

Graphical abstract
Abbreviations: AQP, aquaporin; AVP, arginine vasopressin; AVPR2, V2 receptor; AQP ER, endoplasmic reticulum; GlpF, glycerol facilitator; GpA, glycophorin A; HM, half-membrane-spanning; NDI, nephrogenic diabetes insipidus; TM, transmembrane; wt, wild-type
Keywords: GlpF, Aquaporin, Nephrogenic diabetes insipidus, Activity, Protein oligomerization
Highlights
-
•
The aquaporin 2 mutation V71M causes nephrogenic diabetes insipidus in humans.
-
•
Val71 is highly conserved in aqua(glycero)porins and points into the translocation pore.
-
•
The V71M mutation does not impair the activity and oligomerization of a bacterial homolog.
Abstract
Several point mutations have been identified in human aquaporins, but their effects on the function of the respective aquaporins are mostly enigmatic. We analyzed the impact of the aquaporin 2 mutation V71M, which causes nephrogenic diabetes insipidus in humans, on aquaporin structure and activity, using the bacterial aquaglyceroporin GlpF as a model. Importantly, the sequence and structure around the V71M mutation is highly conserved between aquaporin 2 and GlpF. The V71M mutation neither impairs substrate flux nor oligomerization of the aquaglyceroporin. Therefore, the human aquaporin 2 mutant V71M is most likely active, but cellular trafficking is probably impaired.
1. Introduction
Aquaporins (AQPs) are polytypic transmembrane (TM) channel proteins that selectively facilitate the bidirectional flux of water across cellular membranes in all domains of life [1,2]. While the classical aquaporins are only permeable for water, the subfamily of the aquaglyceroporins additionally enables the diffusion of small, uncharged molecules, such as linear polyalcohols. All AQPs have an extremely conserved tertiary structure [3], and all AQPs assemble into homotetramers within cellular membranes [4,5], with the monomer being the functional unit [4,6]. The monomeric protein consists of six TM helices and two re-entry loops that are each formed by a half-membrane-spanning (HM) helix and a non-helical part [4,7]. The two re-entry loops each contain highly conserved NPA motifs, consisting of the residues Asn, Pro and Ala that provide the intimate interface of the two HM helices and also contribute to substrate conductance.
While in the bacterium Escherichia coli (E. coli) only two AQPs, AqpZ and GlpF, are found, 13 human AQPs (AQP0-12) have been identified until today. These human AQPs differ in substrate selectivity, permeability and their tissue specific expression [8]. While it is intuitively clear that AQPs are directly involved in water conductance in tissues and organs, studies with AQP knockout mice revealed an involvement of human AQPs in physiological processes that are not directly related to water transport. Among those are cell migration, proliferation as well as differentiation and tumor angiogenesis [9]. In human genomes, several variations have been identified in AQP-encoding genes [10,11], and given the diverse physiological functions of human AQPs, mutations or deletions of single amino acids can cause severe human diseases.
The V71M mutation was identified in human AQP2 and is implicated with recessive nephrogenic diabetes insipidus [12]. AQP2 is expressed in the cells of the collecting duct of the kidney where it plays an important role in concentrating urine. Upon binding of arginine vasopressin (AVP) to the V2 receptor (AVPR2), AQP2 is transported to the apical plasma membrane. Binding of AVP to its receptor increases the intracellular cAMP level, which in turn activates the protein kinase A, that phosphorylates AQP2, thereby stimulating the redistribution of AQP2 from intracellular vesicles to the apical plasma membrane [12,13]. A functional analysis of AQP2-V71M in Xenopus laevis oocytes has indicated that the respective mutant renders the human AQP2 non-functional, resulting in retention of AQP2-V71M in the endoplasmatic reticulum (ER) membrane [12]. However, as the measured activity depends on proper trafficking of AQP2-V71M to the plasma membrane, the determined activity might underestimate the remaining channel activity, as cellular trafficking was severely impaired in this mutant. Determining the activity of a mutated protein, with impaired cellular trafficking, might therefore not be straightforward in intact eukaryotic cells.
Here we used the E. coli aquaglyceroporin GlpF as a model to study the impact of the V71M mutation on tetramerization and function of an AQP. The sequence identity between GlpF and the human aquaporins is right up to 35% and is even higher in the region around the two highly conserved NPA motifs, where the respective mutation occurs [14,15]. Given that the tertiary and quaternary structures of all AQPs are remarkably conserved [16,17], the experimentally easily accessible bacterial aquaglyceroporin GlpF has served as an aquaporin model for many years. More importantly, improper cellular trafficking cannot interfere with functional studies when working with the bacterial proteins, which are synthesized directly into the bacterial cell membrane.
