

Abstract
Genome-wide association studies (GWAS) have identified thousands of variants robustly associated with complex traits. However, the biological mechanisms underlying these associations are, in general, not well understood. We propose a gene-based association method called PrediXcan that directly tests the molecular mechanisms through which genetic variation affects phenotype. The approach estimates the component of gene expression determined by an individual’s genetic profile and correlates the “imputed” gene expression with the phenotype under investigation to identify genes involved in the etiology of the phenotype. The genetically regulated gene expression is estimated using whole-genome tissue-dependent prediction models trained with reference transcriptome datasets. PrediXcan enjoys the benefits of gene-based approaches such as reduced multiple testing burden and a principled approach to the design of follow-up experiments. Our results demonstrate that PrediXcan can detect known and novel genes associated with disease traits and provide insights into the mechanism of these associations.
Introduction
Genome-wide association studies (GWAS) have been remarkably successful in identifying susceptibility loci for complex diseases. These studies typically conduct single-variant tests of association to interrogate the genome in an agnostic fashion and, due to modest effect sizes, have come to rely on ever-greater sample sizes1,2 to make meaningful inferences. We have been less successful in developing methods that improve on existing simple approaches. In general, the genetic associations identified as genome-wide significant thus far account for only a modest proportion of variance in disease risk3. Indeed, there is now widespread recognition, if not consensus, that GWAS of disease susceptibility (for which, the relevant genetic effects may be very small) and pharmacologic traits (for which large effect sizes are not unusual)4,5 have resulted in limited conclusive findings on the genetic factors contributing to complex traits. Importantly, the functional significance of most discovered loci, including even those that have been the most reproducibly associated, remains unclear. Assigning a causal link to the nearest gene falls short of elucidating a functional connection, as recently demonstrated by the obesity-associated variants within FTO that form long-range functional connections with IRX36. And while GWAS will no doubt continue to identify many more susceptibility loci, the question of how to advance biological knowledge of the underlying mechanisms of disease risk remains a paramount challenge.
A large portion of phenotypic variability in disease risk for a broad spectrum of disease phenotypes can be explained by regulatory variants, i.e. genetic variants that regulate the expression levels of genes7–10. For example, almost 80% of the chip-based heritability of disease risk for 11 diseases from the WTCCC can be explained by genome variation in DNase I hypersensitivity sites, which are likely to regulate chromatin accessibility and thus transcription11.
Large genomic consortia (e.g., ENCODE12) are generating an unprecedented volume of data on the function of genetic variation. The Genotype-Tissue Expression (GTEx13) project is an NIH Common Fund project that aims to collect a comprehensive set of tissues from 900 deceased donors (for a total of about 20,000 samples) and to provide the scientific community a database of genetic associations with molecular traits such as mRNA levels. (See GTEx main paper14 on Phase 1 data.) Other large-scale transcriptome datasets include Genetic European Variation in Health and Disease15 (GEUVADIS, 460 lymphoblastoid cell lines), Depression Genes and Networks (DGN, 922 whole blood samples)16, and Braineac (130 individuals with multiple brain region samples)17. Yet, effective methods that harness these reference transcriptome datasets for disease mapping are lacking.
Methodologically, gene-based approaches and multi-marker association tests have been developed as alternatives to traditional single-variant tests. By conducting tests of association on biologically informed aggregates of SNPs, such tests seek to evaluate a priori functionally relevant units of the genome and, in many cases, reduce the multiple-testing penalty that plague single-variant approaches, by 10 to 100 fold. The incorporation of -omics data, such as those being generated by high-resolution transcriptome studies, provides a means to extend genome-wide association studies by addressing the functional gap. Technological advances in high-throughput methods have reinforced the important finding that intermediate molecular phenotypes are under significant genetic regulation, with expression quantitative trait loci (eQTLs) as the predominant example. However, approaches that fully leverage the comprehensive regulatory knowledge generated by transcriptome studies are relatively lacking despite the fact that these studies have the potential to dramatically improve our understanding of the genetic basis of complex traits13.
We hypothesized that a SNP aggregation approach that integrates information on whether a SNP regulates the expression of a gene can greatly increase the power to identify trait-associated loci either from a strong functional SNP signal or from a combination of modest signals, the so-called grey area of GWAS. The present study suggests that PrediXcan, a novel method that incorporates information on gene regulation from a set of markers, increases the power to detect associations relative to traditional SNP-based GWAS and known gene-based tests under a broad range of genetic architectures and provides mechanistic insights and more easily interpreted direction of effect into the observed associations.
Results
PrediXcan method
PrediXcan, by design, exploits genetic control of phenotype through the mechanism of gene regulation as a way to identify trait-associated genes. Figure 1 is a schematic diagram of the regulatory mechanism that is tested with PrediXcan. An individual's gene expression level (typically unobserved in a GWAS) is decomposed into a genetically regulated expression (GReX) component, a component altered by the trait itself (i.e., a reverse causal effect that may occur if disease status or other conditions alter expression levels), and the remaining component attributable to environmental and other factors. PrediXcan tests the mediating effect of gene expression by quantifying the association between GReX and the phenotype of interest.
Figure 1. Mechanism tested by the PrediXcan method.

This figure shows the conceptual decomposition of the expression level of a gene into three components: genetically determined component, a component altered by the trait itself, and the remaining factors (including environment). PrediXcan estimates the genetically regulated component of expression (GReX) and correlates it with the trait to identify trait-associated genes.
We use reference transcriptome datasets from studies such as GTEx13, GEUVADIS15, and DGN16 among others, to train additive models of gene expression levels. These models allow us to estimate the genetically regulated expression, GReX. We denote the estimated value with a hat, These estimates constitute multiple-SNP prediction of expression levels. The weights for the estimation are stored in our publicly available database.
The analogy with genotype imputation is relevant here. Genotype imputation uses information from a reference sample to learn how to impute genotypes at the unmeasured SNPs in the test set. Similarly, PrediXcan uses a reference dataset in which both genome variation and gene expression levels have been measured to develop prediction models for gene expression. We use these prediction models to “impute” gene expression (which is unobserved in a typical GWAS), and we do so by estimating the genetically determined component, GReX.
PrediXcan application to a GWAS dataset consists of “imputing” the transcriptome using the weights derived from reference transcriptome datasets and correlating the GReX with the phenotype of interest using regression methods (e.g., linear, logistic, Cox) or non-parametric approaches (e.g., Spearman). (For the specific results on disease phenotypes analyzed here, we used logistic regression with disease status.) We are aware of the attenuation bias that arises because of the error in the estimation of GReX. This is a subject to be investigated in the future, but this bias does not invalidate our analysis since we only use the estimate of GReX as a discovery tool. Figure 2 summarizes the flow of the method development described above.
Figure 2. PrediXcan framework.

