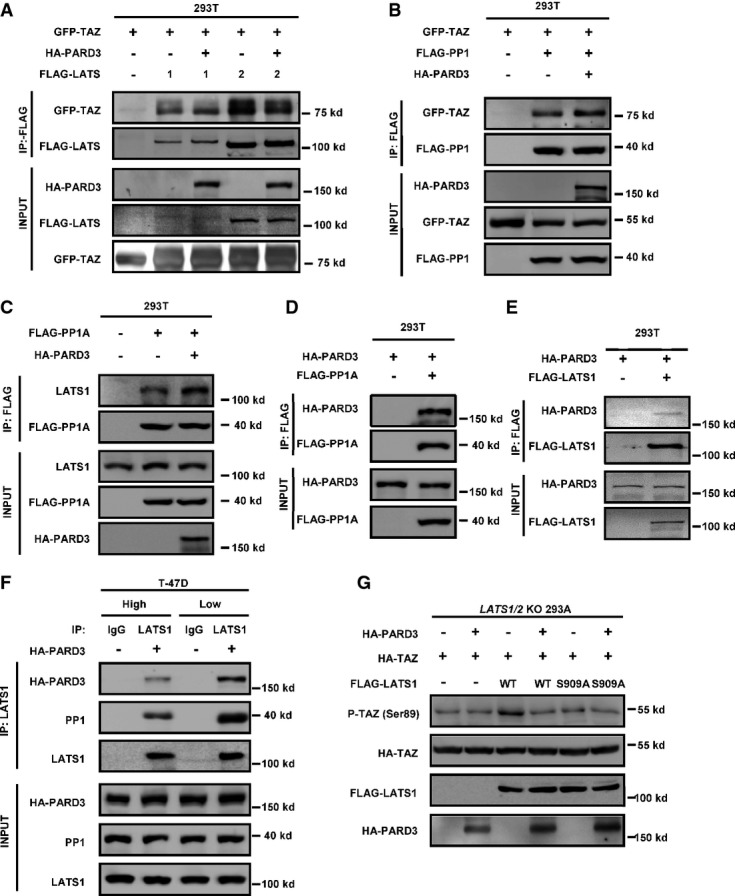
We wanted to explore the mechanism how PARD3 inhibits TAZ, which is phosphorylated by LATS1/2 and dephosphorylated by PP1A 19. We performed Co-IP experiments and detected that PARD3 neither reduced the interaction of TAZ with LATS1 nor increased the TAZ interaction with PP1A (FigEV2A and B). Interestingly, PARD3 enhanced the interaction of endogenous PP1A and LATS1 (Fig3A). Reciprocal Co-IP experiments also showed that PARD3 strengthened the association between PP1A and LATS1 (Fig EV2C). These results suggested that PARD3 may inhibit TAZ phosphorylation by inhibiting LATS1, which is activated by phosphorylation. Moreover, we also found that PARD3 interacted with PP1A and LATS1 (FigEV2D and E), suggesting a possibility that PARD3 may act as a scaffold to facilitate the association between LATS1 and PP1A. To this end, we examined the effect of PP1A on LAST1 phosphorylation at Ser909, which is the site required for LATS1 kinase activation 20. As expected, overexpression of PARD3 reduced the phosphorylation of endogenous LATS1 at Ser909 (Fig3B). In the previous section (Fig1C and D), we noted that the effect of PARD3 on YAP/TAZ phosphorylation was cell density dependent. Interestingly, an increased PP1/PARD3/LATS1 interaction was observed at low cell density in T-47D cells (Fig EV2F). Furthermore, we found that when PARD3 was knocked down, the phosphorylation level of LATS1 on Ser909 was increased under low cell density, while no change was detected at high density under which LATS1 was already activated (Fig3C). Expression of PARD3 is shown in Fig EV1E. In order to directly measure the kinase activity of LATS1, we performed an in vitro kinase assay using immunopurified LATS1 and bacterially purified His-TAZ protein as a substrate. We observed that PARD3 co-expression reduced LATS1 kinase activity toward TAZ (Fig3D), demonstrating that LATS1 kinase activity was inhibited by PARD3 co-expression. The TAZ reporter assay also indicated that PARD3 suppressed the inhibitory effect of LATS1 on TAZ (Fig3E).
PARD3 interacts with LATS1 and PP1A
- A PARD3 has no effect on the interaction between TAZ and its kinase LATS1. HEK293T cells were transfected with GFP-tagged TAZ and FLAG-tagged LATS1 in the presence or absence of HA-PARD3, and proteins were immunoprecipitated with FLAG beads for Western blot analysis.
- B PARD3 does not affect the binding of GFP-TAZ and its phosphatase PP1A. The indicated constructs were transfected into HEK293T cells, and proteins were immunoprecipitated with FLAG beads for Western blot analysis.
- C PARD3 enhances the interaction of PP1A with endogenous LATS1. HEK293T cells were transfected with FLAG-tagged PP1A in the presence or absence of HA-PARD3, and proteins were immunoprecipitated with FLAG beads for Western blot analysis.
- D, E PARD3 interacts with PP1A and LATS1. The indicated constructs were transfected into HEK293T cells, and proteins were immunoprecipitated with FLAG antibody. Cell lysates were analyzed by Western blot as indicated.
