

Abstract
Measuring intracellular metabolism has increasingly led to important insights in biomedical research. 13C tracer analysis, although less information-rich than quantitative 13C flux analysis that requires computational data integration, has been established as a time-efficient method to unravel relative pathway activities, qualitative changes in pathway contributions, and nutrient contributions. Here, we review selected key issues in interpreting 13C metabolite labeling patterns, with the goal of drawing accurate conclusions from steady state and dynamic stable isotopic tracer experiments.
Keywords: 13C tracer analysis, metabolite labeling patterns, metabolic fluxes, metabolite concentrations, steady state labeling patterns, dynamic labeling patterns, mammalian cell metabolism, nutrient contributions, metabolic pathway activities
Introduction
Investigating cellular metabolism has a long-standing history in various research areas such as biochemistry, biotechnology and cellular physiology. A widely applicable toolbox to quantitatively measure intracellular metabolism has been developed in the context of biochemical engineering [1]. In light of the emerging realization that altered cellular metabolism contributes to many diseases including cancer, metabolic syndromes, and neurodegenerative disorders, these approaches are being increasingly applied to address biomedical research questions [2–9].
Cellular metabolism can be characterized by many parameters including nutrient uptake and metabolite secretion rates, intracellular metabolite levels, intracellular metabolic rates (fluxes), nutrient contributions to metabolite and macromolecule synthesis, and pathway activities [2,3,9–12].
Metabolomics, which provides absolute or relative intra- or extracellular metabolite levels, is a broad and sensitive method to detect differences in metabolic states between conditions [13–16]. Changes in intracellular metabolite levels indicate an altered activity of the connected consuming or producing reactions (e.g. enzymatic, non-enzymatic, or transport reactions) [17–21]. However, concentration changes do not readily allow conclusions on metabolic rates (fluxes), or the direction of the flux changes, since an increase in metabolite concentration can both be indicative of increased activity of metabolite producing enzymes, but also decreased activity of metabolite consuming enzymes.
In combination with growth rates (which provide global information on metabolic fluxes to biomass production), metabolite uptake/secretion rates provide a macroscopic picture of overall metabolism. For instance, measuring the rate of glucose depletion from the media reports the rate of glucose used by cells in a culture system. However, these data alone are insufficient to reveal intracellular fluxes throughout the different metabolic pathways.
To examine intracellular fluxes (metabolite amount converted/cell/time), heavy isotope (most frequently 13C) labeled nutrients (tracers) are commonly utilized [22–29]. In formal 13C flux analysis, labeling patterns in intracellular metabolites resulting from metabolizing a 13C labeled nutrient, cellular uptake and secretion rates, and prior knowledge of the biochemical reaction network are combined to computationally estimate metabolic fluxes [11,30–34]. In practice, resolving metabolic fluxes from measured data can be time and data-intensive. In many cases, however, direct interpretation of 13C labeling patterns (without formal 13C flux analysis) is sufficient to provide information on relative pathway activities, qualitative changes in pathway contributions via alternative metabolic routes, and nutrient contribution to the production of different metabolites. We refer to this direct interpretation of 13C labeling patterns as 13C tracer analysis. Here, we discuss selected important aspects to consider when performing 13C tracer analysis to ensure correct data interpretation and to increase the insight obtained by stable isotopic tracer experiments.
Metabolic steady state versus isotopic steady state
Metabolic steady state requires that both, intracellular metabolite levels and intracellular metabolic fluxes of a cell or a cell population are constant (Figure 1a) [35]. Controlled culture systems that ensure metabolic steady state are continuous cultures (known as chemostats), where cell number and nutrient concentrations are maintained constant throughout the experiment [36]. More commonly, experiments are performed at pseudo-steady state, where changes in metabolite concentrations and fluxes are minimal on the timescale over which the measurement is being made. In adherent mammalian cell culture, perfusion bioreactors and nutrostats [37,38], where nutrient concentrations but not cell number are constant over time, are closest to a chemostat. In conventional monolayer culture, the exponential growth phase is often assumed to reflect metabolic pseudo-steady state, because cells in the culture steadily divide at their maximal condition specific rate, given that nutrient supply does not become limiting [39]. So long as biological changes (e.g. differentiation) occur slowly relative to the timescale of metabolic measurement, non-proliferating cells are generally also in metabolic pseudo-steady state. This can be verified by time resolved measurements of metabolic parameters of interest [40]. In case the biological system is not in metabolic pseudo-steady state, e.g. following acute signaling events or nutrient modulations, tracer experiments can still provide qualitative and quantitative information on metabolic pathway fluxes, but interpretation of non-steady state data require different approaches [30,41–43] than the here discussed 13C tracer analysis at metabolic pseudo-steady state.
Figure 1.

Labeling basics. a) Time dependent metabolic changes: metabolism reaches a metabolic steady state when the parameters of interest (e.g. glucose uptake rate) are constant over time. b) Time dependent labeling changes: upon addition of an isotopically labeled carbon source, the isotopic enrichment will increase in the metabolites until the steady state enrichment is reached. c) Mass distribution vector (MDV) (also known as mass distribution (MID) vector): Labeling patterns are MDVs that consist of the fractional abundance of each isotopologue (mass isotopomer). M denotes mass of the unlabeled metabolite. d) Cellular compartmentalization: Most labeling pattern detection methods cannot resolve different cellular compartments, thus the whole cell average labeling pattern is measured.
While metabolic steady state addresses the state of metabolism, isotopic steady state characterizes the enrichment of a stable isotopic tracer in metabolites. When a 13C labeled substrate is added and subsequently metabolized, the metabolites will become with time increasingly enriched for 13C until the point where the 13C enrichment is stable over time (Figure 1b). From a practical perspective, isotopic steady state is reached when 13C enrichment into a given metabolite is stable over time relative to experimental error and/or the desired measurement accuracy. These enrichment dynamics differ depending on the analyzed metabolite and the tracer employed, since the time required to reach isotopic steady state depends on both the fluxes (i.e. rate of conversion) from the nutrient to that metabolite, and the pool sizes of that metabolite and all intermediate metabolites. For example, upon labeling with 13C-glucose, isotopic steady state in glycolytic intermediates typically occurs within minutes, whereas for TCA cycle intermediates it may take several hours. For many amino acids that are both produced by the cell and are supplemented in the media isotopic steady state may never be achieved in standard monolayer culture, due to constant and rapid exchange between the intracellular and the extracellular amino acid pools. In such a situation, qualitative tracer analysis can easily be misleading, and quantitative, formal approaches are required (e.g. [44]).
Key aspects
Proper interpretation of labeling data depends on prior assessment of whether the system is at metabolic pseudo-steady state. If so, interpretation of tracer data is most simple if labeling is allowed to proceed also to isotopic steady-state
The time to reach isotopic steady state depends both on the tracer being employed and the metabolites being analyzed.
Many amino acids are freely exchanged between intracellular and extracellular pools. This can prevent labeling from reaching isotopic steady state and any intracellular metabolite pool that is in rapid exchange with a larger extracellular pool is subject to this complication.