Here we show that neither the in vivo oligomerization nor the in vivo activity of GlpF-V71M were impaired. As the tertiary and quaternary structures of all AQPs are highly conserved, the impact of the V71M mutation on the structure and function of human AQP2 will be similar. Consequently, based on the results obtained with the bacterial model system, the AQP2 mutant V71M is probably active but most likely not properly processed in eukaryotic cells, and the mutated protein likely retards in the ER, as shown in Xenopus laevis oocytes [12]. The V71M mutation can thus be classified as a class-II-mutation.
2. Material and methods
2.1. Mutagenesis
The V71M mutation was introduced into the plasmids pRSET-His-GlpF, pLexA-GlpF and pGlpF [18] by site-directed mutagenesis using the oligonucleotides QCGlpFV71Mfor (5′–CATCTTAATCCCGCTATGACCATTGCATTGTGG–3′) and QCGlpFV71Mrev (5′–CCACAATGCAATGGTCATAGCGGGATTAAGATG–3′) purchased from Eurofins MWG Operon (Ebersberg, Germany). The sequence of the constructs was confirmed by sequencing the plasmid DNA.
2.2. Expression and purification of GlpF by affinity chromatography
Expression of GlpF in E. coli BL21DE3 cells transformed with the plasmids pRSET-His-GlpF or pRSET-His-GlpF(E43A/V71M) and protein purification were conducted as described in detail previously [18]. The protein concentration was determined by measuring the absorption at 280 nm, using a calculated extinction coefficient of 37,930 M−1 cm−1 (ExPasy, ProtParam tool).
2.3. Semi-native SDS–PAGE analysis
In a semi-native SDS–PAGE analysis, no SDS is present in the sample buffer and the native tetrameric state of GlpF is preserved [5,19]. Samples were incubated for 15 min at room temperature with SDS-free sample buffer (50 mM Tris–HCl (pH 6.8), 10% (v/v) glycerol and 0.04% (w/v) Bromphenol blue) and separated on a 10% SDS–PAGE gel. Thereafter, SDS–PAGE gels were stained with Coomassie brilliant blue R250.
2.4. GALLEX measurements
The GlpF interaction propensity was measured with the GALLEX assay [18,20]. LexA-GlpF fusion proteins were expressed in E. coli SU101 cells transformed with the plasmids pLexA-GlpF or pLexA-GlpF (E43A/V71M). For control measurements, cells were also transformed with the empty expression plasmid (pMal-p2 (−)), as well as the plasmids pLexA-GpA and pLexA-GpA-G83I [20]. The plasmid pLexA-GpA encodes the TM domain of human glycophorin A (GpA) that forms very stable TM homodimers, and pLexA-GpA-G83I encodes the G83I-mutated GpA TM domain characterized by a diminished dimerization tendency [20,21]. The wt and the G83I-mutated TM domains are both N-terminally fused to the LexA-DNA binding domain and C-terminally to the MalE domain. Protein expression was performed in LB medium supplemented with 100 μg/mL ampicillin, 30 μg/mL chloramphenicol and either a constant isopropyl-β-d-thiogalactopyranoside (IPTG) concentration of 500 μM or increasing IPTG concentrations (10–500 μM) [20]. Cells grew to an OD600 of 0.6 and β-galactosidase activities were measured as described [22]. Six independent colonies were analyzed for each construct. To obtain the relative β-galactosidase activity (rel. β-gal. act.), the β-galactosidase activities were normalized to the β-galactosidase activity determined for GlpF-wt after induction of the protein expression with 500 μM IPTG.
2.5. GlpF activity measurements
The GlpF activity was assessed by measuring the kinetics of the flux of the linear polyalcohol ribitol over the inner E. coli membrane using a SX20 stopped flow spectrometer (Applied Photophysics, Leatherhead, U.K.) as described in [18]. E. coli SK46 cells were transformed with the plasmids pMal-p2x (empty expression plasmid (−)), pGlpF, pGlpF-E43A, or pGlpF-V71M, and protein expression was induced either in presence of a constant IPTG concentration of 500 μM or at increasing IPTG concentrations (10–500 μM) [18]. Ribitol was chosen as the substrate to reduce the background of the measurements. While the ribitol conductance rate of GlpF is comparable to its glycerol conductance rate, the intrinsic permeability of membranes for ribitol is lower than for glycerol [4].
