

Abstract
Helicobacter pylori genetic variation is a crucial component of colonization and persistence within the inhospitable niche of the gastric mucosa. As such, numerous H. pylori genes have been shown to vary in terms of presence and genomic location within this pathogen. Among the variable factors, the Bab family of outer membrane proteins (OMPs) has been shown to differ within subsets of strains. To better understand genetic variation among the bab genes and to determine whether this variation differed among isolates obtained from different geographic locations, we characterized the distribution of the Bab family members in 80 American H. pylori clinical isolates (AH) and 80 South Korean H. pylori clinical isolates (KH). Overall, we identified 23 different bab genotypes (19 in AH and 11 in KH), but only 5 occurred in greater than 5 isolates. Regardless of strain origin, a strain in which locus A and locus B were both occupied by a bab gene was the most common (85%); locus C was only occupied in those isolates that carried bab paralog at locus A and B. While the babA/babB/- genotype predominated in the KH (78.8%), no single genotype could account for greater than 40% in the AH collection. In addition to basic genotyping, we also identified associations between bab genotype and well known virulence factors cagA and vacA. Specifically, significant associations between babA at locus A and the cagA EPIYA-ABD motif (P<0.0001) and the vacA s1/i1/m1 allele (P<0.0001) were identified. Log-linear modeling further revealed a three-way association between bab carried at locus A, vacA, and number of OMPs from the HOM family (P<0.002). En masse this study provides a detailed characterization of the bab genotypes from two distinct populations. Our analysis suggests greater variability in the AH, perhaps due to adaptation to a more diverse host population. Furthermore, when considering the presence or absence of both the bab and homA/B paralogs at their given loci and the vacA genotype, an association was observed. Our results highlight the multifactorial nature of H. pylori mediated disease and the importance of considering how the specific combinations of H. pylori virulence genes and their multiple interactions with the host will collectively impact disease progression.
Introduction
Helicobacter pylori (H. pylori) is a successful pathogen, colonizing the gastric mucosa of over 50% of the world’s population [1, 2]. This pathogen has gained notoriety for its ability to colonize the inhospitable niche of the stomach and to cause gastric diseases [1, 2]. H. pylori cause persistent, potentially lifelong infections; however, only about 20% of individuals infected will develop symptomatic infection. Although clinical manifestations occur only in a subset of infected individuals, these can be severe and include peptic ulcers and gastric cancer. Rates of H. pylori infection and gastric cancer mortality vary geographically. East Asian countries such as China, Japan, and South Korea have high rates of H. pylori infection, and these populations also have some of the highest rates of gastric cancer [3–6].
Environmental, host, and bacterial factors are known to influence H. pylori-associated disease outcome and also vary geographically [7]. Interestingly, H. pylori is a bacterial species that shows exceptionally high rates of genetic variability and intra-species diversity. Indeed, these polymorphisms have been used to determine the geographic origin of H. pylori isolates [8, 9]. Furthermore, it is highly probable that these genetic differences influence virulence. Previous studies have shown that severe disease outcomes such as gastric cancer and ulcers are associated with polymorphism in H. pylori virulence factors such as the cytotoxin-associated gene A (cagA) and vacuolating cytotoxin (vacA) [4, 6, 10–12]. In addition, disease outcome has been associated with several outer membrane proteins (OMPs). Specifically, OMPs such as AlpA and B (adherence-associated lipoprotein A and B), BabA (blood group antigen binding adhesin), HomB (Helicobacter outer membrane B), HopZ (Helicobacter outer membrane protein Z), OipA (outer membrane inflammatory protein A) and SabA (sialic acid binding adhesion) are all associated with variable disease outcomes [13–18]. In comparison to the polymorphisms within cagA and vacA, genetic variability within the OMP families is based upon presence and absence of different closely related paralogs. For example, the bab-family of genes is made up of three paralogs babA, babB and babC, which can be located at three different H. pylori chromosomal loci referred to as locus A, B and C [19–21]. Recently, we presented detailed epidemiological studies of cagA, vacA, and homB from a collection of 260 H. pylori clinical isolates from South Korea [11, 12, 17, 22]. Our studies showed a significant association between infection with H. pylori strains carrying the EPIYA-ABD cagA genotype and the development of gastric cancer [22]. Moreover, the majority of H. pylori isolates from the South Korean population encoded the most virulent toxins, CagA EPIYA-ABD motif and VacA s1/i1/m1 allele [11]. While, we found no association between the presence of homB and the progression to severe gastric disease [17], other groups working with isolates from different geographic regions have shown that the presence of homB was associated with development of peptic ulcer disease in children and young adults [23, 24] and with gastric cancer development and the presence of cagA [25]. The variations in these results suggest that the impact of the homB gene on disease is geographically dependent [17].
Our studies and others have revealed that certain polymorphisms are more prevalent within specific populations [11, 12, 17, 22, 26–29]. Thus, it is possible that the combination of virulence factor polymorphisms present within a population may influence the rate of severe disease outcome. Therefore, the polymorphism of each virulence factor must be evaluated within various populations to help understand the relationship between these genotypes and their associations with gastric diseases including cancer.
The bab gene family has been shown to have a positive association with gastric cancer and duodenal ulcers [6, 30–32]. BabA, one member of this family, was the first identified adhesin in H. pylori and mediates the binding of bacteria to the fucosylated blood group O antigen Lewis b (Leb), which is highly expressed in gastrointestinal epithelial cells [33, 34]. Similar to several H. pylori OMPs, BabA has two closely related paralogous, BabB (HopT) and BabC (HopU); however, the functions of BabB and BabC are unknown. These OMPs, particularly BabA and BabB, have nearly identical N- and C-terminal domains but a divergent middle region, suggesting that the middle variable region of BabA is most likely responsible for the specific adhesin function [19, 34–36]. Interestingly, it has been shown that BabB expression is modulated by changes in the number of cytosine-thymidine (CT) dinucleotide repeats in the 5′ region of the babB gene resulting in slipped-strand mispairing [37–39]. Changes in the number of CT repeats results in a frame shift; ‘IN’-frame bab genes are translated into a full functional Bab or ‘OUT’-of-frame bab genes into a premature partial Bab [37–39]. Furthermore, bab expression can be modulated by gene conversion between the loci, most likely by intragenomic homologous recombination [35, 40]. This gene conversion results in chimeric bab genes where the CT repeats of the babB 5′ region become associated with babA or babC subjecting these genes to phase variation. These recombination events may play an important role in regulation of adherence. For example, babBA chimera results in the regulation of babA by changes in CT repeats that lead to changes in the binding to Leb on gastric epithelial cells [41].
To gain further insight into genetic diversity of H. pylori isolates within the South Korean population, we analyzed the bab gene family. In addition, we expanded our analysis to include clinical isolates obtained from an American hospital to allow for strain comparisons between different geographic regions.
