

Abstract
Intracellular protein aggregation is the hallmark of several neurodegenerative diseases. Aggregates formed by polyglutamine (polyQ)-expanded proteins, such as Huntingtin, adopt amyloid-like structures that are resistant to denaturation. We used a novel purification strategy to isolate aggregates formed by human Huntingtin N-terminal fragments with expanded polyQ tracts from both yeast and mammalian (PC-12) cells. Using mass spectrometry we identified the protein species that are trapped within these polyQ aggregates. We found that proteins with very long intrinsically-disordered (ID) domains (≥100 amino acids) and RNA-binding proteins were disproportionately recruited into aggregates. The removal of the ID domains from selected proteins was sufficient to eliminate their recruitment into polyQ aggregates. We also observed that several neurodegenerative disease-linked proteins were reproducibly trapped within the polyQ aggregates purified from mammalian cells. Many of these proteins have large ID domains and are found in neuronal inclusions in their respective diseases. Our study indicates that neurodegenerative disease-associated proteins are particularly vulnerable to recruitment into polyQ aggregates via their ID domains. Also, the high frequency of ID domains in RNA-binding proteins may explain why RNA-binding proteins are frequently found in pathological inclusions in various neurodegenerative diseases.
Introduction
Accumulation of intracellular and extracellular protein aggregates—frequently in the form of amyloid—is a common feature of multiple age-associated human disorders, particularly neurodegenerative diseases [1, 2]. Amyloid is a highly-ordered aggregate that consists of polypeptides arranged in filamentous, beta sheet-rich structures with abundant interlocking hydrogen bonds between sheets [3–6]. Multiple proteins can adopt this architecture [7], and once established, they all share extraordinary resistance to proteolysis, chaotropic agents, detergents, and mechanical breakage [8–12]. Amyloid aggregates—or their oligomeric precursors—are believed to contribute to cellular toxicity through a variety of mechanisms, such as physically disrupting membranes or sequestering essential heterologous proteins [13, 14].
Triplet CAG expansions resulting in polyglutamine (polyQ) tracts are characteristic of at least nine neurodegenerative diseases [15, 16], of which the most common is Huntington disease (HD). Proteins with expanded tracts of polyQ, which are natively unstructured, are predisposed to adopt conformations with amyloid-like properties [17, 18]. When polyQ tracts of the Huntingtin protein (Htt) exceed ~35 glutamines, Htt fragments can form intracellular inclusions [17, 19–21] resulting in impaired Htt function and aberrant protein interactions [22]. These inclusions (or their early precursors) may confer a dominant gain-of-function cellular toxicity [23, 24]. Other studies suggest that aggregation caused by polyQ expansion can result in loss of protein function (and thus pathology), either directly via functional loss of the aggregating species [25], or from the sequestration of proteins that tightly interact with the aggregate [26, 27]. Thus, the proteins that interact with aggregates may mediate pathological mechanisms, either by conferring new cytotoxicity or by contributing to a general loss of function. For these reasons, determining how and why specific proteins interact with polyQ aggregates is important to understanding—and ultimately combating—disease mechanisms.
Recently, we established a mass spectrometry-based approach for non-targeted identification of intracellular amyloid-forming and amyloid-associated proteins [9, 28]. Our method, called TAPI (Technique for Amyloid Purification and Identification), exploits the biophysical characteristics of amyloid, namely detergent resistance and high molecular weight, to isolate amyloid aggregates from cell lysates. Stringent nuclease treatment followed by SDS-gel electrophoresis eliminates non-specific or loosely associated proteins. Thus the TAPI protocol differs from antibody-based pull-down protocols by limiting positive hits to those most tightly associated with amyloid-like aggregates.
In this study we applied the TAPI protocol coupled with mass spectrometry to aggregates formed by polyQ-expanded huntingtin fragments in both yeast (S. cerevisiae) and mammalian cells (PC-12, rat neuronal precursor). Previously, various proteins have been shown to interact with Htt or Htt fragments [27, 29–33], but our approach was designed to identify proteins that are directly trapped within the amyloid-like polyQ aggregates to determine the types of proteins most prone to irreversible inclusion. We hypothesized that these aggregates would recruit and/or sequester proteins with common biophysical properties. We observed that inclusion into polyQ aggregates was mediated by long intrinsically-disordered (ID) protein domains (≥ 100 amino acids) in two evolutionary divergent cell models. Also, many proteins normally associated with neuronal aggregation in other degenerative diseases (especially amyotrophic lateral sclerosis (ALS)) were disproportionately recruited into polyQ aggregates in mammalian cells. This study expands the emerging connection between ID domains and neurodegenerative disease [34], and demonstrates that long ID domains predispose proteins to be recruited into amyloid-like aggregates.
Materials and Methods
Cell Lines and Maintenance
Yeast strains BY4741 (MATa his3 leu2 met15 ura3 [PIN+] [psi-]) or W303 (MATa leu2 ade2-1 ura3 can1 trp1 his3 gal+) were transformed with Gal-Inducible Huntingtin Exon 1 polyQ expansion plasmids (Htt-Q25-GFP or Htt-Q103-GFP) [9, 35]. Genes (or truncated variants) were cloned into pFPS261 and pFPS262, which encode single HA tags in-frame with the multiple cloning site, thus adding c-terminal epitope tags to genes cloned into the Xho1 site. Plasmids pFPS261 (CEN LEU2 P GAL1) and pFPS262 (2μ LEU2 P GAL1) are respectively derivatives of previously-described pH316 [36] and pH317 [37]. Truncated SGT2-ΔID is SGT2Δ300–346, and truncated FUS-ΔID is FUSΔ1–135. Human α-synuclein-GFP was expressed from the previously-described plasmid DK258 (2μ LEU2 P GAL1) [38]. All strains were cultured in synthetic defined media with appropriate auxotrophic selection for plasmid maintenance. Protein expression was induced overnight with growth on selective galactose-containing medium.
The PC-12 cell lines were previously described by Wyttenbach et al. [39]. Briefly, the PC12 cells were stably transformed with Doxycycline-inducible GFP-tagged normal Htt-Q23 Exon 1 or expanded Htt-Q74. Cells were cultured on collagen IV (BD Biosciences) coated T-75 flasks and maintained in DMEM with 75 μg/mL hygromycin, 100 U/mL penicillin/streptomycin, 2 mM L-glutamine, 10% heat-inactivated horse serum, 5% Tet-negative fetal bovine serum and 100 μg/mL G418 at 37°C, 10% CO2. Culture reagents were obtained from Corning.
Technique for Amyloid Purification and Identification (TAPI)
The yeast TAPI protocol was performed as previously described [9] with the following alterations: Buffer A- 30mM Tris-HCl pH = 7.5, 5 mM DTT, 40 mM NaCl, 3 mM MgCl2, 5% glycerol, 1x Complete protease inhibitors cocktail (Roche), 20 mM NEM (Sigma), 0.5 μl Benzonase nuclease (250 U/ul; Sigma); RNase A (200 μg/ml; Sigma) treatment for 15’ at 4°C prior to ultracentrifugation at 300,000 g.
Mammalian TAPI samples were prepared as follows: a cell pellet of at least 1x109 cells (~100μl in volume) were lysed in cold modified 300 μl RIPA buffer (1% Triton X-100, 0.1% SDS, 1% Sodium deoxycholate, 150 mM NaCl, 10 mM Na3PO4, 50 mM NaF, 5 mM MgCl2, 5mM DTT, 5mM Na3VO4, with 1x protease inhibitors (Roche) and 33 U DNase 1 (Sigma), 3 mg RNase A (Sigma) and 750 U Benzonase (Sigma)), followed with a 10-minute incubation at room temperature and then 20 minutes of mild rotation at 4°C. Lysates were then spun at low speed (5 minutes centrifugation at 100 g) and the pellets were subjected to a second lysing in modified RIPA buffer with rotation for 20 minutes at 4°C. Combined supernatants were run through a 30% sucrose gradient by ultracentrifugation (2 hours at 45000 rpm at 4°C with a Beckman SW-50A rotor). Some samples were analyzed at this point to determine the presence of specific proteins in the pellet fraction by western blotting. After ultracentrifugation, the pellet was re-suspended in high SDS buffer (1x TBS, 5 mM DTT, 5 mM EDTA, 2–4% SDS, with 1x protease inhibitors (Roche)) and incubated with gentle mixing for at least 20 minutes at 37°C. Samples were run at 200 volts on an acrylamide gel (Any kD, Bio-Rad) in 10% glycerol with 0.1% bromophenol blue to monitor sample migration. The top 3 millimeters of the wells were excised and frozen. Frozen gel fragments were thawed and resuspended in elution buffer (10mM Tris pH 8.0, 0.4% SDS, 5mM DTT), then mixed and incubated at 99°C for several minutes. Sample volume was reduced by half in a speed-vac and applied to a desalting column (Zeba spin, Thermo Scientific), pre-equilibrated with 25mm triethylammonium bicarbonate (TeABC). The flow-through was analyzed for elution efficiency by western blot prior to digestion.
Samples were digested for mass spectrometry (MS) analysis using a previously described method [40] with minor modifications. Briefly, DTT was added to the sample to obtain a 15 mM solution prior to addition of iodoacetamide to a concentration of 50 mM followed by a 20-minute incubation at 30°C in the dark. Next, 9M urea was added to the sample, which was then filtered (30 kDA amcon 30K spin filter—centrifuged at 16000 x g 5–10 min), and washed with 25mM TeABC twice, and finally trypsin digested (5–10μg trypsin) overnight at room temperature. The sample was retrieved from the column by centrifugation (16000 x g for 4 min), and washed with 25mM TeABC prior to lyophilization. Lyophilized samples were analyzed by tandem MS/MS by Johns Hopkins Mass Spectrometry and Proteomics Facility.