2.6. Isolation of GlpF from membranes
To determine the amount of expressed GlpF, membranes were isolated from equal volumes of E. coli NT326 (GALLEX measurements) and SK46 cells (activity measurements) transformed with the plasmids pMal-p2, pLexA-GpA, pLexA-GpA-G83I, pLexA-GlpF, pLexA-E43A, or pLexA-GlpF-V71M and pMal-p2x, pGlpF, pGlpF-E43A, or pGlpF-V71M, respectively. Cells were incubated in LB medium supplemented with 100 μg/mL ampicillin containing either a constant IPTG concentration or increasing IPTG concentrations (10–500 μM). After an OD600 of 0.8 was reached, cells were centrifuged (10 min, 3,220 g, 4 °C) and resuspended in 25 mM Tris–HCl (pH 8.0), 2 mM Na2EDTA × 2H2O and 0.1% (v/v) protease inhibitor cocktail, thereby adjusting the OD600 to 2.0 in a volume of 15 mL. Cells were then disrupted by sonication in an ice-water bath, using a Branson Sonifier 250 (G. Heinemann). Cell debris was separated by centrifugation at 12,000 g and 4 °C for 10 min, GlpF-containing membranes were then collected by ultracentrifugation (165,000 g, 4 °C, 1 h). The membrane pellet was resuspended in 100 μL 50 mM phosphate buffer (pH 8.0), 300 mM NaCl and 10% glycerol. A bicinchoninic acid assay was performed to determine the protein content of the membranes using the BCA protein assay kit (Thermo Fisher Scientific). 2.4 μg protein were incubated in SDS–PAGE sample buffer (2% (w/v) SDS, 50 mM Tris–HCl (pH 6.8), 10% (v/v) glycerol and 0.04% (w/v) bromphenol blue) for 15 min at room temperature. After performing an SDS–PAGE analysis, using 10% SDS–PAGE gels, the separated proteins were transferred to a PVDF membrane, and thereafter GlpF was detected using an antibody directed against the GlpF C-terminus (VVEEKETTTPSEQKASL, Gramsch Laboratories). The LexA-GlpF fusion constructs were detected using an antibody from Novus recognizing the LexA DNA binding domain.
3. Results and discussion
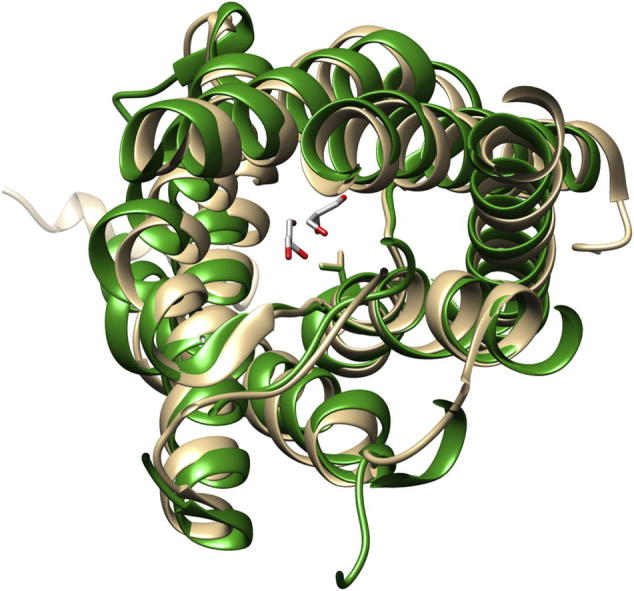
When expressed in Xenopus laevis oocytes, the V71M mutation was suggested to render AQP2 non-functional, resulting in retention of AQP2-V71M in the ER membrane [12]. However, determining the activity of a mutated protein with impaired cellular trafficking might be problematic in intact eukaryotic cells, as only the activity of proteins incorporated into the plasma membrane is assayed. Thus, the question arises whether trafficking of the mutated protein is impaired due to its inactivation, or whether activity measurements failed due to improper trafficking. To answer this question, we analyzed a mutated bacterial homolog, with a conserved structure and function, since in bacteria, proteins are synthesized directly into the cell membrane and thus impaired trafficking is not an issue. However, using the bacterial homolog GlpF as a model for analyzing the impact of the V71M mutation on oligomerization and function of the human AQP2 requires at least that the mutated amino acid is strictly conserved within the primary and tertiary structure of GlpF and AQP2. Superposition of the GlpF and AQP2 crystal structures indicates that the conformation and arrangement of the TM helices is identical in GlpF and AQP2 and that Val71 points into the translocation pore in both proteins (Fig. 1A–C). Furthermore, as can be seen in a sequence alignment, Val71 of AQP2 is conserved in GlpF and within the majority of the human classical AQPs and aquaglyceroporins (Fig. 1D). The sequence alignment further reveals that the primary sequence is also highly conserved in the region bordering the mutation. Thus, GlpF is a suitable and experimentally accessible model to study the impact of the V71M mutation on the activity of an aqua(glycerol)porin. Interestingly, in the classical aquaporins AQP5 and -10, the larger, hydrophobic amino acids Ile and Phe are located at the position typically occupied by V71, indicating that replacement of this amino acid by other residues, possessing bulkier side-chains, might be well tolerated.