- F The interaction of PP1/PARD3/LATS1 is regulated by cell density. HA-PARD3 was transfected into T-47D cells, immunoprecipitation with LATS1 antibody was performed, and the immunoprecipitated samples were used for Western blot analysis as indicated.
- G LATS1 S909A mutant is insensitive to PARD3 regulation. WT and S909A were cotransfected into LATS1/2 265.55 KO 293A cells with HA-TAZ. Western blot analysis was performed as indicated.
Figure 3.

The aPKC binding region of PARD3 dephosphorylates LATS1 at Ser909 to decrease the kinase activity of LATS1
- PARD3 increases the binding of endogenous LATS1 and PP1A. The HA-PARD3 construct was transfected into HEK293T cells. Endogenous LATS1 was immunoprecipitated and subjected to Western blot analysis with indicated antibodies.
- PARD3 reduces the autophosphorylation of endogenous LATS1. HEK293T cells were transfected with the indicated plasmids. Western blot analysis was performed using a phospho-LATS1 (Ser909) antibody after immunoprecipitation with LATS1 antibody.
- Knockdown of PARD3 increases the phosphorylation of LATS1 at Ser909 at low cell density. T-47D cells were transiently transfected with PARD3 siRNAs in different cell densities (4:1), immunoprecipitated with LATS1 antibody for Western blot analysis. The expression of PARD3 is shown in FigEV1E.
- LATS1 activity is inhibited in the absence of PARD3. The indicated plasmids were transfected into HEK293T cells. LATS1 immunoprecipitated with FLAG antibody was subjected to in vitro kinase assay using His-TAZ purified from E. coli as a substrate. The products were analyzed by Western blotting as indicated.
- PARD3 increases the activity of TAZ with ectopically expressed LATS1. Plasmids encoding the indicated proteins were cotransfected in HEK293T cells together with the TEAD reporter plasmids. The luciferase assay showed that even if TAZ was inhibited by LATS1, PARD3 still activated TAZ. The results are average ± SEM of three independent experiments. *P < 0.05, **P < 0.01.
- PARD3 does not dephosphorylate TAZ in LATS1/2 KO cells. The indicated constructs were transfected into LATS1/2 KO 293A cells. The phosphorylation of TAZ was analyzed by Western blot as indicated.
- Schematic illustration of structure and deletion constructs of PARD3. Numbers refer to amino acid positions. aPKC-BR, aPKC binding region; CR1, conserved region 1; PDZ, PSD-95/Dlg/ZO-1.
- 3N construct (the aPKC binding region) is sufficient to dephosphorylate TAZ at Ser89. The indicated constructs were transiently transfected into HEK293T cells. Cell lysates were analyzed by Western blot as indicated.
- The 3N construct increases the interaction between endogenous LATS1 and PP1A. The indicated constructs were transfected into HEK293T cells, after immunoprecipitation with LATS1 antibody, and Western blot analysis was performed as indicated.
- LATS1 activity is inhibited in the absence of WT and 3N constructs of PARD3. LATS1 immunoprecipitated with FLAG antibody was subjected to in vitro kinase assay. The products were analyzed by Western blotting as indicated.
Next, we examined whether LATS is required for PARD3 to modulate TAZ phosphorylation. For this purpose, we examined the LATS1/2 KO (knockout) HEK293 cells that were created by CRISPR/Cas9 method. Consistent with our model, PARD3 expression had no effect on TAZ phosphorylation in the LATS1/2 KO cells (Fig3F), indicating that LATS1/2 are essential in TAZ regulation by PARD3. Furthermore, we observed that PARD3 still induced TAZ dephosphorylation in the wild-type LATS1, but not the LATS1 S909A mutant, rescued LATS1/2 KO cells (Fig EV2G). Together, these results support a model that PARD3 inhibits LATS1 activity through increasing its interaction with PP1A, eventually leading to TAZ dephosphorylation and activation.
PARD3 consists of four domains: CR1 (1N), PDZ domains (2N), aPKC binding region (aPKC-BR, refer to 3N), and coiled-coil region (4N) (Fig3G). Using deletion mutants, we showed that the aPKC-BR domain was sufficient to inhibit TAZ phosphorylation at Ser89, while the other domains had no effect (Fig3H). We further performed co-immunoprecipitation experiments and found that overexpression of the aPKC-BR domain of PARD3 increased the binding of endogenous LATS1 with PP1A and reduced the phosphorylation of LATS1 at Ser909 (Fig3I). The in vitro kinase assays also confirmed that ectopic expression of PARD3 aPKC-BR domain inhibited LATS1 kinase (Fig3J). Collectively, these data indicate that the aPKC binding region of PARD3 is sufficient to inhibit LATS1 by promoting LATS1 and PP1A interaction.