The workflow illustrates the steps used in developing the PrediXcan method. The top panel shows the data used from the reference transcriptome studies: genotype and expression levels (GTEx, GEUVADIS, DGN, etc). The sample size of the study is denoted by n, m is the number of genes considered, M is the total number of SNPs, and p is the number of available tissues. The second panel shows the additive model used to build a database of prediction models, PredictDB. T represents the expression trait, and Xk is the number of reference alleles for SNP k. The coefficients of the models for each tissue are fitted using the reference transcriptome datasets and optimal statistical learning methods chosen among LASSO, Elastic Net, OmicKriging, etc. The bottom panel shows the application of PrediXcan to a GWAS dataset. Using genetic variation data from the GWAS and weights in PredictDB, we “impute” expression levels for the whole transcriptome. These imputed levels are correlated with the trait using regression (e.g., linear, logistic, Cox) or non-parametric (Spearman) approaches. (For the disease phenotypes in the WTCCC datasets and the replication dataset reported here, we used logistic regression with disease status.)
Features of PrediXcan
PrediXcan is, as we have emphasized, particularly focused on a mechanism – gene expression regulation – that has already been established as being contributory to common diseases, including psychiatric and neurodevelopmental disorders7. The test has the potential to identify gene targets for therapeutic applications because it is inherently mechanism-based and provides directionality.
Additional advantages include:
Like other gene-based tests, it has much smaller multiple-testing burden (~20K tests maximum, ~10K genes with high quality prediction in most tissues) compared to single variant tests (~5–10M tests). Moving beyond the stringent Bonferroni correction, priors on genes can be less restrictive than for SNPs.
Informative priors and groupings of functional units (based on known pathways, for example) are much more straightforward to construct for genes than SNPs.
No actual transcriptome data are required since the predicted expression levels are a function of genetic variation alone. Thus, the method can be applied to any existing dataset with large-scale genome interrogation such as those in dbGaP or other repositories. Re-analyses of existing datasets, with a focus on mechanism using PrediXcan, address a gap that has largely characterized GWAS to date.
Reverse causality is not a major concern since disease status or drug treatment does not alter germline genomic variation.
Meta-analysis of gene-based results is simplified since less stringent harmonization between studies is required.
Multiple tissues can be evaluated using a reference transcriptome dataset (such as GTEx). In general, the only limitation is the availability of gene expression data in the given tissue for model building, which need not be, from the same study as that used for phenotype investigation. In cases where transcriptome data are available, separate analyses should be performed to simplify interpretation.
The approach can be applied to common or rare variants. In general, larger sample sizes for the training set will be needed to achieve good prediction models with rare variants.
Database of prediction models and software
We make the prediction models (derived from LASSO18 and elastic net19) and the software to predict the transcriptome (in a variety of tissues) (see Materials and Methods) publicly available.
Predicting the transcriptome
We built prediction models in the DGN whole blood cohort using LASSO, the elastic net (α=0.5), and polygenic score at several p-value thresholds (single top SNP, 1×10−4, 0.001, 0.01, 0.05, 0.5, 1). We assessed predictive performance using 10-fold cross-validation (R2 of estimated GReX vs. observed expression) as well as in an independent set. We found that LASSO performed similarly to the elastic net and that LASSO outperformed the polygenic score at all thresholds, although all methods are highly correlated (see Supplemental Figure 1). For subsequent analyses, we focused on the prediction models using the elastic net because we found it to perform well and to be more robust to slight changes in input SNPs (potentially due to variations in imputation quality between cohorts).
We estimated the heritability of gene expression in DGN attributable to SNPs in the vicinity of each gene using a mixed-effects model (see Materials and Methods) and calculated variances using restricted maximum likelihood as implemented in GCTA20. We use only local SNPs since we found that heritability estimates using all genotyped SNPs were too noisy to make meaningful inferences.
We use heritability estimates as our benchmark for the prediction R2 since this constitutes the upper limit of our prediction performance. For genes for which an elastic net model was available (n=10,427), the average heritability in DGN was 0.153. In comparison, the average 10-fold cross-validated prediction R2 for elastic net was quite close at 0.137; for the polygenic score (P<1×10−4) and top-SNP models, average prediction R2 values were sizably lower at 0.099 and 0.114, respectively. We show the performance R2 for each model in Figure 3, with the corresponding heritability estimate and confidence interval in the background for comparison. We also note that elastic net predictive performance reached or exceeded the lower bound of the heritability estimate for 94% of genes, while polygenic score (P<1×10−4) did so for just 76% of the genes and the top SNP for 80% of the genes (Figure 3), consistent with the performance ranking given by the average (across genes) R2.
Figure 3. Cross-validated prediction performance vs heritability.
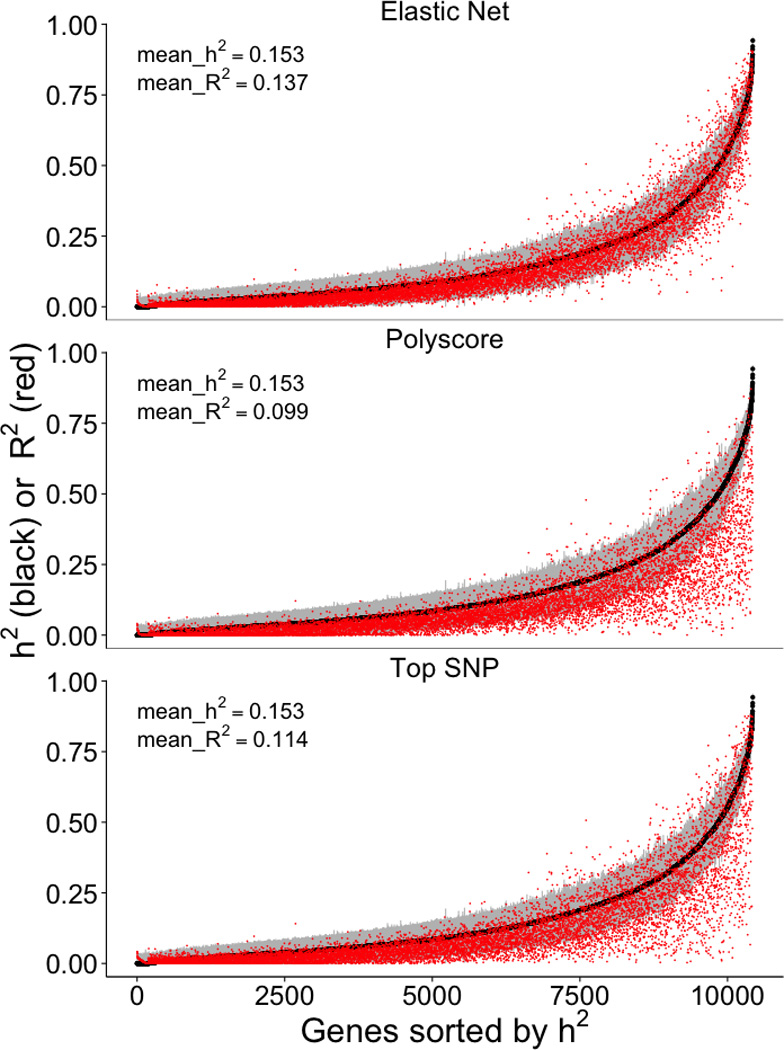
This figure shows the prediction performance (R2 of GReX vs. observed expression in red) compared to gene expression heritability estimates (black with 95% confidence interval in gray). Performance was assessed using 10-fold cross-validation in the DGN whole blood cohort (n=922) with the elastic net, polygenic score (p < 1×10−4), and using the top SNP for prediction.