Mass Spectrometry Analysis
Samples were run on Q-Exactive (Thermo Scientific) or Orbitrap Velos (Thermo Scientific) at 70,000 resolution for MS and 17,500 for MS2, or 30,000 resolution for MS and 15,000 for MS2 respectively. The data were collected in data dependent mode with the top 15 precursors chosen for MS/MS. The peptides were eluted with a 90 minute gradient at 300 nanoliters per minute after trapping and desalting for 5 minutes at 5 microliters per minute. Peptides were fragmented with a normalized collision energy of 27 for Q-Exactive, and 35 for Orbitrap Velos. Target values were 3E6 ions with 60 millisecond maximum injection time for MS and 5E4 with 250 milliseconds for MS2 for Q-Exactive and 1E6 ions for MS with a 100 millisecond maximum injection time for MS and 5e4 with 300 milliseconds for MS2 for LTQ Orbitrap Velos.
All data were searched using Mascot (v2.6 Matrix Science) through Proteome Discoverer (v1.4 Thermo Scientific). The database for Yeast included Htt-Q25-GFP in addition to the RefSeq 2014 Saccharomyces cerevisiae and the database for Rat included GFP-Htt-Q23 in addition to the RefSeq 2014 Rattus norvegicus. Variable modifications included oxidation on Met, deamidation on N and Q, and carbamidomethylation on C. Data were searched with a 30 part per million (ppm) tolerance for precursor mass and 0.03 daltons for fragment masses. Data were searched with and without the MS2 processor node which deisotopes the MS2 spectra to the +1 charge state prior to searching. Data were filtered through the Target Decoy PSM Validator.
The resulting data were filtered through Scaffold software for Total Spectra Count at 5% FDR. Criteria for proteins to be defined as associated with Htt-Q74/103 (Htt-PolyQ aggregate) were as follows: 2 or more total spectra, and present in the expanded Htt-polyQ aggregate while absent in the short Htt-polyQ control sample (henceforth termed binary) in at least 2 of 4 samples examined. Thus, the requirement for a protein to be considered positive is that in at least 2 of the matched samples (containing a pair of Q-short and a Q-long samples) performed at the same time on the same instrument, the protein shows ≥2 spectra in the Q-long and not the Q-short. This resulted in 52 Htt-polyQ aggregate associated proteins identified in Saccharomyces cerevisiae (Table 1; S1 File) and 91 Htt-polyQ aggregate associated proteins identified in Rattus norvegicus (Table 2; S2 File). In the accompanying supplementary files, we also include the proteins that meet a less stringent threshold: present in the Htt-polyQ aggregate sample while absent in the control for at least one sample set (binary) and 2-fold greater spectra number in Htt-Q74/103 (Htt-PolyQ aggregate) than the short Htt-polyQ control in at least one additional sample pair. As a control for the above method of processing, we also used normalized spectral counting (NCS), a label-free quantification method that compares the number of MS/MS spectra assigned to each protein normalized for the total spectral counting among samples [41, 42]. The NCS processing yielded very similar data sets (data not shown).
Table 1. Molecular function of proteins associated with Htt-polyQ aggregates as identified by TAPI (N = 52) in Saccharomyces cerevisiae.
| Functional Category | % of total | Protein Name |
|---|---|---|
| Protein Quality Control/ Chaperone | 12% | Apj1, Bmh1, Def1, Mca1, Sgt2, Ydj1 |
| RNA/DNA Binding | 44% | Ccr4, Cyc8, Dhh1, Eap1, Hrp1, Ixr1, Mbf1, Mcm1, Mot3, Nab3, Nam8, New1, Nrd1, Pbp1, Pin4, Pop2, Puf3, Snf5, Srp54, Taf5, Tup1, Whi3, Ygr250c |
| Mitochondrial | 8% | Apj1, Nam8, Puf3, Ynl208w |
| Endocytosis, Vessicle & Cytoskeletal Transport | 21% | Akl1, Ent1, Ent2, Gts1, Pan1, Scd5, Sla1, Yap1801, Yap1802, Pin3, Sec24 |
| Other | 21% | Cbk1, Epo1, Gal2, Mum2, Nup57, Nup100, Nup116, Pgd1, Sml1, Slm1, Ylr177w |
Molecular function determined by gene ontology and Saccharomyces Genome Database. Proteins in bold represent those with overlapping cellular functions; totals exceed 100% because of multiple categorizations. Yeast ribosomal proteins Rpl30 and Rps6b were excluded.
Table 2. Molecular function of proteins associated with Htt-polyQ aggregates as identified by TAPI (N = 91) in Rattus norvegicus.
| Functional Category | % of total | Gene/Protein Name |
|---|---|---|
| Protein Quality Control/ Chaperone | 26% | ADRM1, CLTC, DDI2, DNAJA2, DNAJA4, DNAJB1, DNAJC7, HSP90AA1, HSPA8, LAP3, PPIA, PSMB2, PSMC1, PSMC2, PSMC3, RAD23B, SGTA, SQSTM1, SUMO2, UBQLN2, UBQLN4, UBXN7, USP7, VPS35 |
| RNA/DNA binding* | 37% | AARS2, AKAP81, ARL6IP4, ATP5A1, ATRX, DDX5, DYNC1H1, EIF4G1, EIF4G2, FASN, FUS, GIGYF2, HNRNPA3, HNRNPF, HNRNPH2, HNRNPM, HNRNPU, HSP90AA1, MATR3, NONO, NR3C1, PCBP1, PPIA, PRPF40A, PRRC2B, RBMS1, SF1, TDP-43, TCERG1, TCF20, TNRC6B, TUFM, XRN2, YTHDF1 |
| Mitochondrial | 18% | AARS2, ACAD9, ACSF2, ATP5A1, CLTC, ETFB, GLS, HADHA, IDH2, NDUFS7, OGDH, PCK2, PDHB, SUCLG2, TUFM |
| Endocytosis, Vessicle & Cytoskeletal Transport | 14% | AAK1, ARL6IP4, ASAP1, CLINT1, CLTC, CNN2, DYNC1H1, MYO1D, NSFL1C, RAB10, SCYL2, TFG, VPS35 |
| Other | 15% | EP300, GNAO1, KPRP, LDHA, MAGED1, MLF2, PHGDH, PLEKHB2, PPP2R1A, SIK3, SFN, TGM3, THY1, YWHAB |
Molecular function assignments were determined by gene ontology, RGD, and NCBI.
* RNA-binding was assigned in some cases on empirical data [99], thus classification does not necessarily imply primary function. Proteins in bold represent those that were placed in multiple categories, thus totals can exceed 100%. Rat ribosomal proteins Rpl6 and Rpl13a were excluded.
Bioinformatic Analysis
The proteins identified to be associated with polyQ aggregates using TAPI and MS were further characterized by molecular function, Q/N content and intrinsic disorder. Gene Ontology, Saccharomyces Genome Database (SGD) and the Rat Genome Database (RGD) were used to determine the molecular function of each protein [43–45]. Q/N-rich regions were defined as 30 or more Q/N in an 80-amino acid stretch [46]. For the cases in which data were not available, we developed a PERL-based algorithm to examine protein sequences for Q/N-rich regions (S3 File).
Long intrinsically-disordered (ID) domains were determined using the IUPred-L structural prediction algorithm; ID domains were defined as 30 or more amino acids with a disorder score of 0.5 or greater [47]. To approximate the percentage of proteins in the yeast and rat genomes with long ID regions, 100 and 200 proteins respectively (~ twice the size of each sample data set; S1 and S2 Files), were randomly selected using a random number generator in alignment with the full proteomes of yeast (S. cerevisiae) and Rat (R. norvegicus) downloaded from uniprot (www.uniprot.org). Domains with intrinsic disorder were evaluated for all proteins by IUPred-L. Chi-Square 2x2 Fisher’s Exact test (Graphpad software) was used to determine if proteins with long ID domains (≥100 amino acids) were significantly enriched in polyQ aggregates. Analysis was also performed to ensure that the TAPI methodology was not biased towards identifying proteins that are abnormally large or abundant. The size (kDa) and cellular abundance (molecules/cell) was determined for each protein in the yeast sample set and compared with the whole proteome (values accumulated from [48–51]; S1 Fig).
Western Blotting
Western blotting of cell lysates (input) and TAPI-purified samples were used to verify high molecular weight protein aggregation (observed as large species that cannot migrate beyond the top of an acrylamide stacking gel) and confirm that specific proteins are trapped in polyQ aggregates. Standard Western blotting techniques were employed using nitrocellulose or PVDF membranes, which were probed with primary antibodies against the following targets at dilutions of ~1:5000: αGFP (Roche), αHA (Santa Cruz and Sigma), αErk (Santa Cruz sc-93), αFUS (Bethyl), αhnRNPA1 (Cell Signaling), αRAD23B (Protein Tech), αTDP43 (Protein Tech), and αUBQLN2 (Novus Bio- 5f5). Appropriate HRP-conjugated secondary antibodies were used at 1:1000 dilutions, followed by HRP chemiluminescent substrate (Pierce ECL) for visualization.
Lysate Partitioning
Analysis of proteins (Bmh1p, Def1p, FUS, Ent2p and Sgt2p) entrapped within aggregates was performed by observing the fraction of protein in the total lysate, supernatant or pellet fraction that partitioned to the stacking well of an acrylamide gel under standard SDS electrophoresis conditions. Briefly, yeast lysates were prepared by mechanical breakage using glass beads in TAPI Buffer A (with RNase A). Pellet and supernatant fractions were prepared as described in the TAPI methodology described above. Cellular fractions were subjected to SDS-PAGE using Any kD gels (Bio-Rad) followed by Western Blotting. The effectiveness of the TAPI buffer to eliminate RNA from the aggregates was examined by RNase-Free Agarose gel electrophoresis with and without nuclease treatment (S1B Fig). The FUS protein is prone to degradation following cell lysis, thus denaturing buffer (10 mM Tris, pH 7.5, 8 M urea) was used to visualize protein levels under conditions in which degradation is greatly inhibited. This enables confirmation that the lysates contain equivalent initial amounts of FUS.