Phosphorylation of Ser144/873 modulates PARD3 activity
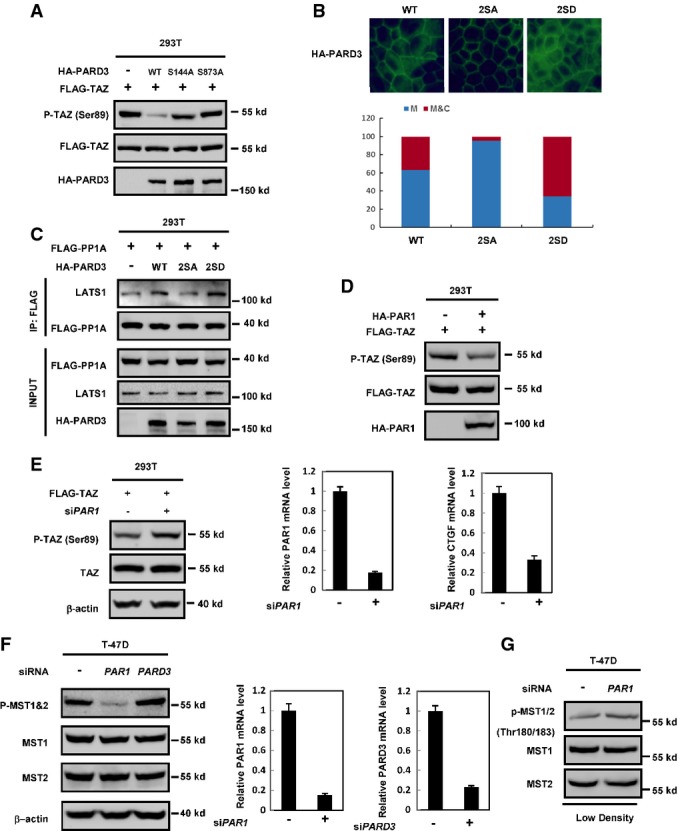
Numerous studies have shown that PARD3 is regulated by phosphorylation. Phosphorylation of PARD3 at Ser144 and Ser873 by the serine/threonine kinase PAR1 disrupts epithelial apical–basal polarity and promotes multilumen cyst formation 21. aPKC phosphorylates PARD3 at Ser827 to regulate epithelial tight junction 22, while RhoA stimulates PARD3 phosphorylation at Thr833 to disrupt PAR complex formation 23. Moreover, PARD3 is also phosphorylated by Aura A at Ser962 to regulate its function in neuronal polarity 24. We investigated whether these phosphorylation events might influence the activity of PARD3 in Hippo pathway regulation. PARD3 with mutations of the various phosphorylation sites were transfected into HEK293T cells, Western blot analysis revealed that the PARD3 S144A or S873A mutant was defective to inhibit TAZ phosphorylation when compared to the wild-type PARD3, while the other PARD3 mutants could still reduce TAZ phosphorylation (Figs4A and EV3A). Ser144 and Ser873 in PARD3 are phosphorylated by PAR1, and phosphorylation of these residues releases PARD3 from the PAR complex at tight junctions into the cytoplasm 21. To further test the function of PARD3 Ser144 and Ser873 phosphorylation, double mutants of phosphomimetic 2SD (S144D/S873D) and non-phosphorylatable 2SA (S144A/S873A) were generated. We first confirmed that wild type and 2SD mutant of PARD3 localized in both membrane and cytoplasm, while 2SA mutant mainly localized to plasma membrane (FigEV3B). We found that PARD3 2SA mutant was inactive to decrease TAZ phosphorylation in the cotransfection experiments, while the PARD3 2SD could strongly inhibit TAZ phosphorylation (Fig4B). In addition, ectopic expression of wild-type PARD3, but not the 2SA mutant, reduced the phosphorylation of endogenous LATS1 at Ser909 and increased the interaction between LATS1 and endogenous PP1A (Figs4C and EV3C). We further determined LATS1 kinase activity and found that wild-type PARD3, but not the 2SA mutant, decreased LATS1 activity (Fig4D).
Figure 4.

Phosphorylation of PARD3 at Ser144/Ser873 diminishes TAZ dephosphorylation
- S144A and S873A mutations of PARD3 diminish TAZ dephosphorylation. PARD3 was reported to be regulated by phosphorylation. The indicated constructs that contain six point mutations of PARD3 were cotransfected with FLAG-TAZ into HEK293T. Western blot analysis was performed as indicated.
- Wild type and 2SD but not 2SA mutant of PARD3 decreases the phosphorylation of TAZ. The indicated constructs were transiently transfected into HEK293T cells. Western blot analysis was performed as indicated.
- The wild type but not 2SA mutant of PARD3 increases the binding of LATS1 and PP1A and decreases the phosphorylation of LATS1 at Ser909. The indicated constructs were transfected into HEK293T cells, and proteins were immunoprecipitated with LATS1 antibody. Cell lysates were analyzed by Western blot as indicated.
- LATS1 activity is not inhibited in the absence of 2SA mutant of PARD3. The indicated plasmids were transfected into HEK293T cells. LATS1 immunoprecipitated with FLAG antibody was subjected to in vitro kinase assay. The products were analyzed by Western blotting as indicated.
Phosphorylation of PARD3 at Ser144/Ser873 diminishes TAZ dephosphorylation
- S144A and S873A of PARD3 diminish phosphorylation of TAZ at Ser89. The indicated constructs were cotransfected with FLAG-TAZ into HEK293T cells. Western blot analysis was performed as indicated.