Labeling patterns
The term ‘labeling pattern’ refers to a mass distribution vector (MDV) (they are also frequently called mass isotopomer distribution (MID) vectors) (Figure 1c). The shift in mass of a metabolite occurs due to the incorporation of isotopes. Metabolites that only differ in the isotope composition are isotopologues (they are frequently also called mass isotopomers). MDVs describe the fractional abundance of each isotopologue normalized to the sum of all possible isotopologues. A metabolite with n carbon atoms can have 0 to n of its carbon atoms labeled with 13C, resulting in isotopologues that increase in mass (M) from M+0 (all carbons unlabeled e.g. 12C) to M+n (all carbons labeled e.g. 13C). Hence, the MDV represents the relative abundances of M+0 to M+n isotopologues for one particular metabolite (Figure 1c). Consequently, the sum of all fractions from M+0 to M+n is 100% or 1. Note that in respect to 13C each isotopologue has isotopomers (same isotope composition but different position of the isotope within the metabolite), when n denotes the number of carbons in a metabolite and k the number of carbons that are 13C (Figure 1c). Isotopomers can only be resolved using a detection method that can assign a specific position to a 13C within a molecule (e.g. nuclear magnetic resonance spectroscopy [45], mass spectrometry analysis of multiple fragments [46] or in specific cases tandem mass spectrometry [47,48]). While information on the position of a the 13C label can increase the information content of labeling data, the MDV is typically sufficient to draw conclusions on nutrient contributions, and also often regarding pathway activities. Notably, while we will discuss 13C tracer analysis, the above-described MDVs can be also applied to other stable isotopes including 15N and 2H.
To apply MDVs to assess nutrient contributions and pathway activities, it is important to first correct for the presence of naturally occurring isotopes, e.g, 13C (1.07% natural abundance (na)), 15N (0.368% na), 2H (0.0115% na), 17O (0.038% na), 18O (0.205% na), 29Si (4.6832% na), or 30Si (3.0872% na) [49–51]. For example, glutamate and α-ketoglutarate, which are normally in complete exchange and share the same carbon backbone, should accordingly have matching MDVs. Yet, since they differ in their molecular formula, uncorrected MDVs of glutamate and α-ketoglutarate will not match because of the natural occurring isotopes in N, H, and O. For analytical methods that require metabolite derivatization to enable chromatographic separation (e.g. gas chromatography - mass spectrometry), the chemical modification adds additional C, H, N, O, and Si atoms to the metabolites [22,52]. Hence, the natural labeling of all atoms in the metabolite and the derivatization agent needs to be taken into account when performing data correction. For analysis of underivatized metabolites (e.g. by liquid chromatography - mass spectrometry), naturally occurring 13C has a much greater effect than other natural isotopes, and it is minimally imperative to correct for it.
A general applicable correction matrix can be formulated based on equation (1).
| (1) |
Here, the vector I denotes the fractional abundances of the measured metabolite ions. M represents the MDV corrected for naturally occurring isotopes. n denotes the number of carbon atoms that are present in the analyzed metabolite ion and are subject to isotope labeling. u denotes additional measured ion abundances beyond n originating from natural isotopes in the metabolite or the derivatization. L denotes the correction matrix and the columns LMk denote the theoretical natural MDV when k (0 to n) carbons are 13C. The correction matrix L can be calculated based on the sum formula of the metabolite ion under consideration of natural isotope abundances [49,53,54]. To solve the linear equation system at least n+1 abundances have to be measured. If more than n+1 abundances are considered, this results in an overdetermined system and provides a more robust solution. Tools for quickly converting raw into corrected MDVs are available [55,56].
When using analytical approaches involving selected ion monitoring (SIM) or selected reaction monitoring (SRM) mass spectrometry, it is important to consider upfront the potential role of naturally occurring isotopes when setting the selected mass range [50]. In cases involving derivatization with Si-containing reagents, inclusion of these higher mass ranges may be important and the required mass range can be estimated based on multinomial expansion (typically a shift of up to 4 amu beyond the mass of the fully labeled metabolite should be considered).
Comparison between labeled and unlabeled samples is sufficient to determine whether an observed mass shift truly reflects labeling (as opposed to merely natural isotope abundance). It is not appropriate, however, to subtract the MDV of an unlabeled sample from the labeled sample. Typically, the main natural abundance peak in the unlabeled sample will be M+1, whereas in labeled samples natural abundance results in peaks at higher masses.
The natural occurring isotopes can be also used to validate the applied mass spectrometry method for its accuracy to measure isotopologue distributions [22]. Specifically, metabolites can be extracted from cells fed with naturally labeled nutrients (commonly referred to as unlabeled nutrients) and consequently the measured MDV of these metabolites should accurately (< ±1.5%) reflect the theoretical distribution of natural occurring isotopes. With this validation the applied mass spectrometry method can be improved or metabolites for which the isotopologue distribution is measured with poor accuracy can be excludes. It is important to be aware of the extent of error in MDV measurements and to interpret resulting labeling data accordingly. Random error in MDV measurement is often significant for metabolites that are low abundance (i.e. measurement signal close to noise). Systematic error in MDV measurement is more serious and can reflect metabolite misannotations or overlaps of the measured metabolite ions with same mass ions from sample matrix compounds. In case the accuracy to measure isotopologue distributions is validated, data variability can be a subject of the experimental procedure (e.g. inadequate metabolism quenching) or the biological system (e.g. rapid metabolic shifts or a continuous metabolic drift).
Key aspects
Correction for natural abundance facilitates proper interpretation of labeling data.
Subtracting the measured MDV of an unlabeled metabolite from the measured MDV of the labeled metabolite is not a valid method to correct for natural abundance.
Labeling patterns must be interpreted in light of the experimental error in MDV measurements of the chosen analytical approach. Measurement error will typically be higher for low abundance compounds.
In case measurement inaccuracy can be excluded, data variability can result from the experimental procedure or the biological system
Cellular compartments
Eukaryotic cells have organelles such as mitochondria and peroxisomes, and these organelles result in intracellular compartmentalization of metabolites and metabolic reactions. Many metabolites are present in multiple intracellular compartments and even spatial distribution within a compartment might occur. This adds a layer of complexity to understanding metabolism. Only the average labeling pattern and metabolite levels from all compartments within a cell can be measured using most current techniques (Figure 1d) [57,58].
Depending on the metabolite of interest, compartment-specific labeling patterns in some cases can be inferred from labeling of metabolites that are produced exclusively in one compartment (Figure 1d). For example, pyruvate is found both in the cytosol and in the mitochondria. Lactate and alanine are both directly produced from pyruvate. Lactate dehydrogenase, the enzyme which interconverts pyruvate and lactate, is a strictly cytosolic enzyme [59], an assumption in agreement with the observation that the deletion of the mitochondrial pyruvate carrier does not affect lactate production [60,61]. The finding that mitochondrial pyruvate carrier deletion drastically affects alanine production [60,61] supports that alanine is produced extensively from mitochondrial pyruvate [62]. Thus, under experimental conditions in which neither exogenous alanine nor lactate is available to cells, lactate labeling likely reflects the labeling pattern of cytosolic pyruvate, while alanine labeling better reflects the labeling pattern of mitochondrial pyruvate. Additionally, engineered compartment-specific production of metabolites in cells can also be used to provide compartment specific information. For example, labeling of NADPH in the mitochondria and the cytosol was determined by compartmentalized transfer of deuterium to the metabolite 2-hydroxyglutarate (2-HG) [63]. Specifically, transient expression of either mutant isocitrate dehydrogenase 1 or 2 results in compartment specific production of 2-HG that utilizes NADPH available in that location. This approach, and a similar approach but without engineered compartment specific production of 2-HG was used to infer compartmentalized serine – glycine interconversion [63,64].