Materials and Methods
Bacterial Strains and Culture Conditions
A total of 160 H. pylori clinical isolates were used in our analysis. The 80 South Korean H. pylori clinical isolates (KH) were a subset of a collection of South Korean strains previously characterized for distribution of cagA EPIYA polymorphism, vacA s/i/m polymorphism, and homA/B paralog [11, 12, 17, 22], while the other 80 isolates were obtained from patients at Vanderbilt University Medical Center, Nashville TN, USA. Written informed consent was received from each patient, and the protocol was approved by the Vanderbilt University and the Nashville Department of Veterans Affairs Institutional Review Board (IRB# 5190). Gastric biopsies were harvested from individuals at the Veterans Affairs Medical Center in Nashville undergoing upper endoscopy, and used for bacterial culture. Isolates from biopsies were confirmed to be H. pylori by positive urease, catalase, and oxidase tests, and typical appearance on Gram stain. These strains are referred to as American H. pylori clinical isolates (AH). For 80 AH, polymorphisms in cagA and vacA, and homB paralog were determined using the strategies previously developed for characterization of KH [11, 12, 17, 22]. The 160 KH and AH are described in detail in S1 Table. All H. pylori clinical isolates were cultured as previously described [22]. Briefly, bacterial isolates preserved at -80°C, were grown and expanded on antibiotic-supplemented horse blood agar plates under microaerophilic conditions created in GasPak jars with Campypak generators (Mitsubishi Gas Chemical Company, Inc, Tokyo, Japan).
bab genes genotyping
Chromosomal DNA was extracted from H. pylori strains G27, J99, and 26695 and from the 160 clinical isolates using the G-spin total DNA extraction kit (Intron Biotechnology, inc., Seoul, South Korea). Primers used for genotyping and sequencing bab genes in this study were based on the genome sequences of H. pylori strains G27, J99 and 26695 (GenBank accession numbers CP001173, AE001439, and CP003904, respectively) (Table 1). PCRs using a locus-specific forward primer and bab-specific reverse primer (babAr1, babBr1 or babCr1) were carried out to investigate the presence or absence of babA, babB or babC, respectively (Fig 1) [20, 42]. If all three PCRs per locus were negative, the locus was considered empty. Sequencing of amplicons was used to identify bab chimeras and whether the bab genes were ‘IN’-frame or ‘OUT’-of-frame according to the number of CT repeats. In addition, to assess the entire babC gene (approximately 2200 bp), PCR amplicons of locusAf/babCr1 and babCf1/locusAr were sequenced using primers babCf1, babCf2, babCf3, babCr1, babCr2, locusAf, and locusAr and the resulting overlapping DNA fragments were aligned. Sanger dideoxy sequencing was performed at Cosmo Genetech Co., Ltd. (Seoul, South Korea) and Geno Tech Corp (Seoul, South Korea), and the resulting DNA sequences were analyzed using Vector NTI version 9.1 (Invitrogen, Carlsbad, CA,USA) and Sequencher 5.1 (Gene Codes Corp., Ann Arbor, MI, USA).
Table 1. Primer sequences for analysis of H. pylori bab genes.
| Primer name | Primer sequence (5’→3’) | Reference |
|---|---|---|
| locusAf | TTTTGAGCCGGTGGATATATTAG | HypDF1 [42] d |
| locusBf | CTTTAATCCCCTACATTGTGGA | S18F1 [42] d |
| locusCf a | ACCCTAGTGGGCATGTGGTA | Hp1-AS [20] d |
| locusAr b | GGAAATGCGCACACGAGGG | this study |
| babAr1 b | TTTGCCGTCTATGGTTTGG | BabAR1 [42] d |
| babAr2 b | GAAAGGATGTGTTTTTTCATG | this study |
| babBr1 b | TCGCTTGTTTTAAAAGCTCTTGA | BabBR1 [42] d |
| babBr2 b | CTGCCAGGACCACAAGCAGT | this study |
| babCf1 b | GCGGCAAACATCATGCAAGTC | this study |
| babCr1 b | GACTTGCATGATGTTTGCCGC | this study |
| babCf2 b | CGGGTTTGCTCAAAGAAAAAAYC c | this study |
| babCr2 b | GRTTTTTTCTTTGAGCAAACCCG c | this study |
| babCf3 b | AAAGAATAACCCCTATAGCC | this study |
a One nucleotide in the primer was modified from the indicated primer in reference.
b Primer was used for sequencing.
c Underlined Y indicates C or T, and underlined R indicates G or A.
d Primer name was used in the indicated reference.
Fig 1. Outline of bab genotyping by PCR.
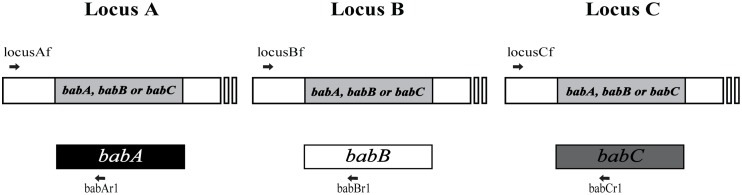
(Top) Schematic representation of the three loci where the bab genes are generally detected: Locus A, B and C. The annealing positions (arrows) and names of each locus-specific forward are shown. (Bottom) Annealing positions (arrows) and names of bab-specific reverse primer are indicated with their respective bab gene: babA depicted by the black box, babB depicted by the white box, babC depicted by the grey box. Primers are listed in Table 1, and a full explanation of the genotyping scheme can be found in the Materials and Methods.
Phylogenetic Analysis
Phylogenetic trees were constructed using Phylip v3.695 (Phylogeny Inference Package) [43]. First, an input file was generated based on gene variation at seven distinct loci: cagA (EPIYA type), vacA (s/i/m type), hom loci A/B (hom type) and bab loci A/B/C (bab type). Each locus was assigned a discrete numerical character corresponding to its type. Consequently, each clinical isolate was distinguished by a seven-number code representing its genotype. For example, strain G27 has the following genotype: cagA EPIYA (ABCC), vacA (s1/i1/m1), hom locus A (homB), hom locus B (empty), bab locus A (babC), bab locus B (babB), and bab locus C (babA). This genotype corresponds to the number 2020321 in the input file. All resulting codes were utilized to generate the strain input files used in subsequent analyses (S3 Table).
We began constructing the trees via the SEQBOOT program to generate multiple data sets. With this program we used a combination of bootstrapping and character permutation to generate 100 data sets. These data sets were then used as the input file in the PARS program, a general parsimony program that carries out the Wagner parsimony method and allows for up to 8 discrete character states and the use of ‘?’ for states that are unknown. PARS was run using the default settings with the following exceptions: species order was randomized using a random number seed of 5, with 10 replicates and the trees were constructed from the 100 data sets generated in SEQBOOT. PARS searches for the best fit tree, saving the 100 best options. As a final step, we used the PARS output trees in the CONSENSE program to generate a majority-rules consensus tree. In doing this, the branch lengths of our final tree represent the number of times a given branching event occurred in the 100 trees generate by PARS. FigTree v1.4.2 is a tree editing program [44].
Statistical Analysis
Statistical analysis was conducted as previously described [12]. Briefly, two-way associations between bab genotype, hom genotype, cagA EPIYA polymorphisms, vacA s/i/m polymorphisms, and disease state were analyzed using the Fisher’s exact test. Log linear modeling was used to assess higher order associations that were significant at a 5% level. Using categorical values to represent our data sets, we fit a saturated model. We then applied a backward selection algorithm that eliminates the least significant associations at each step and then reforms the model to look for associations. Data analysis was conducted using SPSS version 22 software (SPSS Inc., Chicago, IL, USA) or SAS version 9.3 software (SAS Institute Inc., Cary, NC, USA).