Fig. 1.

Alignment of the GlpF and AQP2 structures and sequences. Superposing the structures of GlpF (green) and AQP2 (beige) illustrates the high conservation of the AQP tertiary structures. While structural differences are found in the extra membranous loops in the crystal structures, the conformation of the TM helices is conserved. (A) Superposition of GlpF and AQP2 seen sideways. Both, the N- and the C-terminus, are located in the cytoplasm. (B) Top view onto the translocation pore as seen from the periplasm or extracellular space, respectively. Depicted is the amino acid residue Val71 in GlpF and in AQP2. For clarity, the periplasmatic/extracellular loop regions are not shown. (C) Zoom in on the structure around the first NPA motif. The structural alignment was prepared using the program Chimera [29]. (PDB: 1FX8 und 4NEF). (D) Sequence alignment of GlpF and the classical human AQPs and aquaglyceroporins. The conserved NPA motif is highlighted in gray, and the conserved Val residue (position 71 in AQP2) is highlighted in bold. One dot highlights conserved residues, two dots highly conserved residues and a star indicates identical residues. The sequence alignment was performed using the program ClustalW2 [30]. The sequences were obtained from the UniProtKB/TrEMBL data bank [31].
3.1. Assembly of GlpF-V71M is not impaired
Aquaporins assemble into tetramers already in the ER [23,24], and the tetrameric structure is crucial for proper cellular trafficking of AQP2. Only 25% of monomeric AQPs are glycosylated with complex oligosaccharides in the Golgi apparatus [23,25], and N-glycosylation is essential for trafficking of AQP2 from the Golgi apparatus to the plasma membrane [23]. Therefore, we first assessed a potential impact of the V71M mutation on the stability of the purified GlpF. For comparison, the well characterized GlpF mutant E43A, that exhibits both, a reduced oligomerization propensity and activity, was also analyzed [18]. As depicted in Fig. 2A, the GlpF-wt protein runs as a stable tetramer on SDS-gels, while GlpF-E43A was mainly found as a monomer. The in vitro tetramer stability of GlpF-V71M was also reduced compared to GlpF-wt, however not to the same extent as observed in case of the GlpF-E43A protein. Nevertheless, the observed destabilization of the GlpF-V71M tetramer might be solely relevant in the rather artificial micellar environment. Therefore, the GALLEX assay was applied to next study the in vivo oligomerization behavior of GlpF-V71M in a native membrane [18,20]. In the GALLEX assay, a LexA DNA-binding domain is fused to the GlpF N-terminus. As both termini of GlpF are located on the cytoplasmic side of the inner E. coli membrane (Fig. 1A), the LexA DNA-binding domain of the LexA-GlpF chimeric protein is also located in the cytoplasm. When GlpF oligomerizes, two LexA DNA-binding domains come in close proximity and bind to the promoter/operator region of the lacZ gene, thereby repressing expression of the β-galactosidase [20]. Therefore, the β-galactosidase activity is a direct measure of the GlpF interaction propensity within its native membrane environment. The determined oligomerization propensities indicate no differences in the in vivo oligomerization propensity of GlpF-V71M compared to the GlpF-wt whereas the oligomerization propensity of GlpF-E43A was also reduced in vivo (Fig. 2B). To study the in vivo oligomerization behavior of GlpF-V71M in comparison to the GlpF-wt in greater detail, the expression of the LexA-GlpF fusion proteins was induced by addition of increasing IPTG concentrations. In previous analyses we have noticed that increased GlpF expression can be induced by addition of 10–500 μM IPTG [18], and thus we have determined the β-galactosidase activities in this IPTG concentration range. As can be seen in Fig. 3A, the determined oligomerization propensities at all tested IPTG concentrations again indicate no differences in the in vivo oligomerization propensity of GlpF-V71M compared to GlpF-wt. Additionally, expression of the wt and V71M-mutated GlpF proteins was analyzed at each respective IPTG concentration by Western blot analyses to account for potential differences in the expression level. As expected, the expression level of both, the wt and the V71M-mutated GlpF, increased with increasing IPTG concentrations and the expression levels at a given IPTG concentration were similar for the wt and the V71M-mutated protein (Fig. 3B). It is worth mentioning that the GALLEX measurements do not allow distinguishing between the different oligomeric states that contribute to the signal, and not only a tetrameric but i.e. also a dimeric GlpF could affect the measured β-galactosidase activities. While this cannot be resolved with the GALLEX assay, the signal in the GALLEX assay is most likely based on the GlpF tetramer, which is the most stable GlpF structure [5].