Predictive performance of elastic net was similar whether all SNPs from the 1000 Genomes imputation or the HapMap Phase II subset were included in the model building (Supplemental Figure 2). Models based on imputed data (both the 1000 Genomes and the HapMap subset) substantially outperformed models based on genotyped SNPs in WTCCC (Supplemental Figure 2). Thus, we chose the elastic net models built in the smaller HapMap SNP subset, relative to 1000 Genomes, in our applications of PrediXcan to reduce computation time without sacrificing performance. As reference transcriptome studies increase in sample size, we may need to switch to a more dense imputation to take advantage of increased prediction performance from rare variants.
We also tested the prediction models trained in the DGN whole blood cohort on several independent test cohorts with available whole-genome genotype and transcriptome data. We used weights derived from the DGN whole blood data (“training set”) to predict gene expression levels (treated as quantitative traits) in GEUVADIS LCLs (lymphoblastoid cell lines) and nine GTEx pilot tissues (“test sets”). Figure 4 provides a Q-Q plot showing the expected (under the null, correlation between two independent vectors with the same sample size) and observed R2 (between observed and predicted) from the elastic net prediction in GEUVADIS LCLs. We find a substantial departure from the null distribution indicating that the elastic net model trained in DGN (equation 2 of Materials and Methods, with effect size estimates ) captures a significant proportion of the transcriptome variability. The average prediction R2 is 0.0197 for GEUVADIS LCLs. For GTEx tissues, the prediction R2 values are 0.0367 (adipose), 0.0358 (tibial artery), 0.0356 (left ventricular heart), 0.0359 (lung), 0.0269 (muscle), 0.0422 (tibial nerve), 0.0374 (sun exposed skin), 0.0398 (thyroid), and 0.0458 (whole blood). Interestingly, we also find a substantial departure from the null distribution of expected R2 values for predicted expression using DGN weights in each of the nine GTEx tissues suggesting that models developed in whole blood are still useful for understanding diseases that affect other primary tissues (Supplemental Figure 3). Consistent with this, average prediction R2 is highest for whole blood as expected but the loss in power for other tissues is modest.
Figure 4. Prediction performance of elastic net tested on a separate cohort.
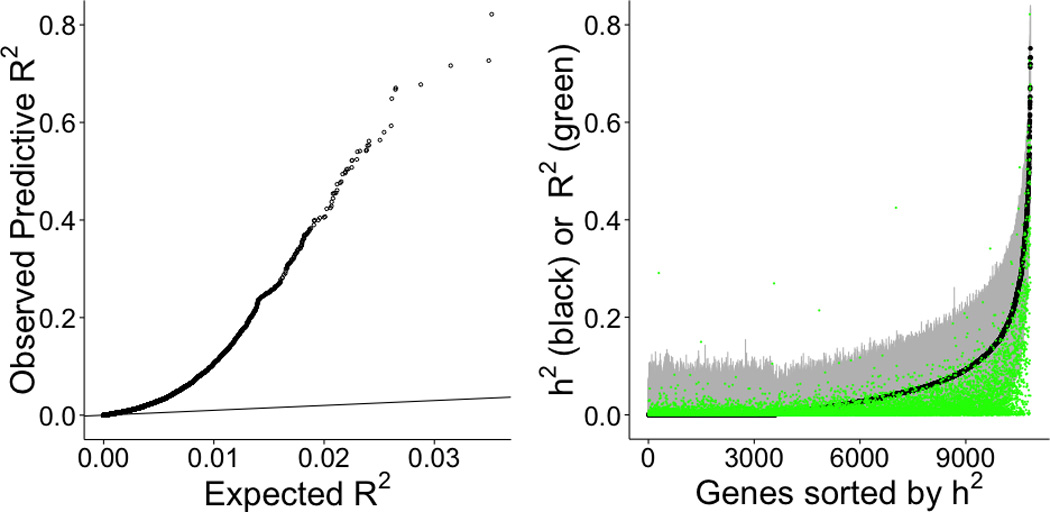
Using whole blood prediction models trained in DGN, we compared predicted levels of expression with observed levels on lymphoblastoid cell lines from the 1000 Genomes project. RNA-sequenced data (n=421) on these cell lines have been made publicly available by the GEUVADIS consortium. Left panel shows the squared correlation, R2, between predicted and observed levels plotted against the null distribution of R2 Right panel shows prediction performance (R2 of GReX vs. observed expression in green) compared to GEUVADIS gene expression heritability (h2) estimates (black with 95% confidence interval in gray).
Figure 5 illustrates the genes with some of the highest correlations from this analysis, providing a comparison of the predicted expression and the observed expression. Among these genes, both ERAP2 and its paralog ERAP1 play fundamental roles in MHC antigen presentation21, immune activation and inflammation.
Figure 5. Examples of well-predicted genes.
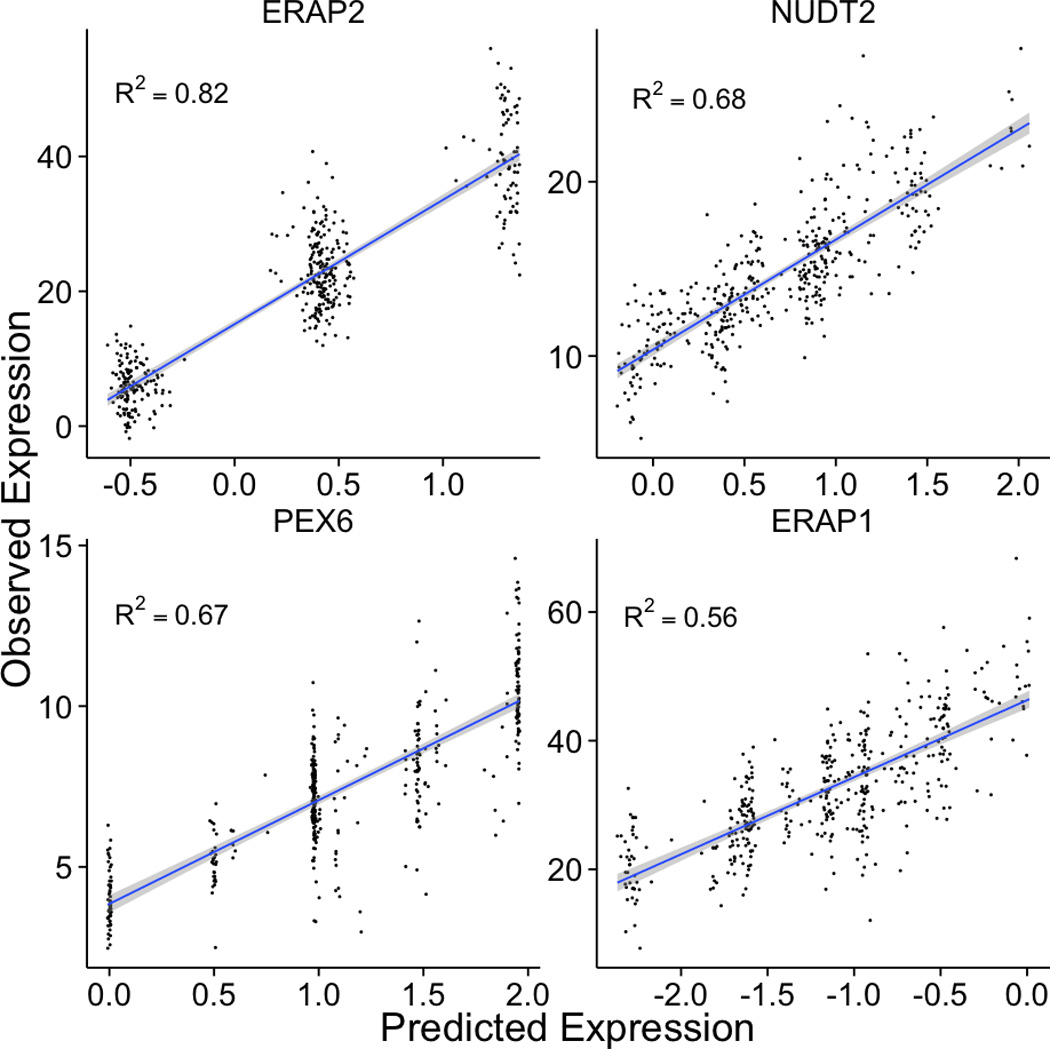
These plots show observed vs. predicted levels of 4 genes. Predicted levels were computed using whole blood elastic net prediction models trained in DGN data. Observed levels were RNA-seq data in lymphoblastoid cell lines generated by the GEUVADIS consortium.