- Immunofluorescence localization of PARD3 WT, 2SD mutant, and 2SA mutant. MDCK cells stably expressing WT, 2SA, or 2SD of PARD3 were stained with HA antibody. The quantification is shown below. 2SA mutant but not the WT and 2SD mutant of PARD3 decreases interaction between PP1A and endogenous LATS1. The indicated constructs were transfected into HEK293T cells, and proteins were immunoprecipitated with FLAG antibody. Cell lysates were analyzed by Western blot.
- Wild type and 2SD but not 2SA mutant of PARD3 increases the interaction between FLAG-PP1A and endogenous LATS1. The indicated constructs were transfected into HEK293T cells, and proteins were immunoprecipitated with FLAG antibody. Cell lysates were analyzed by Western blot as indicated.
- PAR1 induces TAZ dephosphorylation at Ser89. HEK293T cells were transiently transfected with HA-PAR1 and FLAG-TAZ, and the cells were harvested for Western blot analysis.
- Knockdown of PAR1 increases the phosphorylation of TAZ and inhibits the expression of CTGF. 293T cells were transfected with siRNA targeting PAR1, the phosphorylation of TAZ was detected by Western blot, and the expression of CTGF was detected using real-time PCR.
- Knockdown of PAR1 but not PARD3 decreases the phosphorylation of MST1/2 at Thr183/180. T-47D cells were transfected with the indicated siRNAs, and cells were harvested at high density for Western blot analysis. The knockdown efficiencies were determined by real-time PCR on the right.
- Knockdown of PAR1 increases the phosphorylation of MST1/2 at Thr183/180 slightly. T-47D cells were transfected with the indicated siRNAs, and cells were harvested at low density for Western blot analysis.
It was reported that PAR1 inhibited kinase activity of Hippo in Drosophila 25. We examined whether PAR1 also regulates the phosphorylation of TAZ. Overexpression of PAR1 decreased TAZ phosphorylation while silencing of PAR1 increased the TAZ phosphorylation and inhibited the expression of TAZ target gene CTGF (FigEV3D and E). However, PARD3 did not affect MST1/2 phosphorylation (FigEV3F). These data indicate that phosphorylation at S144/S873 promotes PARD3 translocalization from tight junctions to cytosol and the cytoplasmic PARD3 promotes the interaction between LATS1 and PP1A, resulting in LATS1 dephosphorylation and inactivation. Notably, Huang et al 25 reported that overexpression of PAR1 inhibited Hpo-Thr195 phosphorylation and activity in fly cells; however, we detected that knockdown PAR1 reduced MST1/2 Thr180/183 phosphorylation (FigEV3F). Our experiment was performed at high cell density, under which MST should be active. However, if we performed PAR1 knockdown at low cell density, the phosphorylation of MST1/2 at Thr180/183 was increased slightly (FigEV3G). The above results indicate that the effect of PAR1 on MST Thr180/183 phosphorylation is cell density dependent. As PAR1 was reported to regulate several cell contact proteins, we speculate that effect of PAR1 knockdown on MST phosphorylation might be due to a consequence of altered cell contact, especially at high cell density.
PARD3 regulates YAP/TAZ function in gene expression and cell growth
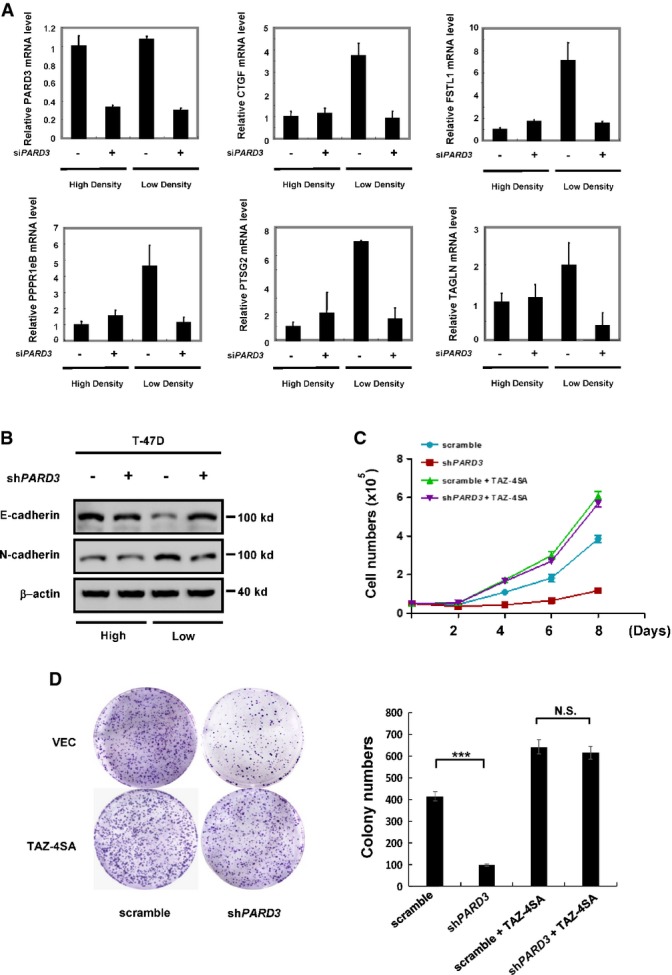
As a transcriptional co-activator, TAZ exerts its biological function by modulating target genes expression. We utilized a PARD3 siRNA oligo that was verified above and determined that knockdown of PARD3 significantly reduced the mRNA level of TAZ target gene CTGF, FSTL1, PPPR1eB, PTSG2, and TAGLN when cells were cultured at low density (Fig5A). However, PARD3 knockdown did not significantly affect the expression of these TAZ target genes under high cell density. Interestingly, WT and 2SD mutant of PARD3 could rescue the expression of TAZ target genes in PARD3 depletion cells, while the 2SA mutant did not (Fig EV4A). Moreover, PARD3 knockdown in T-47D cells enhanced the ability of TAZ to reduce the expression of N-cadherin, a marker for mesenchymal cells, and to enhance E-cadherin, a marker for epithelial cells at low density (Figs5B and EV4B). These results indicate that PARD3 modulates the expression of endogenous TAZ target genes.