Key aspects
In most cases cell average labeling patterns are measured. Because many metabolites are present in more than one subcellular compartment, this can affect the extent and pattern of the metabolite labeling observed.
Steady state labeling
MDVs describe the relative fractions of isotopologues within a metabolite. At isotopic steady state, and in the absence of compartment-specific labeling patterns, MDVs are independent of metabolite levels. Therefore, metabolites that are in complete exchange such as glutamate and α-ketoglutarate have identical MDVs even though their intracellular levels are very different [65–67] (Figure 2a). Consequently, any further analysis of relative pathway activities, qualitative changes in pathway contributions, or nutrient contributions based on isotopic steady state labeling data only requires MDVs and is independent of the metabolite levels. Notably, this simplifying assumption breaks down when compartmentation is significant and results in compartment-specific labeling patterns (see section above).
Figure 2.

Interpretation of labeling data. a) Steady state labeling data are independent from the metabolite levels. b) Fractional contribution quantifies the contribution of a labeled nutrient to the metabolite of interest. c) Exchange fluxes can lead to rapidly labeled metabolites although the net flux of the nutrient to the metabolites is small. d) Dynamic labeling patterns are metabolite level dependent: The flux from glutamine to glutamate is the same in condition A and B, but in condition A the glutamate levels are greater than in condition B. Consequently, the labeling dynamics of glutamate in condition A are slower than in condition B although the flux from glutamine to glutamate is the same in both conditions. e) Relative flux activity between two conditions can be evaluated without kinetic flux calculations if both the labeling dynamics and all metabolite levels of the pathway of interest are altered in the same direction.
Nutrient contribution
To determine which fraction of a metabolite’s carbon is produced from a certain nutrient the fractional contribution (FC) using the fully 13C-labeled nutrient can be calculated based on equation (2). Using positionally labeled nutrients for this analysis is not advised because positionally labeled tracers will not only reflect changes in the FC but also differential pathway usage. For example, the FCfrom glucose in pyruvate calculated from a 1-13C1-glucose tracer can be altered between conditions because of a reduction in the fraction of pyruvate produced from glucose or because the forward flux through the oxidative and non-oxidative pentose phosphate pathway is increased, leading to the incorporation of the 13C labeled carbon into CO2.
| (2) |
Here n is the number of C atoms in the metabolite, i denotes the isotopologues, and m the abundance of an isotopologue. Alternatively, FC can be directly calculated from the MDV by equation (3), which takes advantage of the fact that the sum of all fractions from M+0 to M+n is already normalized to 1.
| (3) |
Here s is the relative fraction of the isotopologues.
If only two carbon sources (e.g. glucose and glutamine) contribute to the formation of a metabolite, the sum of FCfrom glutamine and FCfrom glucose will be 100% or 1 for this metabolite (Figure 2b). Thus, the relative contributions of carbon sources to a metabolite can be determined from FCs. As an example, this approach was applied to reveal a switch from glucose to glutamine-derived tricarboxylic acid (TCA) cycle metabolites during metformin treatment [68]. For any metabolite that is subject to a carboxylation reaction the FC values will be reduced due to incorporation of unlabeled CO2 [69]. Similarly any other incorporation of unlabeled carbon sources will also lead to a reduced FC. For example a low FC of fatty acids from 13C6-glucose and 13C5-glutamine in hypoxia was recently used to reveal a contribution from serum acetate to fatty acid synthesis [70].
Also isotope impurity of the tracer will reduce FC values. Yet, for standard quality of tracers (e.g. 1% for 13C6-glucose), the reduction of FC values based on isotope impurity is marginal. For example, although in 13C6-glucose with 1% isotope impurity only 94% of the glucose molecules carry at each carbon position a 13C, the FC for this 13C6-glucose is 0.99. Thus, normalizing to the FC of the tracer has in this case little effect. However, for tracers with higher isotope impurity a normalization to the FC of the tracer can be useful.
Nutrient contribution indirectly provides some information on flux: it reveals the fraction of the metabolite being formed by the sum of all pathways leading from the labeled nutrient to the metabolite. It does not reveal the activity of specific pathways, nor absolute fluxes. For example, two metabolites can have identical FC although the net flux (Figure 2c) of the labeled nutrient to one of the metabolites is much smaller than to the other, but between both metabolites exists a rapid exchange flux. Thus, rapid exchange fluxes (Figure 2c) can readily label metabolites although the net flux to the metabolite might be marginal.
Key aspects
If the sum of the labeled nutrient contributions to a metabolite do not sum up to 100% or 1, and the labeling in the metabolite is in isotopic steady state, there are additional sources that contribute to the production of that metabolite.
In general, nutrient contributions alone do not reveal specific or absolute fluxes.
Exchange fluxes can lead to labeled metabolites although the net flux to the metabolites is marginal.
Pathway activity
Specific isotopologues do not provide perse information on absolute fluxes, rather they allow conclusions on relative pathway activities and qualitative changes in pathway contributions to the production of a certain metabolite. Thereby, isotopologue patterns can indicate the activity of alternative metabolic routes.
Relative pathway activity
Relative pathway activities can be inferred from a ratio between two alternative and converging pathways. A 13C labeled tracer can be fed to the cells, which is designed to result in different labeling patterns when converted through either of the two alternative metabolic pathways. Calculating a split ratio of the activity between two alternative and converging pathways requires that the labeling patterns of the metabolites are valid surrogates of the two pathways, and that a converged pathway metabolite can be measured [50]. Consequently, the sum of the relative activity of both alternative and converging pathways is 100% or 1. In those cases where additional information can provide the forward flux for one of the pathways, then the forward flux of the other pathway can be calculated based on the split ratio. For example, the pentose phosphate pathway has both oxidative and non-oxidative branches that connect to glycolysis at different locations [71]. Under some conditions, pentoses produced via the oxidative pathway can re-enter glycolysis via the non-oxidative pathway, providing two routes from glucose-6-phosphate to trioses. When 1,2-13C2-glucose is converted through glycolysis, M+0 and M+2 pyruvate will be formed, while conversion of glucose to pyruvate through the oxidative pentose phosphate pathway will lead to M+0, M+1, and M+2 pyruvate. The split ratio (relative pathway activity) between glycolysis and the pentose phosphate pathway can be estimated based on the above-described different labeling patterns [72,73]. Notably, the difference in pyruvate labeling is not informative as to relative pentose phosphate pathway and glycolysis flux if the non-oxidative pentose phosphate pathway flux is directed toward pentose production, as is the case in many cancer cells [74,75]. A more direct measurement of oxidative pentose phosphate pathway flux can be obtained from quantifying 14CO2 production from 1-14C1-glucose versus 6-14C1-glucose [76]. Alternatively, formal 13C flux analysis or isotopomer analysis based on nuclear magnetic resonance can be applied to determine the oxidative pentose phosphate pathway activity from 13C-labeling data (e.g. [77,78]).