We also generated prediction models trained in the DGN whole blood cohort that included trans-eQTLs (>1Mb from gene start or end or on a different chromosome) generated from linear regression (p<10−5). We tested the predictive performance of these models in the GTEx whole blood cohort. While a few genes had higher correlations between predicted and observed expression than expected by chance, the departure from the null distribution was much smaller than that for the prediction models based on local SNPs (Supplemental Figure 4), perhaps due to the low power to map trans SNPs. Based on this result, in this paper we focus primarily on results based on local SNPs.
Application of PrediXcan to WTCCC
We applied PrediXcan to seven complex disease phenotypes from the WTCCC study22. For this purpose, we utilized the DGN whole blood elastic net prediction models. We correlated the estimated genetically regulated gene expression for close to 8700 genes with disease status for each WTCCC dataset and identified 41 significant associations (Bonferroni corrected p < 0.05) with five diseases (Table 1). Notably, we identified 29 genes associated with type 1 diabetes (T1D) risk (Table 1 and Fig. 6), 8 of which were outside of the extended MHC. Complete results for the remaining 6 diseases are shown in Supplemental Figures 5 and 6. Consistent with the original GWAS of WTCCC diseases, our most significant results were for autoimmune diseases22.
Table 1. Top PrediXcan results for WTCCC using DGN whole blood prediction models.
PrediXcan results for bonferroni significant gene associations. To account for multiple testing, we used a significance threshold of 5.76×10−6 for all diseases. Chromosome and gene start position are based on GENCODE version 12. The cross validated prediction R2 between predicted and observed gene expression is based on 10-fold cross validation within the DGN whole blood sample.
| Disease | Gene | Evidence | Chr | TSS | PrediXc an Z- Statistic |
PrediXc an P- value |
No. of SNPs in Predictor |
Cross Validated Prediction R2 |
|---|---|---|---|---|---|---|---|---|
| RA | DCLRE1B | V | 1 | 114,447, 763 | −6.68 | 2.46E-11 | 4 | 0.0388 |
| RA | PTPN22 | G | 1 | 114,356, 433 | 5.67 | 1.44E-08 | 32 | 0.0795 |
| BD | PTPRE | 10 | 129,705, 325 | 4.94 | 7.71E-07 | 38 | 0.0355 | |
| CD | ATG16L1 | G | 2 | 234,118, 697 | 6.37 | 1.94E-10 | 20 | 0.0638 |
| CD | IL23R | G | 1 | 67,632,0 83 | 5.23 | 1.74E-07 | 38 | 0.0378 |
| CD | APEH | V | 3 | 49,711,4 35 | 5.14 | 2.77E-07 | 31 | 0.1164 |
| CD | ZNF300 | G ‡ | 5 | 150,273, 954 | −4.98 | 6.29E-07 | 34 | 0.0387 |
| CD | NKD1 | G | 16 | 50,582,2 41 | −4.91 | 8.91E-07 | 43 | 0.0693 |
| CD | BSN | G ‡ | 3 | 49,591,9 22 | −4.68 | 2.89E-06 | 39 | 0.2336 |
| CD | GPX1 | V | 3 | 49,394,6 09 | −4.62 | 3.87E-06 | 28 | 0.0211 |
| CD | SLC22A5 | G ‡ | 5 | 131,705, 444 | −4.54 | 5.75E-06 | 42 | 0.6356 |
| HT | KCNN4 | 19 | 44,270,6 85 | −4.7 | 2.62E-06 | 81 | 0.4655 | |
| T1D | DCLRE1B | V | 1 | 114,447, 763 | −7.84 | 4.34E-15 | 4 | 0.0388 |
| T1D | ZNF165 | M | 6 | 28,048,7 53 | 7.3 | 2.92E-13 | 19 | 0.0374 |
| T1D | ERBB3 | G | 12 | 56,473,6 41 | −6.81 | 1.01E-11 | 9 | 0.2206 |
| T1D | EGFL8 | H | 6 | 32,132,3 60 | 6.33 | 2.52E-10 | 36 | 0.0558 |
| T1D | C6orf136 | H | 6 | 30,614,8 16 | −6.33 | 2.52E-10 | 15 | 0.0137 |
| T1D | HCG27 | H | 6 | 31,165,5 37 | −6.33 | 2.52E-10 | 81 | 0.3721 |
| T1D | GTF2H4 | H | 6 | 30,875,9 61 | 6.33 | 2.52E-10 | 69 | 0.0982 |
| T1D | DDR1 | H | 6 | 30,844,1 98 | 6.33 | 2.52E-10 | 48 | 0.1427 |
| T1D | AGER | H | 6 | 32,148,7 45 | −6.33 | 2.52E-10 | 39 | 0.0502 |
| T1D | POU5F1 | H | 6 | 31,130,2 53 | 6.33 | 2.52E-10 | 45 | 0.2874 |
| T1D | ATP6V1G2 | H | 6 | 31,512,2 39 | 6.33 | 2.52E-10 | 95 | 0.2543 |
| T1D | TUBB | H | 6 | 30,687,9 78 | 6.33 | 2.52E-10 | 56 | 0.0295 |
| T1D | AIF1 | H | 6 | 31,582,9 61 | 6.33 | 2.52E-10 | 34 | 0.039 |
| T1D | CYP21A2 | H | 6 | 32,006,0 42 | −6.33 | 2.52E-10 | 80 | 0.229 |
| T1D | LSM2 | H | 6 | 31,765,1 73 | 6.33 | 2.52E-10 | 31 | 0.0317 |
| T1D | VARS2 | H | 6 | 30,876,0 19 | 6.33 | 2.52E-10 | 87 | 0.3628 |
| T1D | APOM | H | 6 | 31,620,1 93 | −6.33 | 2.52E-10 | 58 | 0.0699 |
| T1D | DDAH2 | H | 6 | 31,694,8 15 | −6.33 | 2.52E-10 | 32 | 0.1943 |
| T1D | NCR3 | H | 6 | 31,556,6 72 | −6.33 | 2.52E-10 | 79 | 0.2548 |
| T1D | ZSCAN16 | M | 6 | 28,092,3 38 | 6.16 | 7.37E-10 | 34 | 0.0291 |
| T1D | ZKSCAN4 | M | 6 | 28,212,4 01 | 6.15 | 7.73E-10 | 17 | 0.0991 |
| T1D | PTPN22 | G | 1 | 114,356, 433 | 5.83 | 5.41E-09 | 32 | 0.0795 |
| T1D | RPS26 | G ‡ | 12 | 56,435,6 37 | 5.82 | 6.00E-09 | 23 | 0.0719 |
| T1D | GDF11 | V | 12 | 56,137,0 64 | −5.75 | 9.11E-09 | 39 | 0.0341 |
| T1D | SUOX | G ‡ | 12 | 56,390,9 64 | −5.47 | 4.49E-08 | 50 | 0.1339 |
| T1D | BTN3A2 | M | 6 | 26,365,3 87 | −5.11 | 3.30E-07 | 49 | 0.7662 |
| T1D | PRSS16 | M | 6 | 27,215,4 80 | 4.83 | 1.34E-06 | 31 | 0.1639 |
| T1D | FAM109A | V | 12 | 111,798, 339 | −4.76 | 1.94E-06 | 17 | 0.0665 |
| T1D | SH2B3 | G | 12 | 111,843, 752 | 4.67 | 3.05E-06 | 26 | 0.0368 |
Evidence: H= HLA-region genes on chromosome 6p21; M=extended Major Histocompatibility Complex; G=Genes previously reported to be associated with disease risk in the NHGRI GWAS catalog excluding studies with WTCCC samples; G‡= reported in studies including WTCCC samples; V= in vicinity of genes of reported gene (1MB).