Figure 5.
Knockdown of PARD3 reduces the function of TAZ
- Knockdown of PARD3 leads to the decreased expression of the TAZ downstream targets CTGF, FSTL1, PPPR1eB, PTSG2, and TAGLN at low cell density (the ratio of high:low is 4:1). Forty-eight hours after transfection of the indicated siRNAs into T-47D cells, the target genes’ expression levels and the knockdown of PARD3 were analyzed using real-time PCR. The results are average ± SEM of three independent experiments.
- Knockdown of PARD3 increases E-cadherin expression and decreases N-cadherin expression in T-47D cells at low cell density. The lysates were analyzed by Western blot, and all data are normalized to β-actin.
- TAZ-4SA expression blocks the inhibition of cell growth induced by PARD3 knockdown. Growth curves of PARD3-knockdown T-47D cells overexpressing TAZ-4SA were determined. The results are average ± SEM of three independent experiments.
- TAZ-4SA expression blocks the inhibition of colony formation induced by PARD3 knockdown. 5 × 103 PARD3-knockdown T-47D cells overexpressing TAZ-4SA were cultured in 6-well plates for 3 weeks before colonies were counted. Colonies were then visualized by crystal violet staining and counted. The results are average ± SEM of three independent experiments. N.S.: not significant, ***P < 0.001.

Knockdown of PARD3 reduces the function of YAP/TAZ
- A WT and 2SD mutant, but not 2SA mutant, of PARD3 rescue the expression of target genes inhibited by PARD3 knockdown. T-47D cells with stable knockdown and overexpression of WT and 2SA and 2SD mutants of PARD3 were used as indicated. The expression of target genes was analyzed by real-time PCR. The efficiency of endogenous PARD3 knockdown is shown above, and the expression of exogenous HA-PARD3 was verified by Western blot. The results are average ± SEM of three independent experiments.
- B Knockdown of PARD3 increases E-cadherin expression and decreases N-cadherin expression at the mRNA level in T-47D cells at low cell density. PARD3-knockdown T-47D cells were analyzed by qPCR. The results are average ± SEM of three independent experiments.
- C Knockdown of PARD3 inhibits T-47D cell growth which can be rescued by overexpression of YAP2-5SA. The results are average ± SEM of 3 independent experiments.
- D, E Overexpression of TAZ-4SA (D) and YAP2-5SA (E) PARD3-knockdown T-47D cell. It was verified by Western blot and real-time PCR.
- F Knocking down TAZ inhibits the growth of T-47D cells overexpressing PARD3. Growth curves of TAZ–knockdown T-47D cells overexpressing PARD3 were determined. Cell lysates were analyzed by Western blot to verify HA-PARD3 and TAZ expression. The results are average ± SEM of 3 independent experiments.
- G WT and 2SD mutant, but not 2SA mutant, of PARD3 rescue colony formation inhibited by PARD3 knockdown. T-47D stable cell lines (5 × 103 cells) were cultured in 6-well plates for 3 weeks. Colonies were visualized by crystal violet staining and counted. The results are average ± SEM of three independent experiments. **P < 0.01.
YAP/TAZ positively promotes cell proliferation 16,26. We examined whether PARD3 modulates cell growth. Knockdown of PARD3 caused a significant reduction in cell growth in T-47D cells (Fig5C). We also expressed the constitutively active TAZ-4SA, which cannot be phosphorylated and inhibited by LATS. As expected, the expression of TAZ-4SA promoted T-47D cell growth. Importantly, cell growth of the TAZ-4SA-expressing cells was not sensitive to PARD3 knockdown (Fig5C). Similar results were observed when the constitutively active YAP2-5SA was tested (Fig EV4C). PARD3 knockdown and YAP/TAZ expression were verified by real-time PCR and Western blot, respectively (FigEV4D and E). Consistently, knocking down TAZ inhibited the growth of T-47D cells expressing PARD3 (FigEV4F). These data are consistent with a role of PARD3 promoting cell growth by activating TAZ. Moreover, our observations suggest that PARD3 controls cell growth by modulating the phosphorylation status of YAP/TAZ. Finally, we investigated the role of PARD3 in anchorage-independent growth. Knockdown of PARD3 inhibited colony formation of T-47D cells that could be overcome by TAZ-4SA expression (Fig5D). Furthermore, WT and 2SD mutant of PARD3 could rescue colony formation in PARD3-knockdown cells, while 2SA did not (Fig EV4G). Together, our results support a model that PARD3 promotes cell growth by activating YAP/TAZ.