Confocal Microscopy
Confocal microscopy slides were prepared on Poly-L-lysine coated slides with 1x106 cells/spot, fixed with 4% paraformaldehyde in 1x PBS and permeablized with 0.1% Triton X-100 in PBS. Primary antibodies for RAD23B (Bethyl) or FUS (Bethyl) were added at 1:200 dilution, followed by type-specific Alexa Fluor conjugated secondary antibodies (α-rabbit 647 and α-mouse 568, Southern Biotech) at 1:1000 dilution in 1% fetal calf serum in 1x PBS. Sample slides were mounted with DAPI-containing fluoramount (EMS) and viewed on a Zeiss 710 confocal laser scanning microscope and analyzed using Zen software (2009).
Thioflavin-T Analysis
Thioflavin-T (Th-T; Sigma) fluorescence was used to determine if HttQ103-GFP aggregates are in an amyloid-like form. Purified Sup35-NM fibers (amyloid positive control from previous study [52]), HttQ25-GFP, and HttQ103-GFP samples were treated with 1μg Th-T in 50mM Tris pH 8.0, 50mM NaCl buffer in a black 96-well plate. Samples were analyzed on a BioTek Synergy H1 plate reader using an excitation of 440 nm and emission at 490 nm. To determine if Thioflavin-T absorbance was significantly different between HttQ25-GFP and HttQ103-GFP, a two-tailed T-test analysis was used.
Results
Isolation & analysis of polyQ aggregates in yeast
To identify aggregate-associated proteins, human HttQ103-GFP and HttQ25-GFP [35] were expressed under control of a galactose-inducible (GAL1) promoter in the yeast Saccharomyces cerevisiae. Both expression constructs contain the human huntingtin (Htt) exon 1 fragment with polyQ tracts (103 or 25 glutamines, respectively) fused in frame with green fluorescent protein (GFP). As observed previously, HttQ25-GFP is soluble during expression, whereas HttQ103-GFP forms toxic cytoplasmic SDS-resistant aggregates [35] (Fig 1A) that have amyloid-like tinctorial properties and can be trapped at the top of an SDS acrylamide gel [9] (Fig 1B; S1B Fig). Proteins that are specifically associated with Htt amyloid-like aggregates were isolated using the TAPI method [9, 28], which traps the large detergent-resistant species in acrylamide gel matrix for subsequent extraction and identification. As demonstrated in Fig 1B, the TAPI method isolates amyloid-like aggregates of HttQ103-GFP, whereas the non-aggregate-forming HttQ25-GFP does not form species large enough, or sufficiently detergent-resistant, for isolation.
Fig 1. Polyglutamine-expanded Huntingtin exon 1 forms aggregates in yeast that can be isolated by TAPI.

(A) Fluorescence microscopy of yeast expressing GFP-tagged Huntingtin exon 1 (Htt) with a normal (Q25) or an expanded polyglutamine tract (Q103). (B) Western blot of GFP-Htt-Q25 and GFP-Htt-Q103 showing that high molecular weight aggregates can be isolated from Htt-Q103 expressing yeast cells. Lysate = input; TAPI = purified aggregate.
Proteins tightly associated with the isolated Htt-polyQ aggregates were identified using tandem mass spectrometry (MS/MS). Qualitative comparison of all identified proteins from the HttQ103-GFP samples—relative to the HttQ25-GFP samples—reveals a subset that is only associated with the large polyQ aggregates (Table 1; S1 File). In total, 52 proteins were considered polyQ-associated because they were reproducibly found in the expanded HttQ103 aggregate (Table 1) while absent in the HttQ25 control sample (described in methods). To confirm that our approach is not enriching for abundant, large, or charged proteins, the polyQ aggregate-associated proteins were compared against the entire yeast proteome. No obvious differences in size distribution, abundance ([51]; S1 Fig), or charge (the avg. pI of TAPI-identified proteins is 7.1, the same as the approximation for the yeast proteome [53]) were observed between our TAPI-identified proteins and that of the entire yeast proteome.
Molecular functions of polyQ aggregate-associated proteins in S. cerevisiae
HttQ103-GFP aggregate-associated proteins were examined using gene ontology (GO) and Saccharomyces genome database (SGD) to assign their functions and properties [43–45, 54]. Unexpectedly, RNA/DNA-binding (mostly RNA binding; Table 1) proteins make up the largest percentage of HttQ103-GFP aggregate-associated proteins. In fact, more than 1/3rd of the polyQ-associated proteins are specifically characterized as RNA-binding proteins (RBPs). Previous studies suggest that RBPs may localize to aggregates because RNA co-aggregates with amyloid-forming proteins [55]. However the TAPI method involves extensive RNase treatment [9, 28]; while we cannot conclude that RNA is completely absent, the vast majority of RNA is eliminated prior to aggregate isolation (S1B Fig). As most RNA-dependent interactions should be lost, a preponderance of RBPs in the HttQ103-GFP aggregate is likely independent of RNA-mediated interactions.
Yeast proteins recruited into polyQ aggregates share common biophysical properties
PolyQ aggregates have been proposed to induce heterologous protein misfolding via a “cross-seeding” mechanism [56]. Previously, Michelitsch and Weissman observed that Q/N-rich domains have an increased propensity to adapt amyloid structure and concluded that 30 glutamine and/or asparagine residues within an 80-amino acid stretch served as a good predictor of amyloid formation [46]. Assuming that the polyQ aggregates could “cross-seed” such Q/N-rich proteins, we analyzed the 52 identified proteins and found they are significantly more likely to have Q/N-rich domains than the whole of the yeast proteome (54% vs. 2% [46], respectively; S1 File). For comparison, if simply measuring for total glutamine content of the identified proteins, the HttQ103-GFP aggregate-associated proteins have only a two-fold greater total percentage of glutamine content relative to the yeast proteome (respectively, 9.7% vs. 3.8%[57]; S1 File).
An enrichment of Q/N-rich segments also implies an increase in intrinsically unstructured protein domains. When each of the polyQ aggregate-associated proteins was examined for global intrinsic disorder (described here: [58]), the aggregate-associated proteins show a higher average total percent intrinsic disorder relative to the average value for the whole yeast proteome (48% vs. 20%, respectively). However, if the identified proteins are specifically analyzed for containing discrete regions that are intrinsically disordered (using IUPred-L prediction algorithm described in the methods [47]), the polyQ-associated proteins are strongly enriched for the presence of an ID domain (Fig 2). Previous studies have classified ID domains as unstructured regions greater than 20–40 amino acids long [59, 60]. For the yeast proteins associated with polyQ aggregates, almost all have an ID domain of at least ≥30 amino acids in length (92% vs. 31% of the proteome control, respectively; S1 File). However, ~2/3rds of the proteins contain very long ID domains of ≥100 amino acids (63% versus 9%; Fig 2), and strikingly of these proteins, ~1/3rd contain no Q/N-rich domain.
Fig 2. Cellular proteins trapped with Htt polyQ aggregates are disproportionately composed of long intrinsically-disordered (ID) domains.

(A) Comparisons of the percentages of proteins with long ID domains of the 52 yeast proteins in Table 1 (reproducibly found by TAPI to be tightly associated with Htt-Q103-GFP aggregates) versus 100 randomly-selected yeast proteins (S1 File). Most of the identified proteins have long ID domains of at least 100 amino acids. (B) Comparisons of the percentages of proteins with long ID domains of the 91 rat proteins in Table 2 (reproducibly found by TAPI to be tightly associated with GFP-Htt-Q74 aggregates) versus 200 randomly-selected rat proteins (S2 File). ID domains are defined as regions of 30 or more amino acids with IUPred scores of 0.5 or greater [47, 98]. Chi-Square Fisher’s Exact test (Graphpad software) was used to determine significance between TAPI-identified proteins and proteome control sets.
ID domains facilitate protein interactions with RNA, so the large cohort of RBPs we found associated with polyQ aggregates could simply be a result of these proteins disproportionately possessing ID domains. The RBP subset of aggregate-associated yeast proteins was compared to all putative and characterized RBPs in the yeast proteome for ID content (S1 File). A majority (70%) of the HttQ103-GFP aggregate-associated RBPs contain ID domains of ≥100 amino acids, while RBPs in general rarely have such long ID domains (~17%; S1 File). Thus, the high frequency of RBPs in the polyQ aggregates might result from the presence of long ID domains in these proteins, rather than a result of some uncharacterized RNA-binding mechanism of polyQ aggregates.
Biochemical confirmation of protein recruitment to polyQ aggregates in yeast
Def1p, Ent2p, Sgt2p and Bmh1p are among the proteins we found to be specific to polyQ aggregates in yeast. We also found that mammalian homologs of Ent2p, Sgt2p and Bmh1p (CLINT1, SGTA and YWHAB, respectively) co-aggregate with polyQ in mammalian cells (discussed in detail below). Ent2p, Sgt2p and Bmh1p were previously shown to have effects on protein aggregation (or toxicity) in yeast models [61–63], while Def1p has not been shown to influence protein aggregation. We selected Def1p, Ent2p, Sgt2p and Bmh1p for biochemical confirmation of the MS results.
The presence of Def1p, Ent2p, Sgt2p and Bmh1p in polyQ aggregates was tested by immunoblotting following a modified version of our TAPI protocol (Fig 3). HttQ103-GFP, expressed in yeast, forms a high molecular weight aggregate that partitions to the pellet fraction and gets stuck at the top of an SDS-PAGE gel (Fig 3A). When HA-tagged Def1p, Ent2p, Sgt2p and Bmh1p are co-expressed with HttQ103-GFP, they show a similar pattern in their respective western blots, but only when expressed with the long polyQ expansion, not with HttQ25-GFP (Fig 3B). Thus, the interactions of all three proteins with polyQ aggregates are sufficiently strong that they co-fractionate and withstand the conditions of SDS-PAGE, resulting in their retention in the large resistant species that cannot migrate into the gel (Fig 3B). The His3 protein was chosen as a negative control as it was never identified in our TAPI samples. Immunoblotting confirms that unlike Def1p, Sgt2p and Bmh1p, HA-tagged His3p is not entangled within polyQ aggregates (Fig 3B), thus recapitulating our MS results biochemically. To ensure that proteins were not independently forming large detergent-resistant aggregates as a consequence of cellular stress caused by HttQ103-GFP, cells were treated with two alternative stresses: proteasome inhibitor MG132 and over-expressed human α-synuclein protein, which is toxic in yeast cells [64]. Neither condition resulted in Sgt2p getting stuck at the top of an acrylamide gel (S1C Fig).