Importantly, if a single nutrient contributes to a pathway, then steady state labeling data are not informative as to relative or qualitative pathway activity or flux. For instance, in most cases glycolytic intermediates are labeled primarily from glucose and the fact that glycolytic intermediates are labeled from glucose at steady state does not provide any information on the magnitude of the glycolytic flux.
Qualitative changes in pathway contribution
Qualitative changes in pathway contribution are indicative of whether a certain pathway is used to produce a metabolite. Thereby, only the labeling pattern that is indicative for the pathway of interest is analyzed. Consequently, no quantitative split ratio is calculated between the pathway of interest and the alternative conversion routes, and only qualitative information is obtained. Examples are changes in the fractional abundance of M+5 citrate from 13C5-glutamine to suggest reductive glutamine metabolism [79–84], or M+3 malate from 13C6-glucose to suggest contribution of pyruvate via pyruvate carboxylase. The example of pyruvate carboxylase is discussed in full detail in Box 1. Recognizing the limitations of the specific isotopologues to be indicative for relative pathway activities or qualitative changes in pathway contribution is important, and use of more than one tracer as well as investigation of more than one metabolite labeling can increase confidence in conclusions (Box 1).
Box 1. Qualitative changes in pathway contribution: 13C tracer analysis to identify changes in pyruvate anaplerosis.
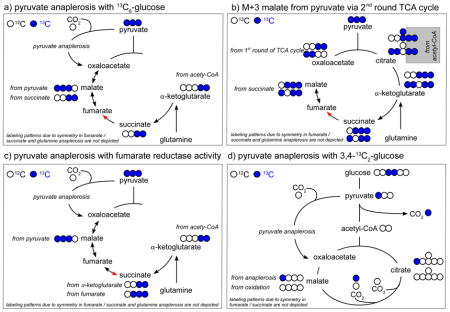
Pyruvate anaplerosis is the counterpart to glutamine anaplerosis and allows the TCA cycle to continuously oxidize acetyl-CoA simultaneously to providing carbon backbones for biomass production. During pyruvate anaplerosis, pyruvate is converted via pyruvate carboxylase to oxaloacetate to compensate for metabolite loss from TCA cycle due to biomass production.
Pyruvate anaplerosis can be identified by measuring M+3 malate, oxaloacetate, or fumarate under conditions when 13C6-glucose is consumed (a). Notably, oxaloacetate is not measurable by many metabolomics protocols because it requires a direct derivatization during quenching due to its chemical instability [96]. However, the labeling pattern of aspartate can serve as a surrogate of oxaloacetate labeling in aspartate-free medium, because if the medium does not contain this amino acid, oxaloacetate is the sole source of aspartate carbon. Notably, if oxaloacetate from pyruvate anaplerosis is further used in the TCA cycle, also a fraction of M+5 citrate from 13C6-glucose will appear, because M+3 oxaloacetate will be combined with M+2 acetyl-CoA.
Under 13C6-glucose labeling conditions M+3 malate, aspartate, and fumarate can also be formed by multiple oxidation rounds in the TCA cycle (b). Hence, to ensure that the M+3 malate, aspartate, and fumarate from 13C6-glucose are indicative of pyruvate anaplerosis, this isotopologue should be compared to the M+3 succinate (b). Comparison between malate, aspartate, and fumarate with succinate is thereby directly possible because they all consist of the same four-carbon backbone. Thus, differences between the M+3 in malate, aspartate, and fumarate and the M+3 succinate represents the pyruvate anaplerosis contribution to the TCA cycle, given that fumarate reductase activity is absent.
Fumarate reductase activity of the succinate dehydrogenase complex converts fumarate to succinate and thereby constitutes anaerobic electron transport. This activity is found in many bacteria and fungi, but it has been also shown to occur in some mammalian cells during starvation, ischemic, or hypoxic conditions [97–99]. When fumarate reductase activity is observed under 13C6-glucose labeling conditions, the M+3 malate, aspartate, and fumarate can match the M+3 succinate although pyruvate anaplerosis is active (c). Therefore, the M+3 malate, aspartate, and fumarate should also be compared to the M+3 and M+4 α-ketoglutarate, because the α-ketoglutarate to succinate reaction can be considered to operate only in forward direction (c). Notably, α-ketoglutarate consists of a five carbon backbone and in the reaction to succinate CO2 is lost. Thus, this difference in the carbon backbone has to be taken into account when the MDVs of malate, aspartate, and fumarate are compared to α-ketoglutarate.
To assess the contribution of pyruvate to TCA cycle via pyruvate carboxylase with an additional tracer, 1-13C1-pyruvate (or 3,4-13C2-glucose, which is converted to 1-13C1-pyruvate) can be used (d) [100,101]. Using these tracers, the 13C labeled carbon is lost in the pyruvate dehydrogenase reaction but is retained when pyruvate enters via pyruvate anaplerosis into the TCA cycle (d). Any further TCA cycle metabolization of oxaloacetate from pyruvate anaplerosis, however, will lead to loss of the the labeled carbon (d). Notably, when using a pyruvate tracer in the presence of glucose as a carbon source, the enrichment of 13C-pyruvate needs to be taken into account when estimating relative pyruvate anaplerosis.
Hence, pyruvate anaplerosis is a good example that it can be helpful to depict not only the isotopologue of interest, but the MDVs of multiple connected metabolites.
Key aspects
Steady state labeling patterns are independent of metabolite levels. Consequently, multiplying MDVs with metabolite levels at isotopic steady- state (or simply reporting the absolute magnitudes of different labeled species) does not reliably provide information on any metabolic changes. More reliable information is obtained by examining the steady-state labeling fractions themselves to infer relative or qualitative pathway activities and nutrient contributions.
Relative pathway activities and qualitative changes in pathway contribution to the production of a metabolite do not allow conclusions on absolute flux magnitudes.
Steady state labeling patterns are information-rich for metabolites in pathways with more than one source of nutrient contribution or alternative metabolic routes for nutrient contribution.
Steady state labeling patterns for linear pathways without alternative nutrient contributions are not informative for pathway activity.
If no formal split ratio of pathway contribution can be calculated, any pathway activity inferred from steady state labeling patterns is qualitative.
It is important to remember that relative contributions may change due to the increased activity of one pathway, or the decreased activity of another pathway.
Analysis of labeling patterns from more than one 13C tracer can increase confidence in data interpretation.
Analysis of labeling patterns in more than one metabolite can increase confidence in data interpretation.