Nucleotide Sequence Accession Numbers
The sequences for bab genotypes in American and South Korean strains were deposited in the NCBI GenBank database with accession numbers KP339308 to KP339491.
Results
Sample Population
Complete demographic data was available for all 80 AH (S1A and S2A Tables). For the American population, the mean patient age was 57 years, with an age range of 27–84 years. 20.0% (16 patients) of the population was black, and 80.0% (64 patients) was white. Finally, 79 of the patients were male; there was one white female patient, age 50. Of these clinical isolates, 7.5% were from patients with cancer/pre-malignant lesions (2.5% with gastric carcinoma and 5.0% with Barrett’s Esophagus), 43.8% were from patients with peptic ulcer disease (31.3% with duodenal ulcers and 12.5% with gastric ulcers), 32.5% were from patients with gastritis, and 16.3% were from patients with esophagitis. Within the South Korean population, age and gender were missing for 2 individuals. Of the remaining 78, the mean age was 48 years, ranging from 20–86 years of age. The South Korean population had a more even distribution between males and females, with 48.7% (38 patients) female and 51.3% (40 patients) male. Within the South Korean female patient group, the mean age was 51, with an age range of 21–86 years. Within the South Korean male patient group, the mean age was 46, with an age range of 20–76 years. Of the 80 KH samples analyzed, 18.8% were from patients with gastric cancer, 33.8% were from patients with peptic ulcer disease (32.5% with duodenal ulcers and 1.3% with gastric ulcers), and 47.5% were from patients with gastritis.
bab genes genotyping
The distribution of identified bab genes at locus A, B and C is shown in Fig 2. Genotypic analysis revealed that in addition to babA, babB and babC, three types of chimeric bab genes were also present: babAB, babBA and babBC. A total of 23 different bab genotypes, including a single strain where bab was absent from the three known loci (-/-/-), were identified within the 160 isolates chosen for this study. While 6.9% of strains (11/160) carried a single bab gene, the majority of the strains (85.0%, 136/160) carried two bab genes, one at locus A and one at locus B (Fig 2). Of note, locus C was only occupied if both other loci also carried a bab gene (Fig 2). In the KH, 11 of the 23 genotypes were identified, and isolates carrying two bab genes were most common, accounting for 87.5% of the isolates. The genotype babA/babB/-, position 1, 2, and 3 correspond to locus A, B and C, respectively, occurred in the 78.8% of the isolates (Fig 2). In comparison, there were many more bab genotypes within the AH. In fact, 19 of the 23 genotypes were identified. Twelve genotypes were unique to the AH while 4 were unique to the KH. Similar to the KH, isolates carrying two bab genes were most common among AH, accounting for 82.5% of the isolates; however, while the most common genotype of babA/babB/- occurred in only 38.8%, genotypes of babAB/babB/- (16.3%) and babC/babB/- (15.0%) showed a higher frequency in this population (Fig 2). Whereas locus A of KH was occupied by babA in greater than 90% of isolates (74/80), most of the diversity within the bab genotypes for the AH was driven by locus A, which was significantly more variable (P<0.0001) than seen in KH. Indeed, babA occurred at locus A only 47.5% of the time (S1 Fig). The next most frequent occupants of locus A were chimeric bab genes (28.8%), followed by babC (15.0%) and babB (7.5%).
Fig 2. Distribution of the bab genotypes in AH and KH (n = 160).

Genotypes of one, two, three, or none bab genes are grouped into I, II, III, and IV, respectively. These classifications were made on the basis of number of bab genes present at locus A, B, and C. A black, white, or grey box indicated babA, babB, and babC, respectively. ‘-’ indicates the absence of bab gene at the designated loci. ‘AH%’ and ‘KH%’ indicate the frequency of each individual genotype within the specified population (n = 80) and ‘Total%’ shows the percent of the respective genotype in the overall population (n = 160). Bracketed numbers indicate the number of isolates possessing each respective genotype out of the population examined. ‘*’ indicates genotypes containing chimeric form(s).
In total, we identified 3 chimeric bab genes, babAB, babBA and babBC that were the result of gene conversion. This gene conversion likely occurs in the 5′region (~60 to 240 bp from the initiation codon) and in the 3′region (~1200 to 2100 bp) but this later event is hard to detect due to the homology in this region. It can be speculated that the characteristic of the chimera would follow that of the second bab gene, which would constitute the majority of the chimeric gene. bab chimeric forms were relatively common in the AH group, occurring at one or more loci in 26/80 (32.5%) of isolates versus 6/80 (7.5%) in the KH group (Fig 2 and S2 Table). The most common chimeric gene was babAB, which accounted for 88.9% of chimeric genes; 90.0% of chimeras in AH (27/30) and 83.3% in KH (5/6) (Fig 2 and S2 Table). Within the babB coding sequence there are variable CT repeats approximately 56 bp downstream from the initiation codon that are responsible for phase variation. In the babAB chimera, this region is displaced with the corresponding part of babA [41], resulting in loss of phase variable babB. Although it appeared much less frequently, babBA and babBC chimeras were also present (Fig 2 and S1 Fig). In these instances, CT repeats from the 5′region of babB were introduced to babA and babC, respectively, resulting in phase variable babA and babC.
Eighty-two bab genes from the AH group and 72 bab genes from the KH group contained a run of CT repeats (5–14 CT repeats) (Table 2). In all cases, the CT repeats were associated with the 5′ region of babB genes; therefore, babBC and babBA also carry CT repeats. CT repeats were found at all three loci within the AH, with the majority (84.2%) occurring at locus B (Table 2). Similarly, in the KH group a majority of the bab genes containing CT repeats were found at locus B (97.2%) (Table 2). Interestingly, when comparing locus B in the AH group to the KH, significantly more babB genes were ‘IN’-frame in the KH group than in the AH (P = 0.0341, data not shown). Although not significant, the trend was similar for all babB genes regardless of loci, where more babB genes were ‘OUT’-of-frame (54.9%) than ‘IN’-frame (45.1%) in the AH group (S2 Fig). The opposite trend was observed in the KH group; babB genes tended to be ‘IN’-frame (59.7%) as compared to ‘OUT’-of-frame (40.3%) (S2 Fig).