Fig 3. Western blotting confirms that TAPI-identified proteins are trapped in large, detergent-resistant Htt-polyQ aggregates.
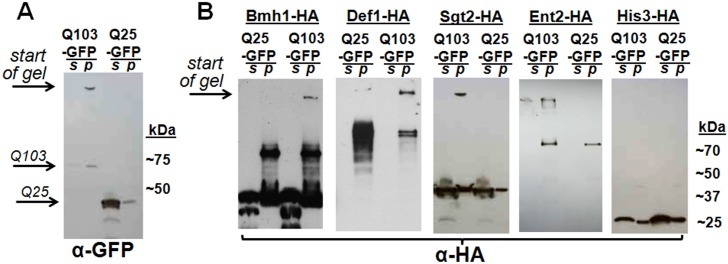
(A) Immunoblotting reveals that Htt-Q103-GFP, but not Htt-Q25-GFP, forms large detergent-resistant aggregates that fractionate in the pellet (partial TAPI purification; see methods) and remain at the top of an acrylamide gel under SDS-PAGE conditions. (B) Immunoblotting confirms that HA-tagged Bmh1p, Def1p, Ent2p, and Sgt2p (proteins identified by mass spec) get trapped in the detergent-resistant aggregates that can be seen stuck at the top of the gels in the pellet fractions. As a negative control, HA-tagged His3p (not identified by mass spec) shows no susceptibility to co-aggregation with Htt-Q103-GFP. Note that Def1p was not easily visualized in the supernatant fraction because it is prone to degradation (data not shown). Samples were spun at 45,000 rpm, except Ent2p (10,000 rpm). S = supernatant; P = pellet fraction.
Analysis of polyQ aggregate-associated proteins in PC-12 cells
With the observation that RBPs and proteins with ID or Q/N-rich domains are bound to HttQ103-GFP aggregates in the yeast model, we asked if the same would be true in a mammalian system. To test this, another Huntington disease model was used: mammalian PC-12 cells stably expressing doxycycline-inducible HttQ23-GFP or HttQ74-GFP developed by David Rubinsztein’s lab [39]. Again, both constructs contain the Htt exon 1 fragment with a polyQ tract (23 or 74) fused in frame with GFP, and only the protein with pathogenic extended polyQ forms SDS-resistant aggregates (Fig 4A). In this model, the polyQ aggregates have an approximately equal distribution in the cytoplasm and nucleus [39], thus may interact with most of the non-secreted cellular proteome. The amyloid-forming HttQ74-GFP, but not HttQ23-GFP, could be successfully purified and detected using the TAPI method, as shown in Fig 4B. MS analysis followed by comparison of all identified proteins showed a subset that was unique to samples with polyQ aggregates. Using the criteria described above, 91 proteins were considered specific to the HttQ74-GFP aggregates (Table 2; S2 File).
Fig 4. Polyglutamine-expanded Huntingtin exon 1 forms aggregates in PC-12 cells that can be isolated by TAPI.

(A) Fluorescence microscopy of PC-12 cells expressing doxycycline inducible transgene GFP-tagged Huntingtin exon 1 (Htt) with normal (Q23) or expanded polygluamine tract (Q74). (B) Western blot of GFP-Htt-Q23 and GFP-Htt-Q74 showing high molecular weight aggregates can be isolated from Htt-Q74 expressing PC-12 cells. Lysate = input; TAPI = purified aggregates.
Molecular functions of polyQ aggregate-associated proteins from PC-12 cells
Characterization of the proteins enriched in polyQ aggregates from PC-12 cells reveals a disproportionate number of RBPs, as similarly observed in yeast (Table 2; S1 and S2 Files). Also, several functional homologs were common to the polyQ aggregates isolated from both the yeast and mammalian cells (S1 Table): RNA-binding proteins DDX5 (yeast Dhh1p) and hnRNPA3 (yeast Hrp1p), 14-3-3 proteins YWHAB and SFN (yeast Bmh1p), endocytosis proteins CLINT1 (yeast Ent1/2p) and AAK1 (yeast Akl1p), and chaperone proteins SGTA (yeast GET pathway protein Sgt2p), DNAJA2 and DNAJA4 (yeast Ydj1p and Apj1p).
Biophysical properties of proteins that associate with polyQ aggregates in PC-12 cells
Biophysical characterization reveals the HttQ74-GFP aggregate-associated proteins from PC-12 cells are enriched for Q/N-rich regions relative to the entire rat proteome (7% versus 0.4%), albeit to a much lesser degree than in yeast. Proteins containing long ID domains (≥100 amino acids) are significantly increased among the TAPI-identified HttQ74-GFP aggregate-associated proteins (31% vs. 18% for proteome control, respectively, p = 0.021; Fig 2; S2 File). As in yeast, this suggests that cellular proteins with long ID domains may be inherently prone to inclusion in polyQ aggregates.
Neurodegenerative disease-linked proteins are recruited into polyQ aggregates
Among the aggregate-specific proteins in PC-12 cells, we also identified a significant subset of proteins that are neurodegenerative disease-associated (Table 3). Surprisingly, these proteins were not limited to huntingtin-interacting proteins; we identified a cadre of ALS-linked proteins. We hypothesized polyQ aggregates could pull in proteins that are prone to aggregation in other pathological contexts. When we probed the purified polyQ aggregates (purified fraction confirmed in Fig 5A) with antibodies specific to proteins that are known to aggregate in the motor neurons of ALS patients (and were identified by MS in this study), we corroborated the specific presence of FUS, TDP-43, and UBQLN2 in the Htt-Q74 aggregates (Fig 5B). We hypothesized that other ALS-linked proteins may be similarly recruited into aggregates but escaped detection by MS. Immunoprobing for the ALS-linked HNRPA1 also revealed its presence in the purified polyQ aggregates (Fig 5B). For a control, we probed for the kinase Erk, which was not identified by MS in our samples, and indeed, it could not be found in the polyQ aggregates (Fig 5A). In total, of the HttQ74-GFP aggregate-associated proteins in PC-12 cells, 21% (19/91) have previously been found in the intraneuronal inclusions of various neurodegenerative diseases (Table 3). Of these disease-linked HttQ74-GFP aggregate-associated proteins, many are RBPs (7/19) and more than half (9/19) contain very long ID domains (≥100 amino acids) (Table 3; S2 File).
Table 3. Htt-polyQ aggregate-associated proteins found in inclusions and aggregates in various neurodegenerative diseases.
| Gene/Protein Name | Disease or Disease Model | ID Domain | RBP | PQC | Reference |
|---|---|---|---|---|---|
| AAK1 | ALS-SOD1 | 209, 81, 50 | [100] | ||
| FUS | ALS | 284, 71, 87 | yes | [101–103] | |
| HNRNPA3 | ALS | 39 | yes | [104] | |
| HSPA8 | HD & SCA1 | 46, 36 | yes | [94, 105] | |
| MATR3 | ALS | 31, 57, 42, 119, 72 | yes | [106] | |
| MLF2 | HD | 136 | [107] | ||
| NONO | ALS-FUS | 66 | yes | [108] | |
| PPIA | ALS-SOD1 | None | yes | yes | [109] |
| RAD23B | HD, SCA3 | 120, 67, 41 | yes | [110, 111] | |
| SQSTM1 | PD & ALS | 153 | yes | [112–115] | |
| SUCLG2 | AD | None | [116] | ||
| SUMO2 | HD & ALS | None | yes | [117, 118] | |
| TARBP (TDP-43) | ALS | 45, 55 | yes | [119, 120] | |
| TCERG1 | HD | 138, 177, 70, 36 | yes | [74] | |
| TCF20 | HD | 87, 739, 50, 32, 709, 34, 121 | yes | [121] | |
| TFG | CMTD | 35, 54, 105 | [122] | ||
| TGM3 | HD | 49 | [123] | ||
| UBQLN2 | ALS | 62, 58, 42, 81, 31, 84 | yes | [124] | |
| YWHAB | ALS | None | [125] |
The numbers for ID Domains indicate the length of distinct unstructured regions ≥30, for the identified rat proteins from PC-12 cells, as determined by IUPred-L. RBP = RNA-binding protein as described in the legend of Table 2; PQC = protein quality control.
Fig 5. Confirmation of polyQ-associated proteins from PC-12 cells identified by TAPI.

(A) Western blotting shows that the addition of doxycycline to the PC-12 cell model induces the expression of HttQ74-GFP, resulting in aggregates that can be purified by TAPI. The kinase ERK is probed as a negative control; ERK was never identified by mass spectrometry, so is not expected to co-fractionate with polyQ aggregates. (B) Western blot analysis of TAPI-purified polyQ aggregates from PC-12 cells confirms the presence of several disease-associated proteins only in the Htt-Q74 samples. All proteins migrated near their predicted molecular weights. For control, the TAPI procedure was conducted in parallel on the induced Htt-Q23 cell line (FUS, TDP-43, UBQLN2, HNRNPA1) or the un-induced Htt-Q74 cell line (CLINT1, HSPA8, RAD23B, SGTA). (C) Confocal microscopy shows localization of identified proteins to Htt-Q74 aggregates in PC12 cells. (left) RAD23B, nominally a DNA repair protein, localizes to nuclear Htt-Q74 inclusions but not cytoplasmic inclusions. (middle) FUS, an RNA-binding protein localizes to nuclear and cytoplasmic Htt-Q74 inclusions. (right) CLINT1, a clatherin-interacting protein, is observed in cytoplasmic Htt-Q74 aggregates. Arrows indicate foci with co-localized proteins. Green = GFP; Magenta = CLINT1, FUS or RAD23B in merge.