Fig. 2.

In vitro and in vivo oligomerization of GlpF-wt, GlpF-E43A and GlpF-V71M. (A) Semi-native SDS–PAGE analysis of purified GlpF-wt and the GlpF mutants E43A and V71M (T: tetramer, Di: dimer; M: monomer). 7.5 μg protein was loaded per lane. (B) The oligomerization tendency of the GlpF-wt, GlpF-E43A, and GlpF-V71M was assessed using the GALLEX assay. As controls, the rel. β-gal. activities of the wt and G83I-mutated GpA TM domains, that possess a strong and weak dimerization propensity, respectively, as well as the rel. β-gal. activity determined in cells transformed with the empty expression plasmid (−) were determined. The determined interaction propensity of GlpF-wt was set at 1.0. For the Western blot analysis shown in the lower panel an antibody recognizing the LexA DNA binding domain was used.
Fig. 3.

Oligomerization of wt and V71M-mutated GlpF at increasing IPTG concentrations. (A) The oligomerization tendency of the wt ( ) and V71M-mutated GlpF (
) and V71M-mutated GlpF ( ) was analyzed in vivo at increasing IPTG concentrations using the GALLEX assay (n = 6 ± SE). Plotted is the rel. β-gal. act. vs. the IPTG concentration. (B) Western blot analyses, probing the expression of GlpF-wt and GlpF-V71M in E. coli membranes, were performed using an antibody directed against the GlpF C-terminus.
) was analyzed in vivo at increasing IPTG concentrations using the GALLEX assay (n = 6 ± SE). Plotted is the rel. β-gal. act. vs. the IPTG concentration. (B) Western blot analyses, probing the expression of GlpF-wt and GlpF-V71M in E. coli membranes, were performed using an antibody directed against the GlpF C-terminus.
Based on our results, the mutation V71M does not influence the in vivo oligomerization propensity of GlpF. As the tertiary structure of AQP2 and GlpF is conserved (Fig. 1), it appears to be reasonable to assume that the impact of the V71M mutation on oligomerization of the human AQP2 is similar. Thus, the V71M mutation does, most likely, not dramatically affect proper tetramerization of the human AQP2. Nevertheless, the increased sensitivity against SDS-induced unfolding (Fig. 2A) indicates slight structural differences between the wt and V71M-mutated proteins, which might be a reason for AQP2-V71M being recognized by the cellular quality control system, resulting in ER-associated degradation.
3.2. Structural rearrangements probably ensure the unimpaired substrate flux through GlpF-V71M
To further investigate a potential impact of the V71M mutation, we measured the GlpF-facilitated water and ribitol flux across the E. coli inner membrane. Rapid mixing of GlpF expressing E. coli SK46 cells with a hypertonic ribitol solution causes initial shrinkage of the cells, owing to water efflux. As ribitol diffuses into the cells via GlpF, water flows back causing the cells to reswell. Shrinkage and reswelling of the cells can be assessed by measuring the light scattering intensity at a 90° angle, and will cause an initial increase of the light scattering signal followed by a decay of the signal (compare Fig. 4A). As can be seen in Fig. 4A, the activities of the wt and V71M-mutated GlpF were nearly identical, whereas the activity of GlpF-E43A was clearly reduced. Noteworthy, the decreased activity of GlpF-E43A cannot be entirely attributed to its slightly decreased expression level (80% compared to the expression level of the GlpF-wt) (Fig. 4B). As conducted in the oligomerization measurements (Fig. 3), the activity of GlpF-V71M was also studied in closer detail by addition of increasing IPTG concentrations. To assess the rate constants of the GlpF-facilitated water and ribitol flux, both the exponential growth and the decay of the light scattering signal were fitted using a double exponential function. As already suggested by the similar course of the light scattering curves (Fig. 4A), the activity measurements performed at increasing protein concentrations indicated no differences, neither in the water nor in the ribitol flux facilitated by GlpF-wt and GlpF-V71M (Fig. 5A, B). To account for possible differences in the expression level of the wt and V71M-mutated GlpF, the amount of GlpF present in the membranes was again assessed immunochemically via Western blot analyses (Fig. 5C). As observed before, the Western blot analyses indicated an increasing expression of both, GlpF-wt and GlpF-V71M, and further revealed a comparable expression level at a given IPTG concentration for the wt and the V71M-mutated protein (Fig. 5C). Therefore, replacement of Val71 by Met does not dramatically interfere with substrate conductance, and hence the V71M mutation does not impair the protein activity. Although Val71 is located in close proximity to the peptide bonds in the non-helical part of the first-HM