Dynamic labeling
Dynamic labeling is a powerful method to infer flux from metabolite labeling data and metabolite levels (Figure 2d) [30,41,51,85]. During dynamic labeling, how fast a metabolite pool becomes labeled is measured. The underlying principle is that the greater a flux the faster a metabolite pool becomes labeled; however, considering the size of the metabolite pool is critical as larger metabolite pools will take longer to be labeled than smaller metabolite pools (Figure 2d). Thus, dynamic labeling patterns are inherently metabolite level dependent, and will also depend on the pool size and labeling rates of upstream metabolites if the labeled intermediate is not directly produced from the 13C tracer.
Integrating dynamic labeling data into metabolic models has mainly been applied to microbial systems, although other systems have been recently investigated as well [34,39,86–89]. For a meaningful direct interpretation of dynamic labeling patterns a suitable time resolution (i.e. multiple time points that cover the labeling dynamics) and measurement of the metabolites that are upstream of the metabolite of interest are essential. This is required to a) obtain reliable curve fits and b) determine when the dynamic profile transitions to steady state labeling. Notably, dynamic labeling is limited by the feasible time resolution. Therefore, low flux pathways such as glutamine anaplerosis to the TCA cycle (conversion of glutamine to α-ketoglutarate) are easier to correctly infer with dynamic labeling data than high flux pathways such as glycolysis. Additionally, the direct interpretation of dynamic labeling patterns (without sophisticated methods such as non-stationary 13C flux analysis) requires that the metabolite levels are constant over time. In practice, this is often achieved by exchanging the medium for a period of time with unlabeled medium prior to adding 13C-labeled media. This allows the intracellular metabolite levels that are in rapid exchange with the medium (e.g. lactate) to equilibrate to a medium without high levels of these metabolites present extracellularly. Importantly, 13C tracer analysis as discussed here is only valid if the media change does not affect the metabolic steady state.
For calculating fluxes from dynamic labeling data two cases have to be considered. Either the direct substrate metabolite of the reaction of interest is 100% labeled and the metabolite of interest is not a product from a condensation reaction, or this is not the case. In the first case flux can be calculated based on equation (4) [51].
| (4) |
Here, XU is the unlabeled metabolite level, fX is the sum of fluxes producing the metabolite X from the tracer substrate and XT is the total metabolite level (sum of all labeling states). Hence, XU/XT is the fraction of M+0 in the metabolite of interest. In the second case isotopically non-stationary 13C flux analysis [30,40,85] or kinetic flux profiling [51,90,91] can be performed. These methods require the same additional data and information as steady state 13C flux analysis and/or measurements of metabolite levels along the pathway of interest. Moreover, to measure biosynthesis and turnover of polymers mass isotopomer distribution analysis (MIDA) [92] or isotopomer spectral analysis (ISA) [93] can be applied.
In some experimental setups, pragmatic simplifications to compare flux between two conditions have been applied [66,94,95]. For example, a pulse of a fully labeled carbon source is added to the medium in a reference and in a perturbed condition (e.g. mutant, or drug treatment). Under the precondition that all metabolite levels along the pathway of interest are lower in the perturbed condition than in the reference condition, a slower or equal decrease in M+0 of the metabolite of interest in the perturbed condition signifies a lower pathway flux in the perturbed condition (Figure 2e). To allow assessment of metabolite levels and labeling dynamics, both parameters should be depicted separately. Notably, such a qualitative analysis of dynamic labeling data and metabolite levels are subject to the assumption that the non-compartment specific metabolite level measurement does not impact the data interpretation and that the metabolites of the pathway of interest are not in rapid exchange with other metabolites outside the pathway. If doubts about these assumptions exist, then conclusions based on the assumptions should be verified with formal non-stationary 13C flux analysis.
Key aspects
Multiple time points are essential to interpret directly dynamic 13C labeling patterns. Interpretations of single time points of dynamic 13C labeling patterns are not reliable.
Dynamic labeling is limited by the feasible time resolution (e.g. glycolytic intermediates are labeled in the second to minute range).
Qualitative and quantitative assessment of dynamic labeling patterns (without formal non-stationary 13C flux analysis) must take metabolite levels into account.
Interpretation of labeling dynamics in a pathway with metabolites that are in rapid exchange with other metabolites outside a pathway require other approaches than the here discussed direct 13C tracer analysis.
Compartment-dependent metabolite production can impact the interpretation of dynamic labeling data.
Concluding remarks
13C- and other isotope-labeled tracers can be powerful tools to interrogate the metabolism of cells. They can determine relative pathway activities, qualitative changes in pathway contribution, nutrient contributions, and help infer metabolic fluxes. Analysis using more than one tracer and examination of multiple metabolites can help to increase the confidence in conclusions from direct 13C tracer analysis. Moreover, integration of labeling data with additional information such as uptake and secretion rates will increase the resulting understanding of cellular metabolism and confidence in the biological conclusions. Importantly, the biological question of interest dictates which metabolic parameters (uptake rates, relative pathway activities, pathway/nutrient contributions, or fluxes) are most important to determine. Taking into account the considerations discussed here will hopefully be useful to the growing set of scientists engaged in metabolic tracer studies.
Highlights.
13C tracer analysis can be used to probe intracellular metabolism in cells
Steady state labeling patterns can quantify nutrient contributions
Steady state labeling patterns can inform about pathway activities
Dynamic labeling patterns combined with metabolite levels can quantify some fluxes
Analysis of multiple metabolites and various tracers enhances information output
Acknowledgments
The authors would like to recognize the breath of outstanding laboratories that perform high-quality tracer analysis, and apologize for the inability to fully cite the associated literature.
We thank Dimitrios Anastasiou, Chi V. Dang, Teresa W.-M. Fan, Richard M. Higashi, Andrew N. Lane, and Hunter Moseley for the support and discussions, and Stefan Christen, Ben Pirongs, Gianmarco Rinaldi, Roberta Schmieder, and Yiping Wang for their feedback on the manuscript. SMF acknowledges funding support from Marie Curie – CIG, FWO – Odysseus, Concern Foundation, FWO – Research Grants, Eugène Yourassowsky Schenking, and Bayer Health Care.