TAZ is a major functional output of the Hippo pathway, and it is regulated by dynamic reversible phosphorylation. In this report, we identified PARD3 as a potential activator of TAZ. It appears that the cytoplasmic PARD3, but not the tight junction associated, stimulates TAZ activity. YAP, the paralog of TAZ, is also regulated by PARD3 in a similar fashion. The scaffold protein PARD3 associates with PAR6 and atypical protein kinase C (aPKC) to regulate junction assembly and polarity 27. Loss of these proteins or their dysregulation might result in disruption of polarity and carcinogenesis 28-30. PARD3 localized at tight junctions between two neighboring epithelial cells when they contact each other. In scattered cells, PARD3 is in cytoplasm 31,32. Our results indicate that PARD3 can activate TAZ by the following mechanism. At low cell density, the cytoplasmic PARD3 recruits the phosphatase PP1A to LATS1, thereby inducing LATS1 dephosphorylation and inactivation. When LATS is inhibited, TAZ phosphorylation is reduced and the dephosphorylated TAZ enters cell nucleus to bind TEAD and induce gene expression, resulting in stimulation of cell growth. At high density, PARD3 forms complex with PAR6 and aPKC and thus associates with the tight junction 4. In addition, PARD3 can be regulated by the PAR1-dependent phosphorylation. PAR1 phosphorylates PARD3 at Ser144 and Ser873 and releases PARD3 from tight junction. The phosphorylated and cytoplasmic PARD3 then could activate TAZ. Our study also indicates that PP1A plays a crucial role in LATS1 dephosphorylation and activity regulation. Overexpression of PARD3 increases the interaction between PP1A and LATS1 as well as reduces LATS1 Ser909 phosphorylation and kinase activity. It has been reported that the tight junction protein AMOT inhibits YAP/TAZ by promoting their phosphorylation 33. We propose that the tight junction might serve as a platform for Hippo pathway regulation in response to cell density and cell polarity. Our study suggests that tight junction proteins not only regulate cell polarity and cell–cell contact but also participate in cellular signaling.
Materials and Methods
Cell culture and transfection
HEK293T, A375, and T47D cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal calf serum (MP Biomedicals) and 100 units/ml penicillin and streptomycin (Invitrogen). Cell transfection was performed using Lipofectamine 2000 (Invitrogen) or calcium phosphate method. Cells were harvested at 24 h post-transfection for protein analyses or luciferase activity assay. To establish stable PARD3 knocking down cells, pMKO.1-shPARD3 retroviruses were generated and used to infect T-47D and A375 cells and stable pools were selected in puromycin (1 μg/ml)-containing media for 7 days. TAZ stable pool cells were seeded in 6-well plates with 5 × 104 cells/well in triplicate. Cell growth was counted every 2 days for 10 days.
Cell fractionation
HEK293T and T-47D cells were transiently transfected with HA-PARD3 constructs and siRNA targeting PARD3. Cells were lysed with subcellular fractionation buffer containing 250 mM sucrose, 20 mM HEPES, pH7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 25 mM NaF, and a mixture of protease inhibitors (Roche). Centrifuged out the nuclear pellet at 3,000 g for 10 min at 4°C, the cytoplasmic fraction was taken from the supernatant. The nuclear pellet was washed with the fractionation buffer and lysed with SDS buffer containing 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, 1 mM EDTA, 1 mM PMSF, 25 mM NaF, and cocktail protease inhibitors (Roche). Both fractions were analyzed by Western blot.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 for 5 min at 4°C. After blocking in 0.5% BSA for 30 min, cells were incubated with the first antibody diluted in 0.5% BSA overnight. After washing with PBS for three times, cells were incubated with Alexa Fluor 488- or 594-conjugated secondary antibodies (1:1,000 dilution) for 1 h. Then, cells were subjected to fluorescence microscopy.
Luciferase assays
A mixture of activator plasmid (pGal4-TEAD), 5× upstream activating sequence (UAS)-luciferase reporter, and Renilla was cotransfected with the indicated plasmids (FLAG-TAZ, HA-PARD3, and control empty vectors) into HEK293T cells in a 24-well plate. Luciferase activity was measured after 24 h using a dual-luciferase reporter assay system (catalog no. E1960; Promega) by a luminometer (model TD-20/20). Transfection efficiency was normalized to thymidine kinase-driven Renilla luciferase activity.