Since one fifth of the proteins we identified have been previously linked to pathological aggregates, perhaps many of the remaining proteins represent novel aggregate-associated proteins that have never been specifically probed in various pathological contexts. We selected HSPA8, CLINT1 and SGTA as candidate proteins that could potentially be recruited into pathological aggregates in neurodegenerative disease. We chose CLINT1 and SGTA because intriguingly their homologs (Sgt2p and Ent2p, respectively) were also identified in our yeast model. The Hsp70 protein HSPA8 was selected because Hsp70s have been previously suspected to play critical roles in neurodegenerative disease [65–67]. We confirmed by immuno-blotting the presence of all three proteins in the highly-purified Htt-Q74-GFP aggregates from PC-12 cells (Fig 5b).
Confirmation of recruitment of identified proteins to polyQ aggregates by immunocytochemistry
The localization of selected polyQ aggregate-associated proteins was also confirmed by confocal microscopy (Fig 5C). The proteins CLINT1, RAD23B and FUS were selected because they appeared to be particularly strong hits based on the initial analysis of the polyQ mass spectrometry data and Western blot results. Using immunocytochemistry, each was observed to be aberrantly recruited to the major sites of HttQ74-GFP aggregation (Fig 5C). However, despite strong over-expression of HttQ74-GFP and the formation of large intracellular aggregates, the recruited proteins did not appear to completely localize to the aggregates. In fact, only a fraction of each protein’s respective total was found at the aggregate.
Intrinsically-disordered domains play a role in the recruitment of proteins to polyQ aggregates
We selected yeast Sgt2p and human FUS to further examine the role of ID domains in localization to Htt-polyQ aggregates. Sgt2p is involved in protein quality control and does not contain a known RNA-binding domain (RBD). Interestingly, we also identified Sgt2p’s mammalian homolog, SGTA, among the proteins associated with polyQ aggregates in PC-12 cells. The FUS protein contains a distinct RBD and a long N-terminal ID domain, and when expressed in yeast, exhibits aggregation and toxicity reminiscent of what is observed in diseased motor neurons [68–71].
To determine the contribution of their respective ID domains toward recruitment to polyQ aggregates, we created expression vectors in which the major ID domain (as determined by IUPred-L) of both FUS and Sgt2p is deleted (FUSΔID = FUSΔ1–134; Sgt2ΔID = Sgt2Δ300–346). The full-length versions of FUS and Sgt2p, or their ΔID counterparts, were co-expressed with either HttQ25-GFP or HttQ103-GFP. We truncated the TAPI protocol to easily evaluate the co-localization of the proteins with polyQ aggregates (lysate partitioning; see methods). When we isolated the Htt-polyQ aggregates by lysate partitioning from the Sgt2-transformed strains (shown in Fig 6A as the resistant species stuck at the top of the gel), we observed an enrichment of full-length Sgt2p in the Htt-polyQ high molecular weight aggregates (Fig 6B, left panel). However, when the major ID domain was deleted, most of the co-localization with the polyQ aggregate is eliminated (Fig 6B, right panel). The exact same pattern was observed for FUS and FUSΔID (Fig 6C and 6D, respectively). For comparison, we also used an engineered variant of FUS, which has leucines in place of four conserved phenylalanines (FUS(4F-L): amino acids 305, 341, 359 and 368) in the RNA recognition motif (RRM) domain. FUS(4F-L) was previously shown to be RNA-binding incompetent [72]. We observed that FUS(4F-L) was recruited to polyQ aggregates as readily as wild-type FUS (Fig 6E and 6F), thus suggesting that RNA binding may not play a significant role in facilitating a protein’s inclusion into polyQ aggregates.
Fig 6. The ID domains of Sgt2p and Fus mediate their localization to Htt-polyQ aggregates in yeast cells.

(A, B) Western blots of lysates from yeast strain W303 expressing HttQ25-GFP or HttQ103-GFP in combination with HA-tagged Sgt2p or Sgt2ΔID (A = αGFP; B = αHA). (C, D) Western blots of cells expressing HttQ25-GFP or HttQ103-GFP in combination with FUS or FUSΔID (C = αGFP; D = FUS & α-β actin). Because FUS is quickly degraded in non-denaturing conditions, input controls using urea lysis of cells were included to show initial protein loads. (E, F) Western blots of FUS or FUS(4FL) in HttQ25-GFP-expressing or HttQ103-GFP-expressing cells.
Discussion
Compositional analysis of polyQ aggregates using TAPI
Models of huntingtin exon 1 mimic truncated versions of huntingtin found in intraneuronal aggregates [73], and thus are helpful for studying intracellular aggregation. Here we analyze the protein species that get recruited into amyloid-like aggregates formed by polyQ-expanded Huntingtin exon 1 in both yeast (Q103) and mammalian cells (Q74). The distinctive tinctorial properties and detergent resistance of the aggregates indicates that they are in an amyloid-like state (S1B Fig).
Proteins that specifically associate with polyQ aggregates are hypothesized to play either positive or negative roles in pathogenic processes. Previous attempts to identify the proteins that interact with huntingtin, have employed yeast-two-hybrid, immuno-precipitation and immuno-histochemical screening [27, 30, 32, 33, 74–78]. The overlap in identified proteins from different methods is often low or the identification of hundreds of proteins limits clarity. Also, antibody-based screening is limited to a predetermined set of functioning antibodies and thus may overlook unexpected interactions. Our mass spectrometry-coupled approach, called TAPI, is specific to the most chemically-resistant forms of protein aggregates and eliminates the need to excise individual bands from acrylamide gels [9, 28]. The high stringency of TAPI—due to DNase, RNase, and detergent treatment along with SDS-gel electrophoresis of aggregates—minimizes the identification of non-specific and loosely-associated proteins. In sum, our data set, in addition to the work of others, helps identify the important factors that make specific proteins most vulnerable to inclusion into polyQ aggregates.
Our analysis in both yeast and mammalian systems revealed a compact group of proteins enriched in polyQ aggregates. In both cell types, the proteins recruited to aggregates belonged to common functional classes (Tables 1 & 2); RNA-binding, endocytosis-related and mitochondrial proteins were disproportionately found in all Htt-polyQ samples. Importantly, the proteins identified by mass spectrometry could be independently confirmed by immuno-blotting. Moreover, our findings are supported by a previous study in Neuro 2A cells, which coupled Sarkosyl treatment with conventional 1D-gel separation and manual band excision, to identify polyQ-associated proteins [67]. Among the twelve proteins identified by Mitsui and coworkers, 8 were also identified by our approach in PC-12 cells (EEF1A1, HSPA8, HSP90AB1, MLF2, PSMC1, RAD23B, UBQLN2, YWHAB; Table 1), suggesting that across cell types, certain proteins consistently have a high propensity for inclusion into polyQ aggregates. Moreover, functional orthologs are common to both the yeast and mammalian samples (YWHAB/Bmh1/2p, DDX5/Dhh1p, SGTA/Sgt2p, CLINT1/Ent1/2p, hnRNPA3/Hrp1 and HSPA8/Ssa1p). The recruitment of SGTA/Sgt2p and CLINT1/Ent2p into polyQ aggregates was confirmed in both the yeast and mammalian systems by immuno-detection methods. This overlap—not only between TAPI samples, but also across divergent organisms—suggests that these specific proteins (or their properties) may play a role in processes linked to pathological polyQ aggregation. This is supported in the literature where Ssa1p [35], HSP70 [79] and Sgt2p [77] have all been directly connected to polyQ aggregates in various models. As for the clathrin-interacting proteins CLINT1 and Ent2p, their presence suggests that aggregates can have important interactions with vesicle-dependent processes.
RNA binding proteins are disproportionately recruited to polyQ aggregates
RNA-binding proteins were highly represented in the Htt-polyQ TAPI data sets from yeast and rat cells. The presence of quality-control proteins, such as chaperones, was not particularly surprising since Htt-polyQ forms toxic intracellular aggregates. However, the enrichment of RNA-binding proteins was unexpected, especially considering the extensive nuclease treatment that is used prior to isolation of the aggregates (S1B Fig). RNA-binding proteins have been shown to contribute to the pathologies of a number of neurodegenerative diseases [55, 80]. The aggregation of RNA-binding proteins is transient in normal cellular homeostasis, but may accumulate in neurodegenerative diseases due to pathological alteration of assembly and clearance pathways [55]. This aberrant accumulation is frequently tied to interactions mediated by “prion-like” domains [81, 82], which are intrinsically unstructured domains that resemble the domains that enable certain yeast proteins to adopt self-propagating amyloid conformations. It is this intrinsically-disordered property that is likely responsible for the abundance of RNA-binding proteins in polyQ aggregates, because nucleic acid-interacting proteins frequently having intrinsically unstructured regions. Thus, our results suggest that a proteins’s ID domain, not the RNA binding per se, may be the major determinant of inclusion into Htt-polyQ aggregates (Fig 6C and 6D). This is corroborated by our observation that disrupting the RNA-binding domain of FUS had no effect on its inclusion in polyQ aggregates (Fig 6E and 6F).
Mitochondrial proteins are found in polyQ aggregates
Mitochondrial proteins represent a significant fraction of the polyQ-associated proteins in yeast and rat cells, 8% and 18% respectively. In yeast, this percentage is less than the proteome representation of mitochondrial proteins (~18[83]), but in rat cells this is an over-representation (5–12%[84]). In yeast cells the polyQ aggregates form primarily in the cytoplasm, and in the rat cells the aggregates are equally in the nucleus and cytoplasm. Mitochondrial proteins are mostly synthesized in the cytoplasm and transported as unfolded polypeptides into mitochondria post-translationally. A probable explanation for the presence of mitochondrial proteins in aggregates is that because of their unfolded conformation they are vulnerable to integration into aggregates or because of their dependence on chaperones may be more sensitive to general problems associated with protein quality control.