Table 2. ‘IN’-frame or ‘OUT’-of-frame status deduced by the number of CT dinucleotide repeats in the babB coding region.
| Group-Locus | Frame Status | Pattern of CT repeats | Sequence of CT-repeats region | Number Identified* |
|---|---|---|---|---|
| AH-A (9) | IN | 8 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCGTTTTTG | 4 |
| IN | 11 | ATGAAAAAAACCCTCCTACTCTCTCTCTCTCTCTCTCTCTCGTTTTTG | 1 | |
| OUT | 10 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCGTTTTTG | 2 | |
| OUT | 9 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCGTTTTTG | 1 | |
| OUT | 7 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCGTTTTTG | 1 | |
| AH-B (69) | IN | 8 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCGTTTTTG | 17 |
| IN | 11 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCTCGTTTTTG | 10 | |
| IN | 14 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCTCTCTCTCGTTTTTG | 2 | |
| OUT | 9 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCGTTTTTG | 15 | |
| OUT | 10 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCGTTTTTG | 10 | |
| OUT | 7 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCGTTTTTG | 8 | |
| OUT | 6 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCGTTTTTG | 4 | |
| OUT | 12 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCTCTCGTTTTTG | 2 | |
| OUT | 13 | ATGAAAAAAACCCTTTTA CTCTCTCTCTCTCTCTCTCTCTCTCT CGTTTTTG | 1 | |
| AH-C (4) | IN | 8 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCGTTTTTG | 3 |
| OUT | 10 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCGTTTTTG | 1 | |
| KH-A (2) | IN | 8 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCGTTTTTG | 1 |
| OUT | 10 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCGTTTTTG | 1 | |
| KH-B (70) | IN | 8 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCGTTTTTG | 31 |
| IN | 11 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCTCGCTTTTG | 7 | |
| IN | 5 | ATGAAAAAAACCCTTTTACTCTCTCTCTCGTTTTTG | 4 | |
| OUT | 10 | ATGAAAAAAATCCTTTTACTCTCTCTCTCTCTCTCTCTCGTTTTTG | 10 | |
| OUT | 9 | ATGAAAAAACCCCTTTTTCTCTCTCTCTCTCTCTCTCGTTTTTG | 7 | |
| OUT | 7 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCGTTTTTG | 6 | |
| OUT | TT+6 | ATGAAAAAAACCCTTTTATTCTCTCTCTCTCTCGTTTTTG | 3 | |
| OUT | TT+8 | ATGAAAAAAACCCTTTTATTCTCTCTCTCTCTCTCTCGTTTTTG | 1 | |
| OUT | 12 | ATGAAAAAAACCCTTTTACTCTCTCTCTCTCTCTCTCTCTCTCGTTTTTA | 1 |
* This number corresponds to the number of times the specific CT-repeat pattern was identified at the given locus.
() The bracketed number indicates the number of times a bab gene carrying CT repeats was identified.
Since very little is known about babC, we took this opportunity to sequence 15 babC genes from our collection and compare the deduced BabCs. As shown in S3 Fig we defined a BabC consensus sequence by comparing 14 BabCs and one BabBC from this study along with the BabC from G27. According to the babC sequences, we identified only one chimeric babBC gene (AH-J68) and no babAC gene. Furthermore, we found that the N-terminus of BabC was most similar to that of BabA, and the C-terminus of BabC was homologous to that of both BabA and BabB (Fig 3). The amino acid sequence of BabC contained two variable regions, VR1 spanning from amino acid 212 to 246 and VR2 spanning from amino acid 409 to 417. The BabC sequences, including VR1 and VR2, shared over 70% identities (Fig 3 and S3 Fig).
Fig 3. Schematic comparison of BabA, BabB, and BabC.

The representative structures of BabA and BabB are based on the J99 amino acid sequence, whereas the structure of BabC is based on the consensus sequence defined in this study. The N-HR indicates N-terminal homology region, M-VR indicates middle variable region, which is characterized by sequence difference among the three Bab proteins. Note that the M-VR is conserved for each of the Bab proteins. The C-HR indicates C-terminal homologous region that shows >90% identity. The CT-R present at the N-terminus of BabB refers to the CT repeat region. VR-1 and VR-2 in BabC indicate regions of variability among the BabC amino acid sequences analyzed in this study (n = 15).
To further understand the bab genotypes in these populations, we next sought to determine if carrying a specific bab gene at one locus was associated with the presence of bab genes at the other loci. Given the difference in bab genotypes between the two populations, we first examined each population separately. A significant association was found in the KH group between the genotype at locus A and locus C (P = 0.0027) (Table 3). Conversely, in the AH group a significant association was found between locus A and locus B (P = 0.0028). When KH and AH were combined, there was also a significant association between locus A and locus B (P = 0.0131) (Table 3). Of note, in the AH/KH-combined analysis babC was more likely to occur at locus A when babB at locus B was ‘OUT’-of-frame (78.6%, 11/14). Furthermore, if BabB was being expressed from locus A, then babB at locus B was more likely to be ‘OUT’-of-frame (60.0%, 15/25). Yet interestingly, if babA occupied locus A, then babB at locus B was more likely to be ‘IN’-frame (58.9%, 66/112).
Table 3. Significant two-way comparisons of bab genotype and other factors in both populations a .
| bab genotype comparison | P value b for distribution within | ||
|---|---|---|---|
| KH | AH | Combined c | |
| babA,B,C or empty at Locus A vs babA,B,C or empty at Locus B | 0.1046 | 0.0028 | 0.0131 |
| babA,B,C or empty at Locus A vs Full or empty bab at Locus C | 0.0027 | 0.4111 | 0.4500 |
| babA,B,C or empty at Locus A vs one or two hom loci occupied | 0.0167 | <0.0001 | 0.0029 |
| babB ‘IN’ or Other at Locus B vs one or two hom loci occupied | 0.0210 | 1.0000 | 0.6070 |
| Full or empty bab at Locus C vs one or two hom loci occupied | 1.0000 | 0.0390 | 0.1750 |
| babA,B,C or empty at Locus A vs homA or homB | 0.6390 | <0.0001 | <0.0001 |
| babA,B,C or empty at Locus B vs homA or homB | 0.7340 | 0.0160 | 0.0260 |
| babA,B,C or empty at Locus A vs vacA s1 or vacA s2 | 1.0000 | <0.0001 | <0.0001 |
| babA,B,C or empty at Locus A vs vacA i1 or vacA i2 | 0.9999 | <0.0001 | <0.0001 |
| babA,B,C or empty at Locus A vs vacA m1 or vacA m2 | 0.9999 | <0.0001 | <0.0001 |
| babA,B,C or empty at Locus A vs cagA EPIYA-ABD or Other | 0.5010 | N/A* | <0.0001 |
| babA,B,C or empty at Locus B vs Cancer/Gastric ulcer or Duodenal ulcer/Gastritis | 0.0101 | 0.828 # | 0.483 # |
| babB or other at Locus B vs Cancer or Gastric ulcer or Duodenal ulcer or Gastritis | 0.0012 | 0.806 # | 0.447 # |
a For simplicity, Table 3 only contains associations for which a statistically significant association was found in at least one grouping; however, an exhaustive analysis was conducted on numerous other permutations of the data.
b Statistically significant P values are in boldface type.
c All isolates (n = 160) analyzed as a single group
*No ABD in the AH
#Also includes a category for Esophogitis/Barrett's Esophagus.