Figure 6. PrediXcan results for type 1 diabetes.

Complete results for our analysis of type 1 diabetes from the WTCCC using gene expression predicted with the DGN whole blood predictors. Panel (a) shows association p-values based on gene position across the genome. Panel (b) shows the same results plotted against the null expectation in a q–q plot. The red line in panel (b) shows the null expected distribution of p-values. In panels (a) and (b), the blue line represents the bonferroni corrected genome-wide significance threshold. The top 3 genes are labeled. Panel (c) shows the results of our GWAS enrichment analysis. The histogram shows the expected number of genes with a p-value < 0.01 based on 10,000 random permutations. The large point shows the observed number of previously known T1D genes that fall below this threshold.
As has been previously reported for complex autoimmune diseases23, we observed genes that were associated with multiple autoimmune diseases, namely T1D, Crohn’s disease (CD), and rheumatoid arthritis (RA). Interestingly, the top (genome-wide significant) PrediXcan gene for both T1D and RA, DCLRE1B, has not been previously reported (in the NHGRI catalog) in either disease, but has been linked to CD, ulcerative colitis and inflammatory bowel disease24. Lower predicted expression of DCLRE1B was associated with increased disease risk for both RA and T1D. Interestingly, higher predicted expression of DCLRE1B was nominally associated with increased Crohn’s disease risk in our PrediXcan analysis (p = 0.001). Similarly, PTPN22 was significantly (positively) associated with RA and T1D (table 1), and nominally (negatively) associated with CD (p-value = 0.017). Previous single variant analyses implicated PTPN22 with multiple autoimmune diseases including RA, T1D, CD, myasthenia gravis, and vitiligo according to the NHGRI catalog25. These results highlight the known overlap in genetic risk factors for autoimmune diseases.
All genes in table 1, excluding PTPRE and KCNN4 (discussed below), have been either previously reported with GWAS studies, or are located in the vicinity of reported genes (within 1MB). About 35% of all GENCODE protein-coding genes are reported (in the NHGRI catalog) or within 1 MB of a reported gene as associated with a WTCCC disease. For T1D, 5 out of the 29 genome-wide significant genes have been reported via conventional single variant analyses (as curated by the NHGRI25 repository of GWAS results). Furthermore, 21 of the genes associated with T1D in our analysis lie within the extended MHC (Table 1), a region that is known to be associated with disease risk26. Additionally, ERBB3, which contains SNPs previously associated with T1D in GWAS27, showed a negative correlation with disease risk in PrediXcan (p < 10−11), which is consistent with a prior study that showed risk genotypes associated with lower expression of ERBB3 in PBMCs28. Furthermore, it has been reported that subjects with protective genotypes had higher percentages of ERBB3+ monocytes and dendritic cells leading to greater T cell proliferation28. These results highlight one of the key advantages of PrediXcan, which is to provide the direction of effect.
The results described above highlight gene associations that attain genome-wide significance. Additionally, we tested for enrichment of reported disease genes among our PrediXcan results using less stringent significance thresholds. Reported genes were derived from the comprehensive NHGRI catalog of disease-associated variants identified using GWAS25. Five of the seven diseases (bipolar disorder (BD), coronary artery disease (CAD), CD, RA, T1D) had a significant enrichment of reported genes in the PrediXcan results (Figure 6C, Supplemental Figure 7). Results for other p-value thresholds were similar (results not shown). These enrichment analyses on the PrediXcan findings suggest that among the genes that fail to meet strict genome-wide significance, there are likely to be true disease associations.
In addition to the results described above for autoimmune diseases, we identified two potentially novel disease-associated genes. Lower predicted expression of KCNN4 was associated with an increased risk of hypertension (p-value = 2.62 × 10−6, Table 1) and high predicted PTPRE expression was associated with increased risk of bipolar disorder (p-value = 7.71 × 10−7, Table 1). Interestingly, an intronic SNP in PTPRE was previously found to associate with response to the stimulant amphetamine29,30. In contrast to the original WTCCC single-variant analyses22, the PrediXcan analysis for bipolar disorder and hypertension produced genome-wide significant results. Additional studies of these genes are warranted.
Using publically available meta-analysis results, we summarized the single-variant association results for SNPs that are included in the prediction models for the top disease-associated PrediXcan genes. See Supplemental Note and Supplemental Table 1 for the results of this analysis.
We applied PrediXcan and two widely-used gene-based tests (VEGAS and SKAT) to WTCCC. In a Q-Q plot showing all three distributions of p-values, for genes outside of the HLA region, from these gene-based tests (Figure 7), SKAT had improved performance relative to VEGAS, and PrediXcan showed the most extreme departure from the null at the tail end of the distribution.
Figure 7. Comparison of gene-based methods.

Q-Q plot showing distribution of p-values derived from each method (VEGAS, SKAT, and PrediXcan) for genes outside of the HLA region for Rheumatoid Arthritis.
To replicate our findings, we applied the DGN elastic net whole blood prediction models to an independent rheumatoid arthritis GWAS from Vanderbilt University’s BioVU repository (see Materials and Methods). Both genes (DCLRE1B and PTPN22) that were found to be genome-wide significant in the WTCCC rheumatoid arthritis data were also significant, with concordant direction of effect, in the replication samples (p = 0.012 and p = 0.036, respectively).