Intrinsically-disordered domains facilitate protein recruitment to polyQ aggregates
Why are proteins with ID domains tightly and disproportionately associated with Htt-polyQ aggregates? Not only did we observe an enrichment of ID domain-containing proteins, but previous proteomic studies of polyQ also reveal data sets that are rich in such proteins [32, 78, 85]. Ratovitski et al. observed that proteins with ID domains of ≥30 amino acids were enriched in aggregates formed by Htt-50Q in HEK293 cells. This is consistent with our results, although we found significant enrichment with very long ID domains (≥100aa). Similarly, Raychaudhuri et al performed a bioinformatic analysis of intrinsic disorder in neurodegenerative disease-associated proteins. Their bioinformatics dataset (obtained from Entrez Gene database keyword search) when compared to control datasets indicates an increase in intrinsic disorder (using FoldIndex) for Huntington disease-associated proteins with ID domains up to 100 amino acids in length [86]. Intrinsically unstructured regions frequently facilitate molecular interactions or serve as sites of post-translational modifications [60], as well as being prominent features of many nucleic acid-binding proteins and chaperones [87, 88]. These domains generally lack hydrophobic residues sufficient for adopting a folded structure in aqueous environment [89] and thus may be more accessible to aggregation simply by virtue of accessibility. We demonstrated that elimination of the major ID domains of two proteins eliminated their co-aggregation with polyQ (Fig 5).
While we do not assert that ID domains are solely responsible for association with Htt-polyQ aggregates, it is clear that ID domain content plays a prominent role in recruiting secondary proteins to aggregates. In some cases, quality-control proteins could even employ intrinsic disorder within a specific domain to facilitate the functional recognition of misfolded protein aggregates. However, because of the large number of quality-control proteins that we identified it cannot be concluded that there is a single mechanism by which all such proteins are tightly associated with polyQ aggregates; some proteins, such as UBQLN2, likely only have specific affinity for aggregates following ubiquitination.
Why are neurodegenerative disease-associated proteins recruited to polyQ aggregates?
Neurodegenerative disease-linked proteins that have been identified in cellular inclusions in their respective diseases represent a fifth (19/91) of the total TAPI-identified Htt-polyQ aggregate-associated proteins (Table 3). Examining this sub-set reveals that nearly all of them contain ID domains. We observed that some ALS-associated proteins were trapped in polyQ aggregates. Three of these proteins—FUS, HNRPA1 and TDP-43 –are RNA-binding proteins that form pathological inclusions in certain forms of ALS and frontotemporal lobar dementia (FTLD) [90–92]. These proteins have ID domains that resemble the domains of yeast prion proteins due to similar amino-acid composition. It has been concluded that these prion-like domains may be primary-aggregating species, but the fact that these ALS-associated proteins are pulled into polyQ aggregates suggests that in some long-lived cells, such as neurons, there could be underlying protein quality control problems with proteins like FUS and TDP-43 getting preferentially recruited into pre-existing primary aggregates [93]. For example, it is possible that over decades, intermediate-length polyQ expansions in various proteins lead to persistent aggregates that recruit proteins with ID domains; these inclusions would be marked (i.e. immuno-positive) by specific aggregation-prone proteins. Alternatively, the diminution of protein-quality control with aging may create a cellular environment where proteins with long ID domains are increasingly susceptible to aggregation, thus polyQ models and their induced stress may be good tools for identifying proteins that are most vulnerable to aggregation (or functionally localize to aggregates) under conditions of compromised protein-quality control. We hypothesize SGTA and CLINT1—proteins we confirmed to co-aggregate with polyQ—may be two such candidate proteins with the potential to localize to multiple types of pathological neuronal inclusions. However, just because proteins co-localize with polyQ aggregates in cell models does not mean they will similarly co-localize in animal models or in diseased neurons. HSPA8, whose presence we confirmed in polyQ aggregates in PC-12 cells was not observed in cytoplasmic polyQ aggregates in a transgenic mouse model of Huntington’s disease [94].
Mechanisms of toxicity
Both the yeast and mammalian model systems show a correlation between polyQ-expanded huntingtin aggregation and cellular toxicity [35, 39], however much debate still persists as to the mechanism by which aggregation leads to cell death [95]. One possible mechanism is that sequestration of proteins to an aggregate may impair cellular function [96], which is arguably an indirect loss of function. Such sequestration by aggregates of polyQ-expanded Ataxin3 (spinocerebellar ataxia-causing protein) was proposed to cause a loss-of-function toxicity [26]. Our observations suggest this indirect loss-of-function toxicity due to sequestration of essential proteins could occur with huntingtin aggregation as well. We observe altered cellular localization for a subset of proteins when Htt-polyQ aggregates are present. It is possible that these proteins may maintain some function while associated with the Htt-polyQ aggregate. If the ID domain of a protein becomes embedded in the polyQ aggregate, while globular or functional domains remain peripheral, function in the wrong place at the wrong time could be a gain-of-function toxicity associated with aggregates. Stoichiometrically, this may be more plausible than loss-of-function because we observe only a fraction of any given co-aggregating species mis-localized to the polyQ aggregate, which itself is quite abundant due to over-expression of Htt (Fig 5). Of course, a combination of gain-of-function and loss-of-function mechanisms could contribute to the overall cellular toxicity.
Although many techniques have been employed to identify huntingtin-interacting proteins, few examine specifically the amyloid form. Our results show that a select group of proteins are trapped by polyQ amyloid-like aggregates. Proteins with long ID domains are disproportionately prone to inclusion, as are many proteins that are associated with other neurodegenerative diseases. The enrichment in ID domain-containing proteins in polyQ aggregates, and the elimination of this enrichment when the ID domains are deleted, reveals the significant role of protein structure in determining if a protein gets secondarily recruited into certain types of aggregates. Thus, while some proteins might be predicted to be recruited into aggregates because of their function (i.e. quality-control proteins or proteins that interact with the soluble form of an aggregating species), many proteins may be recruited simply as a consequence of their secondary and tertiary structural elements. The metastable structure and accessibility of long ID domains may render proteins particularly susceptible to aberrant inclusion in amyloid-like aggregates. Recently, Habch and colleagues put forth the idea that ID proteins represent a class of pharmacological targets [97]. As our results suggest, if the recruitment of specific ID domain-containing proteins into pathological aggregates is critical to cellular degeneration, then targeting ID domains to reduce their sequestration may have therapeutic potential in a variety of neurodegenerative diseases.
Supporting Information
A—Comparison of protein size and abundance for TAPI proteins versus the yeast proteome. B—Efficacy of Rnase treatment in the TAPI procedure (left panel) and Th-T fluorescence of crude aggregates isolated from yeast expressing Htt-Q103-GFP or HttQ25-GFP (right panel). C—Proteasomal inhibition or proteo-toxic stress is not sufficient to cause Sgt2p to be trapped in (or form) detergent-resistant high-molecular weight aggregates.
(PDF)
This Excel file contains tables that list initial mass spectrometry results for multiple yeast samples and the values for ID domains for all identified proteins, as well as controls. S1A includes the proteins identified by our stringent binary analysis (see methods) and contains biochemical characterization of the identified proteins. S1B contains an expanded list of identified proteins using a less stringent arbitrary threshold of the mass spec data. S1C includes the list of 100 random yeast proteins used for comparison purposes. S1D includes the RBPs identified by TAPI as well as the proteomic RBPs, identified by Gene Ontology search, and their ID domains as predicted by IUPred-L.
(XLSX)
This Excel file contains tables that list initial mass spectrometry results for multiple rat samples and the values for ID domains for all identified proteins, as well as controls. S2A includes the proteins identified by the stringent binary analysis (see methods) and contains biochemical characterization of the identified proteins. S2B is an expanded list of identified proteins using a less stringent arbitrary threshold of the mass spec data. S2C includes the list of 200 random rat proteins and their ID domains as predicted by IUPred-L.
(XLSX)
(PL)
(DOCX)
Acknowledgments
We would like to thank Barrington Burnett for experimental suggestions, Rachel Cox for help with microscopy and editing the manuscript, Cara Olsen for help with statistical analysis, Christopher Grunseich for assistance with cell culture, David Rubinsztein for the PC-12 cell lines, and Jesse Guidry for mass spectrometry advice.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Uniformed Services University New Investigator Award, Grant # RO75QF.