Co-immunoprecipitation
HEK293T cells were transiently transfected using the calcium phosphate method, and the total amount of DNA was always adjusted with empty vectors. Lysed cells 24 h post-transfection with lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.3% NP-40, 50 mM NaF, 1.5 mM Na3VO4, protease inhibitor cocktail [Roche], 1 mM PMSF) for 30 min on ice and centrifuged at 12,000 g for 15 min at 4°C. The supernatant was incubated at 4°C for 3 h with the anti-FLAG (M2) antibody covalently coupled to agarose beads (Sigma-Aldrich) or with the appropriate first antibody and 10 μl protein A agarose (Millipore). Before the addition of antibodies, a small aliquot of each supernatant was preserved and diluted with 5× SDS–PAGE sample buffer for later Western blot analysis (input). The beads were washed extensively with lysis buffer three times and centrifuged at 5,000 g for 5 min between each wash. Protein was eluted from beads with SDS sample buffer. Lysates were resolved on 8–10% SDS–polyacrylamide gels and transferred onto nitrocellulose (Bio-Rad) for Western blotting.
In vitro kinase assay
For the LATS1 kinase assays, HEK293T cells were transfected with the indicated constructs. Thirty-six hours post-transfection, cells were lysed with lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 50 mM NaF, 1.5 mM Na3VO4, protease inhibitor cocktail [Roche], 1 mM dithiothreitol, 1 mM PMSF) and immunoprecipitated with anti-FLAG antibodies. The immunoprecipitates were washed three times with lysis buffer, followed by a single wash with wash buffer (40 mM HEPES, 200 mM NaCl) and a single wash with kinase assay buffer (30 mM HEPES, 50 mM potassium acetate, 5 mM MgCl2). The immunoprecipitated LATS1 was subjected to a kinase assay in the presence of 500 μM cold ATP and 1 μg His-TAZ expressed and purified from Escherichia coli as the substrate. The reaction mixtures were incubated at 30°C for 30 min, terminated with sodium dodecyl sulfate (SDS) sample buffer. Phosphorylation was detected by Western blotting with the phospho-TAZ-specific antibody.
RNA isolation and real-time PCR
Total RNA was isolated from cultured cells using Trizol reagent (Invitrogen). cDNA was synthesized by reverse transcription using oligo (dT) (TransGen Biotech) as the primer and proceeded to real-time PCR with gene-specific primers in the presence of SYBR Premix Ex Taq (TaKaRa). The relative abundance of mRNA was calculated by normalization to β-actin mRNA.
Plasmids and antibodies
The constructs of TAZ, YAP2, LATS1, 14-3-3, β-TRCP, and PP1A were obtained as described previously. cDNA coding PARD3 was a gift from Dr.Tony Pawson. Point mutations and truncation of PARD3 were generated by site-directed mutagenesis and PCR. The short hairpin RNA sequence against PARD3 is GCCATCGACAAATCTTATGAT.
Antibodies specific to FLAG (Sigma, F7425), HA (Santa Cruz, sc-7392), MYC (Huabio, H1208-1), GFP (Abmart, M20004) (CST#3195) YAP (Cell Signaling Technology, #4912), pYAP (S127) (CST#4911S), E-cadherin (CST#3195), N-cadherin (CST#4061), TEAD (BD Transduction Laboratories, 610922), LATS1 (Bethyl laboratory, A300-477A), pLATS1 (CST#9157), His (CST#2366), tubulin (NeoMarkers, #581P), LaminA/C (GenScript, A01455), and β-actin (GenScript, A00702) were purchased from commercial sources. Antibodies to TAZ and to phospho-TAZ (S89) were generated by immunizing rabbits with indicated antigen peptides at Shanghai Genomic Inc. Other materials and chemicals were obtained from commercial sources.
Colony formation assay
Colony formation assay was performed with T-47D breast cancer cell line. PARD3 knocking down stable cells (5 × 103) were seeded on 6-well plates and maintained in DMEM supplemented with 10% fetal bovine serum for 2–3 weeks. Cells were fixed with 4% paraformaldehyde, and then, colonies were stained with 0.005% crystal violet.
Acknowledgments
We thank members of the Fudan Molecular and Cell Biology laboratory for discussion throughout this study. We also thank Biomedical Core Facility, Fudan University for technical support. This work was supported by MOST 973 (2015CB910401, 2011CB910601, 2012CB910103), NSFC (Grant No. 81430057, 81225016, 31271454), Shanghai Key basic research program (12JC1401100), and Shanghai Outstanding Academic Leader (Grant No. 13XD1400600) to Q.Y.L. This work was also supported by the Youth Science and Technology Leading Talent by MOST to Q.Y.L. This work was also supported and NIH grants (K.L.G. and Y.X.).