Discussion
Gene expression, as an intermediate phenotype between genetic variation and higher-level phenotypes, is an important mechanism underlying disease susceptibility and drug response. Studies of the transcriptome in several tissues13 have shown that variation in gene expression is heritable32,33 and can be mapped to the genome. Particularly, eQTL mapping provides an immediate view of the effects of genetic variants on the phenotype closest to genetic variation, namely transcript abundance, and thus promises to enable the discovery of the molecular mechanisms underlying human phenotypic variation34. Furthermore, transcriptome regulation studies facilitate the consideration of thousands of gene expression phenotypes in parallel, thereby enabling a comprehensive approach to understanding the genetic basis of complex traits35. In this study, we developed a method that explicitly utilizes the wealth of regulatory information derived from transcriptome regulation studies to map trait-associated loci.
Our PrediXcan method tests the mediating effects of gene expression levels by quantifying the association between the genetically regulated levels of expression and the phenotype of interest. To implement this, we developed prediction models of gene expression using large-scale transcriptome study datasets (DGN, GEUVADIS, GTEx). (Summary statistics on samples per tissue for every data release are available from the GTEx portal.) After extensive testing, we chose to use the elastic net model, which performed similarly to LASSO, but substantially outperformed simple polygenic approaches. Manor and Segal36 have published results on robust prediction of expression levels using K nearest neighbor (KNN) and elastic net approaches. Based on their conclusion that a combination of elastic net and KNN along with the use of genomic annotation such as GC content can improve prediction performance, it is reasonable to hypothesize that the incorporation of a more comprehensive functional annotation approach into the PrediXcan framework can yield additional performance gain.
Application of the method to WTCCC data recapitulated many known loci but also identified novel genome-wide significant genes. We believe that a systematic reanalyses of GWAS datasets in comprehensive repositories such as dbGAP and the European Genome-phenome Archive (EGA) could provide a cost-effective approach to uncovering novel disease mechanisms using only existing genomic resources.
In contrast to other gene-based tests, PrediXcan provides the direction of effect, which may yield opportunities for therapeutic development. The development of therapeutics that down-regulate a gene is generally easier to achieve than therapeutics that up-regulate a gene; thus, genes with expression levels that are positively correlated with disease risk may be more favorable drug targets for novel therapies. The direction of effect may also provide information to elucidate pathways and the opportunity to explore systems-based approaches to the development of disease. The prediction models can be applied to genotype data of subjects in large biobanks to investigate potential side-effects of drugs with specific gene targets. Finally, direction of effect can be used to improve the interpretation of sequence analyses of genes showing significant correlation of predicted expression with phenotype, since phenotypes associated with reduced expression of genes are more likely to show a relative excess of rare variants. Indeed, we believe that PrediXcan offers intriguing opportunities to combine results of rare and common variant association tests within whole genome sequencing studies, and more generally, to combine results of rare variant gene-based tests from sequencing studies with results of PrediXcan gene-based tests from the large body of existing GWAS for the same phenotypes. Thus, PrediXcan is a method developed to integrate –omics data that can facilitate integration of results from common and rare variant studies.
Regarding the multiple testing correction approach, here we have used Bonferroni correction using the total number of genes tested. In general, both single-variant and PrediXcan analyses will be performed; thus the question that arises is how to address the issue of multiple testing adjustment. The prior probability for a SNP to be causal is much smaller than the prior probability of causality for a gene so it would not be fair to subject SNP tests and gene-based tests to the same level of adjustment. Since we are presenting only gene-based results in our application and given the highly conservative nature of Bonferroni correction, there is no need to further adjust our results. A more conservative approach would be to divide the significance threshold used by a factor of two for the multiple testing using gene-based and SNP-based approaches.
Given the large contribution of regulatory variants on complex traits9,10,37, our method is likely to identify causal genes. However, we do not claim causality since SNPs that contribute to the expression of a gene can also act through other mechanisms to determine the phenotype of interest. Replication and experimental validations are needed to determine causality.
In conclusion, we presented a novel gene-based test, PrediXcan that incorporates functional information with regard to gene regulation to identify genes associated with disease traits in large GWAS or whole genome sequence datasets. Our method has the advantage of providing biological insights into the mechanism, namely regulation of gene expression, and direction of effect. This approach can be readily applied to existing GWAS datasets through the use of our publically available PredictDB resource. We further show the utility of our approach by identifying and replicating a number of novel candidate associations within the previously analyzed WTCCC dataset.
Methods
Genomic and Transcriptomic Data
DGN RNA-Seq Dataset
We obtained whole blood RNA-Seq38 and genome-wide genotype data for 922 individuals from the Depression Genes and Networks cohort16, all of European ancestry. For our analyses, we used the HCP (hidden covariates with prior) normalized gene-level expression data used for the trans-eQTL analysis in Battle et al.16 and downloaded from the NIMH repository. Approximately 650K SNPs (minor allele frequency [MAF] > 0.05, Hardy-Weinberg Equilibrium [P > 0.05], non-ambiguous strand [no A/T or C/G SNPs]) comprised the input set of SNPs for imputation, which was performed on the University of Michigan Imputation-Server39,40 with the following parameters: 1000G Phase 1 v3 ShapeIt2 (no singletons) reference panel, SHAPEIT phasing, and EUR population. Non-ambiguous strand SNPs with MAF > 0.05, imputation R2 > 0.8 were retained for subsequent analysis. To reduce computational burden in the application to WTCCC, we used models developed on the HapMap Phase II subset of SNPs.
GEUVADIS RNA-Seq Dataset
We obtained freely available RNA-Seq data from 421 lymphoblastoid cell lines (LCLs) generated by the GEUVADIS consortium15 and genotype data generated by the 1000 Genomes project. We used GEUVADIS as a validation dataset to test the gene prediction models generated in the DGN cohort.
GTEx RNA-Seq Datasets
We used the nine tissues with the largest sample size in the Genotype-Tissue Expression (GTEx) Pilot Project14 to test the gene prediction models generated in the DGN cohort. Tissue samples included subcutaneous adipose (n=115), tibial artery (n=122), left ventricle heart (n=88), lung (n=126), skeletal muscle (n=143), tibial nerve (n=98), skin from the sun-exposed portion of the lower leg (n=114), thyroid (n=112), and whole blood (n=162). In each tissue, normalized gene expression was adjusted for gender, the top 3 principal components (derived from genotype data), and the top 15 PEER factorsh.2p2csry (to quantify batch effects and experimental confounders)41. We used GTEx to test the portability of predictors developed in whole blood (from the DGN cohort) across a wide variety of tissues.
Additive model for gene expression traits
We use an additive genetic model to characterize gene expression traits:
| (1) |
where Yg is the expression trait of gene g, wk,g is the effect size of marker k for gene g, Xk is the number of reference alleles of marker k, and ϵ is the contribution of other factors that determine the expression trait assumed to be independent of the genetic component. We note that the summation in model (1) is the genetically determined component of gene expression (i.e., GReX).
Effect sizes (wk,g) in model (1) can be estimated using multiple approaches. In this paper we compare penalized approaches such as LASSO (Least Absolute Shrinkage and Selection Operator)18 and the elastic net19 as well as the more naive simple polygenic score estimates. However, other statistical machine learning approaches42, such as Random Forest43 or OmicKriging44, can be used within the PrediXcan framework to develop prediction models.