Associations between bab and hom genes
Given the associations among bab loci, we next assessed whether bab genotype was associated with other outer membrane proteins. We have previously looked at the association between homB and disease in the South Korean population [17]. hom paralog (homA and homB) from the Hom protein family, which shares over 90% identity, are known to occupy two possible loci (also called locus A and B) [45]. Therefore, we chose to evaluate any association between the bab genes, and homA and homB. Once again, given the differences in the two populations, each was analyzed individually as well as combined. There were no significant associations when comparing strains for presence of homA versus homB. Given that homA and homB are homologous and share two conserved loci, locus A and locus B, we also considered hom gene number in which we looked at whether or not both hom locus A and locus B were occupied regardless of if it was homA or homB. We found a significant association with the bab paralog present at locus A and number of hom loci occupied. This association was seen in both the AH and KH groups (P<0.0001 and P = 0.0167, respectively) (Table 3). In the AH group, presence of a babB at locus A was more often associated with a single hom gene (61.5%, S1A and S2A Tables). Conversely, in the KH babA at locus A occurred most often with a single hom (97.3%, S1B and S2B Tables). When the groups were combined, the association between number of hom loci occupied and the bab gene at locus A remained significant (P = 0.0029) (Table 3). One factor that stands out when the groups were combined was that although babC was rare (n = 14), it occurred only in isolates with a single hom gene. Also of note in the AH group, in strains where locus C was occupied there was a significant difference between carrying a single hom gene versus two hom genes (P = 0.039). Although the finding wasn’t significant in the KH group, the same trend was seen; locus C was only occupied in strains with a single hom gene (Table 3 and data not shown).
Associations between bab, cagA, and vacA
It has been well established that particular cagA and vacA polymorphisms are associated with more severe disease outcomes [12, 22, 27, 46]. Due to their association with disease outcome, we next assessed whether particular bab genotypes were found in association with known cagA or vacA polymorphisms. There were no significant associations between cagA and bab when the groups were looked at separately; however, when considering the clinical isolates as a whole, there was a significant association between the bab gene at locus A and the cagA EPIYA polymorphism (P<0.0001) (Table 3). The cagA EPIYA-ABD polymorphism occurred more frequently in strains that carry a babA at locus A (92.9%, 65/70), whereas strains carrying a babB or babC at locus A were most likely to have a non EPIYA-ABD (93.1%, 27/29 and 85.7%, 12/14, respectively) (S1 and S2 Tables). Similar to cagA, vacA associations were only apparent when KH and AH were combined. When we analyzed all 160 isolates, there were significant associations between bab at locus A and the s, i, and m regions of vacA (P<0.0001 for each) (Table 3). Further analysis of the associations between the vacA polymorphic regions and bab at locus A uncovered a three-way association between bab at locus A, the vacA s, i, m regions, and occupation of a single or both hom loci (Table 4). If we first consider strains carrying vacA s1 polymorphisms, these strains were most likely to harbor a babA at locus A. Interestingly, babB occurs almost equally with the s1 and s2 vacA polymorphisms; however, 100.0% of strains containing babB at locus A and s2 carried a single hom gene compared to strains carrying s1, which were more likely to carry two hom genes. The association between s2/babB/single hom and s1/babB/two hom genes were both significant (P = 0.00062 and P = 0.01846, respectively, data not shown). The i and m regions of vacA clustered together in that i1 compared to i2 and m1 compared to m2 resulted in the same distributions. babA at locus A was more frequently associated with i1 and m1 as compared to babC, which was much more commonly associated with i2 and m2. Once again, babB appeared to be found in relatively equal frequencies with i1m1 and i2m2; however, when we analyzed the vacA genotype of strains carrying babB at locus A, the strains with only an i2m2 vacA type were more likely to have a single hom gene (87.5%, 14/16, P = 0.00744) and strains with an i1m1 vacA classification were more likely to carry two hom genes (76.9%, 10/13, P = 0.00166) (data not shown).
Table 4. Three-way comparisons of bab genotype and other factors in both populations.
| bab genotype comparison | P value a for distribution within | ||
|---|---|---|---|
| KH | AH | Combined b | |
| babA, B or C at Locus A vs vacA i1/i2 vs one or two hom loci occupied | N/A | 0.097 | 0.001 |
| babA, B or C at Locus A vs vacA s1/s2 vs one or two hom loci occupied | N/A | 0.173 | 0.002 |
| babA, B or C at Locus A vs vacA m1/m2 vs one or two hom loci occupied | N/A | 0.882 | 0.001 |
| babA, B or C at Locus A vs vacA s1i1m1/other vs one or two hom loci occupied | 1.000 | N/A # | N/A # |
| babA, B or C at Locus A vs cagA (AB &Other $ /ABCs/ABD) vs vacA m1/m2 | N/A | 1.0000 | 0.158 |
| babA, B or C at Locus A vs cagA (AB &Other $ /ABCs/ABD) vs vacA s1/s2 | N/A | 0.963 | 0.881 |
| babA, B or C at Locus A vs cagA (AB &Other $ /ABCs/ABD) vs vacA i1/i2 | N/A | 0.916 | 0.158 |
| babA, B or C at Locus A vs cagA (Other/ABD) vs vacA s1i1m1/other | 1.0000 | N/A # | N/A # |
| babA, B or C at Locus A vs cagA (AB &Other $ /ABCs/ABD) vs one or two hom loci occupied | 0.976 | 1.000 | 0.044 |
| babA, B or C at Locus B vs one or two hom loci occupied vs vacA s1/s2 | N/A* | 0.823 | 0.061 |
a Statistically significant P values are in boldface type.
b All isolates (n = 160) analyzed as a single group.
#These comparison were only done in KH; for AH and combined we looked at i, s, and m regions of vacA separately, which wasn’t feasible with KH since was overwhelmingly s1/i1/m1.
*All KH strains are vacA s1.
$AB&Other refers to any cagA EPIYA motif that is not ABC(1–4), or ABD.
Associations between bab genotype and disease
Given the association between the virulence factors, cagA and vacA and the bab genes, particularly at locus A, we asked if there was an association between bab genotype and disease. The only significant association between bab and disease was seen in the KH group. In this group, there was a significant association between the bab genes present at locus B and disease outcome (P = 0.0101) (Table 3). Specifically, this association appeared to be driven by the fact that isolates that lacked a bab gene at locus B were more likely (66.7%) to come from individuals suffering from more severe disease outcome (gastric cancer and gastric ulcers) than from individuals suffering from gastritis and duodenal ulcers (33.3%). It was also interesting to note, although the finding was not significant (P = 0.176), that individuals with cancer (gastric carcinoma) or pre-malignant lesions (Barrett’s Esophagus) were more likely to have a babB gene that was ‘IN’-frame’ (82.4%, 14/17) compared to ‘OUT’-of-frame’ (17.6%, 3/17) (data not shown).
Phylogenetic Analysis of AH and KH
To complement our statistical analysis, we also conducted a phylogenetic analysis based on gene variation at seven loci: cagA (EPIYA type), vacA (s/i/m type), hom locus A and B (hom type), and bab locus A, B, and C (bab type). As shown in Fig 4A, using these parameters led to a distinct geographical separation between the KH and AH. Only, 6 KH (7.5%) cluster with the AH: K47, K78, K262, K57, K24, and K197. Interestingly, while most of these strains have the predominant KH cagA ABD EPIYA motif and vacA s1/i1/m1 type, their bab and hom genotypes more closely resembled the AH. Given this observation, we next constructed a separate consensus tree using only the hom and bab genes (Fig 4B). Remarkably, this tree was very similar to the tree generated using all 7 loci; once again, the majority of the KH and AH segregated, and the same 6 KH grouped more closely with the AH. Closer inspection of the OMP genotypes revealed that the hom genotypes of the 6 KH that group among the AH, deviate from the single hom at locus B genotype that predominates in the KH.