Bibliography
- 1.Toya Y, Shimizu H. Flux analysis and metabolomics for systematic metabolic engineering of microorganisms. Biotechnology Advances. 2013;31:818–826. doi: 10.1016/j.biotechadv.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Keibler MA, Fendt SM, Stephanopoulos G. Expanding the concepts and tools of metabolic engineering to elucidate cancer metabolism. Biotechnol Prog. 2012;28:1409–1418. doi: 10.1002/btpr.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiller K, Metallo C, Stephanopoulos G. Elucidation of Cellular Metabolism Via Metabolomics and Stable-Isotope Assisted Metabolomics. Current Pharmaceutical Biotechnology. 2011:12. doi: 10.2174/138920111795909096. [DOI] [PubMed] [Google Scholar]
- 4.Young JD. Metabolic flux rewiring in mammalian cell cultures. Current Opinion in Biotechnology. 2013;24:1108–1115. doi: 10.1016/j.copbio.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiller K, Metallo CM. Profiling metabolic networks to study cancer metabolism. Current Opinion in Biotechnology. 2013;24:60–68. doi: 10.1016/j.copbio.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 7.DeBerardinis RJ, Thompson CB. Cellular Metabolism and Disease. What Do Metabolic Outliers Teach Us? Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Communication and Signaling. 2013:11. doi: 10.1186/1478-811X-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Molecular Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. This review discusses different mechanisms that cells use to adapt their metabolism to specific physiological states and how metabolic flux analysis can be applied to identify important regulatory nodes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sévin DC, Kuehne A, Zamboni N, Sauer U. Biological insights through nontargeted metabolomics. Current Opinion in Biotechnology. 2015;34:1–8. doi: 10.1016/j.copbio.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat Rev Cancer. 2004;4:551–561. doi: 10.1038/nrc1390. [DOI] [PubMed] [Google Scholar]
- 13.Roberts LD, Souza AL, Gerszten RE, Clish CB. John Wiley Sons, Inc, editor. Current Protocols in Molecular Biology. 2001. Targeted Metabolomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zamboni N, Sauer U. Novel biological insights through metabolomics and 13C-flux analysis. Curr Opin Microbiol. 2009;12:553–558. doi: 10.1016/j.mib.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Reaves ML, Rabinowitz JD. Metabolomics in systems microbiology. Current Opinion in Biotechnology. 2011;22:17–25. doi: 10.1016/j.copbio.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Ser Z, Locasale JW. Development and Quantitative Evaluation of a High-Resolution Metabolomics Technology. Analytical Chemistry. 2014;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Fendt SM, Buescher JM, Rudroff F, Picotti P, Zamboni N, Sauer U. Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol Syst Biol. 2010;6:356. doi: 10.1038/msb.2010.11. Similar to 18 this research article identifies a functional relation between metabolite concentrations and enzyme activities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18*.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. Similar to 17 this reaseach article highlights the identification of organizing metabolic principles from systems-level absolute metabolite concentration data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Link H, Kochanowski K, Sauer U. Systematic identification of allosteric protein-metabolite interactions that control enzyme activity in vivo. Nat Biotech. 2013;31:357–361. doi: 10.1038/nbt.2489. [DOI] [PubMed] [Google Scholar]
- 20.Buescher JM, Liebermeister W, Jules M, Uhr M, Muntel J, Botella E, Hessling B, Kleijn RJ, Le Chat L, Lecointe F, et al. Global Network Reorganization During Dynamic Adaptations of Bacillus subtilis Metabolism. Science. 2012;335:1099–1103. doi: 10.1126/science.1206871. [DOI] [PubMed] [Google Scholar]
- 21.Keller MA, Piedrafita G, Ralser M. The widespread role of non-enzymatic reactions in cellulare metabolism. Current Opinion in Biotechnology. 2015:34. doi: 10.1016/j.copbio.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamboni N, Fendt SM, Ruhl M, Sauer U. (13)C-based metabolic flux analysis. Nat Protoc. 2009;4:878–892. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
- 23.Walther JL, Metallo CM, Zhang J, Stephanopoulos G. Optimization of 13C isotopic tracers for metabolic flux analysis in mammalian cells. Metab Eng. 2012;14:162–171. doi: 10.1016/j.ymben.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crown SB, Antoniewicz MR. Publishing 13C metabolic flux analysis studies: a review and future perspectives. Metab Eng. 2013;20:42–48. doi: 10.1016/j.ymben.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crown SB, Antoniewicz MR. Selection of tracers for 13C-metabolic flux analysis using elementary metabolite units (EMU) basis vector methodology. Metab Eng. 2012;14:150–161. doi: 10.1016/j.ymben.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wittmann C. Fluxome analysis using GC-MS. Microbial Cell Factories. 2007;6:6. doi: 10.1186/1475-2859-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wittmann C, Heinzle E. Application of MALDI-TOF MS to lysine-producing Corynebacterium glutamicum: a novel approach for metabolic flux analysis. Eur J Biochem. 2001;268:2441–2455. doi: 10.1046/j.1432-1327.2001.02129.x. [DOI] [PubMed] [Google Scholar]
- 28.Adler P, Bolten CJ, Dohnt K, Hansen CE, Wittmann C. Core Fluxome and Metafluxome of Lactic Acid Bacteria under Simulated Cocoa Pulp Fermentation Conditions. Applied and Environmental Microbiology. 2013;79:5670–5681. doi: 10.1128/AEM.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiechert W. 13C metabolic flux analysis. Metab Eng. 2001;3:195–206. doi: 10.1006/mben.2001.0187. [DOI] [PubMed] [Google Scholar]
- 30**.Wiechert W, Nöh K. Isotopically non-stationary metabolic flux analysis: complex yet highly informative. Current Opinion in Biotechnology. 2013;24:979–986. doi: 10.1016/j.copbio.2013.03.024. This review article discusses formal approaches for non-startionary 13C flux analysis. [DOI] [PubMed] [Google Scholar]
- 31.Antoniewicz MR. 13C metabolic flux analysis: optimal design of isotopic labeling experiments. Current Opinion in Biotechnology. 2013;24:1116–1121. doi: 10.1016/j.copbio.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Zamboni N. 13C metabolic flux analysis in complex systems. Current Opinion in Biotechnology. 2011;22:103–108. doi: 10.1016/j.copbio.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 33.Crown SB, Antoniewicz MR. Parallel labeling experiments and metabolic flux analysis: Past, present and future methodologies. Metab Eng. 2013;16:21–32. doi: 10.1016/j.ymben.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34**.Niedenführ S, Wiechert W, Nöh K. How to Measure Metabolic Fluxes: A Taxonomic Guide for 13C Fluxomics. Current Opinion in Biotechnology. 2015;34:82–90. doi: 10.1016/j.copbio.2014.12.003. This review article is the counterpart to the here described 13C tracer analysis as they describe formal 13C flux anaysis approaches. [DOI] [PubMed] [Google Scholar]
- 35.Leighty RW, Antoniewicz MR. Dynamic metabolic flux analysis (DMFA). A framework for determining fluxes at metabolic non-steady state. Metab Eng. 2011;13:745–755. doi: 10.1016/j.ymben.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Stephanopoulos GN, Aristidou AA, Nielsen J. In: Metabolic Engineering: Principles and Methodologies. Stephanopoulos GN, Aristidou AA, Nielsen J, editors. Academic Press; 1998. pp. 1–694. [Google Scholar]