loop that interacts with the permeating substrate via hydrogen bond formation (Fig. 6A), the mutation to the larger Met did not impair substrate diffusion through GlpF, as shown in our activity measurements (Fig. 5). Apparently, the V71M mutation does not reduce the translocation pore diameter drastically, since otherwise a significant impact on the GlpF function would have been detected. As the conformation of Val71 is highly similar in GlpF and AQP2 (Fig. 1B, C), analogous conformational adjustments of the introduced Met residue are likely to occur in both, GlpF and AQP2. Nevertheless, the diameter of the translocation pore is approximately 1 Å smaller in the region of the NPA motifs in classical AQPs compared to the translocation pore of aquaglyceroporins [26]. Thus, one could argue that the mutation of Val to the larger amino acid Met could be more critical in classical AQPs, despite the smaller substrate. However, in the classical aquaporins AQP5 and -10 the larger, hydrophobic amino acids Ile and Phe are located at the respective positions (Fig. 1D). Comparing the structures of AQP5 and -2 reveals that the HM helix 3 in AQP5 is slightly stretched (Fig. 6B), possibly to accommodate the additional methyl group without reducing the diameter of the translocation pore. Thus, conformational adjustments according to this mechanism likely maintain the unimpaired substrate flux through the V71M-mutated AQP2 translocation pore.
Fig. 4.

Activity of GlpF-wt, GlpF-E43A and GlpF-V71M. (A) Typical light scattering curves are shown, observed using E. coli SK46 cells expressing GlpF-wt (green), GlpF-E43A (gray), GlpF-V71M (beige) or E. coli cells transformed with an empty vector (black). (B) Western blot analyses, probing the expression of GlpF-wt, GlpF-E43A and GlpF-V71M in E. coli SK46 membranes at an IPTG concentration of 500 μM, were performed using an antibody directed against the GlpF C-terminus.
Fig. 5.

Water and ribitol conductance rates. Kinetics of the water (A) and ribitol (B) conductance facilitated by GlpF-wt () and GlpF-V71M () are plotted vs. the IPTG concentration (n = 5 ± SD). (B) Western blot analyses, probing the expression of GlpF-wt and GlpF-V71M in E. coli SK46 membranes, were performed using an antibody directed against the GlpF C-terminus.
Fig. 6.

The aquaporin translocation pore. (A) The GlpF translocation pore as seen from the periplasm with one molecule glycerol in the pore. (PDB: 1FX8). The aquaporin translocation pore is defined by six TM helices and two re-entry loops that are formed by a HM helix and a non-helical part [4,7]. Depicted is the first re-entry loop (loop B) consisting of the HM helix 3 and a non-helical part (G64-L67). Hydrogen bonds between the carbonyl oxygen atoms (red) of the peptide bonds in the non-helical parts of the two HM loops and the passing water and linear polyalcohols, respectively, contribute to substrate conductance [32,33]. (B) Superposition of the AQP2 (beige) and AQP5 (blue) translocation pore, as seen from the extracellular site. HM helix 3 is slightly stretched in AQP5 compared to AQP2, possibly to accommodate the additional methyl group of Ile72. The structural alignment was prepared using the program Chimera [29]. (PDB: 4NEF and 3D9S).
4. Conclusion
Taken together, the V71M mutation in AQP2 that causes nephrogenic diabetes insipidus likely solely affects cellular trafficking but not the activity of AQP2. While the results of the GALLEX measurements show that the bacterial homolog GlpF forms stable oligomers, the SDS–PAGE analysis indicates that the mutation might slightly destabilize the protein structure. In case of AQP2, such destabilization might be recognized by the ER quality control system resulting in protein degradation rather than in trafficking to the plasma membrane. Consequently, the results obtained using the V71M-mutated bacterial homolog together with the impaired cellular trafficking observed in Xenopus laevis oocytes [12] indicate that the mutation V71M can be classified as a class II mutation. The mutation thus causes conformational changes that impair AQP2 trafficking but hardly affect the properties of the translocation pore. An altered conformation affecting the structure but not the activity of an AQP, was also described for the AQP2 mutant T126M [27]. In case of the AQP2 mutant T126M, the defects in cellular routing were already partially rescued by inhibition of Hsp90 that is believed to interact with and promote ER-dependent degradation [28]. Thus, a possible pharmaceutical strategy for treatment of nephrogenic diabetes insipidus caused by a V71M-mutated AQP2 is to correct the defective transport of the otherwise functional protein from the ER into the membranes of the collecting duct of the kidney.