Fig 4. Phylogenetic analysis of AH and KH.

(A) An unrooted consensus tree based on cagA EPIYA type, vacA s/i/m type, homA/B genotype, and bab genotype. (B) An unrooted consensus tree based on homA/B and bab genotypes only. Trees were created in PHYLIP with Wagner parsimony followed by the majority-rules consensus method. KH branches/nodes are red and AH branches/nodes are black. The six KH grouped with AH are identified by numbers 1–6 in both trees and correspond to the following strains:(1) K47, (2) K78, (3) K262, (4) K57, (5) K24, and (6) K197.
Discussion
The bab loci have been investigated in detail in several human populations as well as in long term animal colonization studies [21, 39, 47–49]; however, in this study we not only asked what the bab genotypes were in South Korean and American populations, but also which bab genes were phase variable, which of the phase variable bab genes were ‘IN’-frame or ‘OUT’-of-frame, and how the bab genotypes were associated with other genetic markers. Furthermore, we sought to compare these findings between two distinct populations. The KH came from a population with high levels of H. pylori infection and a large burden of stomach cancer, while the AH came from a population known to have lower levels of infection and stomach cancer. Although higher rates of H. pylori infection in the KH group can partially explain the increased rates of stomach cancer, it is also possible that genetic differences between the H. pylori circulating in the South Korean compared to the strains circulating in the United States may also play a role. For our study we began by genotyping the three bab loci in 80 clinical isolates obtained from South Korea and 80 clinical isolates from the United States. Overall, we identified 23 different bab genotypes. Of these 23, 19 were found in the AH and 11 in the KH group (Fig 2). Interestingly, although there are a thousand potential bab genotypes, our work and previous studies have shown that not all combinations are observed [21, 42, 47, 50]. For example, consistent with previous studies [21, 42, 50], we found that carrying two bab genes at locus A and locus B was the most common genotype. In the KH group, close to 80% of isolates showed babA/babB/- genotype. Conversely, even though the AH group did not have one genotype that was found in greater than 40% of the population, the majority of isolates still carried two bab genes: babA/babB/-, babAB/babB/-, or babC/babB/-. Our data suggest that while there are a large number of possible combinations of the bab genes at different loci, certain genotypes predominate and the others are rare. Although we identified 23 genotypes, only 5 genotypes were found in at least 5 of the isolates, and 4 genotypes were found only within the KH group while 12 genotypes were exclusive to the AH group. Also of note, in both the KH and AH collections, locus A was most frequently occupied by babA, and locus B by babB. Furthermore, locus C was only occupied when locus A and B carried a bab gene (Fig 2 and S1 Fig). Taken together, these data show that isolates carrying two bab genes are most common regardless of geographic origin of the strain. Based upon these observations, one might speculate that in an ancestral strain locus A was occupied by babA and locus B by babB, and as H. pylori adapted, gene conversion events have resulted in varying bab genotypes. Since the bab genes encode OMPs, and since BabA is known to act as an adhesin, it is likely that differences in the bab genotype affect how the bacteria interacts with the host. Therefore, perhaps the genotypes that appear to occur in only 1 or 2 individuals reflect adaptation of H. pylori to an individual host, or across gastric regions within a host, and also within an individual over time [42].
In addition to bab locus genotyping, we also examined which bab genes were subject to phase variation. As previously shown, CT repeats associated with the 5′ region of babB can result in slipped-strand mispairing leading to a premature stop in translation [37–39]. In the KH group, babB at locus B was significantly more likely to be ‘IN’ frame’ compared to the AH which may reflect a difference in the host populations since H. pylori likely uses phase variation as a response to host changes. Probably through gene conversion, CT repeats were also identified in association with babA and babC at locus A. These data suggest that gene conversion occurred in these strains and resulted in phase variable babA and babC, imparting H. pylori with additional means of regulation over these genes. Interestingly, the most common gene conversion of babAB among 3 types of chimeric bab genes deprived babB of the regulation of phase variation.
Evidence from this study and others suggest that varying the bab genotype is a mechanism of H. pylori adaptation [33, 41]. To try and gain a better understanding of these adaptations, we asked if there were associations among the bab loci. In both populations, babB was more likely to be expressed from locus B when babA occupied locus A. Conversely, if babB was expressed from locus A then the babB at locus B was more likely to be ‘OUT’ of frame. Through these analyses we also noted that the babC gene was far more likely to occur in strains that also carry a babB. Furthermore, in strains containing both babB and babC, babB was more likely to be ‘OUT’ of frame. Taken together, these data suggest that H. pylori actively adjusts its repertoire of bab genes, not only through gene replacement, but also through the formation of chimeric proteins and phase variation supporting the finding of other authors [35, 42]. Although the function of babB and babC are unknown, it could be speculated that the Bab OMPs share overlapping functions, perhaps acting as adhesins similar to BabA. The differences between babA, babB, and babC may affect their function. For instance, if they are functioning as adhesins, these differences may result from different binding targets. Alternatively, the difference between these three genes may also make them antigenically distinct and allow them to escape the immune system.
To this end, we expanded our analysis to include other OMPs from the Hom family. There were no statistically significant differences when we compared strains carrying homA to those carrying homB; however, given that homA and homB are greater than 90% identical we decided to ask whether or not we saw an difference in vacA, cagA and bab genotypes between strains carrying a single hom gene at locus A or locus B versus strains in which both loci were occupied, regardless of whether it was homA or homB. We analyzed each population individually as well as combined, and observed an association between the bab gene carried at locus A and number of homA and/or B genes. In both population, babA occurred most frequently with a single homA or homB gene, babB occurred relatively equally whether a single or both hom loci were occupied, and babC was only found in strains with a single homA or homB gene. If one considers the environment in which H. pylori persist, having a variable repertoire of OMPs is likely paramount to the success of the organism. While H. pylori has greater than 50 OMPs, to our knowledge this is the first evidence showing an association between the Hom and Bab families [16, 37, 45]. Since only the function of babA is known, it is difficult to speculate how these proteins may interact functionally [33]; however, one can surmise that H. pylori finds the right combination of OMPs as a means to balance between the adherent population, which is close to nutrients, and a free population, which is less susceptible to immune clearance. Through mechanisms such as phase variation and gene conversion H. pylori is able to fine tune its OMP repertoire in response to changes in the environment.