Author contributions
X-BL carried out most of the experiments and wrote the manuscript; C-YL contributed to some Western blot experiments; ZW helped with the RNAi and cell culture; Y-PS contributed to reagents’ preparation and cell culture; YX, Q-YL, and K-LG supervised the entire project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Expanded View Figures PDF
Review Process File
References
- Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9:534–546. doi: 10.1101/gad.9.5.534. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Guan KL. Hippo signaling at a glance. J Cell Sci. 2010;123:4001–4006. doi: 10.1242/jcs.069070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, Guan KL. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. 2009;284:13355–13362. doi: 10.1074/jbc.M900843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- Mertens AE, Rygiel TP, Olivo C, van der Kammen R, Collard JG. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J Cell Biol. 2005;170:1029–1037. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T, Karasawa M, Sugitani Y, Fujisawa M, Akimoto K, Ohno S, Noda T. PAR3 is essential for cyst-mediated epicardial development by establishing apical cortical domains. Development. 2006;133:1389–1398. doi: 10.1242/dev.02294. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Macara IG. The PAR proteins: fundamental players in animal cell polarization. Dev Cell. 2007;13:609–622. doi: 10.1016/j.devcel.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegtel DM, Ellenbroek SI, Mertens AE, van der Kammen RA, de Rooij J, Collard JG. The Par-Tiam1 complex controls persistent migration by stabilizing microtubule-dependent front-rear polarity. Curr Biol. 2007;17:1623–1634. doi: 10.1016/j.cub.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Costa MR, Wen G, Lepier A, Schroeder T, Gotz M. Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development. 2008;135:11–22. doi: 10.1242/dev.009951. [DOI] [PubMed] [Google Scholar]
- Chen X, Macara IG. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol. 2005;7:262–269. doi: 10.1038/ncb1226. [DOI] [PubMed] [Google Scholar]
- Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat Cell Biol. 2013;15:189–200. doi: 10.1038/ncb2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey LM, Montalbano J, Mihai C, Macara IG. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell. 2012;22:601–614. doi: 10.1016/j.ccr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iden S, van Riel WE, Schafer R, Song JY, Hirose T, Ohno S, Collard JG. Tumor type-dependent function of the par3 polarity protein in skin tumorigenesis. Cancer Cell. 2012;22:389–403. doi: 10.1016/j.ccr.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Hauri S, Wepf A, van Drogen A, Varjosalo M, Tapon N, Aebersold R, Gstaiger M. Interaction proteome of human Hippo signaling: modular control of the co-activator YAP1. Mol Syst Biol. 2013;9:713. doi: 10.1002/msb.201304750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Intercellular junctions and cellular polarity: the PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr Opin Cell Biol. 2001;13:641–648. doi: 10.1016/s0955-0674(00)00264-7. [DOI] [PubMed] [Google Scholar]
- Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, Li T, Chan SW, Lim CJ, Hong W, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem. 2010;285:37159–37169. doi: 10.1074/jbc.M110.152942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Lv X, Li T, Xu Y, Zhou X, Zhao S, Xiong Y, Lei QY, Guan KL. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J Biol Chem. 2011;286:5558–5566. doi: 10.1074/jbc.M110.194019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- Yang Z, Xue B, Umitsu M, Ikura M, Muthuswamy SK, Neel BG. The signaling adaptor GAB1 regulates cell polarity by acting as a PAR protein scaffold. Mol Cell. 2012;47:469–483. doi: 10.1016/j.molcel.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T. Involvement of ASIP_PAR-3 in the promotion of epithelial tight junction formation.pdf. J Cell Sci. 2002;115:2485–2495. doi: 10.1242/jcs.115.12.2485. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Goto TM, Sugimoto M, Nishimura T, Shinagawa T, Ohno S, Amano M, Kaibuchi K. Rho-kinase phosphorylates PAR-3 and disrupts PAR complex formation. Dev Cell. 2008;14:205–215. doi: 10.1016/j.devcel.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Khazaei MR, Puschel AW. Phosphorylation of the par polarity complex protein Par3 at serine 962 is mediated by aurora a and regulates its function in neuronal polarity. J Biol Chem. 2009;284:33571–33579. doi: 10.1074/jbc.M109.055897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HL, Wang S, Yin MX, Dong L, Wang C, Wu W, Lu Y, Feng M, Dai C, Guo X, et al. Par-1 regulates tissue growth by influencing hippo phosphorylation status and hippo-salvador association. PLoS Biol. 2013;11:e1001620. doi: 10.1371/journal.pbio.1001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, Zeng Q, Hong W. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68:2592–2598. doi: 10.1158/0008-5472.CAN-07-2696. [DOI] [PubMed] [Google Scholar]
- Assemat E, Bazellieres E, Pallesi-Pocachard E, Le Bivic A, Massey-Harroche D. Polarity complex proteins. Biochim Biophys Acta. 2008;1778:614–630. doi: 10.1016/j.bbamem.2007.08.029. [DOI] [PubMed] [Google Scholar]
- Feigin ME, Muthuswamy SK. Polarity proteins regulate mammalian cell-cell junctions and cancer pathogenesis. Curr Opin Cell Biol. 2009;21:694–700. doi: 10.1016/j.ceb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey LM, Macara IG. Epithelial organization, cell polarity and tumorigenesis. Trends Cell Biol. 2011;21:727–735. doi: 10.1016/j.tcb.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011;18:1470–1477. doi: 10.1038/cdd.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Hirose T, Tamai Y, Hirai S, Nagashima Y, Fujimoto T, Tabuse Y, Kemphues KJ, Ohno S. An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of Caenorhabditis elegans polarity protein PAR-3. J Cell Biol. 1998;143:95–106. doi: 10.1083/jcb.143.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y, Hamada S, Ohno S, Suetsugu S. Phosphoinositide binding by par-3 involved in par-3 localization. Cell Struct Funct. 2011;36:97–102. doi: 10.1247/csf.11005. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011;25:51–63. doi: 10.1101/gad.2000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File