References
- 1. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nature medicine. 2004. July;10 Suppl:S10–7. [DOI] [PubMed] [Google Scholar]
- 2. Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science (New York, NY. 2002. June 14;296(5575):1991–5. [DOI] [PubMed] [Google Scholar]
- 3. Baxa U. Structural basis of infectious and non-infectious amyloids. Curr Alzheimer Res. 2008. June;5(3):308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Quarterly reviews of biophysics. 2008. November;41(3–4):265–97. 10.1017/S0033583508004733 [DOI] [PubMed] [Google Scholar]
- 5. Shewmaker F, McGlinchey RP, Wickner RB. Structural insights into functional and pathological amyloid. The Journal of biological chemistry. 2011. May 13;286(19):16533–40. 10.1074/jbc.R111.227108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tycko R. Solid-state NMR studies of amyloid fibril structure. Annual review of physical chemistry. 2011;62:279–99. 10.1146/annurev-physchem-032210-103539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vendruscolo M, Knowles TP, Dobson CM. Protein solubility and protein homeostasis: a generic view of protein misfolding disorders. Cold Spring Harbor perspectives in biology. 2014. December;3(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dong J, Castro CE, Boyce MC, Lang MJ, Lindquist S. Optical trapping with high forces reveals unexpected behaviors of prion fibrils. Nature structural & molecular biology. 2010. December;17(12):1422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kryndushkin D, Pripuzova N, Burnett B, Shewmaker F. Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. The Journal of biological chemistry. 2013. September 20 288(38):27100–11 10.1074/jbc.M113.485359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. The Journal of biological chemistry. 2003. December 5;278(49):49636–43. [DOI] [PubMed] [Google Scholar]
- 11. MacPhee CE, Dobson CM. Chemical dissection and reassembly of amyloid fibrils formed by a peptide fragment of transthyretin. Journal of molecular biology. 2000. April 14;297(5):1203–15. [DOI] [PubMed] [Google Scholar]
- 12. Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science (New York, NY. 2000. August 25;289(5483):1317–21. [DOI] [PubMed] [Google Scholar]
- 13. Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002. April 4;416(6880):507–11. [DOI] [PubMed] [Google Scholar]
- 14. Hoffner G, Djian P. Monomeric, oligomeric and polymeric proteins in huntington disease and other diseases of polyglutamine expansion. Brain sciences. 2014;4(1):91–122. 10.3390/brainsci4010091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual review of neuroscience. 2007;30:575–621. [DOI] [PubMed] [Google Scholar]
- 16. Nelson DL, Orr HT, Warren ST. The unstable repeats—three evolving faces of neurological disease. Neuron. 2013. March 6;77(5):825–43. 10.1016/j.neuron.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997. August 8;90(3):549–58. [DOI] [PubMed] [Google Scholar]
- 18. Chen S, Berthelier V, Hamilton JB, O'Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002. June 11;41(23):7391–9. [DOI] [PubMed] [Google Scholar]
- 19. Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Experimental neurology. 1999. March;156(1):92–9. [DOI] [PubMed] [Google Scholar]
- 20. Gourfinkel-An I, Cancel G, Duyckaerts C, Faucheux B, Hauw JJ, Trottier Y, et al. Neuronal distribution of intranuclear inclusions in Huntington's disease with adult onset. Neuroreport. 1998. June 1;9(8):1823–6. [DOI] [PubMed] [Google Scholar]
- 21. Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997. August 8;90(3):537–48. [DOI] [PubMed] [Google Scholar]
- 22. Davranche A, Aviolat H, Zeder-Lutz G, Busso D, Altschuh D, Trottier Y, et al. Huntingtin affinity for partners is not changed by polyglutamine length: aggregation itself triggers aberrant interactions. Human molecular genetics. 2011. July 15;20(14):2795–806. 10.1093/hmg/ddr178 [DOI] [PubMed] [Google Scholar]
- 23. Rubinsztein DC, Carmichael J. Huntington's disease: molecular basis of neurodegeneration. Expert reviews in molecular medicine. 2003. August;5(20):1–21. [DOI] [PubMed] [Google Scholar]
- 24. Jacobsen JC, Gregory GC, Woda JM, Thompson MN, Coser KR, Murthy V, et al. HD CAG-correlated gene expression changes support a simple dominant gain of function. Human molecular genetics. 2011. July 15;20(14):2846–60. 10.1093/hmg/ddr195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henshall TL, Tucker B, Lumsden AL, Nornes S, Lardelli MT, Richards RI. Selective neuronal requirement for huntingtin in the developing zebrafish. Human molecular genetics. 2009. December 15;18(24):4830–42. 10.1093/hmg/ddp455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang H, Li JJ, Liu S, Zhao J, Jiang YJ, Song AX, et al. Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: implication in a loss-of-function pathology. Scientific reports. 2014;4:6410 10.1038/srep06410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013. July 3;154(1):134–45. 10.1016/j.cell.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 28. Kryndushkin D, Wear MP, Shewmaker F. Amyloid cannot resist identification. Prion. 2013. December 23;7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004. March;20(3):146–54. [DOI] [PubMed] [Google Scholar]
- 30. Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends in biochemical sciences. 2003. August;28(8):425–33. [DOI] [PubMed] [Google Scholar]
- 31. Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Molecular biology of the cell. 2001. May;12(5):1393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS genetics. 2007. May 11;3(5):e82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tourette C, Li B, Bell R, O'Hare S, Kaltenbach LS, Mooney SD, et al. A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. The Journal of biological chemistry. 2014. March 7;289(10):6709–26. 10.1074/jbc.M113.523696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uversky VN. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Frontiers in aging neuroscience. 2015;7:18 10.3389/fnagi.2015.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. The Journal of cell biology. 2002. June 10;157(6):997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edskes HK, Wickner RB. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full-length protein. Proceedings of the National Academy of Sciences of the United States of America. 2002. December 10;99 Suppl 4:16384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Edskes HK, Wickner RB. A protein required for prion generation: [URE3] induction requires the Ras-regulated Mks1 protein. Proceedings of the National Academy of Sciences of the United States of America. 2000. June 6;97(12):6625–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kryndushkin D, Wickner RB, Shewmaker F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein & cell. 2011. March;2(3):223–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Human molecular genetics. 2001. August 15;10(17):1829–45. [DOI] [PubMed] [Google Scholar]
- 40. Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods. 2009. May;6(5):359–62. 10.1038/nmeth.1322 [DOI] [PubMed] [Google Scholar]
- 41. Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, et al. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005. October;4(10):1487–502. [DOI] [PubMed] [Google Scholar]
- 42. Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. Journal of proteome research. 2006. September;5(9):2339–47. [DOI] [PubMed] [Google Scholar]
- 43. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000. May;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic acids research. 2012. January;40(Database issue):D700–5. 10.1093/nar/gkr1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laulederkind SJ, Hayman GT, Wang SJ, Smith JR, Lowry TF, Nigam R, et al. The Rat Genome Database 2013—data, tools and users. Briefings in bioinformatics. 2013. July;14(4):520–6. 10.1093/bib/bbt007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proceedings of the National Academy of Sciences of the United States of America. 2000. October 24;97(22):11910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics (Oxford, England). 2005. August 15;21(16):3433–4. [DOI] [PubMed] [Google Scholar]
- 48. Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006. June 15;441(7095):840–6. [DOI] [PubMed] [Google Scholar]
- 49. Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, et al. Global analysis of protein expression in yeast. Nature. 2003. October 16;425(6959):737–41. [DOI] [PubMed] [Google Scholar]
- 50. Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI. A sampling of the yeast proteome. Molecular and cellular biology. 1999. November;19(11):7357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nature biotechnology. 2007. January;25(1):117–24. [DOI] [PubMed] [Google Scholar]
- 52. Shewmaker F, Kryndushkin D, Chen B, Tycko R, Wickner RB. Two prion variants of Sup35p have in-register parallel beta-sheet structures, independent of hydration. Biochemistry. 2009. June 16;48(23):5074–82. 10.1021/bi900345q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schwartz R, Ting CS, King J. Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome research. 2001. May;11(5):703–9. [DOI] [PubMed] [Google Scholar]
- 54. Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Molecular cell. 2012. June 8;46(5):674–90. 10.1016/j.molcel.2012.05.021 [DOI] [PubMed] [Google Scholar]
- 55. Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013. August 15;154(4):727–36. 10.1016/j.cell.2013.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, Nukina N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci. 2009. April 22;29(16):5153–62. 10.1523/JNEUROSCI.0783-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Karlin S, Brocchieri L, Bergman A, Mrazek J, Gentles AJ. Amino acid runs in eukaryotic proteomes and disease associations. Proceedings of the National Academy of Sciences of the United States of America. 2002. January 8;99(1):333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics (Oxford, England). 2004. September 1;20(13):2138–9. [DOI] [PubMed] [Google Scholar]
- 59. Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, et al. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. Journal of proteome research. 2007. May;6(5):1899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tompa P, Fuxreiter M, Oldfield CJ, Simon I, Dunker AK, Uversky VN. Close encounters of the third kind: disordered domains and the interactions of proteins. Bioessays. 2009. March;31(3):328–35. 10.1002/bies.200800151 [DOI] [PubMed] [Google Scholar]
- 61. Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2006. July 18;103(29):11051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kiktev DA, Patterson JC, Muller S, Bariar B, Pan T, Chernoff YO. Regulation of chaperone effects on a yeast prion by cochaperone Sgt2. Molecular and cellular biology. 2012. December;32(24):4960–70. 10.1128/MCB.00875-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, et al. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. Faseb J. 2009. February;23(2):451–63. 10.1096/fj.08-117614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science (New York, NY. 2003. December 5;302(5651):1772–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005. March;62(6):670–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bonini NM. Chaperoning brain degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2002. December 10;99 Suppl 4:16407–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mitsui K, Doi H, Nukina N. Proteomics of polyglutamine aggregates. Methods in enzymology. 2006;412:63–76. [DOI] [PubMed] [Google Scholar]
- 68. Kryndushkin D, Wickner RB, Shewmaker F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein & cell. 2011. March 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fushimi K, Long C, Jayaram N, Chen X, Li L, Wu JY. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein & cell. 2011. February 14;2(2):141–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS biology. 2011. April;9(4):e1001052 10.1371/journal.pbio.1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS biology. 2011. April;9(4):e1000614 10.1371/journal.pbio.1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Daigle JG, Lanson NA Jr, Smith RB, Casci I, Maltare A, Monaghan J, et al. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Human molecular genetics. 2013. March 15;22(6):1193–205. 10.1093/hmg/dds526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nature genetics. 1998. February;18(2):150–4. [DOI] [PubMed] [Google Scholar]