Acknowledgements
This work was supported by Grants from the “Stiftung Rheinland-Pfalz für Innovation”, the Research Center “Complex Materials” (COMATT) and the German Chemical Industrial Fund. Author contributions: N.Kl. and D.S. conceived and designed the project, N.Kl., N.Kü. and D.H. acquired the data, N.Kl., N.Kü., and D.S. analyzed and interpreted the data, N.Kl. and D.S. wrote the paper.
References
- 1.Preston G.M., Carroll T.P., Guggino W.B., Agre P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]
- 2.Fujiyoshi Y., Mitsuoka K., de Groot B.L., Philippsen A., Grubmuller H., Agre P., Engel A. Structure and function of water channels. Curr. Opin. Struct. Biol. 2002;12:509–515. doi: 10.1016/s0959-440x(02)00355-x. [DOI] [PubMed] [Google Scholar]
- 3.Jung J.S., Preston G.M., Smith B.L., Guggino W.B., Agre P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model. J. Biol. Chem. 1994;269:14648–14654. [PubMed] [Google Scholar]
- 4.Fu D., Libson A., Miercke L.J., Weitzman C., Nollert P., Krucinski J., Stroud R.M. Structure of a glycerol-conducting channel and the basis for its selectivity. Science. 2000;290:481–486. doi: 10.1126/science.290.5491.481. [DOI] [PubMed] [Google Scholar]
- 5.Veerappan A., Cymer F., Klein N., Schneider D. The tetrameric alpha-helical membrane protein GlpF unfolds via a dimeric folding intermediate. Biochemistry. 2011;50:10223–10230. doi: 10.1021/bi201266m. [DOI] [PubMed] [Google Scholar]
- 6.Lagree V., Froger A., Deschamps S., Pellerin I., Delamarche C., Bonnec G., Gouranton J., Thomas D., Hubert J.F. Oligomerization state of water channels and glycerol facilitators – involvement of loop E. J. Biol. Chem. 1998;273:33949–33953. doi: 10.1074/jbc.273.51.33949. [DOI] [PubMed] [Google Scholar]
- 7.Murata K., Mitsuoka K., Hirai T., Walz T., Agre P., Heymann J.B., Engel A., Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 8.Verkman A.S., Anderson M.O., Papadopoulos M.C. Aquaporins: important but elusive drug targets. Nat. Rev. Drug Discov. 2014;13:259–277. doi: 10.1038/nrd4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verkman A.S. Aquaporins. Curr. Biol. 2013;23:R52–R55. doi: 10.1016/j.cub.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorani M.D., Manley G.T., Giacomini K.M. Genetic variation in human aquaporins and effects on phenotypes of water homeostasis. Hum. Mutat. 2008;29:1108–1117. doi: 10.1002/humu.20762. [DOI] [PubMed] [Google Scholar]
- 11.Klein N., Neumann J., O’Neil J.D., Schneider D. Folding and stability of the aquaglyceroporin GlpF: implications for human aqua(glycero)porin diseases. Biochim. Biophys. Acta. 1848;2015:622–633. doi: 10.1016/j.bbamem.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Marr N., Bichet D.G., Hoefs S., Savelkoul P.J., Konings I.B., De Mattia F., Graat M.P., Arthus M.F., Lonergan M., Fujiwara T.M., Knoers N.V., Landau D., Balfe W.J., Oksche A., Rosenthal W., Muller D., Van Os C.H., Deen P.M. Cell-biologic and functional analyses of five new aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2002;13:2267–2277. doi: 10.1097/01.asn.0000027355.41663.14. [DOI] [PubMed] [Google Scholar]
- 13.Kamsteeg E.J., Heijnen I., van Os C.H., Deen P.M. The subcellular localization of an aquaporin-2 tetramer depends on the stoichiometry of phosphorylated and nonphosphorylated monomers. J. Cell Biol. 2000;151:919–930. doi: 10.1083/jcb.151.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonen T., Walz T. The structure of aquaporins. Q. Rev. Biophys. 2006;39:361–396. doi: 10.1017/S0033583506004458. [DOI] [PubMed] [Google Scholar]