The heritability of gene expression defines an upper bound to how well we can predict the trait. We estimated the narrow-sense heritability for each gene using a variance component model with a genetic relationship matrix (GRM) estimated from genotype data, as implemented in GCTA20. No pair of subjects from the 922 individuals in DGN shared genetic relatedness (π̂) in excess of 5% and thus all were included in the narrow-sense heritability estimation. SNPs in the vicinity of each gene (within 1Mb of gene start or end, as defined by the GENCODE45 version 12 gene annotation), with MAF > 0.05, and in Hardy-Weinberg Equilibrium (P > 0.05) were used to construct the GRM for each gene. We calculated the proportion of the variance of gene expression explained by these local SNPs using the following mixed-effects model37:
where Y is a gene expression trait and b a vector of fixed effects. Here Alocal is the GRM calculated from the local SNPs, and (the random effect) Glocal denotes the genetic effect attributable to the set of local SNPs with var In this paper we focus on the component of heritability driven by SNPs in the vicinity of each gene since the component based on distal SNPs could not be estimated with enough accuracy to make meaningful inferences.
Estimation of the genetic component of gene expression levels (GReX)
In the simple polygenic score approach, we estimate wk as the single-variant coefficient derived from regressing the gene expression trait Y on variant Xk (as implemented in the eQTL analysis software Matrix eQTL46) using the reference transcriptome data. This yields an estimate, , for a GWAS study sample, of the (unobserved) genetically determined expression of each gene g:
| (2) |
In this implementation of polygenic score, we include all SNPs (regardless of linkage disequilibrium [LD]) that are associated with the expression level of the gene at a chosen p-value threshold in the prediction model.
In contrast, LASSO uses an L1 penalty as a variable selection method to select a sparse set of (uncorrelated) predictors18 while the elastic net linearly combines the L1 and L2 penalties of LASSO and ridge regression respectively to perform variable selection19. We used the R package glmnet to implement LASSO and elastic net with α=0.5.
For each gene, LASSO, the elastic net and the simple polygenic score were used to provide an estimate of GReX̂ (using equation 2, with the effect size estimates , and , respectively). We included only local SNPs (within 1Mb of the gene start or end). In order to determine the optimal modeling method, we compared the 10-fold cross-validated prediction R2 (the square of the correlation between predicted and observed expression) for the simple polygenic score () at several p-value thresholds (single top SNP, 1×10−4, 0.001, 0.01, 0.05, 0.5, 1) with that from LASSO () and elastic net ().
We also compared the 10-fold cross-validated prediction R2 from elastic net models with different starting SNP sets from the DGN genotype imputation (4.6M 1000 Genomes Project SNPs (MAF>0.05, R2>0.8, non-ambiguous strand), the 1.9M of these SNPs that are also in HapMap Phase II, and the 300K of these SNPs that were genotyped in the WTCCC).
Performance of transcriptome prediction in independent cohorts
We tested the feasibility of predicting the transcriptome (i.e., estimating the genetic component of each gene expression trait, in an independent test transcriptome dataset) using the elastic net effect sizes trained in the DGN whole blood data (n=922). For the test sets, we used independent RNA-Seq datasets from 421 LCL cell lines from the 1000 Genomes project generated by the GEUVADIS consortium15 and the nine tissues from the GTEx pilot project14 (see Supplemental Figure 3). To assess performance, we used the square of the Pearson correlation, R2, between predicted and observed expression levels.
PrediXcan in the WTCCC GWAS Datasets
To illustrate the method, we applied gene prediction models (derived from whole blood) consisting of DGN elastic net predictors to the seven WTCCC disease studies -- bipolar disorder (BD), coronary artery disease (CAD), hypertension (HT), type 1 diabetes (T1D), type 2 diabetes (T2D), Crohn’s disease (CD), and rheumatoid arthritis (RA)22 . Genotypes imputed to the 1000G reference sets were used. Imputation was done using the University of Michigan Imputation-Server and the same parameters as described for the imputation of DGN data. For each disease, cases and controls (1958 Birth Cohort and the UK Blood Service Cohort) were jointly imputed to avoid subtle differences between cases and controls not attributable to disease risk. We excluded all SNPs with an imputation R2 < 0.8 and for computational speed we kept only the HapMap Phase II subset of SNPs.
For each WTCCC disease, we estimated , and tested it for association with disease risk using logistic regression in R (R-project.org). We restricted our PrediXcan analysis to include genes with a cross-validated prediction R2 > 0.01 (10% correlation) in the DGN sample. Because the WTCCC studies use shared controls, pleiotropy analyses using these datasets would not be straightforward, and comparison of results across diseases was avoided.
GWAS Enrichment analysis
Relative to recent association studies, the WTCCC has a small sample size (~2,000 cases and ~3,000 controls per disease). Thus, even with our novel method and a reduced multiple testing burden, our ability to detect numerous novel gene associations may be limited. Alternatively, we tested each disease for an enrichment of known disease genes identified from the NHGRI GWAS Catalog25. For each disease, we used the reported genes from the GWAS catalog as the set of known disease genes. We excluded studies listed in the NHGRI GWAS catalog that included the WTCCC samples in order to make sure our known gene lists were independent from the current analysis. We then counted the number of known disease genes that had a PrediXcan p-value below a given threshold. We compared this to the null expectation based on 10,000 randomly drawn gene sets of similar size to the known disease gene set to derive an enrichment p-value. We tested enrichment using PrediXcan p-value thresholds of 0.05 and 0.01.
Comparison to large single variant meta-analyses
For the top PrediXcan results in the WTCCC, we cross-referenced the SNPs in the prediction models for these genes with the publically available single-SNP meta-analysis summary results. We excluded T1D from this analysis because, to our knowledge, there are no publically available meta-analysis studies of this disease. We used meta-analyses results for systolic and diastolic blood pressure as a proxy for hypertension. For CD, RA, and BD we were able to use meta-analyses for the same diseases (CD47, RA48, and BD31).
Comparison of gene-based tests (PrediXcan, SKAT, VEGAS)
We compared the results derived from PrediXcan with those from two widely-used gene-based tests, namely VEGAS49 and SKAT50,51. VEGAS aggregates information from the full set of SNPs within a gene and accounts for LD using simulations from the multivariate normal distribution. SKAT is a kernel-based association test that evaluates the regression coefficients of the SNPs within a gene by a variance component score test in a mixed model framework. We generated BED-formatted files for SNPs and genes (as defined by GENCODE v12) and mapped SNPs that met post-imputation QC parameters to gene regions using bedtools. The use of an offline Perl implementation for VEGAS allowed us to examine the dependence of the results from this approach on LD information through the use of the actual genotype data (versus the default HapMap CEU reference panel data). We developed an R-based pipeline that invokes the SKAT package (version 1.0.1) that is publicly available from CRAN. We generated a Q-Q plot showing the distribution of gene-level p-values for association with RA (for genes outside the HLA region) derived from each gene-based test to test for systematic departure from the null expectation (of uniform p-values).