In addition to investigating the relationship between the bab genes and other OMPs, we were also interested in how bab genotypes related to known virulence factors such as cagA and vacA in KH [11, 12, 22, 46] and AH (S1A Table). In case of cagA genotypes, interestingly the AH group contained none of the EPIYA-D motif while KH included 7 strains possessing the EPIYA-C motifs. Importantly, we discovered several associations. A previous study found that strains with babA were more likely to have a cagA gene than strains that lacked babA [20]. While all of the strains in our collection contained a cagA gene [22], we did find that there was a positive association between having babA at locus A and having a cagA EPIYA-ABD motif. Strains that carried babB or babC at locus A were significantly more likely to have a non EPIYA-ABD motif. Carrying a babA at locus A was also significantly more likely to occur with vacA type s1/i1/m1. Our group has previously shown that cagA EPIYA-ABD and vacA s1/i1/m1 are associated with more severe disease in the KH [11]. Our current data suggest that carriage of babA at locus A may also be associated with these more virulent genotypes. Furthermore, we observed a three-way association between bab genotype at locus A, occupation of one or both hom loci and the vacA s1/i1/m1 genotype. Based on these three-way associations, we observed some interesting patterns. For instance, strains harboring a single homA or homB gene were more likely to be associated with vacA s1, i1 and m1 if there was a babA at locus A; however, if there was a babB at locus A, then strains with a single homA or homB gene were more likely to be found with a strain possessing s2/i2/m2, supporting the idea that babA may be associated with more virulent genotypes. However, if both hom loci are occupied and babB is at locus A, then this strain was more likely to be associated with s1, i1, and m1. These findings perhaps demonstrate that it is a specific combination of virulence factors that combine to result in more severe disease outcomes. Although we did not see higher order associations with disease, it could be that our sample size was not adequate to obtain statistical significance over the number of variables that were included.
Taken en masse, the H. pylori isolates obtained from the American population showed a larger degree of diversity in bab genotypes compared to the South Korean population. This may reflect adaptions to a more diverse human population. Furthermore, difference seen between the South Korean and American isolates due to babB phase variation may also be due to differences in the populations. Given that difference in host genetics are known to exist, perhaps babB is beneficial to H. pylori in the South Korean population but not in the American population. Even though the populations were distinct, we identified associations that were seen in both populations, as well as associations that became more apparent when we combined the populations and increased the sample size. Based on our analyses it appears that babA may be associated with more virulent genotypes of cagA and vacA. We also identified an association between OMPs from the Hom family of proteins with the bab genes, suggesting a potential coordination between OMP families. Interestingly, our phylogenetic analysis suggests that the using only the hom and bab genotypes segregated strains as well as the full genotyping scheme. Specifically the hom genotype appears to influence whether the strains cluster with AH or KH. Previous studies have demonstrated that strains with the same cagA EPIYA type cluster geographically [46]; however, to our knowledge this is the first report in which the number and type of OMP genes have been shown to segregate strains geographically. Although homA and homB sequence variation has been shown to cluster strains [51], the prior work focused on strains with a single homA or homB gene, whereas our study focused on which hom loci were occupied and not sequence differences. Thus, our observations suggest that perhaps in addition to geographic variation of homA and homB sequence, there is also a geographic difference in location of the hom genes, as well as number of loci occupied (1 vs. 2). Furthermore, these data suggest that OMPs may be a valuable tool to help develop a comprehensive classification system to identify H. pylori populations and sub-populations. In all, this study has expanded the profile of bab possible genotypes and characterized a new population. Furthermore, it highlights several associations that should be investigated next at a functional level.
Supporting Information
(TIF)
(A) Percentage of ‘IN’ and ‘OUT’ babB gene reading frame in each population. (B) Number of ‘IN’ and ‘OUT’ babB gene reading frame at each locus.
(TIF)
Variable regions 1 and 2 are indicated, VR1 and VR2, respectively, above the BabC amino acid sequence.
(PDF)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank Dr. In-Sik Chung for the collection of Korean H. pylori clinical strain and miss him deeply.
Data Availability
The sequences for bab genotypes in American and South Korean strains were deposited in the NCBI GenBank database with accession numbers KP339308 to KP33949.
Funding Statement
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A2A1A11051054).
References
- 1. Ahuja V, Sharma MP. High recurrence rate of Helicobacter pylori infection in developing countries. Gastroenterology. 2002;123: 653–654. [DOI] [PubMed] [Google Scholar]
- 2. Suerbaum S, Michetti P. Medical progress: Helicobacter pylori infection. New England Journal of Medicine. 2002;347: 1175–1186. [DOI] [PubMed] [Google Scholar]
- 3. Shin A, Shin HR, Kang D, Park SK, Kim CS, Yoo KY. A nested case-control study of the association of Helicobacter pylori infection with gastric adenocarcinoma in Korea. Br J Cancer. 2005;92: 1273–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gwack J, Shin A, Kim CS, Ko KP, Kim Y, Jun JK, et al. CagA-producing Helicobacter pylori and increased risk of gastric cancer: a nested case-control study in Korea. Br J Cancer. 2006;95: 639–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tokudome S, Ando R, Ghadimi R, Tanaka T, Hattori N, Yang Z, et al. Are there any real Helicobacter pylori infection-negative gastric cancers in Asia? Asian Pac J Cancer Prev. 2007;8: 462–463. [PubMed] [Google Scholar]
- 6. Torres LE, Melian K, Moreno A, Alonso J, Sabatier CA, Hernandez M, et al. Prevalence of vacA, cagA and babA2 genes in Cuban Helicobacter pylori isolates. World J Gastroenterol. 2009;15: 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beswick EJ, Suarez G, Reyes VE. H pylori and host interactions that influence pathogenesis. World J Gastroenterol. 2006;12: 5599–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N, et al. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 2011;11: 104 10.1186/1471-2180-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nagasawa S, Motani-Saitoh H, Inoue H, Iwase H. Geographic diversity of Helicobacter pylori in cadavers: forensic estimation of geographical origin. Forensic Sci Int. 2013;229: 7–12. 10.1016/j.forsciint.2013.02.028 [DOI] [PubMed] [Google Scholar]
- 10. Wada A, Yamasaki E, Hirayama T. Helicobacter pylori vacuolating cytotoxin, VacA, is responsible for gastric ulceration. J Biochem. 2004;136: 741–746. [DOI] [PubMed] [Google Scholar]
- 11. Jang S, Jones KR, Olsen CH, Joo YM, Yoo YJ, Chung IS, et al. Epidemiological link between gastric disease and polymorphisms in VacA and CagA. J Clin Microbiol. 2010;48: 559–567. 10.1128/JCM.01501-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jones KR, Jang S, Chang JY, Kim J, Chung IS, Olsen CH, et al. Polymorphisms in the intermediate region of VacA impact Helicobacter pylori-induced disease development. J Clin Microbiol. 2011;49: 101–110. 10.1128/JCM.01782-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297: 573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamaoka Y, Kita M, Kodama T, Imamura S, Ohno T, Sawai N, et al. Helicobacter pylori infection in mice: Role of outer membrane proteins in colonization and inflammation. Gastroenterology. 2002;123: 1992–2004. [DOI] [PubMed] [Google Scholar]