- 15.King L.S., Kozono D., Agre P. From structure to disease: the evolving tale of aquaporin biology. Nat. Rev. Mol. Cell. Biol. 2004;5:687–698. doi: 10.1038/nrm1469. [DOI] [PubMed] [Google Scholar]
- 16.Savage D.F., Egea P.F., Robles-Colmenares Y., O’Connell J.D., 3rd., Stroud R.M. Architecture and selectivity in aquaporins: 2.5 a X-ray structure of aquaporin Z. PLoS Biol. 2003;1:E72. doi: 10.1371/journal.pbio.0000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y., Tajkhorshid E. Molecular mechanisms of conduction and selectivity in aquaporin water channels. J. Nutr. 2007;137:1509S–1515S. doi: 10.1093/jn/137.6.1509S. [DOI] [PubMed] [Google Scholar]
- 18.Cymer F., Schneider D. A single glutamate residue controls the oligomerization, function, and stability of the aquaglyceroporin GlpF. Biochemistry. 2010;49:279–286. doi: 10.1021/bi901660t. [DOI] [PubMed] [Google Scholar]
- 19.Galka J.J., Baturin S.J., Manley D.M., Kehler A.J., O’Neil J.D. Stability of the glycerol facilitator in detergent solutions. Biochemistry. 2008;47:3513–3524. doi: 10.1021/bi7021409. [DOI] [PubMed] [Google Scholar]
- 20.Schneider D., Engelman D.M. GALLEX, a measurement of heterologous association of transmembrane helices in a biological membrane. J. Biol. Chem. 2003;278:3105–3111. doi: 10.1074/jbc.M206287200. [DOI] [PubMed] [Google Scholar]
- 21.Lemmon M.A., Flanagan J.M., Treutlein H.R., Zhang J., Engelman D.M. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 22.Miller J.H. Cold Spring Harbor Laboratory Press; Plainview, New York: 1992. A Short Course in Bacterial Genetics. [Google Scholar]
- 23.Hendriks G., Koudijs M., van Balkom B.W., Oorschot V., Klumperman J., Deen P.M., van der Sluijs P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J. Biol. Chem. 2004;279:2975–2983. doi: 10.1074/jbc.M310767200. [DOI] [PubMed] [Google Scholar]
- 24.Pitonzo D., Skach W.R. Molecular mechanisms of aquaporin biogenesis by the endoplasmic reticulum Sec61 translocon. Biochim. Biophys. Acta. 2006;1758:976–988. doi: 10.1016/j.bbamem.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 25.Baumgarten R., Van De Pol M.H., Wetzels J.F., Van Os C.H., Deen P.M. Glycosylation is not essential for vasopressin-dependent routing of aquaporin-2 in transfected Madin–Darby canine kidney cells. J. Am. Soc. Nephrol. 1998;9:1553–1559. doi: 10.1681/ASN.V991553. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y., Schulten K., Tajkhorshid E. What makes an aquaporin a glycerol channel? A comparative study of AqpZ and GlpF. Structure. 2005;13:1107–1118. doi: 10.1016/j.str.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Tamarappoo B.K., Yang B., Verkman A.S. Misfolding of mutant aquaporin-2 water channels in nephrogenic diabetes insipidus. J. Biol. Chem. 1999;274:34825–34831. doi: 10.1074/jbc.274.49.34825. [DOI] [PubMed] [Google Scholar]
- 28.Yang B., Zhao D., Verkman A.S. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidus in a conditional knock-in mouse model of aquaporin-2 mutation. FASEB J. 2009;23:503–512. doi: 10.1096/fj.08-118422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 30.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apweiler R., Bairoch A., Wu C.H. Protein sequence databases. Curr. Opin. Chem. Biol. 2004;8:76–80. doi: 10.1016/j.cbpa.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Jensen M.O., Tajkhorshid E., Schulten K. The mechanism of glycerol conduction in aquaglyceroporins. Structure. 2001;9:1083–1093. doi: 10.1016/s0969-2126(01)00668-2. [DOI] [PubMed] [Google Scholar]
- 33.Jensen M.O., Mouritsen O.G. Single-channel water permeabilities of Escherichia coli aquaporins AqpZ and GlpF. Biophys. J. 2006;90:2270–2284. doi: 10.1529/biophysj.105.073965. [DOI] [PMC free article] [PubMed] [Google Scholar]