- 74. Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, et al. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2001. February 13;98(4):1811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rutherford NJ, Lewis J, Clippinger AK, Thomas MA, Adamson J, Cruz PE, et al. Unbiased screen reveals ubiquilin-1 and -2 highly associated with huntingtin inclusions. Brain research. 2013. August 2;1524:62–73. 10.1016/j.brainres.2013.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y, Nukina N. Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS letters. 2004. July 30;571(1–3):171–6. [DOI] [PubMed] [Google Scholar]
- 77. Wang Y, Meriin AB, Costello CE, Sherman MY. Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of aggregates from protein conformation disorders. Prion. 2007. Apr-Jun;1(2):128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, et al. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell cycle (Georgetown, Tex. 2012. May 15;11(10):2006–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Novoselova TV, Margulis BA, Novoselov SS, Sapozhnikov AM, van der Spuy J, Cheetham ME, et al. Treatment with extracellular HSP70/HSC70 protein can reduce polyglutamine toxicity and aggregation. Journal of neurochemistry. 2005. August;94(3):597–606. [DOI] [PubMed] [Google Scholar]
- 80. Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, et al. A yeast functional screen predicts new candidate ALS disease genes. Proceedings of the National Academy of Sciences of the United States of America. 2011. December 27;108(52):20881–90. 10.1073/pnas.1109434108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain research. 2012. June 26;1462:61–80. 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. The Journal of cell biology. 2013. April 29;201(3):361–72. 10.1083/jcb.201302044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wiederhold E, Veenhoff LM, Poolman B, Slotboom DJ. Proteomics of Saccharomyces cerevisiae Organelles. Mol Cell Proteomics. 2010. March;9(3):431–45. 10.1074/mcp.R900002-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annual review of genomics and human genetics. 2010;11:25–44. 10.1146/annurev-genom-082509-141720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yao J, Ong SE, Bajjalieh S. Huntingtin is associated with cytomatrix proteins at the presynaptic terminal. Molecular and cellular neurosciences. 2014. November;63:96–100. [DOI] [PubMed] [Google Scholar]
- 86. Raychaudhuri S, Dey S, Bhattacharyya NP, Mukhopadhyay D. The role of intrinsically unstructured proteins in neurodegenerative diseases. PloS one. 2009;4(5):e5566 10.1371/journal.pone.0005566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. Faseb J. 2004. August;18(11):1169–75. [DOI] [PubMed] [Google Scholar]
- 88. Ivanyi-Nagy R, Davidovic L, Khandjian EW, Darlix JL. Disordered RNA chaperone proteins: from functions to disease. Cell Mol Life Sci. 2005. July;62(13):1409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Uversky VN, Gillespie JR, Fink AL. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins. 2000. November 15;41(3):415–27. [DOI] [PubMed] [Google Scholar]
- 90. Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010. April;6(4):211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Current opinion in neurobiology. 2011. December;21(6):904–19. 10.1016/j.conb.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. The Lancet. 2010. October;9(10):995–1007. 10.1016/S1474-4422(10)70195-2 [DOI] [PubMed] [Google Scholar]
- 93. Fuentealba RA, Udan M, Bell S, Wegorzewska I, Shao J, Diamond MI, et al. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. The Journal of biological chemistry. 2010. August 20;285(34):26304–14. 10.1074/jbc.M110.125039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Human molecular genetics. 2000. August 12;9(13):2009–18. [DOI] [PubMed] [Google Scholar]
- 95. Hands SL, Wyttenbach A. Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta neuropathologica. 2010. October;120(4):419–37. 10.1007/s00401-010-0703-0 [DOI] [PubMed] [Google Scholar]
- 96. Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2002. December 10;99 Suppl 4:16412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chemical reviews. 2014. July 9;114(13):6561–88. 10.1021/cr400514h [DOI] [PubMed] [Google Scholar]
- 98. Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. Journal of molecular biology. 2005. April 8;347(4):827–39. [DOI] [PubMed] [Google Scholar]
- 99. Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012. June 8;149(6):1393–406. 10.1016/j.cell.2012.04.031 [DOI] [PubMed] [Google Scholar]
- 100. Shi B, Conner SD, Liu J. Dysfunction of endocytic kinase AAK1 in ALS. International journal of molecular sciences. 2014;15(12):22918–32. 10.3390/ijms151222918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, NY. 2009. February 27;323(5918):1205–8. [DOI] [PubMed] [Google Scholar]
- 102. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (New York, NY. 2009. February 27;323(5918):1208–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Doi H, Okamura K, Bauer PO, Furukawa Y, Shimizu H, Kurosawa M, et al. RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. The Journal of biological chemistry. 2008. March 7;283(10):6489–500. 10.1074/jbc.M705306200 [DOI] [PubMed] [Google Scholar]
- 104. Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta neuropathologica. 2013. March;125(3):413–23. 10.1007/s00401-013-1088-7 [DOI] [PubMed] [Google Scholar]
- 105. Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nature genetics. 1998. June;19(2):148–54. [DOI] [PubMed] [Google Scholar]
- 106. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nature neuroscience. 2014. May;17(5):664–6. 10.1038/nn.3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kim WY, Fayazi Z, Bao X, Higgins D, Kazemi-Esfarjani P. Evidence for sequestration of polyglutamine inclusions by Drosophila myeloid leukemia factor. Molecular and cellular neurosciences. 2005. August;29(4):536–44. [DOI] [PubMed] [Google Scholar]
- 108. Shelkovnikova TA, Robinson HK, Troakes C, Ninkina N, Buchman VL. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Human molecular genetics. 2014. May 1;23(9):2298–312. 10.1093/hmg/ddt622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Basso M, Massignan T, Samengo G, Cheroni C, De Biasi S, Salmona M, et al. Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. The Journal of biological chemistry. 2006. November 3;281(44):33325–35. [DOI] [PubMed] [Google Scholar]
- 110. Bergink S, Severijnen LA, Wijgers N, Sugasawa K, Yousaf H, Kros JM, et al. The DNA repair-ubiquitin-associated HR23 proteins are constituents of neuronal inclusions in specific neurodegenerative disorders without hampering DNA repair. Neurobiology of disease. 2006. September;23(3):708–16. [DOI] [PubMed] [Google Scholar]
- 111. Wang G, Sawai N, Kotliarova S, Kanazawa I, Nukina N. Ataxin-3, the MJD1 gene product, interacts with the two human homologs of yeast DNA repair protein RAD23, HHR23A and HHR23B. Human molecular genetics. 2000. July 22;9(12):1795–803. [DOI] [PubMed] [Google Scholar]
- 112. Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. Journal of the neurological sciences. 2006. November 1;249(1):13–8. [DOI] [PubMed] [Google Scholar]
- 113. Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001. July 20;12(10):2085–90. [DOI] [PubMed] [Google Scholar]
- 114. Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. The American journal of pathology. 2002. January;160(1):255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhou L, Wang H, Ren H, Hu Q, Ying Z, Wang G. Bcl-2 Decreases the Affinity of SQSTM1/p62 to Poly-Ubiquitin Chains and Suppresses the Aggregation of Misfolded Protein in Neurodegenerative Disease. Molecular neurobiology. 2014. October 14. [DOI] [PubMed] [Google Scholar]
- 116. Ramirez A, van der Flier WM, Herold C, Ramonet D, Heilmann S, Lewczuk P, et al. SUCLG2 identified as both a determinator of CSF Abeta1-42 levels and an attenuator of cognitive decline in Alzheimer's disease. Human molecular genetics. 2014. December 15;23(24):6644–58. 10.1093/hmg/ddu372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. O'Rourke JG, Gareau JR, Ochaba J, Song W, Rasko T, Reverter D, et al. SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell reports. 2013. July 25;4(2):362–75. 10.1016/j.celrep.2013.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Niikura T, Kita Y, Abe Y. SUMO3 modification accelerates the aggregation of ALS-linked SOD1 mutants. PloS one. 2014;9(6):e101080 10.1371/journal.pone.0101080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature genetics. 2008. May;40(5):572–4. 10.1038/ng.132 [DOI] [PubMed] [Google Scholar]
- 120. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science (New York, NY. 2008. March 21;319(5870):1668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yamanaka T, Wong HK, Tosaki A, Bauer PO, Wada K, Kurosawa M, et al. Large-scale RNA interference screening in Mammalian cells identifies novel regulators of mutant huntingtin aggregation. PloS one. 2014;9(4):e93891 10.1371/journal.pone.0093891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ishiura H, Sako W, Yoshida M, Kawarai T, Tanabe O, Goto J, et al. The TRK-fused gene is mutated in hereditary motor and sensory neuropathy with proximal dominant involvement. American journal of human genetics. 2012. August 10;91(2):320–9. 10.1016/j.ajhg.2012.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zainelli GM, Dudek NL, Ross CA, Kim SY, Muma NA. Mutant huntingtin protein: a substrate for transglutaminase 1, 2, and 3. Journal of neuropathology and experimental neurology. 2005. January;64(1):58–65. [DOI] [PubMed] [Google Scholar]
- 124. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011. September 8;477(7363):211–5. 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kawamoto Y, Akiguchi I, Fujimura H, Shirakashi Y, Honjo Y, Sakoda S. 14-3-3 proteins in Lewy body-like hyaline inclusions in a patient with familial amyotrophic lateral sclerosis with a two-base pair deletion in the Cu/Zn superoxide dismutase (SOD1) gene. Acta neuropathologica. 2005. August;110(2):203–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A—Comparison of protein size and abundance for TAPI proteins versus the yeast proteome. B—Efficacy of Rnase treatment in the TAPI procedure (left panel) and Th-T fluorescence of crude aggregates isolated from yeast expressing Htt-Q103-GFP or HttQ25-GFP (right panel). C—Proteasomal inhibition or proteo-toxic stress is not sufficient to cause Sgt2p to be trapped in (or form) detergent-resistant high-molecular weight aggregates.
(PDF)
This Excel file contains tables that list initial mass spectrometry results for multiple yeast samples and the values for ID domains for all identified proteins, as well as controls. S1A includes the proteins identified by our stringent binary analysis (see methods) and contains biochemical characterization of the identified proteins. S1B contains an expanded list of identified proteins using a less stringent arbitrary threshold of the mass spec data. S1C includes the list of 100 random yeast proteins used for comparison purposes. S1D includes the RBPs identified by TAPI as well as the proteomic RBPs, identified by Gene Ontology search, and their ID domains as predicted by IUPred-L.
(XLSX)
This Excel file contains tables that list initial mass spectrometry results for multiple rat samples and the values for ID domains for all identified proteins, as well as controls. S2A includes the proteins identified by the stringent binary analysis (see methods) and contains biochemical characterization of the identified proteins. S2B is an expanded list of identified proteins using a less stringent arbitrary threshold of the mass spec data. S2C includes the list of 200 random rat proteins and their ID domains as predicted by IUPred-L.
(XLSX)
(PL)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.