- 37.Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahn WS, Antoniewicz MR. Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab Eng. 2011;13:598–609. doi: 10.1016/j.ymben.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Ahn WS, Antoniewicz MR. Towards dynamic metabolic flux analysis in CHO cell cultures. Biotechnol J. 2012;7:61–74. doi: 10.1002/biot.201100052. [DOI] [PubMed] [Google Scholar]
- 41.Young JD, Walther JL, Antoniewicz MR, Yoo H, Stephanopoulos G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol Bioeng. 2008;99:686–699. doi: 10.1002/bit.21632. [DOI] [PubMed] [Google Scholar]
- 42.Wahl S, Noh K, Wiechert W. 13C labeling experiments at metabolic nonstationary conditions: An exploratory study. BMC Bioinformatics. 2008;9:152. doi: 10.1186/1471-2105-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Y-F, Amador-Noguez D, Reaves ML, Feng X-J, Rabinowitz JD. Ultrasensitive regulation of anapleurosis via allosteric activation of PEP carboxylase. Nat Chem Biol. 2012;8:562–568. doi: 10.1038/nchembio.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Shlomi T, Fan J, Tang B, Kruger WD, Rabinowitz JD. Quantitation of Cellular Metabolic Fluxes of Methionine. Analytical Chemistry. 2014;86:1583–1591. doi: 10.1021/ac4032093. This paper describes how to infer fluxes conneted to synthesis and degradation of amino acids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szyperski T, Glaser RW, Hochuli M, Fiaux J, Sauer U, Bailey JE, Wuthrich K. Bioreaction network topology and metabolic flux ratio analysis by biosynthetic fractional 13C labeling and two-dimensional NMR spectroscopy. Metab Eng. 1999;1:189–197. doi: 10.1006/mben.1999.0116. [DOI] [PubMed] [Google Scholar]
- 46.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Measuring deuterium enrichment of glucose hydrogen atoms by gas chromatography/mass spectrometry. Anal Chem. 2011;83:3211–3216. doi: 10.1021/ac200012p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antoniewicz MR. Tandem mass spectrometry for measuring stable-isotope labeling. Curr Opin Biotechnol. 2013;24:48–53. doi: 10.1016/j.copbio.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 48.Choi J, Grossbach MT, Antoniewicz MR. Measuring complete isotopomer distribution of aspartate using gas chromatography/tandem mass spectrometry. Anal Chem. 2012;84:4628–4632. doi: 10.1021/ac300611n. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H. Correction of 13C Mass Isotopomer Distributions for Natural Stable Isotope Abundance. Journal of Mass Spectrometry. 1996;31:255–262. doi: 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 50*.Nanchen A, Fuhrer T, Sauer U. Determination of Metabolic Flux Ratios From 13C-Experiments and Gas Chromatography-Mass Spectrometry Data. In: Weckwerth W, editor. Metabolomics. Vol. 358. Humana Press; 2007. pp. 177–197. Methods in Molecular Biology™. This book chapter provides detailed insights for how to process labeling data from derivatized metabolites. [DOI] [PubMed] [Google Scholar]
- 51**.Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protocols. 2008;3:1328–1340. doi: 10.1038/nprot.2008.131. This protocol describes how to quantitatively infer flux from dynamic labeling patterns and metabolite concentrations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal Chem. 2007;79:7554–7559. doi: 10.1021/ac0708893. [DOI] [PubMed] [Google Scholar]
- 53.Lee WNP, Byerley LO, Bergner EA, Edmond J. Mass isotopomer analysis: Theoretical and practical considerations. Biological Mass Spectrometry. 1991;20:451–458. doi: 10.1002/bms.1200200804. [DOI] [PubMed] [Google Scholar]
- 54.van Winden WA, Wittmann C, Heinzle E, Heijnen JJ. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol Bioeng. 2002;80:477–479. doi: 10.1002/bit.10393. [DOI] [PubMed] [Google Scholar]
- 55.Moseley H. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinformatics. 2010;11:139. doi: 10.1186/1471-2105-11-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wahl SA, Dauner M, Wiechert W. New tools for mass isotopomer data evaluation in 13C flux analysis: mass isotope correction, data consistency checking, and precursor relationships. Biotechnol Bioeng. 2004;85:259–268. doi: 10.1002/bit.10909. [DOI] [PubMed] [Google Scholar]
- 57.Niklas J, Schneider K, Heinzle E. Metabolic flux analysis in eukaryotes. Curr Opin Biotechnol. 2010;21:63–69. doi: 10.1016/j.copbio.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 58.Wahrheit J, Nicolae A, Heinzle E. Eukaryotic metabolism: measuring compartment fluxes. Biotechnol J. 2011;6:1071–1085. doi: 10.1002/biot.201100032. [DOI] [PubMed] [Google Scholar]
- 59.Adeva M, González-Lucán M, Seco M, Donapetry C. Enzymes involved in l-lactate metabolism in humans. Mitochondrion. 2013;13:615–629. doi: 10.1016/j.mito.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 60.Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, Metallo CM. Regulation of Substrate Utilization by the Mitochondrial Pyruvate Carrier. Molecular Cell. 56:425–435. doi: 10.1016/j.molcel.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang C, Ko B, Hensley Christopher T, Jiang L, Wasti Ajla T, Kim J, Sudderth J, Calvaruso Maria A, Lumata L, Mitsche M, et al. Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Molecular Cell. 56:414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Groen AK, Sips HJ, Vervoorn RC, Tager JM. Intracellular Compartment ation and Control of Alanine Metabolism in Rat Liver Parenchymal Cells. European Journal of Biochemistry. 1982;122:87–93. doi: 10.1111/j.1432-1033.1982.tb05851.x. [DOI] [PubMed] [Google Scholar]
- 63*.Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, Vokes NI, Feist AM, Vander Heiden MG, Metallo CM. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Molecular Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. This reasearch paper describes a method to infer compartimentalized metabolism based on genetically engineered cells expressing a sensor metabolite. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64*.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. This reasearch paper elucidates compartment specific serine-glycine interconversion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Molecular Systems Biology. 2013:9. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zupke C, Sinskey AJ, Stephanopoulos G. Intracellular flux analysis applied to the effect of dissolved oxygen on hybridomas. Applied Microbiology and Biotechnology. 1995;44:27–36. doi: 10.1007/BF00164476. [DOI] [PubMed] [Google Scholar]
- 68.Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, et al. Metformin Decreases Glucose Oxidation and Increases the Dependency of Prostate Cancer Cells on Reductive Glutamine Metabolism. Cancer Res. 2013;73:4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leighty RW, Antoniewicz MR. Parallel labeling experiments with [U-13C]glucose validate E. coli metabolic network model for 13C metabolic flux analysis. Metab Eng. 2012;14:533–541. doi: 10.1016/j.ymben.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 70.Kamphorst J, Murphy D. The 2014 Beatson International Cancer Conference: Powering the Cancer Machine. Cancer Metabolism. 2014;2:25. [Google Scholar]