Replication of PrediXcan findings
We selected individuals from Vanderbilt University’s BioVU repository with a diagnosis of rheumatoid arthritis48 using a previously validated algorithm for identification of RA cases with a reported positive predictive value of 0.94 and sensitivity of 0.87, as previously described52. This trained machine learning classifier was applied to records with at least one International Classification of Diseases, 9th edition code for rheumatoid arthritis to identify true RA cases. RA positive individuals identified by this algorithm were genotyped on two platforms: 833 using the Illumina OmniExpress + Exome chip and 1408 using the Illumina Omni 2.5 BeadChip. A total of 2650 samples from the Illumina Genotype Control set genotyped on Illumina HumanMap550v1/v3 were used for controls. We used the following QC thresholds: sample call rate > 0.98, SNP call rate > 0.99, MAF > 0.05, HWE p-value > 10−3. Imputation was performed using IMPUTE2 with the 1000 Genomes phase 1 v3 European samples as the reference panel, phasing was done with SHAPEIT, and SNPs with imputation quality score (“INFO”) > 0.50 were retained. To replicate the PrediXcan RA findings that meet genome-wide significance, we utilized the DGN whole blood elastic net prediction models (as we had done in the discovery WTCCC data). We estimated the genetically regulated gene expression level in the replication samples and performed logistic regression with disease status.
Supplementary Material
Acknowledgements
We thank Anuar Konkashbaev and Christian Fuchsberger for outstanding technical support and Nicholas Knoblauch for assistance in performing the QC pipeline. We acknowledge the following grants: K12CA139160 (HKI), T32MH020065 (KS), F32CA165823 (HEW), R01 MH101820 and R01 MH090937 (GTEx), P30 DK20595 and P60 DK20595 (DRTC), P50DA037844 (Rat Genomics), UO1GM61393 (PAAR), P50MH094267 (Conte), U01 GM092691 (JCD), U19 HL065962 (P-STAR).
Additional acknowledgements can be found in the Supplementary Note.
Footnotes
URLs
PrediXcan software, https://github.com/hakyimlab/PrediXcan
University of Michigan Imputation-Server, https://imputationserver.sph.umich.edu/start.html
GEUVADIS RNA-Seq data, http://www.geuvadis.org/web/geuvadis/RNAseq-project glmnet package, http://www.jstatsoft.org/v33/i01
International Inflammatory Bowel Disease Genetics Consortium Crohn’s disease meta-analysis data, http://www.ibdgenetics.org/downloads.html
Psychiatric Genomics Consortium bipolar disorder data, http://www.med.unc.edu/pgc/downloads
Open Science Data Cloud, https://www.opensciencedatacloud.org
GTEx Portal, http://www.gtexportal.org/
Author contributions
H.K.I., H.E.W., E.R.G., K.P.S., S.V.M., and K.A. performed the analyses. J.C.D., R.J.C., and A.E.E. provided replication data. E.R.G., H.E.W., K.P.S., and H.K.I. wrote the manuscript. D.L.N., N.J.C., and H.K.I. provided intellectual input and supervised the study. H.K.I. designed the study. All authors reviewed and contributed to the final manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Spencer CC, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477. doi: 10.1371/journal.pgen.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perera MA, et al. The missing association: sequencing-based discovery of novel SNPs in VKORC1 and CYP2C9 that affect warfarin dose in African Americans. Clin Pharmacol Ther. 2011;89:408–415. doi: 10.1038/clpt.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ritchie MD. The success of pharmacogenomics in moving genetic association studies from bench to bedside: study design and implementation of precision medicine in the post-GWAS era. Hum Genet. 2012;131:1615–1626. doi: 10.1007/s00439-012-1221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smemo S, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamazon ER, Huang RS, Cox NJ, Dolan ME. Chemotherapeutic drug susceptibility associated SNPs are enriched in expression quantitative trait loci. Proc Natl Acad Sci U S A. 2010;107:9287–9292. doi: 10.1073/pnas.1001827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis LK, et al. Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet. 2013;9:e1003864. doi: 10.1371/journal.pgen.1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamazon ERIH, Liu C, Members of the Bipolar Disorder Genome Study (BiGS) Consortium. Nicolae DL, Cox NJ. The Convergence of eQTL Mapping, Heritability Estimation and Polygenic Modeling: Emerging Spectrum of Risk Variation in Bipolar Disorder. arxiv. 2013 [Google Scholar]
- 11.Gusev ALS, Neale BM, Trynka G, Vilhjalmsson BJ, Finucane H, Xu H, Zang C, et al. Regulatory variants explain much more heritability than coding variants across 11 common diseases. bioRxiv. 2014 [Google Scholar]
- 12.An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Consortium TG. The Genotype-Tissue Expression (GTEx) pilot analysis: multi-tissue gene regulation in humans. Science. 2015 doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lappalainen T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battle A, et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramasamy A, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17:1418–1428. doi: 10.1038/nn.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tibshirani R. Regression shrinkage and selection via the Lasso. Journal of the Royal Statistical Society Series B-Methodological. 1996;58:267–288. [Google Scholar]
- 19.Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2005;67:301–320. [Google Scholar]
- 20.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammer GE, Kanaseki T, Shastri N. The final touches make perfect the peptide-MHC class I repertoire. Immunity. 2007;26:397–406. doi: 10.1016/j.immuni.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 22.WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotsapas C, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diab Rep. 2011;11:533–542. doi: 10.1007/s11892-011-0223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hakonarson H, et al. A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes. 2008;57:1143–1146. doi: 10.2337/db07-1305. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, et al. Genetically dependent ERBB3 expression modulates antigen presenting cell function and type 1 diabetes risk. PLoS One. 2010;5:e11789. doi: 10.1371/journal.pone.0011789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart AB, et al. Genome-wide association study of d-amphetamine response in healthy volunteers identifies putative associations, including cadherin 13 (CDH13) PLoS One. 2012;7:e42646. doi: 10.1371/journal.pone.0042646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hart AB, et al. Genetic variation associated with euphorigenic effects of d-amphetamine is associated with diminished risk for schizophrenia and attention deficit hyperactivity disorder. Proc Natl Acad Sci U S A. 2014;111:5968–5973. doi: 10.1073/pnas.1318810111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Psychiatric GCBDWG. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morley M, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price AL, et al. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet. 2011;7:e1001317. doi: 10.1371/journal.pgen.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 2008;24:408–415. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manor O, Segal E. Robust prediction of expression differences among human individuals using only genotype information. PLoS Genet. 2013;9:e1003396. doi: 10.1371/journal.pgen.1003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres JM, et al. Cross-tissue and tissue-specific eQTLs: partitioning the heritability of a complex trait. Am J Hum Genet. 2014;95:521–534. doi: 10.1016/j.ajhg.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 38.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–784. doi: 10.1093/bioinformatics/btu704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7:500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hastie T, Tibshirani R, Friedman JH. The elements of statistical learning : data mining, inference, and prediction. xxii. New York, NY: Springer; 2009. p. 745. [Google Scholar]
- 43.Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 44.Wheeler HE, et al. Poly-omic prediction of complex traits: OmicKriging. Genet Epidemiol. 2014;38:402–415. doi: 10.1002/gepi.21808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrow J, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okada Y, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu JZ, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu MC, et al. Powerful SNP-set analysis for case-control genome-wide association studies. Am J Hum Genet. 2010;86:929–942. doi: 10.1016/j.ajhg.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu MC, et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carroll RJ, Eyler AE, Denny JC. Naive Electronic Health Record phenotype identification for Rheumatoid arthritis. AMIA Annu Symp Proc. 2011:189–196. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.