- 15. de Jonge R, Durrani Z, Rijpkema SG, Kuipers EJ, van Vliet AH, Kusters JG. Role of the Helicobacter pylori outer-membrane proteins AlpA and AlpB in colonization of the guinea pig stomach. J Med Microbiol. 2004;53: 375–379. [DOI] [PubMed] [Google Scholar]
- 16. Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, et al. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut. 2006;55: 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang J, Jones KR, Jang S, Olsen CH, Yoo YJ, Merrell DS, et al. The geographic origin of Helicobacter pylori influences the association of the homB gene with gastric cancer. J Clin Microbiol. 2012;50: 1082–1085. 10.1128/JCM.06293-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talebi Bezmin Abadi A, Taghvaei T, Mohabbati Mobarez A, Vaira G, Vaira D. High correlation of babA 2-positive strains of Helicobacter pylori with the presence of gastric cancer. Intern Emerg Med. 2013;8: 497–501. 10.1007/s11739-011-0631-6 [DOI] [PubMed] [Google Scholar]
- 19. Pride DT, Meinersmann RJ, Blaser MJ. Allelic Variation within Helicobacter pylori babA and babB . Infect Immun. 2001;69: 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hennig EE, Allen JM, Cover TL. Multiple chromosomal loci for the babA gene in Helicobacter pylori . Infect Immun. 2006;74: 3046–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Armitano RI, Matteo MJ, Goldman C, Wonaga A, Viola LA, De Palma GZ, et al. Helicobacter pylori heterogeneity in patients with gastritis and peptic ulcer disease. Infect Genet Evol. 2013;16: 377–385. 10.1016/j.meegid.2013.02.024 [DOI] [PubMed] [Google Scholar]
- 22. Jones KR, Joo YM, Jang S, Yoo YJ, Lee HS, Chung IS, et al. Polymorphism in the CagA EPIYA motif impacts development of gastric cancer. J Clin Microbiol. 2009;47: 959–968. 10.1128/JCM.02330-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oleastro M, Monteiro L, Lehours P, Megraud F, Menard A. Identification of markers for Helicobacter pylori strains isolated from children with peptic ulcer disease by suppressive subtractive hybridization. Infect Immun. 2006;74: 4064–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oleastro M, Cordeiro R, Ferrand J, Nunes B, Lehours P, Carvalho-Oliveira I, et al. Evaluation of the clinical significance of homB, a novel candidate marker of Helicobacter pylori strains associated with peptic ulcer disease. J Infect Dis. 2008;198: 1379–1387. 10.1086/592166 [DOI] [PubMed] [Google Scholar]
- 25. Jung SW, Sugimoto M, Graham DY, Yamaoka Y. homB status of Helicobacter pylori as a novel marker to distinguish gastric cancer from duodenal ulcer. J Clin Microbiol. 2009;47: 3241–3245. 10.1128/JCM.00293-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ando T, Peek RM, Pride D, Levine SM, Takata T, Lee YC, et al. Polymorphisms of Helicobacter pylori HP0638 reflect geographic origin and correlate with cagA status. J Clin Microbiol. 2002;40: 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alfizah H, Ramelah M, Rizal AM, Anwar AS, Isa MR. Association of Malaysian Helicobacter pylori virulence polymorphisms with severity of gastritis and patients' ethnicity. Helicobacter. 2012;17: 340–349. 10.1111/j.1523-5378.2012.00956.x [DOI] [PubMed] [Google Scholar]
- 28. Kalaf EA, Al-Khafaji ZM, Yassen NY, Al-Abbudi FA, Sadwen SN. Study of the cytoxin-associated gene a (CagA gene) in Helicobacter pylori using gastric biopsies of Iraqi patients. Saudi J Gastroenterol. 2013;19: 69–74. 10.4103/1319-3767.108474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang XS, Tegtmeyer N, Traube L, Jindal S, Perez-Perez G, Sticht H, et al. A Specific A/T Polymorphism in Western Tyrosine Phosphorylation B-Motifs Regulates Helicobacter pylori CagA Epithelial Cell Interactions. PLoS Pathog. 2015;11: e1004621 10.1371/journal.ppat.1004621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A. 1999;96: 12778–12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abdullah SM, Hussein NR, Salih AM, Merza MA, Goreal AA, Odeesh OY, et al. Infection with Helicobacter pylori strains carrying babA2 and cagA is associated with an increased risk of peptic ulcer disease development in Iraq. Arab J Gastroenterol. 2012;13: 166–169. 10.1016/j.ajg.2012.12.001 [DOI] [PubMed] [Google Scholar]
- 32. Sheu SM, Sheu BS, Chiang WC, Kao CY, Wu HM, Yang HB, et al. H. pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol. 2012;12: 89 10.1186/1471-2180-12-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262: 1892–1895. [DOI] [PubMed] [Google Scholar]
- 34. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279: 373–377. [DOI] [PubMed] [Google Scholar]
- 35. Pride DT, Blaser MJ. Concerted evolution between duplicated genetic elements in Helicobacter pylori . J Mol Biol. 2002;316: 629–642. [DOI] [PubMed] [Google Scholar]
- 36. Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305: 519–522. [DOI] [PubMed] [Google Scholar]
- 37. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori . Nature. 1997;388: 539–547. [DOI] [PubMed] [Google Scholar]
- 38. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori . Nature. 1999;397: 176–180. [DOI] [PubMed] [Google Scholar]
- 39. Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci U S A. 2004;101: 2106–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oleastro M, Menard A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology (Basel). 2013;2: 1110–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Backstrom A, Lundberg C, Kersulyte D, Berg DE, Boren T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci U S A. 2004;101: 16923–16928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Colbeck JC, Hansen LM, Fong JM, Solnick JV. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori . Infect Immun. 2006;74: 4375–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Felsenstein J. PHYLIP (Phylogeny Inference Package). Version 3.7a Distributed by the author. Department of Genome Sciences, University of Washington, Seattle: 2009. [Google Scholar]
- 44.Rambaut A. 2014. Available: http://tree.bio.ed.ac.uk/software/figtree.
- 45. Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun. 2000;68: 4155–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bridge DR, Merrell DS. Polymorphism in the Helicobacter pylori CagA and VacA toxins and disease. Gut Microbes. 2013;4: 101–117. 10.4161/gmic.23797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matteo MJ, Armitano RI, Romeo M, Wonaga A, Olmos M, Catalano M. Helicobacter pylori bab genes during chronic colonization. Int J Mol Epidemiol Genet. 2011;2: 286–291. [PMC free article] [PubMed] [Google Scholar]
- 48. Ohno T, Vallstrom A, Rugge M, Ota H, Graham DY, Arnqvist A, et al. Effects of blood group antigen-binding adhesin expression during Helicobacter pylori infection of Mongolian gerbils. J Infect Dis. 2011;203: 726–735. 10.1093/infdis/jiq090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu H, Fero JB, Mendez M, Carpenter BM, Servetas SL, Rahman A, et al. Analysis of a single Helicobacter pylori strain over a 10-year period in a primate model. Int J Med Microbiol. 2015. 10.1016/j.ijmm.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nell S, Kennemann L, Schwarz S, Josenhans C, Suerbaum S. Dynamics of Lewis b binding and sequence variation of the babA adhesin gene during chronic Helicobacter pylori infection in humans. MBio. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oleastro M, Cordeiro R, Menard A, Yamaoka Y, Queiroz D, Megraud F, et al. Allelic diversity and phylogeny of homB, a novel co-virulence marker of Helicobacter pylori . BMC Microbiol. 2009;9: 248 10.1186/1471-2180-9-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(A) Percentage of ‘IN’ and ‘OUT’ babB gene reading frame in each population. (B) Number of ‘IN’ and ‘OUT’ babB gene reading frame at each locus.
(TIF)
Variable regions 1 and 2 are indicated, VR1 and VR2, respectively, above the BabC amino acid sequence.
(PDF)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
The sequences for bab genotypes in American and South Korean strains were deposited in the NCBI GenBank database with accession numbers KP339308 to KP33949.