- 71*.Stincone A, Prigione A, Cramer T, Wamelink MMC, Campbell K, Cheung E, Olin-Sandoval V, Gruening N-M, Krueger A, Tauqeer Alam M, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biological Reviews. 2014 doi: 10.1111/brv.12140. This review article provides a summary of the latest insights into how the pentose phosphate pathway is regulated and contribute to diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brekke EMF, Walls AB, Schousboe A, Waagepetersen HS, Sonnewald U. Quantitative importance of the pentose phosphate pathway determined by incorporation of 13C from [lsqb]2-13C[rsqb]- and [lsqb]3-13C[rsqb]glucose into TCA cycle intermediates and neurotransmitter amino acids in functionally intact neurons. J Cereb Blood Flow Metab. 2012;32:1788–1799. doi: 10.1038/jcbfm.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morken T, Brekke E, Håberg A, Widerøe M, Brubakk A-M, Sonnewald U. Neuron–Astrocyte Interactions, Pyruvate Carboxylation and the Pentose Phosphate Pathway in the Neonatal Rat Brain. Neurochemical Research. 2014;39:556–569. doi: 10.1007/s11064-013-1014-3. [DOI] [PubMed] [Google Scholar]
- 74.Liu H, Huang D, McArthur DL, Boros LG, Nissen N, Heaney AP. Fructose Induces Transketolase Flux to Promote Pancreatic Cancer Growth. Cancer Research. 2010;70:6368–6376. doi: 10.1158/0008-5472.CAN-09-4615. [DOI] [PubMed] [Google Scholar]
- 75.Ying H, Kimmelman Alec C, Lyssiotis Costas A, Hua S, Chu Gerald C, Fletcher-Sananikone E, Locasale Jason W, Son J, Zhang H, Coloff Jonathan L, et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ashcroft S, Weerasinghe L, Bassett J, Randle P. The pentose cycle and insulin release in mouse pancreatic islets. Biochem J. 1972;126:525–532. doi: 10.1042/bj1260525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahn WS, Antoniewicz MR. Parallel labeling experiments with [1,2-(13)C]glucose and [U-(13)C]glutamine provide new insights into CHO cell metabolism. Metab Eng. 2013;15:34–47. doi: 10.1016/j.ymben.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 78.Marx A, de Graaf AA, Wiechert W, Eggeling L, Sahm H. Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnology and Bioengineering. 1996;49:111–129. doi: 10.1002/(SICI)1097-0290(19960120)49:2<111::AID-BIT1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 79.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mullen AR, Wheaton WW, Jin ES, Chen P-H, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proceedings of the National Academy of Sciences. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, Stephanopoulos G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun. 2013;4:2236. doi: 10.1038/ncomms3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corbet C, Draoui N, Polet F, Pinto A, Drozak X, Riant O, Feron O. The SIRT1/HIF2α Axis Drives Reductive Glutamine Metabolism under Chronic Acidosis and Alters Tumor Response to Therapy. Cancer Research. 2014;74:5507–5519. doi: 10.1158/0008-5472.CAN-14-0705. [DOI] [PubMed] [Google Scholar]
- 84.Fan J, Kamphorst JJ, Rabinowitz JD, Shlomi T. Fatty Acid Labeling from Glutamine in Hypoxia Can Be Explained by Isotope Exchange without Net Reductive Isocitrate Dehydrogenase (IDH) Flux. Journal of Biological Chemistry. 2013;288:31363–31369. doi: 10.1074/jbc.M113.502740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85*.Hörl M, Schnidder J, Sauer U, Zamboni N. Non-stationary 13C-metabolic flux ratio analysis. Biotechnology and Bioengineering. 2013;110:3164–3176. doi: 10.1002/bit.25004. This research article describes a method how to infer local flux ratios from dynamic labeling data. [DOI] [PubMed] [Google Scholar]
- 86.Ma F, Jazmin LJ, Young JD, Allen DK. Isotopically nonstationary 13C flux analysis of changes in Arabidopsis thaliana leaf metabolism due to high light acclimation. Proceedings of the National Academy of Sciences. 2014;111:16967–16972. doi: 10.1073/pnas.1319485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Munger J, Bennett BD, Parikh A, Feng X-J, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotech. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nöh K, Wiechert W. The benefits of being transient: isotope-based metabolic flux analysis at the short time scale. Applied Microbiology and Biotechnology. 2011;91:1247–1265. doi: 10.1007/s00253-011-3390-4. [DOI] [PubMed] [Google Scholar]
- 89.Nicolae A, Wahrheit J, Bahnemann J, Zeng A-P, Heinzle E. Non-stationary 13C metabolic flux analysis of Chinese hamster ovary cells in batch culture using extracellular labeling highlights metabolic reversibility and compartmentation. BMC Systems Biology. 2014;8:50. doi: 10.1186/1752-0509-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang L, Kim D, Liu X, Myers CR, Locasale JW. Estimating Relative Changes of Metabolic Fluxes. PLoS Comput Biol. 2014;10:e1003958. doi: 10.1371/journal.pcbi.1003958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91**.Heise R, Arrivault S, Szecowka M, Tohge T, Nunes-Nesi A, Stitt M, Nikoloski Z, Fernie AR. Flux profiling of photosynthetic carbon metabolism in intact plants. Nat Protocols. 2014;9:1803–1824. doi: 10.1038/nprot.2014.115. This protocol is an extension of 51 to infer fluxes from dynamic labeling data. [DOI] [PubMed] [Google Scholar]
- 92*.Hellerstein MK, Neese RA. Mass isotopomer distribution analysis: a technique for measuring biosynthesis and turnover of polymers. American Journal of Physiology - Endocrinology and Metabolism. 1992;263:E988–1001. doi: 10.1152/ajpendo.1992.263.5.E988. This research paper describes an approach on how to infer biosynthetic pathway fluxes with stable isotopic tracers. [DOI] [PubMed] [Google Scholar]
- 93*.Kharroubi AT, Masterson TM, Aldaghlas TA, Kennedy KA, Kelleher JK. Isotopomer spectral analysis of triglyceride fatty acid synthesis in 3T3-L1 cells. American Journal of Physiology - Endocrinology and Metabolism. 1992;263:E667–E675. doi: 10.1152/ajpendo.1992.263.4.E667. This reasearch paper describes an approach on how to infer fatty acid synthesis with stable isotopic tracers. [DOI] [PubMed] [Google Scholar]
- 94.Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative Phosphoproteomics Reveal mTORC1 Activates de Novo Pyrimidine Synthesis. Science. 2013;339:1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 95.Lunt Sophia Y, Muralidhar V, Hosios Aaron M, Israelsen William J, Gui Dan Y, Newhouse L, Ogrodzinski M, Hecht V, Xu K, Acevedo Paula NM, et al. Pyruvate Kinase Isoform Expression Alters Nucleotide Synthesis to Impact Cell Proliferation. Molecular Cell. 2015;57(1):95–107. doi: 10.1016/j.molcel.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zimmermann M, Sauer U, Zamboni N. Quantification and Mass Isotopomer Profiling of α-Keto Acids in Central Carbon Metabolism. Analytical Chemistry. 2014;86:3232–3237. doi: 10.1021/ac500472c. [DOI] [PubMed] [Google Scholar]
- 97.Tomitsuka E, Kita K, Esumi H. The NADH-fumarate reductase system, a novel mitochondrial energy metabolism, is a new target for anticancer therapy in tumor microenvironments. Annals of the New York Academy of Sciences. 2010;1201:44–49. doi: 10.1111/j.1749-6632.2010.05620.x. [DOI] [PubMed] [Google Scholar]
- 98.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Des Rosiers C, Fernandez CA, David F, Brunengraber H. Reversibility of the mitochondrial isocitrate dehydrogenase reaction in the perfused rat liver. Evidence from isotopomer analysis of citric acid cycle intermediates. Journal of Biological Chemistry. 1994;269:27179–27182. [PubMed] [Google Scholar]
- 100.Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Matés JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crown S, Ahn W, Antoniewicz M. Rational design of 13C-labeling experiments for metabolic flux analysis in mammalian cells. BMC Systems Biology. 2012;6:43. doi: 10.1186/1752-0509-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]